

Abstract
Amyloid β (Aβ) is involved in the etiology of Alzheimer's disease (AD) and may contribute to cognitive deficits by increasing internalization of ionotropic glutamate receptors. Striatal-enriched protein tyrosine phosphatase 61 (STEP61), which is targeted in part to the postsynaptic terminal, has been implicated in this process. Here we show that STEP61 levels are progressively increased in the cortex of Tg2576 mice over the first year, as well as in prefrontal cortex of human AD brains. The increased STEP61 was associated with greater STEP activity, dephosphorylation of phospho-tyr1472 of the NR2B subunit, and decreased NR1 and NR2B subunits on neuronal membranes. Treatment with Aβ-enriched medium also increased STEP61 levels and decreased NR1/NR2B abundance in mouse cortical cultures as determined by biotinylation experiments. In STEP knock-out cultures, Aβ treatment failed to induce NMDA receptor internalization. The mechanism for the increase in STEP61 levels appears to involve the ubiquitin proteasome system. Blocking the proteasome resulted in elevated levels of STEP61. Moreover, STEP61–ubiquitin conjugates were increased in wild-type cortical slices upon Aβ treatment as well as in 12 month Tg2576 cortex. These findings reveal a novel mechanism by which Aβ-mediated accumulation of STEP61 results in increased internalization of NR1/NR2B receptor that may contribute to the cognitive deficits in AD.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder associated with memory loss. A hallmark of AD is the accumulation of amyloid β (Aβ) peptide in brains, a process that has been implicated in the progression of the disease (Haass and Selkoe, 2007). Accumulation of Aβ results from the sequential cleavage of amyloid precursor protein by β-secretase and γ-secretase enzymes (Turner et al., 2003). One hypothesis in the pathophysiology of AD is that soluble forms of Aβ disrupt synaptic function (Hardy and Selkoe, 2002; Venkitaramani et al., 2007). This notion is supported by the correlation between cognitive deficits and loss of synaptic structures (Terry et al., 1991; Masliah et al., 1991). Amyloid plaque formations occur subsequent to loss of synaptic function (Hsiao et al., 1996; Jacobsen et al., 2006), suggesting that synaptic perturbations are an earlier target of Aβ. Notably, exogenous application of soluble Aβ inhibits long-term potentiation (LTP), induces synaptic loss, and blocks cognitive function in rodent models (Walsh et al., 2002; Lacor et al., 2007; Shankar et al., 2008).
Striatal-enriched protein tyrosine phosphatase 61 (STEP61), the only isoform of this brain-specific family of phosphatases expressed in the cortex, localizes to postsynaptic terminals and the endoplasmic reticulum (Boulanger et al., 1995; Oyama et al., 1995). The current model of STEP function is that it opposes the development of synaptic strengthening (Braithwaite et al., 2006a). STEP61 associates with the NMDA receptor (NMDAR) complex, reduces NMDAR activity, and opposes the induction of LTP through a process whereby STEP dephosphorylates a regulatory tyrosine site (tyr1472) on the NR2B subunit, leading to internalization of NR1/NR2B receptor complexes (Pelkey et al., 2002; Braithwaite et al., 2006b). A previous study demonstrated that Aβ activates STEP through a calcineurin-dependent pathway (Snyder et al., 2005). In addition, another study using a mouse model of AD (J20) found that STEP levels are increased (Chin et al., 2005). Together, these results suggest that Aβ may regulate STEP level and activity through several mechanisms.
The ubiquitin proteasome system (UPS) is a major pathway for protein degradation. Ubiquitin is a 7.6 kDa polypeptide that is covalently attached to substrates and typically targets these proteins to the 26S proteasome for degradation. The UPS regulates a wide variety of cellular processes, including synaptic plasticity (Yi and Ehlers, 2007). This pathway is impaired in human AD brains, mouse AD models, and neuronal cultures treated with Aβ (Keller et al., 2000; Lam et al., 2000; Oh et al., 2005; Almeida et al., 2006). A previous study demonstrated that synaptic NMDAR activation normally promotes ubiquitination and degradation of STEP61 (Xu et al., 2009). The current study determines that STEP61 levels are elevated in aged transgenic AD model mice (Tg2576) and in human AD brains, and that Aβ is sufficient to increase STEP61 levels. The increase in STEP61 involves inhibition of the UPS and is associated with excessive internalization of membrane-associated NR1/NR2B subunits. These findings suggest a novel mechanism by which synaptic function is disrupted in AD.
Materials and Methods
Reagents and animals.
All antibodies used in this study are listed in Table 1. The chemicals, cycloheximide, actinomycin, carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG-132), para-nitrophenyl phosphate (pNPP), and orthovanadate were from Sigma-Aldrich. The γ-secretase inhibitor [N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT), lactacystin, epoxymycin, and chloroquine were from Calbiochem. Active Fyn was purchased from Millipore.
Table 1.
Primary and secondary antibodies used in Western blots
| Antibody | Format | Immunogen | Host | Dilution | Source |
|---|---|---|---|---|---|
| Anti-ERK2 | Whole IgG, unconjugated | C-terminus of rat sequence | Rabbit | 1:5000 | Santa Cruz Biotechnology |
| Anti-Aβ (6E10) | Ascites (IgG1) | Peptide corresponding to 3–8 aa of beta amyloid | Mouse | 1:1000 | Covance |
| Anti-β-actin | Whole IgG, unconjugated | Synthetic peptide | Rabbit | 1:5000 | Affinity Bioreagents |
| Anti-pY1472 NR2B | Whole IgG, unconjugated | Synthetic phosphopeptide | Rabbit | 1:1000 | Phosphosolutions |
| Anti-NR2B | Whole IgG, unconjugated | C-terminal of mouse NR2B | Rabbit | 1:1000 | Millipore |
| Anti-GABAA (β2/3) | IgG1, unconjugated | Extracellular domain of β2/3 | Mouse | 1:1000 | Millipore |
| Anti-NR1 | Whole IgG, unconjugated | C-terminus | Mouse | 1:600 | Millipore |
| Anti-synapsin 1 | Whole IgG, unconjugated | Bovine synapsin I | Rabbit | 1:200 | Millipore |
| Anti-STEP | IgG1, unconjugated | Rat synthetic peptide | Mouse | 1:1000 | Boulanger et al., 1995 |
| Anti-human STEP | Whole IgG, unconjugated | Human synthetic peptide | Rabbit | 1:500 | Abgent |
| Anti-pS221 STEP | Whole IgG, unconjugated | Synthetic phosphopeptide | Rabbit | 1:1000 | Snyder et al., 2005 |
| Anti-rabbit | Whole IgG, peroxidase-conjugated | Rabbit Fc | Donkey | 1:10,000 | Amersham Biosciences |
| Anti-mouse | Whole IgG, peroxidase-conjugated | Mouse Fc | Sheep | 1:10,000 | Amersham Biosciences |
The Institutional Animal Care and Use Committee of Yale University approved all experiments. Two strains of mice were used in this study: Tg2576 mice (Charles River Laboratories) (Hsiao et al., 1996) and the STEP knock-out (KO) mouse (Venkitaramani et al., 2009). Rats were purchased from Charles River Laboratories.
