

Abstract
NFkappaB transcription factor regulates gene expression in response to extracellular stimuli such as TNF alpha. The genes regulated by NFkappaB encode for proteins which control cell growth and survival. Met is the tyrosine kinase receptor for hepatocyte growth factor, and it too promotes cell mitogenesis and survival. Previously, we showed that Met gene expression is regulated by TNF alpha. In this report, we identify and characterize a TNF alpha response element in the Met promoter. This element contains tandem C/EBP sites adjacent to an NFkappaB site. Binding of the NFkappaB p65 subunit and C/EBP beta to this element is induced by TNF alpha. To examine the interplay of NFkappaB and Met in vivo, we determined that Met mRNA and protein levels are reduced in the livers of p65-/- mice as compared to controls. In p65-/- mouse embryonic fibroblasts (MEFs), Met induction by TNF alpha is abrogated while Met's basal gene expression is reduced by half as compared to controls. When overexpressed in p65-/- MEFs, Met confers resistance to TNF alpha mediated cell death. Conversely, expression of dominant negative Met in wildtype MEFs renders them sensitive to cell death induced by TNF alpha. A similar response following TNF alpha challenge was observed in hepatocytic cells treated with siRNA to knockdown endogenous Met. Together, these results indicate that the Met gene is a direct target of NFkappaB and that Met participates in NFkappaB-mediated cell survival.
Introduction
The hepatocyte growth factor receptor (also known as Met) is a transmembrane tyrosine kinase that elicits a variety of biological responses including cell growth and survival (for review, see (Birchmeier et al., 2003; Trusolino and Comoglio, 2002)). Met induces cell survival by inhibiting both the intrinsic and extrinsic apoptotic pathways (Bardelli et al., 1996; Fan et al., 1998; Graziani et al., 1991; Wang et al., 2002; Zou et al., 2007). Due to Met's pleotropic effects on cell function, it is not surprising that the Met gene is overexpressed in a significant percentage of human cancers and that its expression is upregulated in remnant tissues following responses to tissue injury or loss (for example, during liver regeneration (Hoshino et al., 1993)). Thus, regulated expression of the Met gene appears to contribute importantly to normal and aberrant growth and survival. It is unclear, however, which factors govern Met gene promoter activity. In order to gain a better understanding of how Met's gene expression is controlled, we cloned and partially characterized the mouse Met promoter (Seol and Zarnegar, 1998) while others cloned the human Met promoter region (Gambarotta et al., 1994). Through analysis of the promoter sequences and functional experimentation, it was determined that transcription factors such as ets1 (Gambarotta et al., 1996), Pax3 (Epstein et al., 1996), Sp1 (Seol and Zarnegar, 1998), p53 (Seol et al., 1999), and AP1 (Seol et al., 2000) regulate the transcription of the Met gene. In other studies, we determined that Met gene expression is highly inducible in human and mouse cell lines following treatment with various inflammatory cytokines such as tumor necrosis factor alpha (TNF alpha) and Interleukin-1 alpha (IL-1 alpha) (Chen et al., 1996; Chen et al., 1997; Moghul et al., 1994). These findings suggest that extracellular cues dictate Met gene activity and that proper Met gene promoter function requires a specific transcription factor repertoire.
Apoptosis is a critical physiological process for organ development, tissue homeostasis, and removal of potentially unsavory cell types. The Rel/NFkappaB transcription factors block apoptosis in a wide range of cells types. NFkappaB (Nuclear Factor kappa B) was originally identified as a nuclear factor that bound to an enhancer element in the kappa light chain immunoglobulin gene (Sen and Baltimore, 1986). It has subsequently been shown to control the inducible expression of numerous genes and to actively participate in processes such as cell growth, inflammation and cancer in organs like the liver (Karin and Greten, 2005). To date, five proteins belonging to the NFkappaB family have been identified in mammalian cells: RelA (also known as p65), c-Rel, RelB, p105/p50 (NFkappaB1), and p100/p52 (NFkappaB2). In unstimulated cells, NFkappaB hetero- and homodimeric complexes are sequestered in the cytoplasm in an inactive form due to association with members of the Inhibitor of NFkappaB (IkB) family (For review, see (Li and Stark, 2002)). Stimuli such as inflammatory cytokines IL-1 beta and TNF alpha lead to phosphorylation of IkB and its disengagement from the NFkappaB dimeric complex (For review, see (Lentsch and Ward, 1999)). NFkappaB dimers then translocate to the nucleus and activate a variety of genes that carry a specific ten basepair DNA consensus binding site (Henkel et al., 1992). Several NFkappaB targets are genes involved in cell survival such as Bcl-2 family members Bcl-XL (Lee et al., 1999) and A1/Bfl1 (Grumont et al., 1999), adaptor molecules TRAF1 and TRAF2, and apoptosis inhibitors IAP-1 and IAP-2 (Wang et al., 1998).
It is well known that both NFkappaB and Met inhibit apoptosis in the liver. For example, the major cause of embryonic death of RelA (p65) knock out mice is massive apoptosis of hepatocytes by day e15 - 16 (Beg et al., 1995), suggesting a role for NFkappaB in protecting cells from pro-apoptotic stimuli produced in the embryonic liver, released perhaps by resident hematopoietic cells (Beg and Baltimore, 1996). Notably, a similar phenotype is seen in Met global knock out embryos (Bladt et al., 1995) indicating that loss of the Met signaling axis blocks efficient hepatocyte growth and survival during the prenatal period. Given the similarity of the liver histology of knock out animals lacking Met or NFkappaB, it is tempting to speculate that their functions in hepatic development are interdependent and inextricably linked.
In this report, we present evidence that NFkappaB upregulates the promoter activity of the Met gene to facilitate cellular resistance to apoptosis. Our findings identify Met as an important NFkappaB transcriptional target and mediator of NFkappaB-induced cell survival.
Materials and Methods
Cell Culture and Plasmids
Hepa1-6, HepG2 and NIH3T3 cells (mouse and human hepatocellular carcinoma cells and normal mouse fibroblasts, respectively) were obtained from the American Type Culture Collection (Rockville, MD). RelA knock out mouse embryonic fibroblasts and wildtype counterpart controls were kindly provided by Dr. Amer Beg (H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL). pCMV–RelA expression vector was kindly provided by Dr. David Brenner (Columbia University, NY, NY). pMEX-C/EBP beta was obtained from Dr. Steven McKnight (University of Texas Southwestern, Dallas, TX). The serially deleted forms of the Met promoter fused to the CAT reporter gene were reported earlier (Seol and Zarnegar, 1998). A dominant negative version of mouse Met (lacking the entire cytoplasmic region of Met) was described previously (Monga et al., 2002) and then subcloned into various vectors including a GFP bicistronic expression vector according to the manufacturer's recommendations (Clontech, Mountain View, CA). The wildtype (S1) and mutant (M1) forms of the composite NFkappaB-C/EBP site in the Met promoter (depicted in Figure 2A) were cloned into the Bgl II site of the pGL2 luciferase reporter gene (Promega, Madison WI).
Figure 2. TNF alpha induces binding of NFkappaB and C/EBP to the Met promoter.