7PA2-conditioned medium preparation.
7PA2-conditioned medium (CM; Aβ-enriched) was prepared as described previously with minor modifications (Walsh et al., 2002). Briefly, 7PA2 and untransfected Chinese hamster ovary (CHO) cell lines were grown to 95% confluency and conditioned in DMEM without serum for 16 h. The γ-secretase inhibitor DAPT inhibits Aβ oligomer production in 7PA2 cells. For the preparation of DAPT-7PA2-CM, 250 nm of DAPT (EMD Biosciences) was added to the DMEM before the 16 h growth. CM was centrifuged (200 × g for 10 min) to remove debris and concentrated 15-fold using a YM-3 column (Millipore). CM was aliquoted into 60 μl aliquots and stored at −20°C. For Western blots, 60 or 120 μl of CM was lyophilized, resuspended in 2× Novex Tricine SDS sample buffer, and loaded onto a tricine gel (10–20%) (Invitrogen). Proteins were transferred to polyvinylidene difluoride membranes, and Aβ was detected with 6E10 antibody.
We used a second technique to remove Aβ from 7PA2-CM (Aβ-enriched) by performing four rounds of immunodepletion using an anti-Aβ antibody (6E10) coupled to protein G-Sepharose beads (GE Healthcare). Concentrated 7PA2-CM (300 μl) was incubated with 6E10 antibody (5 μg) conjugated with protein G-agarose beads (50 μl) overnight at 4°C. The supernatant was then subjected to three additional rounds of immunodepletion by incubating it with 6E10 antibody (5 μg) conjugated with protein G-agarose beads (50 μl) for 2 h at 4°C. The Aβ immunodepleted supernatant was then used in cell culture assays. The beads (20 μl) after each round of immunodepletion were mixed with SDS sample buffer (2×) and subjected to electrophoresis in a 16.5% Tris-glycine gel (Bio-Rad), followed by immunoblotting with 6E10 antibody to determine the extent of immunodepletion.
Cortical cultures and slices.
Cortical neurons were prepared from embryonic day 18 (E18) Sprague Dawley rats (Charles River Laboratories). Pregnant dams were killed with CO2, and cortices were removed from embryos by dissection and triturated with 0.025% trypsin-EDTA. Dissociated neurons were seeded in six-well plates (1 × 106 cells/well) containing Neurobasal medium supplemented with B27. Cultures were maintained for 18 d (37°C, 5% CO2). For conditioned medium treatment, concentrated CHO-CM, 7PA2-CM, or DAPT-7PA2-CM was reconstituted (60 μl/ml or 120 μl/ml) in fresh neurobasal medium. Cells were treated for an additional 2 h before processing to obtain membrane fractions.
Rat cortical slices were prepared as described previously (Hu et al., 2007). After decapitation, cortical slices (300 μm) were recovered in oxygenated artificial CSF for 30 min and treated with CHO-CM or 7PA2-CM (60 μl/ml) for 2 h at 30°C. After treatment, tissue was immediately processed for subcellular fractionation as described below.
Surface biotinylation assay.
Cortical cultures from mouse (E15) embryos were grown as described previously (Hu et al., 2007). After 7PA2-CM (Aβ-enriched) treatment for 1 h, the cells were incubated in PBS containing 1.5 mg/ml sulfo-NHS-LC-biotin (Pierce) for 20 min at 4°C. Neurons were rinsed twice in PBS and lysed in 200 μl PBS with protease inhibitors, 0.1% SDS, and 1% Triton X-100. Ten percent of the lysate was used for protein determination, whereas the remainder was incubated with NeutrAvidin agarose to purify biotinylated proteins (50 μl; Pierce). The ratio of biotinylated receptor to total receptor was measured.
Subcellular fractionation.
Homogenates were prepared from brain tissue or slices using glass homogenizers in homogenization buffer containing the following (in mm): 10 Tris-HCl, pH 7.6, 320 sucrose, 150 NaCl, 5 EDTA, 5 EGTA, 20 NaF, 1 Na3VO4, and protease inhibitors (TEVP). Homogenates were centrifuged at 800 × g to remove nuclei and large debris (P1). The P2 fraction was prepared from S1 by centrifugation at 9200 × g for 15 min. The P2 fraction was resuspended in TEVP buffer containing 35.6 mm sucrose and placed on ice for 30 min, and centrifuged at 25,000 × g for 20 min to obtain LP1 fractions. Cell membrane fractions from primary neuronal cultures were prepared as described previously (Botto et al., 2007). Briefly, neurons were washed with 1× PBS and scraped off in extraction buffer containing the following (in mm): 20 Tris-HCl, pH 7.4, 2 EDTA, 0.5 EGTA, 20 NaF, 1 Na3VO4, and protease inhibitors. After homogenization, tissues were centrifuged at 100,000 × g for 1 h at 4°C, and the pellet was dissolved in extraction buffer containing 1% Triton X-100, sonicated at room temperature, placed on ice for 30 min. The extract was centrifuged at 15,000 × g for 20 min at 4°C, and the supernatant was taken as the membrane fraction.
Transfection and immunoprecipitation.
Human embryonic kidney 293T (HEK-293T) cells (ATCC) were seeded at a density of 1 × 106 cells per well in six-well plates. Cells were transfected with STEP61 or hemagglutinin (HA)-ubiquitin cDNA (1 μg), or both constructs using Lipofectamine 2000 (Invitrogen). After 36 h, cells were treated with MG-132 (10 μm), chloroquine (500 μm), or 0.1% DMSO for 4 h. Cells were lysed in buffer containing (in mm) 50 Tris-HCl, pH 7.5, 1% NP-40, 2 EDTA, 100 NaCl, and 10 sodium orthovanadate supplemented with 10 mm N-ethylmaleimide and protease inhibitors (TNESV) (Roche), and sonicated for three 10 s pulses. The lysates were spun at 12,000 × g for 10 min to obtain supernatants. For immunoprecipitation, supernatants (250 μg) were precleared with protein G-Sepharose beads and mixed with anti-STEP antibody (2 μg) overnight at 4°C (Boulanger et al., 1995). The antibody complex was mixed with protein G-Sepharose (40 μl) and incubated for 2 h at 4°C. Beads were washed four times with TNESV buffer, and bound complexes were eluted using 2× SDS sample buffer and processed for Western blotting.
Ubiquitinated protein pull-down.
Ubiquitinated proteins from mouse cortical tissue and cortical slices were isolated using a ubiquitin enrichment kit (Pierce) according to the manufacturer's protocol. Briefly, cortical tissue or cortical slices after conditioned medium treatment were homogenized in TNESV buffer and sonicated for three 10 s pulses. Four hundred micrograms of total lysate were diluted 1:1 with TBS (supplied in the kit) to a final volume of 400 μl. The samples were incubated overnight with 20 μl of polyubiquitin affinity resin in a column (Pierce) at 4°C, then washed four times (10 min intervals each) in the buffer containing TNSEV and TBS (1:10). The polyubiquitin affinity proteins in the column were eluted with 50 μl of 2XSDS sample buffer and then subjected to SDS-PAGE. The gels were incubated with gel soaking buffer (63 mm Tris-HCl pH 6.8, 2.3% SDS, 5.0 β-mercaptoethanol) for 30 min to aid the transfer of higher molecular weight (HMW) proteins. After transfer, the membrane was probed with anti-STEP (23E5) antibody.
Western blots.
Total proteins (30–50 μg) or immunoprecipitated samples were separated by 8% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies overnight at 4°C followed by secondary antibody for 2 h at room temperature. All antibodies and dilutions used in this study are listed in Table 1. Bands were visualized with a chemiluminescence detection system, analyzed using a G:BOX with the GeneSnap image program (Syngene), and quantified by ImageJ 1.33 supplied by NIH.
Human samples.
Human brain samples were obtained from the Alzheimer's Disease and Schizophrenia Brain Bank (Mount Sinai School of Medicine, New York) and approved by the Institutional Board for use in this study. The control and AD brain samples were from Brodmann area 8 of the frontal cortex. The control samples included in the study were examined and were insufficient to meet Consortium to Establish a Registry of AD (CERAD) diagnostic criteria for AD (Mirra et al., 1991). Frozen tissue samples were homogenized (20 strokes) using a glass homogenizer in buffer containing (in mm) 10 Tris-HCl, pH 7.6, 1% NP-40, 150 NaCl, 2 EDTA, 1 DTT, 20 NaF, 1 Na3VO4, and protease inhibitors, sonicated (three times for 10 s with 10 s intervals), and clarified at 14,000 × g for 20 min at 4°C. The homogenates were collected, protein concentrations were determined by BCA kit (Pierce), and total protein (50 μg) was subjected to Western blotting and probed with anti-human STEP antibody. The blots were normalized with anti-β-actin antibody.
Statistics.
All data were presented as means ± SEM. Differences among multiple groups were evaluated either by two-way ANOVA or one-way ANOVA with Tukey's post hoc test. In cases where comparisons between only two groups were of interest, unpaired t tests were performed. For all analyses, a p value of <0.05 indicated a statistically significant difference.
Results
STEP61 protein levels are increased in cortical tissue from Tg2576 mice and human AD patients
A previous study found that STEP protein was increased in a mouse model of AD, although the mechanism involved was unknown (Chin et al., 2005). STEP61 is the only STEP isoform present in cortical and hippocampal cultures, slices, or tissue (Pelkey et al., 2002; Gurd et al., 1999; Hu et al., 2007; Zhang et al., 2008). We examined STEP61 levels in synaptosomal membrane fractions (LP1) of cortices from 3-, 6-, 9-, and 12-month-old Tg2576 and wild-type (WT) mice (Fig. 1A). The results indicated both transgene- and age-dependent increases in STEP61 levels. STEP61 was significantly elevated at 6 months (141 ± 6.9%), 9 months (144 ± 6.8%), and 12 months (144.3 ± 5.3%) compared to littermate controls (p < 0.05) (Fig. 1B). There was no statistically significant difference in STEP61 levels in 3-month-old Tg2576 compared to WT littermates (108.4 ± 13.7%; p > 0.05). As shown previously (Hsiao et al., 1996), we confirmed that these younger mice produce low levels of Aβ, whereas these levels are increased at older ages [total Aβ (Fig. 1A); soluble Aβ (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material)].
Figure 1.

STEP61 levels are increased in Tg2576 mice and human AD brains. A, Immunoblot of STEP61 and total Aβ levels in WT and Tg2576 mice cortex at 3, 6, 9, and 12 months (MO) of age. B, Quantitation of STEP61 protein levels in WT and Tg2576 mice normalized to ERK2 levels. STEP61 levels were increased in 6-, 9-, and 12-month-old Tg2576 mice compared to age matched WT controls (*p < 0.05; two-way ANOVA with post hoc Tukey's test; n = 4;). C, STEP61 is increased in human AD brain samples. Representative immunoblot of prefrontal cortex from control and AD brains probed with anti-human STEP antibody. D, Quantitation of STEP61 control (n = 10) and AD (n = 18) groups normalized to β-actin levels. Data are expressed as percentage increase in STEP61 compared to controls.
We next examined STEP61 levels in postmortem cortical brain tissue homogenates from patients with clinical and pathological symptoms of AD (Fig. 1C). AD was diagnosed according to CERAD criteria using previously described methods (Haroutunian et al., 1998; Mirra et al., 1991). AD samples [range: clinical dementia rating (CDR), 4–5; postmortem interval (PMI), 3.5–20 h; age, 73–102 d] were compared with control samples with no history of dementia and no pathological evidence of AD (range: CDR, 0; PMI, 3–23 h; age, 62–99 d). There was no significant difference in PMI between AD and control groups (t(26) = 1.16; p > 0.05). The samples were analyzed for STEP61 levels using anti-human STEP antibody. Results indicated an increase in STEP61 immunoreactivity in samples from AD patients (n = 18) compared to control samples (n = 10) (Fig. 1D). Quantitation showed a significant increase in STEP61 levels in AD samples compared to the control samples (AD, 195.2 ± 18.4%; control, 100 ± 12.2%; p < 0.01, Student's t test). Detailed information of individual clinical samples used in the study can be found in supplemental Table 1 (available at www.jneurosci.org as supplemental material). These results indicate that STEP61 levels are increased in both human patients with AD and Tg2576 cortex.