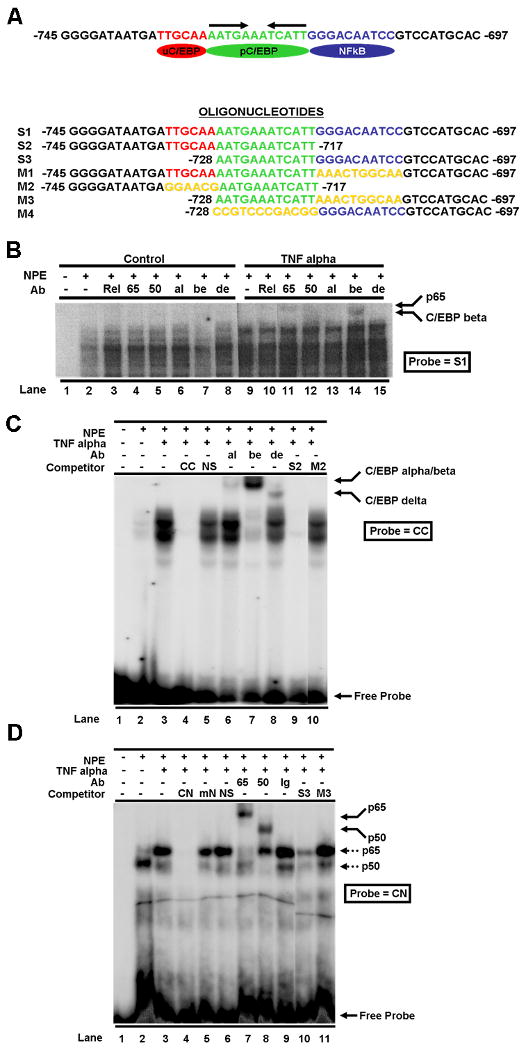
(A) The nucleotide sequence of the Met promoter region corresponding to −745 to -697 bp from the transcription start site is shown (top). It contains a putative NFkappaB site (blue oval/sequence in blue font) and tandem C/EBP sites (upstream C/EBP site [uC/EBP] – red oval/sequence in red font; palindromic C/EBP site [pC/EBP] – green oval/sequence in green font). The arrows over the pC/EBP site indicate the palindromic repeats. Oligonucleotides used in various experiments are shown (below). Yellow font indicates sequence that was mutated in mutant oligomers. (B) The S1 oligonucleotide (a 50-mer) corresponding to the Met promoter composite site was used as a probe in EMSAs. Nuclear protein extracts (NPE) from untreated control (lanes 2-8) or TNF alpha treated Hepa1-6 cells (lanes 9-15) were added as indicated. For supershift assays, various antibodies (Ab) against the NFkappaB family (p65 [65], p50 [50], and c-Rel [Rel]) as well as anti-C/EBP alpha (al), C/EBP beta (be), and C/EBP delta (de) antibodies were utilized as indicated in the figure. Oligonucleotides corresponding to the published consensus C/EBP (C) and NFkappaB (D) binding sites (labeled CC and CN in Figures 2C and D, respectively) were used as probes in gel shift assays with nuclear protein extracts (NPE) from Hepa1-6 cells that were treated with or without TNF alpha. Various unlabeled oligos derived from the Met promoter composite element (i.e. S2, M2, S3, M3 – see [A] for sequence) were added as competitors as indicated in the figures. A variety of antibodies (Ab) as described in [B] were used in supershift studies as indicated. NS – non-specific negative control oligomer; mN – mutant concensus NFkappaB site oligomer; Ig – IgG negative control antibody; p65 – binding complex containing p65; p50 – binding complex containing p50.
Assurances
Appropriate University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) and Recombinant DNA (rDNA) approvals were obtained prior to commencing these studies.
Animals
Snap frozen liver tissue and unstained histological tissue sections of p65-/- or control mice were obtained from Dr. Amer Beg (H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL). For partial hepatectomy experiments, mice were subjected to liver resection as described (Sakamoto et al., 1999).
Semi-quantitative RT-PCR
Total RNA was extracted from frozen liver tissues of p65-/- or control mice using RNAzolB and subjected to semi-quantitative RT-PCR (25, 30, and 40 cycles) following reverse transcription using AMV Reverse Transcriptase (Promega, Madison WI) and Random Primer (Roche Applied Science, Indianapolis IN). The following Met primers were synthesized and used in PCR: Forward: 5′-GCAGAAACCCCCATCCAGAATGTC -3′ and Reverse: 5′-GGCCCCTGGTTTACTGACATACGC-3′. Products were analyzed by agarose gel electrophoresis. Beta-actin primers were purchased from Clontech and added at the recommended concentration as an internal control. Negative controls for PCR contamination included addition of no template and/or no RT: no product was detected by gel analysis.
Western Blot Analysis
Protein lysate was made from liver tissues or cell lines as indicated. Equal amounts of protein (50 ug) were separated by 8% SDS-polyacrylamide under reducing conditions. Protein was transferred to nitrocellulose membrane, and Ponceau S staining was performed to confirm proper transfer and equal loading of samples. The membrane was blocked for 1 h at room temperature with 5% non-fat milk in TBS-T buffer (20mM Tris-HCl, 150 mM NaCl, and 0.1% Tween 20), and then the membrane was incubated with anti-Met antibody (SP-260; Santa Cruz Biotechnology, Santa Cruz, CA) typically at a dilution of 1:500 to 1:1000 at 4°C overnight. The secondary antibody was goat anti-rabbit horseradish peroxidase conjugated from Chemicon and typically used at a 1:20,000 dilution. The signals were visualized by the enhanced chemiluminescence system (ECL solution, Amersham Corp.).
Preparation of nuclear extracts and electrophoretic gel mobility shift assays
Hepa1-6 cells were grown to approximate 80% confluence, serum-starved for overnight and then treated with or without TNF alpha at 50 ng/ml for 3 h. Cells were washed twice with cold phosphate-buffered saline and scraped with a rubber policeman in the same buffer. Nuclear protein extracts were prepared as we previously described (Seol et al., 2000). Double-stranded oligonucleotides used in electrophoretic gel mobility shift assays (EMSAs) were labeled with [alpha-32P] dCTP by end labeling with Klenow fragment of DNA polymerase and used (0.2-0.4 ng) in gel mobility shift. One ug of poly(dI-dC) (Pharmacia) was used as the non-specific competitor in 10 ul of reaction mixture. Ten ug of nuclear protein extract was used in each binding reaction. For competition assays, a 50-fold molar excess of unlabeled oligonucleotides was incubated in reaction mixtures. When antibodies (1 ul) were used in supershift assays, they were incubated with nuclear extracts for 20 minutes before adding other reagents. Gels were run in 0.5 × TBE buffer at a constant voltage of 190 V, dried, and autoradiographed with intensifying screens.
Oligonucleotides and antibodies
The following oligonucleotides were synthesized to performed EMSAs: S1: 5′-GGGGATAATGATTGCAAAATGAAATCATTGGGACAATCCGTCCATGCAC-3′; M1: 5′-GGGGATAATGATTGCAAAATGAAATCATTAACTGGCAAGTCCATG-CAC-3′; S2: 5′-GGGGATAATGATTGCAAAATGAAATCATT-3′; M2: 5′-GGGGATAATGAGGAACGAATGAAATCATT; S3: 5′-AATGAAATCATTGGGAC-AATCCGTCCATGCAC-3′; M3: 5′-AATGAAATCATTAAACTGGCAAGTCC-ATGCAC-3′; M4: 5′-CCGTCCCGACGGGGGACAATCCGTCCATGCAC-3′. The following oligonucleotdides and antibodies were purchased from Santa Cruz Biotechnology, Inc.: consensus C/EBP (CC) site: 5′- TGCAGATTGGGCAAT CTGCA-3′; consensus NFkappaB (CN) site: 5′-AGTTGAGGGGACTTTCCCAGG-3′; antibodies against C/EBP alpha, beta and delta as well as against c-Rel, p65 and p50 were used in gel supershift assays.
CAT/Luciferase Assays
Hepa1-6 cells were cultured in 6-well plates for 24 h and then transfected with various mouse met-CAT chimeric plasmids using lipofectamin (Life Technology Inc.). Five ug DNA and 5 ul of lipofectamin were mixed at room temperature for 30 min and incubated with serum-free DMEM overnight. Cells were then cultured in DMEM for 48 h. After cells were washed twice with PBS, pelleted, resuspended in 150 ul of 0.25 M Tris-HCl (pH 7.5), and disrupted by 5 freeze-thaw cycles, the protein suspension was clarified by centrifugation at 15,000 × g for 5 min at 4°C. The supernatant was collected and assayed for CAT or luciferase activity. CAT activity was determined as follows: reaction mixtures containing the cell extract, 0.2 μCi of [14C] chloramphenicol, and 4.4 mM acetyl coenzyme A were incubated for 2 h at 37°C. The reaction products were separated by thin-layer chromatography (Kodak, Rochester, N.Y.) and visualized by autoradiography. Experimental results were normalized by beta-galactosidase activity. Luciferase activity was measured using Luciferase Assay Reagent (Promega, Madison, WI) according to the manufacturer's recommendations and a Lumat luminometer (Lumat LB 9507, EG&G Berthold, Bad Wildbad, Germany).
Immunohistochemistry
Immunohistochemistry was performed on unstained histologic sections of p65-/- and wildtype mouse embryos. Anti-mouse Met antibody was purchased from Santa Cruz Biotechnology (B-2; Cat# sc-8057) and used at 1:50 dilution. Anti-mouse secondary antibody (Cat# BA-9200) from Vector Laboratories (Burlingame, CA) was used at 1:200 dilution. IgG primary antibody was used as a negative control. Histologic sections were photographed using a Nikon (Melville, NY) Eclipse E600W microscope equipped with a Nikon Coolpix 5700 digital camera.
DN-Met, Met and GFP expression vectors, transfection and cell viability assays
DN-Met and full length mouse Met cDNAs were subcloned into GFP bicistronic or pCMV expression vectors, respectively. GFP was subcloned into the pCMV expression vector. p65-/- or control MEFs were plated in 6-well plates 24 h before transfection. Cells were grown to 50-60% confluence and transfected with the GFP-DN-Met expression vector (p65+/+ MEFs) or pCMV-Met +/- pCMV-GFP expression vector (p65-/- MEFs) by the lipofectamin method for 8 h. Cells were serum starved for 36 h; TNF alpha at a concentration of 50 ng/ml was added for 24 additional h under serum-free conditions. The number of viable (as determined by trypan blue dye exclusion) fluorescent green cells was counted using an inverted fluorescent microscope. Vectors lacking Met cDNA were included as controls where indicated.
siRNA mediated knockdown of Met
Human Met double-stranded RNA oligos were purchased from Ambion, Inc. (Austin, TX) The sequence of the Met3 SiRNA oligo which was used in these studies is 5′-GGTGAAGTGTTAAAAGTTGTT-3′. The control siRNA oligo is GAPDH and was supplied in the siRNA kit. RNA oligos were introduced into the cells by the Silencer™ siRNA Transfection Kit (Applied Biosystems/Ambion, Inc., Austin, TX) following the manufacturer's instructions. Briefly, HepG2 cells were seeded into 12-well plates in a concentration of 105 cells/ml and grown 24 hr before transfection. SiPORT Lipid was mixed thoroughly with OPTI-MEM in reduced serum medium and transfection was carried out as recommended by the supplier. The efficiency of knockdown by RNAi was examined by western blotting after 48 hr. using antibodies to Met, TNFR-I or TNFR-II (purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, CA) After siRNA transfection, cells were serum starved overnight and subjected to TNF alpha mediated apoptosis. Actinomycin-D was added at a concentration of 100 ng/ml while that of recombinant human TNF alpha (R&D Systems, Minneapolis, MN) was added at 20 ng/ml.
Cell Death Assays
The Cytotoxicity Detection Assay (Lactate Dehydrogenase [LDH] Kit, Roche Applied Science, #1644793) was used to determine the extent of cell death. Briefly, cells were grown to 50% confluence in 96 well plates and serum starved overnight. The cells were washed 2× in serum-free medium and were exposed to TNF alpha and actinomycin-D as described above. After incubating the cells at 37°C in 5% CO2, the plates were centrifuged at 250 × g for 10 min. Supernatants were carefully removed (100 ul/well) and transferred into corresponding wells of an optically clear 96-well flat bottom MTP plate. To determine the LDH activity in these supernatants, 100 ul of reaction mixture was added to each well and incubated for 30 min. at RT. During this incubation period, the MTP was protected from light. The absorbance of the samples was measured at 492 nm using a microtiter plate (ELISA) reader.
The second method used to examine the extent of cell death was western blot for caspase 3 activation. Protein lysates were made from cells treated with TNF alpha and actinomycin-D as described above. Anti-cleaved caspase-3 (Asp175) antibody (Cell Signaling Technology, Beverly, MA) was used in Western blot analysis at the dilution recommended by the supplier.
Statistical Analysis
Stasticial analysis was carried out using the Two tailed Student's t-test. Values were considered to be statistically different when the p-value was less than 0.05, and such values are indicated in the figures with symbols.
Results
TNF alpha induces Met protein expression
Previously our laboratory reported that TNF alpha strongly induces Met expression at the mRNA level in ovarian carcinoma cell lines (Moghul et al., 1994) and in a human hepatocellular carcinoma (HepG2) cell line (Chen et al., 1997). To further confirm this induction pattern at the protein level using western blot, we treated Hepa1-6 cells (a mouse hepatocellular carcinoma cell line) and NIH3T3 cells (a mouse fibroblast cell line) with various amounts of TNF alpha for 24 h. As shown in Figure 1A, Met expression was induced by TNF alpha in a dose-dependent manner in both Hepa1-6 and NIH3T3 cells. TNF alpha at 40 ng/ml induced maximal Met expression in Hepa1-6 cells. Interestingly, in NIH3T3 cells which showed lower basal Met expression, TNF alpha at 5 ng/ml induced a dramatic increase in the level of Met protein. At high levels of TNF alpha, we noted a drop in Met protein abundance in both Hepa1-6 and NIH3T3 cells; this observation likely reflects dose-dependent TNF alpha-induced cellular toxicity. In time course studies, Met protein abundance was induced by TNF alpha in Hepa1-6 cells by 3 h of treatment (Figure 1B).
Figure 1. TNF alpha upregulates Met in mouse epithelial and stromal cells.
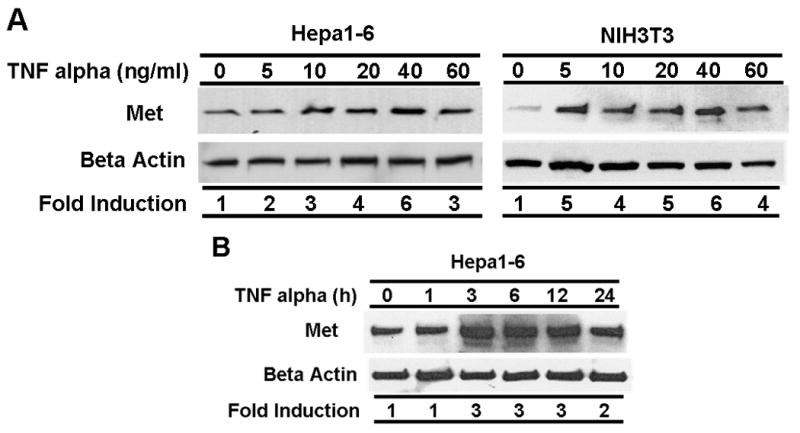
(A) Hepa1-6 and NIH3T3 cells were serum-starved for 24 h and then treated with TNF alpha for different doses as indicated. Protein extracts were prepared and subjected to western immunoblot analysis using an anti-Met antibody. Blots were stripped and probed with an anti-beta actin antibody to document equal protein loading and transfer. The data were normalized using the beta actin signal as an internal control following densitometric analysis. Fold induction over untreated control (0) is shown. (B) TNF alpha (40 ng/ml) was added to serum starved Hepa1-6 cells for varying amounts of time as indicated. Western blot analysis of Met and beta actin protein expression was performed, and data were normalized as described in [A]. Fold induction over untreated control (0) is shown.
TNF alpha induces p65 and C/EBP beta binding to a cis-element in the Met promoter region
Nucleotide analysis of the mouse Met gene promoter region identified a potential NFkappaB binding site at −745 to −697. A putative C/EBP binding site was also found within this region and is located immediately upstream of the NFkappaB site (Figure 2A). It has been reported that members of the C/EBP family and the NFkappaB family can physically interact (Stein et al., 1993) and synergistically activate several cytokine genes (For review, see (Richmond, 2002)). To determine if the putative sites in the Met promoter are functional and can bind to the NFkappaB and C/EBP transcription factors, electrophoretic mobility band shift assays (EMSAs) were performed using 32P labeled 50-mer oligonucleotides corresponding to this cis-element (S1 – Figure 2A). Nuclear protein extracts from TNF alpha-treated or untreated Hepa1-6 cells were incubated with the probe, and the resulting complexes were identified by EMSAs. Various antibodies against NFkappaB and C/EBP family members were used in gel supershift assays to identify the binding factors. As shown in Figure 2B, this element (S1) binds in a specific manner to several proteins as evidenced by formation of multiple complexes. Comparing the pattern of complex formation in the control and TNF alpha treated extracts, binding complexes are seen in the TNF alpha treated extracts. Supershift assays showed no significant reactivity with antibodies against c-Rel, p65, p50, C/EBP alpha, C/EBP beta or C/EBP delta in the nuclear extracts of untreated control cells. On the other hand, p65 and C/EBP beta transcription factors were readily detectable in the TNF alpha-treated cells as indicated in Figure 2B (lanes 11 and 14). Other members of the NFkappaB and C/EBP families were not detectable in treated cells. These results indicate that TNF alpha induces p65 and C/EBP beta binding to the cis-element in the Met promoter region.
The putative NFkappaB and C/EBP sites in the composite cis-element of the Met promoter are functional and bind to p65 and C/EBP beta
To determine the exact binding site of p65 and C/EBP beta within the composite element in the Met promoter, we synthesized various oligomers corresponding to different regions of the composite site. (The nucleotide sequences of these oligos are shown in Figure 2A.) Oligonucleotide S2 corresponds to the Met promoter composite site (-745 to -717) and contains the upstream C/EBP site but lacks the NFkappaB site. A mutant version of this oligo designated M2 was also synthesized and used as a negative control. In this mutant, the TTGCAA which is the core C/EBP site was mutated as indicated in Figure 2A. Oligonucleotide S3 (-728 to -697) contains the putative NFkappaB site in the Met promoter and its mutated version is named M3. The NFkappaB site has the sequence of GGGACAATCC. These oligos were used as competitors in EMSAs and consensus published C/EBP (CC) and NFkappaB (CN) oligonucleotides were used as probes. As shown in Figure 2C, TNF alpha treatment of Hepa1-6 cells induced C/EBP binding activity to the consensus C/EBP binding site (CC – lane 3). The induced binding complex was mostly composed of C/EBP beta as determined by supershift assays (lane 7). Minor amounts of C/EBP alpha and delta were also detected (lanes 6 and 8, respectively). Oligo S2 containing the putative upstream C/EBP binding site in the Met promoter efficiently competed with the probe and abrogated binding (lane 9). On the other hand, the M2 oligomer harboring a mutant upstream C/EBP site did not compete indicating that the competition is site-specific (lane 10). This result agreed with our previous data shown in Figure 2B that TNF alpha mostly induces C/EBP beta binding activity. Subsequently a consensus NFkappaB (CN) oligonucleotide was labeled and used in similar experiments. We found that the binding of the NFkappaB p65 subunit was markedly induced by TNF alpha treatment (Figure 2D, lane 2 vs. lane 1). As expected, specific and efficient binding of the p50 subunit to this site was also detected (lane 8). The Met oligonucleotide containing the NFkappaB site (S3) competed but its mutated version (M3) did not suggesting again that the competition is site-specific (Figure 2D).