Elevated STEP61 levels in aged Tg2576 mice are associated with increased catalytic activity
We next assessed whether the increase in STEP61 was associated with an increase in phosphatase activity. We immunoprecipitated STEP61 from cortical membrane fractions (P2) of 12-month-old Tg2576 and wild-type controls, and determined the phosphatase activity using pNPP as substrate (Fig. 2A). There was a significant increase in phosphatase activity in Tg2576 compared to wild-type control samples (133.0 ± 1.7%; p < 0.01). The increase in activity was blocked by the tyrosine phosphatase inhibitor sodium orthovanadate (10 μm) (data not shown). We also assessed the catalytic activity of immunoprecipitated STEP61 against a glutathione S-transferase (GST)-NR2B fusion protein phosphorylated by Fyn at tyr1472 using a phosphospecific antibody to this site (Fig. 2B,C). There was a significant decrease in phosphorylation of p-tyr1472 by STEP61 immunoprecipitated from Tg2576 mice compared to control mice (WT, 54.2 ± 3.3% of input; Tg, 29.4 ± 1.6% of input; p < 0.01). These results demonstrate that the increased STEP61 was active and dephosphorylated tyr1472 on NR2B.
Figure 2.

Increased STEP61 activity is associated with decreased synaptosomal membrane-associated NR1/NR2B receptor subunits in 12-month-old Tg2576 mice. A, Immunoprecipitated STEP61 from cortical brain lysates of 12-month-old (12 MO) Tg2576 mice has more phosphatase activity against pNPP than STEP61 from age-matched WT mice (**p < 0.01; Student's t test; n = 3). B, C, Representative immunoblot (B) and quantitative analysis (C) show greater dephosphorylation of Fyn-phosphorylated GST-NR2B with immunoprecipitated STEP61 from Tg2576 mice relative to WT controls. Phospho-(pY14721472)-NR2B levels were detected using a specific antibody against the pY1472 site of NR2B. Quantified pY1472 NR2B (pNR2B) levels were normalized relative to Fyn-phosphorylated GST-NR2B levels (input; **p < 0.01; Student's t test; n = 3). D, F, Representative immunoblots showing NR1, pY1472 NR2B, and NR2B levels in LP1 fractions of 3 month (D) and 12 month (F) Tg2576 mice. E, G, Quantitative analysis revealed no significant decrease in NR1, pY1472 NR2B and NR2B in 3-month-old Tg2576 mice (E), but significant decreases at 12 months compared to age-matched WT mice (G; **p < 0.01; Student's t test; n = 6).
There were no significant differences in cortical NR1 and NR2B receptor levels in synaptosomal-associated membrane fractions (LP1) in 3-month-old mice that secrete low levels of Aβ (Fig. 2D,E). In contrast, there was a significant decrease in the expression of these subunits in LP1 fractions at 12 months of age (p < 0.01) (Fig. 2F,G). We also detected a significant decrease in p-Y1472 on the NR2B subunit in Tg2576 compared to littermate controls (p < 0.01) in 12-month-old mice. There was no change in the total amount of receptors between the Tg2576 mice and wild-type littermates at this age (data not shown). Together, the results indicate that the increased levels of STEP61 in older Tg2576 mice had increased phosphatase activity and were associated with a concomitant decrease in p-Y1472 NR2B and a decrease in NR1 and NR2B subunits in synaptosomal membrane fractions.
Treatment of cortical slices and cortical cultures with 7PA2-CM increases STEP61 levels
We found that treatment of cortical cultures with Aβ-enriched 7PA2-CM significantly increased total STEP61 level in a dose-dependent manner compared to untreated cultures or cells treated with control medium (7PA2-CM, 60 μl, 138 ± 9.2%; 120 μl, 176 ± 11.3%; p < 0.05) (Fig. 3A,B). There was no increase in STEP61 after treatment of cortical cultures with conditioned medium from 7PA2 cells pretreated with the γ-secretase inhibitor DAPT to inhibit Aβ production (Fig. 3C,D). DAPT treatment significantly reduced Aβ concentration in 7PA2-CM as determined by immunoblotting with 6E10 antibody or by ELISA (from 5714 ± 415.3 to 1760 ± 24.8 pg/ml) (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material). A complementary approach immunodepleted Aβ from 7PA2-CM with 6E10 antibody, and levels of immunoprecipitated Aβ after each of four rounds of immunodepletion were analyzed by Western blot using 6E10 antibody (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Treatment of cortical cultures with medium in which Aβ was immunodepleted did not result in a significant change of STEP61 levels (Fig. 3C,D). These results demonstrate that Aβ was directly involved in the increase in STEP61 levels in cortical cultures.
Figure 3.

Aβ-enriched, but not control, medium increases STEP61 abundance in cortical neurons. A, Primary cortical cultures were treated with normal medium (untreated), with different amounts of 7PA2-CM (Aβ-enriched), or with CHO-CM (no Aβ) for 2 h. Representative immunoblot shows increased STEP61 levels with 7PA2-CM treatment but not with CHO-CM treatment. B, Quantitative analysis reveals a dose-dependent increase of STEP61 levels after treatment with 7PA2-CM but not with CHO-CM after normalization to total ERK2 levels (*p < 0.05; 7PA2-CM vs untreated; one-way ANOVA with post hoc Tukey's test; n = 5). C, Cortical cultures were treated with normal medium (untreated), with 7PA2-CM with or without the γ-secretase inhibitor DAPT (250 nm), or with 7PA2-CM immunodepleted of Aβ using 6E10 antibody. A representative immunoblot is shown. D, Quantitative analysis shows that DAPT treatment and immunodepleted medium blocks the increase in STEP61. Normalization was performed using total ERK2 (**p < 0.01; one-way ANOVA with post hoc Tukey's test; n = 3). E, Surface biotinylation of cortical cultures is shown after treatment with CHO-CM or 7PA2-CM. Western blots performed on membrane-associated fractions. F, Surface and total NR1, NR2A, NR2B, and GABAAβ 2/3 receptor levels were quantified (**p < 0.01; 7PA2-CM vs CHO-CM; Student's t test; n = 3).
The increase in STEP61 level in cortical neurons was associated with a decrease in surface expression of NMDARs as determined by biotinylation experiments: NR1 (35.2 ± 5.3%; p < 0.01) and NR2B (40.4 ± 3.4%; p < 0.01) (Fig. 3C). As a control, there was no significant change observed in the surface expression of the GABAAβ 2/3 or NR2A receptors (Fig. 3C).
Using a complementary technique, we found that STEP61 level was significantly increased in membrane fractions (LP1) of slices treated with 7PA2-CM compared to control samples (131 ± 6.7%; p < 0.01). In addition, there was a significant reduction in the LP1 membrane enrichment of NR1 (52 ± 5.3%; p < 0.01), NR2B (49.6 ± 3.4%; p < 0.01), and phosphorylation of tyr1472 NR2B (42.8 ± 6.1%; p < 0.01) (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). Similar results were obtained with primary cortical cultures (supplemental Fig. 1F, available at www.jneurosci.org as supplemental material). 7PA2-CM treatment resulted in a decrease in NR1 (35 ± 7.1%; p < 0.01) and NR2B subunits (26.2 ± 8.1%; p < 0.01) in the LP1 fraction compared to CHO-CM treated cultures. Moreover, 7PA2-CM treatment reduced p-tyr1472 NR2B immunoreactivity (38.9 ± 8.7%; p < 0.01) and increased STEP61 levels (145.8 ± 9.3%; p < 0.01).
Aβ-enriched medium increases STEP abundance and STEP dephosphorylation, and decreases NR2B abundance in membrane fractions of cortical slices
STEP61 is implicated in the Aβ-induced endocytosis of NMDARs through activation of calcineurin and dephosphorylation and activation of STEP61 (Snyder et al., 2005). We therefore compared the relative effects of Aβ on STEP level and phosphorylation by examining the time course of the effects of 7PA2-CM treatment of cortical slices (Fig. 4A–C). 7PA2-CM treatment led to an increase in STEP61 levels over the 120 min time course. This increase was paralleled by decreased phospho-STEP levels (Ser221) using a phosphospecific antibody (Paul et al., 2003), and was associated with decreased surface NR2B levels. These results indicated that exogenous application of Aβ-containing CM to cortical slices resulted in the accumulation of active STEP61 and a concomitant decrease in the surface expression of NR1 and NR2B subunits. Decreased phosphorylation of STEP parallels the increase in STEP protein. Thus, changes in STEP protein level as well as phosphorylation appear likely to both be involved in the increased dephosphorylation and endocytosis of NMDAR.
Figure 4.
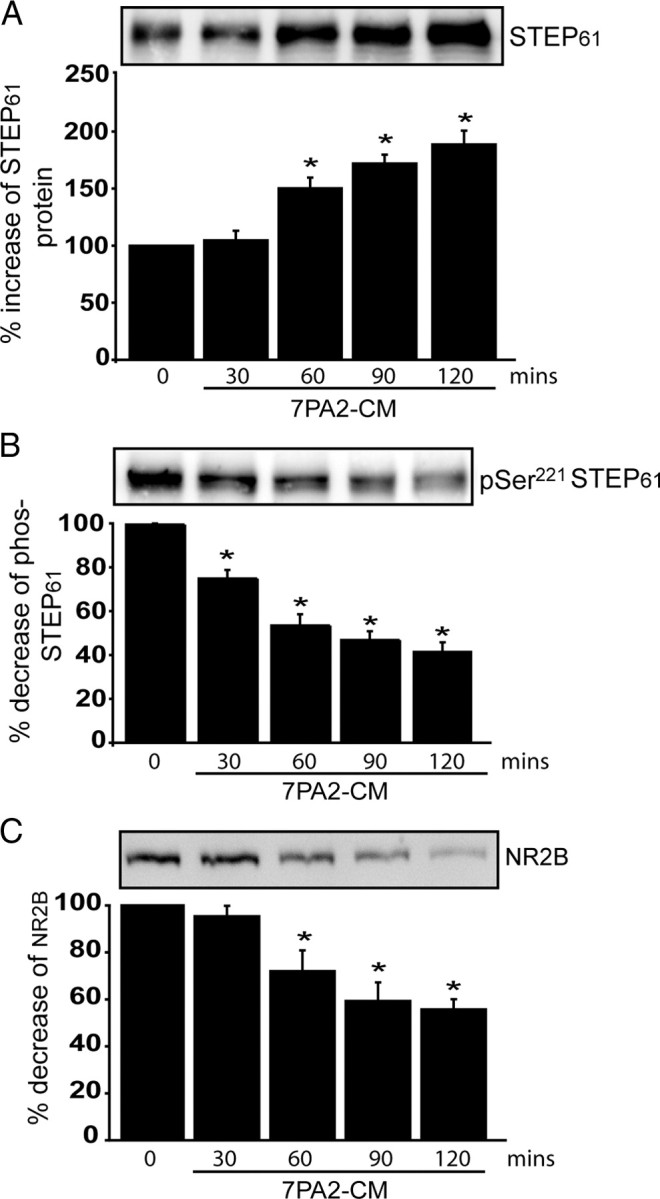
Aβ-enriched medium increases STEP61 levels and STEP61 dephosphorylation, and decreases NR2B abundance in membrane fractions of cortical slices. Cortical slices were incubated with 7PA2-CM (Aβ-enriched) at different time points and processed to obtain membrane fractions (P2). A–C, Samples were analyzed by immunoblotting with anti-STEP antibody (A), anti-pSer221 STEP antibody (B), or anti-NR2B antibody (C). Representative immunoblots are shown along with quantitation of cumulative data. Normalization was performed using total ERK2 (*p < 0.05; one-way ANOVA with post hoc Tukey's test; n = 3).
Aβ-induced glutamate receptor surface expression is abolished in STEP KO cultures
We next examined the role of STEP in Aβ-induced NR1/NR2B receptor endocytosis using biotinylation of surface proteins in cortical cultures from wild-type and STEP KO mice. 7PA2-CM treatment resulted in decreased surface expression of NR1 and NR2B subunits in WT mouse cortical cultures (Fig. 5A,B, lane 1 vs 2) (NR1, 75.2 ± 2.9%; NR2B, 65.5 ± 4.8%; p < 0.01; n = 5). In contrast, there was no significant 7PA2-CM-induced decrease in the surface expression of these receptors in STEP KO cultures (Fig. 5A,B, lanes 4 vs 5) (NR1, 128.6 ± 4.3%; NR2B, 131.7 ± 11.1%; p > 0.05 compared to STEP KO control levels). Higher baseline surface expression of glutamate receptors was detected in STEP KO cultures, consistent with the role of STEP in glutamate receptor internalization (Fig. 5A,B, lanes 1 vs 4) (NR1, 137.5 ± 3.2%; NR2B, 131.7 ± 4.8%; p < 0.01).
Figure 5.