The 12 bp palindrome sequence adjacent to the NFkappaB site in the Met promoter composite element is also a functional C/EBP binding site
In promoter studies, a palindrome sequence usually suggests the presence of a transcription factor binding region. In the case of the Met promoter, a 12 bp palindrome sequence lies between the upstream C/EBP site and the NFkappaB site. Interestingly, this sequence consists of a pentameric inverted repeat of AATGAAATCATT (indicated by arrows in Figure 2A) which may be the binding site for a transcription factor(s) having functional effects on the Met promoter. To further characterize the palindromic sequence, we used different truncated and mutated forms of the composite element in EMSAs. Oligo S3 (corresponding to -728 to −697 of the Met composite site) was labeled and used as a probe with the nuclear protein extracts from TNF alpha treated Hepa1-6 cells. As shown in Figure 3A (lane 3), binding of a prominent complex to S3 was induced upon treatment with TNF alpha. This band supershifted with anti-p65 antibody (lane 5) indicating that it contains NFkappaB. Interestingly, this band also showed impeded migration with anti-C/EBP beta antibody (lane 4), and addition of both antibodies totally supershifted this complex indicating that it most likely contains both p65 and C/EBP beta (lane 6). Based on these observations, we suspected that C/EBP beta binds to the palindromic sequence adjacent to the NFkappaB site since the probe used in these studies (S2) lacks the upstream C/EBP site but contains the palindrome.
Figure 3. C/EBP beta binds to the palindromic sequence in the Met composite element.
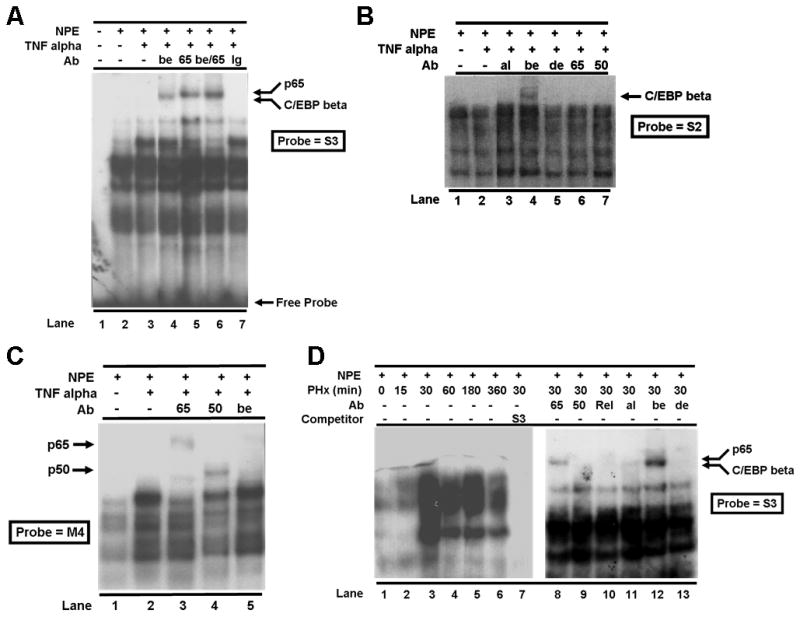
The S3 oligonucleotide (see Fig. 2A for oligomer sequences) containing the NFkappaB site and the palindrome (A), the S2 oligomer containing the upstream C/EBP site and the palindromic sequence (B), or the M4 oligomer containing the NFkappaB site and a mutated palindromic sequence (C) was used as a probe in gel shift assays with nuclear protein extracts (NPE) from Hepa1-6 cells that were treated with or without TNF alpha. A variety of antibodies (Ab) as described in Fig. 2B were used alone or in combination in supershift studies. Ig – IgG negative control antibody. (D) The S3 oligomer containing the NFkappaB site and the palindrome was used as a probe in gel shift assays with nuclear protein extracts (NPE) from mouse livers collected at various times (min. as indicated) after partial hepatectomy (PHx). Antibodies (Ab) as defined in Fig. 2B were used in supershift studies as indicated.
In experiments carried out in Figure 3B, the S2 oligo was used as a probe. This oligo lacks the NFkappaB site but contains the upstream C/EBP site and the palindromic sequence. As shown in the figure, this oligo binds to C/EBP beta (lane 4) but not to NFkappaB p65 or p50 subunits (lanes 6 and 7) as expected in supershift studies. Other C/EBP isoforms (alpha or delta) did not associate with the oligo (lanes 3 and 5, respectively). The mutated form of this probe lacking the upstream C/EBP site (M2) did not compete (data not shown). The studies show that C/EBP beta binds to the upstream C/EBP site; however, they also suggest that binding of C/EBP beta to the palindromic site is dependent on NFkappaB binding to the NFkappaB site.
To further define the NFkappaB site and the adjacent C/EBP palindromic site, oligos M3 and M4 which are mutant versions of S3 having either the core NFkappaB or the C/EBP palindromic site mutated, respectively, were labeled and used in EMSAs. When the NFkappaB site was mutated (M3), neither NFkappaB nor C/EBP beta could bind to this oligo as determined by supershift assays (data not shown). On the other hand, the binding of the NFkappaB p65 subunit to the M4 oligo which has a mutated palindromic C/EBP site was not affected (Figure 3C, lane 3) but, as anticipated, C/EBP beta binding to M4 was abrogated (lane 5). These data confirm our observations discussed above that C/EBP beta binding to the palindromic sequence is dependent on NFkappaB binding to its adjacent cis-element. In the same experiment, we observed binding of the NFkappaB p50 subunit to the M4 oligo (lane 4). This suggests that C/EBP beta binding to the adjacent C/EBP palindromic site blocks the NFkappaB p50 subunit from forming a heterodimeric complex with the NFkappaB p65 subunit.
Induction of p65 and C/EBP beta binding activities to the Met promoter composite site in mouse liver after two-thirds partial hepatectomy
The amount of Met mRNA is altered in the remnant liver after two-thirds partial hepatectomy (PHx) in the rat (Hoshino et al., 1993). It is also reported that p65 (Cressman et al., 1994; FitzGerald et al., 1995) and C/EBP (Yamada et al., 1997; Yamada et al., 1998) binding activities are induced in rodent liver after PHx. To demonstrate whether changes occur in the binding of these transcription factors to the Met composite NFkappaB-C/EBP site during liver regeneration and to demonstrate in vivo relevance to our findings, we made nuclear protein extracts from mouse liver tissue at different time points after partial hepatectomy. We then performed gel shift assays using radiolabeled S3 oligonucleotide as a probe. Results in Figure 3D show that the overall binding activities of NFkappaB and C/EBP beta to the Met composite element are markedly increased 30 min after surgery and remain high during the early phase of liver regeneration (lanes 2-6). The binding complex contains p65 and C/EBP beta (Figure 3D, lanes 8 and 12).
The Met promoter composite element confers synergistic activation by p65 and C/EBP beta to a heterologous promoter
To test the function of the Met promoter composite site, we subcloned it into the pGL2 reporter vector upstream of the SV40 basal promoter. This site (S1) was cloned in both sense (S) and anti-sense (AS) orientations. We also subcloned as control a mutated version (M1) of the composite site (i.e. containing a mutant NFkappaB site) in the sense orientation. These heteropromoter constructs were transfected into Hepa1-6 cells along with the proper negative and positive controls as shown in Figure 4A. Luciferase activities were measured. An approximate 2 to 3 fold upregulation of the reporter gene was found when the composite element was cloned in sense or antisense orientiations as compared to the basal activity of the pGL2 plasmid with the minimal SV40 promoter (SV/L). As expected, orientation did not affect the heterologous promoter suggesting that the Met composite element is a bona fide enhancer. In addition, the fold activation of the reporter gene containing the Met composite element was nearly similar to that achieved by the SV40 enhancer (Figure 4A). The NFkappaB-mutant (M1) composite element had substantially lower activity than the wildtype (S1) composite element which was slightly above the control (SV/L) indicating that the NFkappaB site in this element is critical to its enhancer function.
Figure 4. Met promoter is activated by NFkappaB and C/EBP beta.
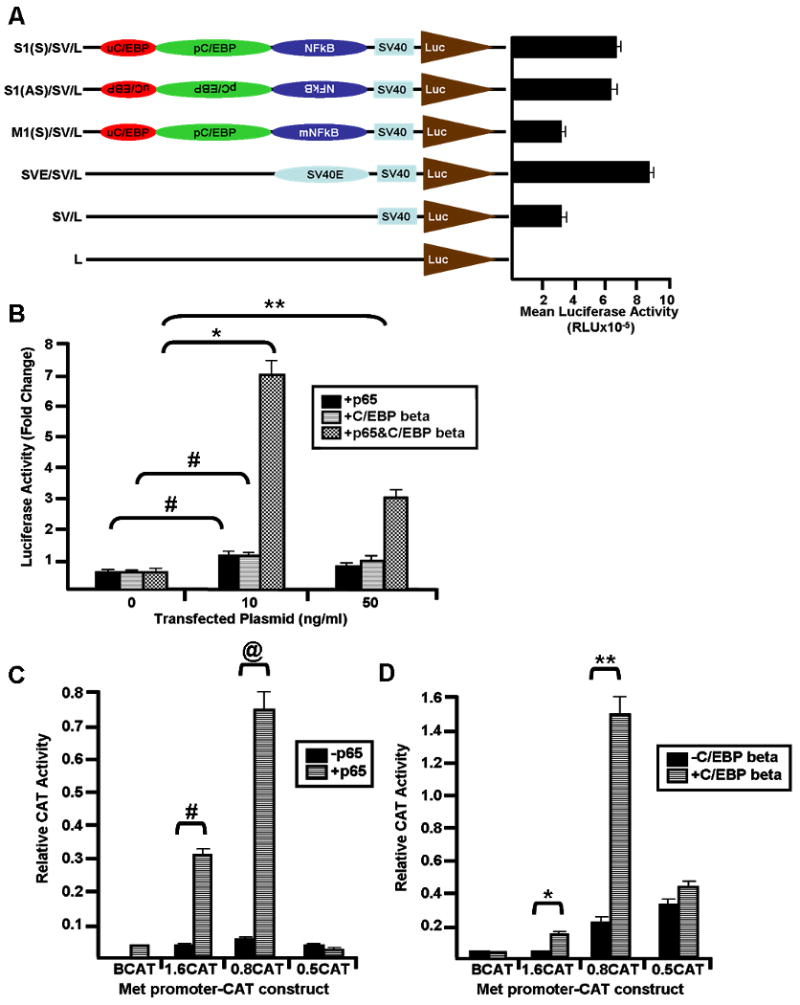
(A) Shown to the left are diagrams of the chimeric luciferase constructs. The S1 oligonucleotide containing the Met promoter composite element (red, green, and blue ovals) or its mutant version (M1) containing a mutated NFkappaB site (mNFkappaB) (see Figure 2A for sequence) was cloned into the pGL2 luciferase vector upstream of an SV40 promoter (light blue box – SV40 or SV). Constructs were transiently transfected into Hepa1-6 cells, and luciferase activity (relative light units – RLU) was measured using a luminometer. Mean luciferase activity (+/- standard deviation) of the various contructs is graphed to the right. Luciferase – brown triangle (Luc or L); SV40 enhancer – light blue oval (SV40E or SVE); (S) – sense orientation; (AS) – antisense orientation. (B) The S1(S)/SV/L construct was transiently transfected into Hepa1-6 cells in the presence or absence of various concentrations (ng/ml) of p65 and/or C/EBP beta expression vectors. Luciferase activity (relative light units – RLU) was measured using a luminometer. Fold change in luciferase activity (+/- standard deviation) was calculated and graphed. Two tailed Student's t-test was performed on the data, and the statistical differences in fold change luciferase activity in the presence (as indicated) or absence (0 ng/ml) of expression vector are shown with symbols. *p=0.0031; **p=0.014; #p=0.008. 5′-deleted Met-CAT constructs containing various lengths (1.6, 0.8, or 0.5 kb – 1.6CAT, 0.8CAT and 0.5CAT, respectively) of the mouse Met promoter were transiently cotransfected with or without p65 (C) or C/EBP beta (D) expression vector into Hepa1-6 cells, and CAT activity was measured. Promoter activity is expressed as relative CAT activity after normalization for beta galactosidase. BCAT – CAT construct containing no promoter region as negative control. Two tailed Student's t-test was performed comparing relative CAT activity in the absence or presence of p65 or C/EBP beta expression vector, respectively, and the statistical differences are indicated with symbols. #p=0.0018; @p=0.0019; *p=0.0003; **p=0.0022.
Transfection of expression vectors for NFkappaB or C/EBP beta also resulted in activation of the heterologous promoter via the Met composite element (S1). As shown in Figure 4B, transfection with both expression vectors synergistically activated the heterologous promoter in a dose dependent manner. We observed maximal synergistic effects (7 fold) when approximately 10 ng/ml of each expression vector were used. Taken together, these results demonstrate that the Met composite site is a functional enhancer and that the p65 and C/EBP beta transcription factors synergistically activate the heteropromoter containing this site.
To determine if p65 and C/EBP beta regulate the activity of the Met promoter, Hepa1-6 cells were cotransfected with CAT reporter vectors containing serial 5′-deleted forms (1.6, 0.8 and 0.5 kb) of the mouse Met promoter region along with expression vectors for p65 or C/EBP beta. As shown in Figure 4C, the 1.6CAT and 0.8CAT constructs exhibited the highest responsiveness to p65 NFkappaB. However, responsiveness was lost in the 0.5CAT construct which lacks the composite Met NFkappaB-C/EBP site. The negative control construct without the Met promoter region showed no response to p65 (BCAT). Similar experiments were performed using the C/EBP beta expression vector with various Met-CAT constructs (Figure 4D); again, the 0.8CAT construct responded strongly while the 0.5CAT showed decreased responsiveness. This proves that the NFkappaB and the C/EBP response elements are located within the 0.8-0.5 kb region of the Met gene promoter where the composite NFkappaB-C/EBP site resides.
Expression of Met is reduced in the livers of p65 knock out mice
To explore the in vivo relationship between p65 and Met, we obtained embryonic tissue from p65-/- and wildtype mice and analyzed it for Met mRNA and protein expression by semi-quantitative RT-PCR and immunohistochemistry, respectively. We found that both Met mRNA (data not shown) and protein (Figure 5A) levels are downregulated in the livers of p65-/- mice as compared to littermate controls.
Figure 5. NFkappaB is essential for optimal Met expression.
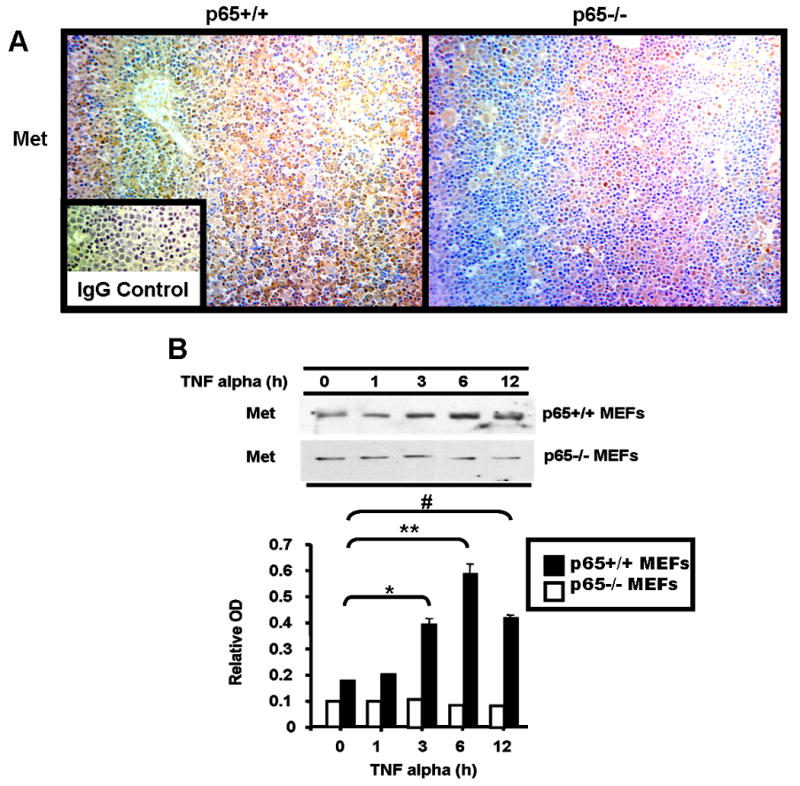
(A) Met protein levels were assessed in p65-/- and contol (p65+/+) mouse fetal liver by immunohistochemistry using anti-Met antibody. Histologic images were photographed at 100× magnification. Inset: IgG control antibody (B) p65+/+ and p65-/- MEFs were treated with TNF alpha for various times as indicated and described in Materials and Methods. Cell protein extract was subjected to western blot analysis for Met protein expression using anti-Met antibody. The western signals were assessed by densitometric analysis (measured in relative OD units) and are graphed below. Two tailed Student's t-test was performed on the data comparing values to the 0 hr. time point, and the statistical differences are indicated with symbols. *p=0.003; **p=0.0018; #p=0.006.