Aβ treatment in STEP KO cultures does not alter surface NR1/NR2B levels. Cortical cultures from WT and STEP KO mice were treated with 7PA2-CM (Aβ-enriched), and biotinylated surface proteins were analyzed by immunoblotting. A, Representative Western blots of NR1 and quantitation are shown. The surface NR1 receptors are normalized to total NR1 levels, and WT CHO (lane 1) served as control for all other treatments (*p < 0.05; **p < 0.01; one-way ANOVA with post hoc Tukey's test; n = 5). STEP KO cultures have higher baseline levels of surface NR1 receptors (KO CHO vs WT CHO; **p < 0.01; n = 5). Aβ treatment decreased surface NR1 in WT cultures (**p < 0.01; Aβ vs CHO in WT; n = 5), but not in STEP KO cultures (p > 0.05; Aβ vs CHO in STEP KO; n = 5). B, Representative Western blots of NR2B and quantitation are shown. The surface NR2B receptors are normalized to total NR2B levels, and WT CHO (lane 1) served as control for all other treatments (*p < 0.05; **p < 0.01; one-way ANOVA with post hoc Tukey's test; n = 5). STEP KO cultures have higher baseline levels of surface NR2B receptors (KO CHO vs WT CHO; **p < 0.01; n = 5). Aβ treatment decreased surface NR2B in WT cultures (**p < 0.01; Aβ vs CHO in WT; n = 5), but not in STEP KO cultures (p > 0.05; Aβ vs CHO in STEP KO; n = 5).
STEP61 is a target for ubiquitination
To investigate the mechanism involved in the regulation of STEP expression, we first analyzed whether translational or transcriptional inhibitors could block the increase in STEP61. Primary cortical cultures were treated with actinomycin D (20 μm) or cycloheximide (100 μm) 20 min before the addition of 7PA2-CM. Neither treatment blocked the increase in STEP61 (Fig. 6A). We also performed semiquantitative reverse transcriptase PCR from treated and untreated cells, and found no significant change in STEP mRNA levels (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Moreover, there were no significant changes in STEP mRNA levels between Tg2576 and wild-type littermates at 3, 6, 9, and 12 months (supplementary Fig. 2B, available at www.jneurosci.org as supplemental material). These results suggest that the increase in STEP61 in cultured neurons or in the AD mouse model was independent of translational and transcriptional mechanisms.
Figure 6.

Increased STEP61 levels do not require transcription or translation but require inhibition of the ubiquitin proteasome system. A, Cortical cultures were pretreated with actinomycin D (20 μm) or cycloheximide (100 μm) for 20 min and then incubated with 7PA2-CM (Aβ-enriched) for 2 h. Normalization was performed using total ERK2 (*p < 0.05, CHO-CM vs other groups; one-way ANOVA with post hoc Tukey's test; n = 3). B, HEK-293T cells were transfected with STEP61 cDNA and incubated for 36 h. Cells were treated with vehicle (0.1% DMSO), the proteasome inhibitor MG-132 (10 μm), or the lysosomal inhibitor chloroquine (500 μm) for 4 h. Representative immunoblot shows an increase in STEP immunoreactivity in the presence of MG-132. C, Cortical cultures were incubated with two structurally different proteasome inhibitors: MG-132 (10 μm and 20 μm) and lactacystin (5 μm and 10 μm) for 2 h. Quantification showed a dose-dependent increase in STEP61 after exposure to both inhibitors and normalization to ERK2 (*p < 0.05, treatment vs control; one-way ANOVA with post hoc Tukey's test; n = 3). D, HEK cells were transfected with STEP61, STEP61 plus HA-tagged ubiquitin cDNAs, or HA-tagged ubiquitin alone. After 36 h, cells were treated with MG-132, immunoprecipitated with STEP antibody, and probed with anti-HA (left) or anti-STEP antibody (right). Higher molecular weight HA and STEP immunoreactivity was observed from cells that coexpressed STEP61 and HA-ubiquitin.
We next examined the possibility that STEP61 expression is regulated at the level of protein degradation. To examine this possibility, STEP61 was expressed in HEK cells, and we studied the effect of proteasome inhibition. Treatment with the proteasome inhibitor MG-132 (10 μm) for 4 h markedly increased STEP61 levels, whereas treatment with the lysosomal inhibitor chloroquine (500 μm) or vehicle alone (0.1% DMSO) did not (Fig. 6B).
To determine whether a similar mechanism occurs in neurons, we treated cortical cultures with the proteasome inhibitors MG-132 (10 and 20 μm) or lactacystin (5 and 10 μm) for 2 h. There was a dose-dependent increase in STEP61 compared to vehicle-treated cultures (Fig. 6C) (p < 0.001). Epoxymycin (10 μm), a structurally different proteasome inhibitor, also resulted in significant increases in STEP61 (p < 0.05; data not shown). These results suggested that the UPS regulates STEP61 in cortical cultures. We then coexpressed STEP61 without or with HA-ubiquitin in HEK cells to address whether STEP61 is a direct target of ubiquitination. Cells were treated with MG-132 (4 h; 10 μm), and STEP61 was immunoprecipitated and probed with anti-HA antibody. Anti-HA recognized a ladder of HMW bands that is typically found with ubiquitin-conjugated proteins (Fig. 6D, left). We then probed with anti-STEP antibody to demonstrate that the HMW proteins were STEP immunoreactive (Fig. 6D, right). Additional studies in HEK cells showed that upon coexpression of STEP61 and HA-ubiquitin, treatment with MG-132 resulted in an increase in nonubiquitinated STEP61, as well as an intense ladder of anti-STEP and anti-HA antibody immunoreactive HMW bands compared to cells not treated with MG-132 (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). These results suggest that inhibition of proteasomal degradation leads to the accumulation of STEP61 presumably as a result of the failure of the UPS to clear STEP-ubiquitin conjugates.
Ubiquitin-conjugated STEP61 accumulates after Aβ treatment of wild-type brain slices and in Tg2576 mice
We treated rat cortical slices with 7PA2-CM or CHO-CM, purified polyubiquitinated proteins, and probed with anti-STEP antibody. The 7PA2-CM-treated slices showed an increase in HMW STEP61 immunoreactive bands (Fig. 7A). As STEP61 is the only STEP isoform present in cortex, the results indicated that ubiquitinated STEP61 is increased in the presence of elevated Aβ levels. As a control, we looked at the ubiquitination of the epidermal growth factor receptor (EGFR), which has been demonstrated previously to be a UPS substrate and is elevated in primary neuronal cultures derived from AD mouse model (Almeida et al., 2006). We found a similar increase in EGFR ubiquitin conjugates after 7PA2-CM treatment compared to CHO-CM (data not shown).