Embryonic fibroblasts from p65 knock out mice lose Met induction by TNF alpha
To prove that p65 is critical for Met gene induction, we obtained p65-/- mouse embryonic fibroblasts (MEFs) and wildtype controls. These cells were treated with TNF alpha for various times (Figure 5B). Their lysates were analyzed by western blot for Met protein expression. The induction of Met by TNF alpha was totally abolished in the p65-/- MEFs. On the other hand, in the wildtype MEFs, Met was induced by 3 to 4 fold at 6 h post treatment. Interestingly, as shown in Figure 5B, the basal level of Met protein in p65-/- MEFs was also lower. Met protein abundance in p65 knock out MEFs was consistently half that seen in the wildtype MEFs. These data strongly suggest that Met is a target gene of the NFkappaB transcription factor, and that p65 controls Met's basal as well as its inducible expression.
Overexpression of Met in p65 knock out MEFs confers resistance to TNF alpha-mediated cell death
In a delicate balance of pro- and anti-apoptotic signaling, TNF alpha can, on the one hand, induce cell death by simultaneously activating the extrinsic (death receptor mediated) and intrinsic (mitochondrial release of cytochrome C mediated by the JNK) cell death pathways via TNF receptor type-I (TNFR-I) activation. However, TNF alpha can also lead to cell survival by stimulating the NFkappaB pathway on the other. Our data led us to hypothesize that one of the reasons why p65-/- MEFs are hypersensitive to TNF alpha mediated cell death is because they lack adequate basal Met gene expression as well as Met expression induced by p65. To test this hypothesis, we overexpressed the mouse Met cDNA construct in p65-/- MEFs to restore Met expression and then exposed these cells to TNF alpha. A mouse Met expression vector was cotransfected with a GFP expression vector in a 10:1 ratio into p65-/- MEFs. Viable green fluorescent cells were counted in duplicate wells after TNF alpha treatment. The results are shown in Figure 6A. Restoring Met expression to p65-/- MEFs increased their survival when they were challenged by TNF alpha treatment as compared to cells transfected with the empty control vector. In other words, reconstitution of p65-/- MEFs with Met rescues them from TNF alpha induced cell death.
Figure 6. Met participates in NFkappaB-mediated cell survival.
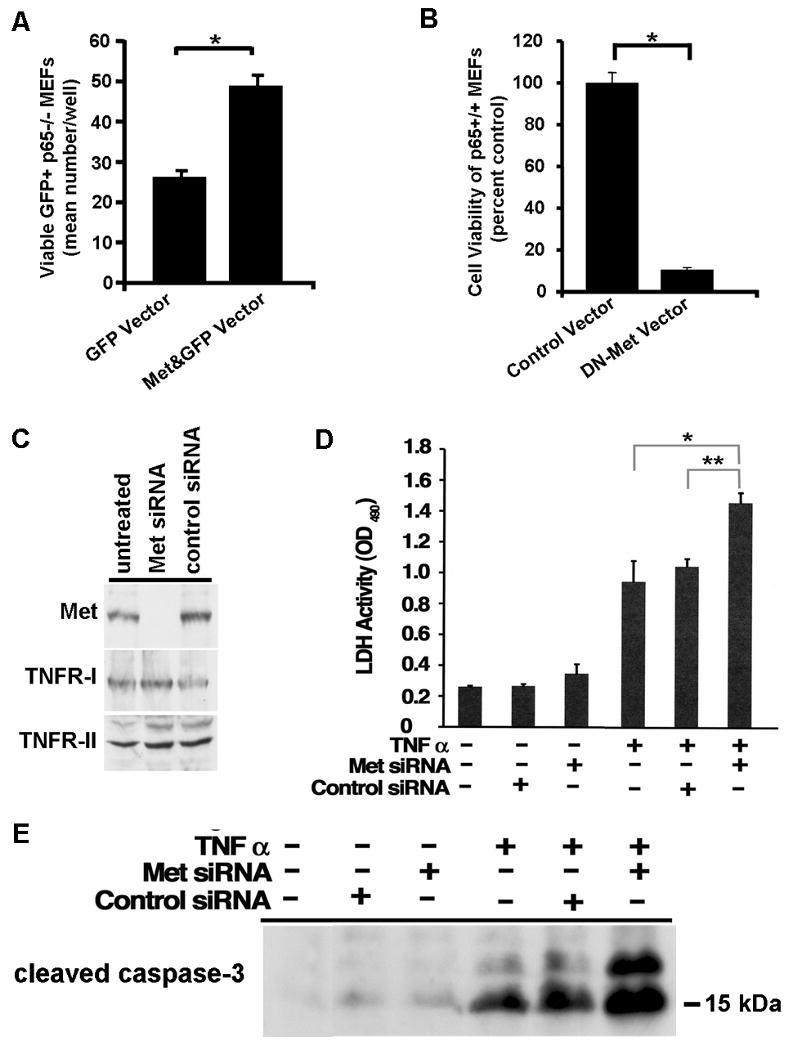
(A) A GFP expression vector was contransfected with (Met&GFP Vector) or without (GFP Vector) a mouse Met expression vector at a 1:10 ratio into p65-/- MEFs. Viable green fluorescent cells were counted after 12 h of TNF alpha treatment as described in Materials and Methods. Mean number of viable GFP+ cells per well was determined for each condition. Two tailed Student's t-test was performed on the data, and the statistical difference is indicated with an asterisk. *p<0.007 (B) p65+/+ MEFs were transfected with a bicistronic GFP expression vector either containing (DN-Met Vector) or lacking (Control Vector) a dominant negative form of mouse Met (DN-Met) and challenged by TNF alpha treatment as described in (A). The extent of cell viability was determined and calculated as percentage of viable cells as compared to control. Two tailed Student's t-test was performed on the data, and the statistical difference is indicated with an asterisk. *p<0.009 (C) HepG2 cells were subjected to siRNA mediated knock down of Met (Met siRNA). Total protein lysates were subjected to western blot analysis using anti-Met, anti-TNFR-I or anti-TNFR-II antibody. Levels of TNFR-I or –II did not change indicating the specificity of the Met siRNA for its target. Control – siRNA to GAPDH. Untreated – cells receiving no siRNA. (D) HepG2 cells treated with siRNA to Met or GAPDH (control siRNA) or with no siRNA received TNF alpha in the culture medium as described in Materials and Methods. LDH cell death assay was performed and optical density at 490 nm was measured. Two tailed Student's t-test was performed on the data, and the statistical differences between cells receiving TNF alpha in the presence or absence of the indicated siRNA are shown with asterisks. *p=0.005; **p=0.001 (E) Total protein lysates were prepared from HepG2 cells treated as in (D). Western blot analysis was performed using anti-cleaved caspase 3 antibody.
Blocking Met signaling by a dominant negative approach decreases cell viability in wildtype MEFs following TNF alpha treatment
Our data to this point suggested to us that Met induction by TNF alpha is functionally important for promoting cell survival and combating apoptosis. To explore this notion more fully, we cloned a dominant negative form of mouse Met (DN-Met) into a bicistronic GFP expression vector. This DN-Met-GFP coexpression plasmid (DN-Met Vector) or its empty control (Control Vector) was transiently transfected into wildtype MEFs (p65+/+). The transfected cells were then challenged by TNF alpha treatment, and the extent of cell viability was determined. TNF alpha treatment profoundly reduced cell viability in cells expressing DN-Met as compared to control (Figure 6B) emphasizing the survival benefit Met confers to cells in the face of TNF alpha exposure.
Blocking Met signaling in hepatocytic cells by Met siRNA-mediated knockdown enhances cell death following TNF alpha treatment
We next wanted to test the effect of inhibiting endogenous Met in a hepatocytic HepG2 cells. We carried out siRNA mediated knock down of Met and observed a dramatic decrease in Met protein by western blot analysis (Figure 6C) in cells receiving specific siRNA as compared to the control siRNA. The expression levels of proteins such as TNFR-I and TNFR-II did not change indicating the specificity of the knockdown effect. When the cells were challenged with TNF alpha, we observed enhanced cell death in cells treated with Met siRNA as compared to cells treated with control siRNA or cells not receiving siRNA (Figures 6D&E). These data indicate that loss of Met sensitizes hepatocytic cells to TNF alpha killing.
Altogether, we demonstrate that, upon challenge with TNF alpha, blocking endogenous Met promotes cell death while restoring Met expression in cells lacking p65 improves survival. These observations highlight the importance of the Met tyrosine kinase to the pro-survival cascade needed to counteract the cell death pathway unleashed by TNF alpha.
Discussion
Previous studies from our laboratory have shown that Met is strongly induced at the mRNA and protein levels in different cell types by several cytokines, especially TNF alpha and IL-1 alpha. The molecular mechanisms mediating the induction of Met gene expression remain largely unknown. In the present report, we have identified and characterized a composite site in the upstream region of the Met promoter which is responsible for the activation of this gene by TNF alpha. This composite site contains tandem C/EBP sites adjacent to one NFkappaB site. Using gel shift and supershift assays and functional analysis, we identified the p65 subunit of the NFkappaB family and the C/EBP beta isoform of the C/EBP family as the transactivating factors which bind to this region and mediate Met gene induction in response to TNF alpha in hepatic cells. We showed that these two transcription factors cooperate with each other to maximally induce the Met gene and that NFkappaB is critically important for Met gene induction. To understand the biological relevance of these observations, we performed functional assays in p65-/- and control mouse embryonic fibroblasts (MEFs). We first observed that in p65-/- MEFs, induction of Met protein by TNF alpha is abolished supporting the notion that p65 plays an essential role in the enhancement of Met gene transcription. We also noted that basal Met protein level is approximately half that seen in the control MEFs.
It is well known that TNF alpha unleashes both pro-survival (i.e. NFkappaB signaling) and pro-apoptotic (via JNK and caspase activation) cues in cells. To experimentally promote TNF alpha's death axis, cells expressing NFkappaB must be treated with chemical agents such actinomycin-D to negate NFkappaB's induction of anti-apoptotic genes. In the absence of NFkappaB signaling, death signals are unopposed and cell death prevails. In light of these facts coupled with our observations, we hypothesized that upregulation of Met protein, a potent pro-survival factor, by the TNF alpha-NFkappaB axis is likely to be important for promotion of cell survival and inhibition of TNF alpha-induced cell death. To test this idea, we carried out two different approaches: gain-of-function experiments (i.e. restoring Met expression to p65-/- cells) and loss-of function experiments (i.e. blocking endogenous Met signaling in wildtype p65+/+ MEFs by dominant negative Met or in HepG2 cells by Met siRNA). We determined that inhibition of endogenous Met in wildtype MEFs or HepG2 cells augmented cell death induced by TNF alpha treatment; on the other hand, enforcing Met expression in p65 knock out MEFs promoted cell survival under the same conditions. Therefore, our current results shed light on the regulation and function of the Met gene and its importance to TNF alpha-NFkappaB mediated cell survival: Met is a bona fide target of NFkappaB that confers survival advantage to cells under conditions that promote NFkappaB activity, such as TNF alpha treatment.
Our laboratory has cloned and partially characterized the mouse Met promoter region (Seol and Zarnegar, 1998). Nucleotide sequence analysis of the mouse Met promoter revealed that it lacks a TATA box, and it has a high GC-content. These features resemble those found in housekeeping and constitutively expressed gene promoters. Interestingly, the Met promoter region also contains several cis-acting regulatory elements that confer inducibility to this gene in response to various growth and stress-related cues. Work in our laboratory has characterized functional AP1, p53, and Sp1 sites within the upstream region (i.e. within 500 bp) from the core promoter. We have found that HGF induces Met expression, at least in part, via the AP1 site (Seol et al., 2000). Moreover, we have found that Met is induced in response to UV irradiation and that p53 contributes to this induction through a p53 response element in the promoter region (Seol et al., 1999). We have reported that the Sp1 sites within the Met proximal promoter are essential for the basal activity of the promoter (Seol and Zarnegar, 1998). In the current studies, we found that NFkappaB and C/EBP beta cooperate to induce Met gene expression via the composite NFkappaB-C/EBP binding site in the mouse Met promoter, a site this is conserved in the human Met gene as well. Synergism between the NFkappaB and C/EBP families in gene activation have been shown to occur in immune response genes such as amyloid (A1, A2, and A3) and cytokines (IL-6, IL-8, and G-CSF) (For review, see (Richmond, 2002)). Although the mechanisms responsible for the cooperative effects have not yet been entirely clarified, productive interaction requires the integrity of both the NFkappaB Rel domain and the C/EBP leucine zipper motif (Stein et al., 1993). Our mutagenesis and gel shift studies indicated that a pair of tandem C/EBP elements lie adjacent to an NFkappaB site in the Met composite site. Furthermore, we observed that p65 occupation of the NFkappaB cis-element is necessary for C/EBP beta binding to the palindromic sequence in the Met composite site, perhaps due to p65-mediated stabilization of C/EBP beta:DNA association through protein-protein interactions. On the other hand, we noted that preventing C/EBP beta engagement of the palindrome promotes p50/p65 NFkappaB heterodimerization thus effectively altering the NFkappaB repertoire acting at the Met promoter composite site. Functional analysis of the Met promoter composite site in a heterologous promoter suggested that the NFkappaB site is particularly crucial for enhancer function. Overall, our data showed that TNF alpha simultaneously induced both p65 and C/EBP beta binding activity to the Met composite element; evidently, for the reasons just stated, p65 is the more critical transactivator, while C/EBP beta partners with p65 to boost Met gene transcription.
Several in vitro and in vivo studies have suggested that Met is crucial for cell survival. Similarly, it has been shown that TNF receptor type-I (TNFR-I) also promotes cell survival through the NFkappaB axis. In the present work, we linked the pro-survival effects of the TNF alpha-NFkappaB axis to Met by identifying Met as a key transcriptional target of this pathway. Studies of p65-/- mouse embryos demonstrate hepatocyte apoptosis, liver degeneration and embryonic lethality at day e15 - 16 (Beg et al., 1995). Similarly, Met-/- mouse embryos die due to multiple defects, including lack of hepatocyte growth and survival (Bladt et al., 1995). We found that Met mRNA levels are lower in p65 knock out livers as compared to control livers using semi-quantitative RT-PCR. However, because embryonic liver is composed of a mixture of cell types thus preventing us from ascertaining Met mRNA expression specifically in the hepatocyte compartment coupled with the fact that a reduction in mRNA abundance may not necessarily be reflected in lower protein levels, we used the complementary approach of immunohistochemistry to examine Met protein abundance in situ in hepatocytes. We observed that Met protein level is substantially lower in the p65 knock out livers as compared to controls. We also determined that siRNA mediated knockdown of Met enhances TNF alpha induced killing in hepatocytic cells (HepG2). It is puzzling why p65-/- knock out mice die of liver apoptosis. Our results may provide a clue as to the reason only liver development is affected in these mice, namely because their hepatocytes lack sufficient expression, and therefore cytoprotection, from Met which makes them susceptible to death (Schematic shown in Figures 7A&B). Since the liver is the major site of hematopoiesis during gestation, it has been postulated that parenchymal liver cells must withstand an onslaught of pro-apoptotic cues (such as TNF alpha and FasL, to name a few) generated to fine tune the hematopoietic cell compartment (Beg and Baltimore, 1996). The role of Met in liver organogenesis and hepatocyte survival has been well established by global (Bladt et al., 1995) and liver specific (Borowiak et al., 2004; Huh et al., 2004) knock out studies in mice. During development, Met is expressed in rodent liver (DeFrances et al., 1992). Thus, we speculate that during the gestational period, ample expression of Met is demanded of the liver to promote hepatocyte growth as well as survival in the presence of the harsher cytokine microenvironment needed to kill unwanted hematopoietic cells.
Figure 7. Schematic depicting that NFkappaB-mediated cell survival is dependent at least in part on Met.
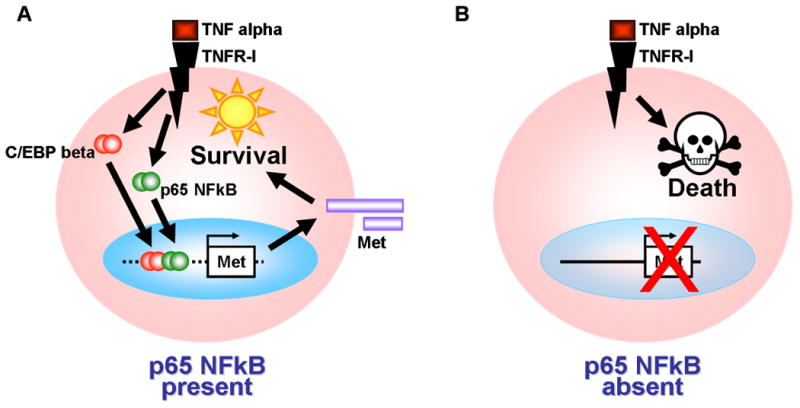
(A) In the presence of NFkappaB (i.e. p65+/+), cells such as hepatocytes are protected from pro-death signals mediated by the TNF alpha/TNFR-I axis. This is due in part to the upregulated expression of Met tyrosine kinase receptor gene, which encodes a potent pro-survival factor, by NFkappaB, and to lesser extent, by C/EBP beta. (B) In cells lacking NFkappaB (i.e. p65-/-), Met expression, and therefore Met-mediated cytoprotection, are reduced. The pro-apoptotic arm of death signals such as TNF alpha is insufficiently countered, and death pathways prevail.