Figure 7.

Ubiquitin-conjugated STEP61 levels are increased in 7PA2-CM-treated cortical slices and in 12-month Tg2576 brains. A, Ubiquitin-conjugated proteins were isolated by affinity chromatography from rat cortical slices after treatment with control CHO-CM or 7PA2-CM (Aβ-enriched) for 2 h. Immunoblotting shows that ubiquitinated STEP61 levels are increased after treatment with 7PA2-CM. B, Ubiquitin-conjugated proteins were isolated from cortical lysates of WT and Tg2576 mice (3 and 12 months) and probed with an anti-STEP antibody. Immunoblot shows that increased levels of ubiquitinated STEP61 at 12 months in Tg2576 cortex. C, HEK cells were transfected with STEP61 (ser221 to ala) cDNA and treated with or without MG-132 (4 h; 10 μm) and immunoprecipitated with anti-STEP antibody. The immunoprecipitated complex was assayed in vitro for phosphatase activity using Fyn-phosphorylated GST-phospho-NR2B as a substrate. Dephosphorylation of pY1472 NR2B (pNR2B) was assessed by immunoblotting with a p-tyr1472-specific antibody and normalized to the initial Fyn-phosphorylated GST-phospho-NR2B levels (input). Inhibition of the proteasome with MG-132 results in greater dephosphorylation of Fyn-phosphorylated GST-phospho-NR2B relative to control (*p < 0.05, Student's t test; n = 3).
We next asked whether there was an accumulation of ubiquitin-conjugated STEP61 in Tg2576 mice. To this end, we affinity purified ubiquitinated proteins from 3- and 12-month-old Tg2576 mice cortex and probed with STEP antibody. Ubiquitinated STEP61 was increased in 12-month-old mice compared to wild-type controls (Fig. 7B). In contrast, 3-month Tg2576 and wild-type controls showed no significant difference in ubiquitin conjugates.
With regard to the above set of results, one question that remained was whether the increased STEP61 levels that resulted from impairment of ubiquitin-mediated turnover was directly associated with the increase in STEP activity. This is difficult to assess in neuronal tissue because STEP phosphorylation/dephosphorylation also plays a role in control of activity. We therefore transfected the nonphosphorylatable form of STEP61 with a serine-to-alanine (S–A) mutation at the Ser221 PKA site into HEK cells and treated without or with MG-132 (4 h; 10 μm). STEP was then immunoprecipitated and activity measured using Fyn-phosphorylated GST-pNR2B as substrate. There was a significant increase in the dephosphorylation of p-Y1472 by STEP61 immunoprecipitated from MG-132-treated cells compared to that from untreated cells (Fig. 7C) (STEP61 (S–A), 75.0 ± 10.3% of input; STEP61 (S–A) plus MG-132, 47.4 ± 9.4% of input; p < 0.05). As this regulatory serine is a target of calcineurin dephosphorylation, these results demonstrate that an additional mechanism other than STEP61 dephosphorylation is involved in the regulation of Y1472 NR2B dephosphorylation. These results support the model that the increase in STEP61 levels after proteasomal blockade is involved in the increased tyr-dephosphorylation of pNR2B.
Discussion
The primary findings from this study indicate that Aβ-mediated NMDAR surface expression requires STEP protein, that the UPS normally processes STEP61, and that there is an increase in STEP levels caused by impairment of the UPS in response to Aβ or in AD model mice. We show that Aβ-mediated reduction in surface expression of NMDARs is absent in STEP KO cultures. In support of a role for the UPS in regulation of STEP61, we find that inhibition of the proteasomal system leads to an accumulation of STEP61 levels in cortical cultures, and that increased STEP61 ubiquitination, protein, and activity are found in Tg2576 mice. Increased STEP protein levels are also found in human AD cortex. Results from our previous work found that the Aβ-mediated reduced surface expression of NMDA receptor subunits is caused by increased endocytosis (Snyder et al., 2005). These previous results, together with the present findings, indicate that STEP61 dephosphorylates the NR2B subunit at its regulatory tyr1472 site, and dephosphorylation of this site leads to internalization of the NMDAR complex from neuronal surface membranes. Moreover, the findings support the hypothesis that regulation of STEP61 by the UPS plays an important role in the control of NMDAR trafficking. The results also suggest that disruption of normal NMDAR trafficking as a result of Aβ-mediated impairment of the UPS contributes to the synaptic dysfunction that occurs in AD.
The elevated levels of STEP61 found in Tg2576 cortex are associated with an increase in tyrosine phosphatase activity compared to wild-type mice. Moreover, there is a significant decrease in the phosphorylation of the regulatory tyr1472 of NR2B subunit of the NMDAR that accompanies the increase in STEP levels. Previous studies have shown that the tyrosine kinase Fyn phosphorylates tyr1472, leading to exocytosis of NMDARs to surface membranes (Dunah et al., 2004; Hallett et al., 2006). It is known that STEP61 dephosphorylates and inactivates Fyn, thus suggesting that STEP61 works through two pathways to promote internalization of NMDARs: direct dephosphorylation of tyr1472 of the NR2B subunit and tyr420 of Fyn (Snyder et al., 2005; Nguyen et al., 2002). A previous study demonstrated that Fyn kinase is downregulated in an AD mouse model, and a possible mechanism was an increase in STEP activity (Chin et al., 2005), a suggestion supported by the present findings.
Increasing evidence suggests that UPS dysfunction plays an important role in the pathogenesis of AD (de Vrij et al., 2004; Hegde and Upadhya, 2007). In human AD brains, ubiquitin immunoreactive inclusion bodies accumulate and proteasomal activity is decreased (Lam et al., 2000; Mori et al., 1987). Proteasomal inhibition results in the accumulation of ubiquitinated proteins, a decrease in free ubiquitin, and increased levels of several proteins involved in AD pathology, including Tau, BACE1, and the regulatory subunit of protein kinase A (Qing et al., 2004; David et al., 2002; Tseng et al., 2007; Gong et al., 2006). Deubiquitinating enzymes that promote the degradation of accumulated proteins are downregulated in brains of AD mouse models and patients (Choi et al., 2004), whereas restoring ubiquitin-recycling enzymes rescues memory deficits and dendritic spine alterations in the AD mouse models (Gong et al., 2006; Smith et al., 2009). These findings support the model that a defective UPS contributes to the progression of AD.
Our results indicate that decreased STEP61 turnover is an important consequence of a defective UPS, and directly links to an impairment of normal NMDAR trafficking at synapses. STEP61 contains two PEST sequences (Bult et al., 1996), which are often present in proteins degraded by the UPS (Rogers et al., 1986), and whether these sequences contribute to STEP61 degradation warrants further investigation. The present results indicate that exposure to Aβ leads to increased levels of STEP protein and activity, and we propose that this increase is involved in dephosphorylation of NR2B leading to NMDAR endocytosis. Our previous studies have also suggested that STEP61 participates in Aβ-mediated NMDAR endocytosis through a mechanism involving Aβ binding to the α7 nicotinic receptor, activation of calcineurin, and dephosphorylation of STEP61 at an inhibitory site within the kinase interacting motif (Snyder et al., 2005). Moreover, PKA activity is reduced and calcineurin activity is increased in AD conditions (Gong et al., 2006; Kuchibhotla et al., 2008), and both are mechanisms that promote STEP61 activation (Paul et al., 2000, 2003). Examination of the kinetics of the changes in STEP protein levels and phosphorylation support the conclusion that increased STEP level and increased activity as a result of dephosphorylation are both likely responsible for the dephosphorylation of NR2B. Indeed, the combination of these two processes would lead to a synergistic effect on STEP activity. Notably, in preliminary studies of α7 nicotinic receptor KO mice, we found only a 30% reduction in the Aβ-mediated NMDAR endocytosis in these mice, supporting the idea that additional mechanisms exist to increase STEP61 activity (Y. Zhang, M. R. Picciotto, and P. J. Lombroso, unpublished data).
To extend our studies to human Alzheimer's patients, we examined STEP levels in postmortem cortical tissue from patients diagnosed with AD. STEP levels were significantly increased in patients with clinical symptoms of AD. Although the samples used are likely cases of sporadic AD, the observed increase in STEP61 levels in human AD brains is an important finding, and supports the hypothesis that altered expression of STEP61 is involved in the pathogenesis of AD. In addition, reduction in NR2B subunits and a decrease in NR2B tyrosine phosphorylation have been reported in human AD brains (Sze et al., 2001). Although the data strongly suggest that Aβ leads to an accumulation of STEP61 levels by inhibiting the UPS, future studies are necessary to provide the molecular details of how STEP ubiquitination is controlled. The UPS system is disrupted in several other CNS disorders including Parkinson's disease, schizophrenia, and Huntington's disease (Altar et al., 2005; Lim and Tan, 2007; Dunah et al., 2000; Lau and Zukin, 2007). As disrupted glutamate signaling is implicated in these disorders, future studies will examine the role of ubiquitination of STEP61 and regulation of NMDAR trafficking in these illnesses. Finally, these results suggest that reducing STEP61 activity through small molecule inhibitors may have therapeutic benefit in AD.
Footnotes
This work was supported by a Brown-Coxe Fellowship to D.V.V.; the National Association of Research on Schizophrenia and Depression; National Institutes of Health Grants MH01527 and MH52711 to P.J.L., AG02219 and AG05138 to V.H., and AG09464 to P.G. and A.C.N.; and a grant from the Cure Alzheimer's Fund to P.G. We thank Drs. Steven Braithwaite, Marilee Ogren, Stephanie Fernandez, and members of our lab for helpful discussions and critical reading of this manuscript. We thank Dr. Strittmatter (Yale University School of Medicine) and Dr. Selkoe (Harvard Medical School, Boston) for providing CHO cells stably transfected with amyloid precursor protein V698F, Dr. Bohmann (University of Rochester Medical Center, Rochester, NY) for the HA-ubiquitin construct, and Richard Huganir (John Hopkins University, Baltimore) for full-length NR2B DNA.
References
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, Young TA, Bullard J, Yokoe H, Webster MJ, Knable MB, Brockman JA. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry. 2005;58:85–96. doi: 10.1016/j.biopsych.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Botto L, Masserini M, Palestini P. Changes in the composition of detergent-resistant membrane domains of cultured neurons following protein kinase C activation. J Neurosci Res. 2007;85:443–450. doi: 10.1002/jnr.21111. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: one STEP at a time. Trends Neurosci. 2006a;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, Oksenberg D, Nikolich K. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006b;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Jr, Sharma E, Lukacsi E, Solimena M, Naegele JR, Lombroso PJ. STEP61: a member of a family of brain-enriched PTPs is localized to the endoplasmic reticulum. J Neurosci. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. J Neurochem. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Fischer DF, van Leeuwen FW, Hol EM. Protein quality control in Alzheimer's disease by the ubiquitin proteasome system. Prog Neurobiol. 2004;74:249–270. doi: 10.1016/j.pneurobio.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Wang Y, Yasuda RP, Kameyama K, Huganir RL, Wolfe BB, Standaert DG. Alterations in subunit expression, composition, and phosphorylation of striatal N-methyl-d-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson's disease. Mol Pharmacol. 2000;57:342–352. [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-d-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Gurd JW, Bissoon N, Nguyen TH, Lombroso PJ, Rider CC, Beesley PW, Vannucci SJ. Hypoxia-ischemia in perinatal rat brain induces the formation of a low molecular weight isoform of striatal enriched tyrosine phosphatase (STEP) J Neurochem. 1999;73:1990–1994. [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Upadhya SC. The ubiquitin-proteasome pathway in health and disease of the nervous system. Trends Neurosci. 2007;30:587–595. doi: 10.1016/j.tins.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu Y, Zhang Y, Venkitaramani DV, Lombroso PJ. Translation of striatal-enriched protein tyrosine phosphatase (STEP) after beta1-adrenergic receptor stimulation. J Neurochem. 2007;103:531–541. doi: 10.1111/j.1471-4159.2007.04749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, Mayer RJ, Layfield R. Inhibition of the ubiquitin-proteasome system in Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:9902–9906. doi: 10.1073/pnas.170173897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lim KL, Tan JM. Role of the ubiquitin proteasome system in Parkinson's disease. BMC Biochem. 2007;8(Suppl 1) doi: 10.1186/1471-2091-8-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer's disease. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev. 2005;126:1292–1299. doi: 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, Ushio Y. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: a light and electron microscopic study with a complementary DNA-generated polyclonal antibody. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004;18:1571–1573. doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. N-Methyl-d-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182:151–159. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, Laferla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2007;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, O'Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Paul S, Zhang Y, Kurup P, Ding L, Tressler L, Allen M, Sacca R, Picciotto MR, Lombroso PJ. Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse. 2009;63:69–81. doi: 10.1002/syn.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Venkitaramani DV, Gladding CM, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]