Additional experimental evidence has been gathered on the role of NFkappaB, C/EBP and Met in promoting hepatocyte survival and growth. For proper liver regeneration to proceed, a balance of pro-growth/pro-survival signals and mitoinhibitors must be maintained (Michalopoulos and DeFrances, 2005). It has been documented that NFkappaB and C/EBP activity (Cressman et al., 1994; Diehl and Yang, 1994; FitzGerald et al., 1995; Greenbaum et al., 1995) as well as Met mRNA expression (Hoshino et al., 1993) are altered during liver regeneration. In support of this, our findings show that NFkappaB and C/EBP binding to the Met composite site is enhanced following partial hepatectomy. Experimental in vivo inhibition (Iimuro et al., 1998) or knock out (Borowiak et al., 2004; Geisler et al., 2007; Huh et al., 2004) of either NFkappaB or Met in the liver renders hepatocytes sensitive to apoptosis and delays liver regeneration in most experimental models. While liver regeneration following surgery or toxin exposure restores the liver to its original homeostatic state, uncontrolled replication of initiated hepatocytes leads to liver cell dysplasia and hepatocellular carcinoma in humans and rodents. The inciting factors of liver tumorigenesis range from infectious agents to inborn errors of metabolism to carcinogen exposure which cause chronic and deleterious regenerative and survival responses in the liver (For review, see (DeFrances, 2005)). Several studies in humans and rodents link hepatocarcinogenesis with aberrant Met signaling (Bell et al., 1999; Park et al., 1999; Sakata et al., 1996; Suzuki et al., 1994; Tavian et al., 2000; Trusolino and Comoglio, 2002). It is becoming clear that the NFkappaB survival pathway is also active in chronic hepatic inflammation, survival of initiated hepatocytes and frank tumorigenesis in the liver (For review, see (Karin and Greten, 2005)).
The mechanisms by which Met regulates apoptosis are only partially understood. Upon activation, Met engages the anti-apoptotic pathway of the PI3-kinase-Akt axis (Graziani et al., 1991). The cytoplasmic domain of Met associates with Bag1, a Bcl2-interacting molecule (Bardelli et al., 1996), and Met signaling leads to upregulated expression of the anti-apoptotic molecule BclXL (Fan et al., 1998). In addition, we have demonstrated that Met's extracellular alpha chain binds to and sequesters the death receptor Fas preventing its activation (Wang et al., 2002; Zou et al., 2007). The net results are cytoprotection against apoptotic stimuli. Therefore, it is a compelling notion that NFkappaB upregulates Met, a potent multi-functional anti-apoptotic signaling molecule, to help stave off cell death. Along with TNF alpha, they comprise a newly described ‘cell survival circuit’: TNF alpha→NFkappaB→Met→survival.
Acknowledgments
We'd like to thank Dr. Amer Beg (H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL) for providing us with tissue and histological tissue sections from p65-/- and control mice. This work was supported in part by an award from the NIH/NCI (R01-CA95782) to RZ and an award from the Rangos Fund for Enhancement of Pathology Research to MCD.
References
- Bardelli A, Longati P, Albero D, Goruppi S, Schneider C, Ponzetto C, Comoglio PM. HGF receptor associates with the anti-apoptotic protein BAG-1 and prevents cell death. EMBO Journal. 1996;15:6205–6212. [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. see comment. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Bell A, Chen Q, DeFrances MC, Michalopoulos GK, Zarnegar R. The five amino acid-deleted isoform of hepatocyte growth factor promotes carcinogenesis in transgenic mice. Oncogene. 1999;18:887–895. doi: 10.1038/sj.onc.1202379. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature Reviews Molecular Cell Biology. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, DeFrances MC, Zarnegar R. Induction of met proto-oncogene (hepatocyte growth factor receptor) expression during human monocyte-macrophage differentiation. Cell Growth & Differentiation. 1996;7:821–832. [PubMed] [Google Scholar]
- Chen Q, Seol DW, Carr B, Zarnegar R. Co-expression and regulation of Met and Ron proto-oncogenes in human hepatocellular carcinoma tissues and cell lines. Hepatology. 1997;26:59–66. doi: 10.1002/hep.510260108. [DOI] [PubMed] [Google Scholar]
- Cressman DE, Greenbaum LE, Haber BA, Taub R. Rapid activation of post-hepatectomy factor/nuclear factor kappa B in hepatocytes, a primary response in the regenerating liver. Journal of Biological Chemistry. 1994;269:30429–30435. [PubMed] [Google Scholar]
- DeFrances MC, Wolf HK, Michalopoulos GK, Zarnegar R. The presence of hepatocyte growth factor in the developing rat. Development. 1992;116:387–395. doi: 10.1242/dev.116.2.387. [DOI] [PubMed] [Google Scholar]
- DeFrances MC, Michalopoulos GK. Molecular Mechanisms of Hepatocellular Carcinoma: Insights to Therapy. Humana Press; Totowa, NJ: 2005. pp. 23–58. [Google Scholar]
- Diehl AM, Yang SQ. Regenerative changes in C/EBP alpha and C/EBP beta expression modulate binding to the C/EBP site in the c-fos promoter. Hepatology. 1994;19:447–456. [PubMed] [Google Scholar]
- Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:4213–4218. doi: 10.1073/pnas.93.9.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Wang JA, Yuan RQ, Rockwell S, Andres J, Zlatapolskiy A, Goldberg ID, Rosen EM. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene. 1998;17:131–141. doi: 10.1038/sj.onc.1201943. [DOI] [PubMed] [Google Scholar]
- FitzGerald MJ, Webber EM, Donovan JR, Fausto N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth & Differentiation. 1995;6:417–427. [PubMed] [Google Scholar]
- Gambarotta G, Boccaccio C, Giordano S, Ando M, Stella MC, Comoglio PM. Ets up-regulates MET transcription. Oncogene. 1996;13:1911–1917. [PubMed] [Google Scholar]
- Gambarotta G, Pistoi S, Giordano S, Comoglio PM, Santoro C. Structure and inducible regulation of the human MET promoter. Journal of Biological Chemistry. 1994;269:12852–12857. [PubMed] [Google Scholar]
- Geisler F, Algul H, Paxian S, Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–2503. doi: 10.1053/j.gastro.2007.03.033. see comment. [DOI] [PubMed] [Google Scholar]
- Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. Journal of Biological Chemistry. 1991;266:22087–22090. [PubMed] [Google Scholar]
- Greenbaum LE, Cressman DE, Haber BA, Taub R. Coexistence of C/EBP alpha, beta, growth-induced proteins and DNA synthesis in hepatocytes during liver regeneration. Implications for maintenance of the differentiated state during liver growth. Journal of Clinical Investigation. 1995;96:1351–1365. doi: 10.1172/JCI118170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes & Development. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel T, Zabel U, van Zee K, Muller JM, Fanning E, Baeuerle PA. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell. 1992;68:1121–1133. doi: 10.1016/0092-8674(92)90083-o. [DOI] [PubMed] [Google Scholar]
- Hoshino Y, Enomoto N, Sakamoto N, Kurosaki M, Ikeda T, Marumo F, Sato C. Expression of the hepatocyte growth factor receptor in the regenerating rat liver. Cancer Letters. 1993;71:119–123. doi: 10.1016/0304-3835(93)90106-j. [DOI] [PubMed] [Google Scholar]
- Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. Journal of Clinical Investigation. 1998;101:802–811. doi: 10.1172/JCI483. erratum appears in. [DOI] [PMC free article] [PubMed] [Google Scholar]; J Clin Invest. 1998 Apr 1;101(7):1541. [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nature Reviews Immunology. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentsch AB, Ward PA. Activation and regulation of NFkappaB during acute inflammation. Clinical Chemistry & Laboratory Medicine. 1999;37:205–208. doi: 10.1515/CCLM.1999.038. [DOI] [PubMed] [Google Scholar]
- Li X, Stark GR. NF[kappa]B-dependent signaling pathways. Experimental Hematology. 2002;30:285–296. doi: 10.1016/s0301-472x(02)00777-4. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK, DeFrances MC. Liver regeneration. Advances in Biochemical Engineering-Biotechnology. 2005;93:101–134. doi: 10.1007/b99968. [DOI] [PubMed] [Google Scholar]
- Moghul A, Lin L, Beedle A, Kanbour-Shakir A, DeFrances MC, Liu Y, Zarnegar R. Modulation of c-MET proto-oncogene (HGF receptor) mRNA abundance by cytokines and hormones: evidence for rapid decay of the 8 kb c-MET transcript. Oncogene. 1994;9:2045–2052. [PubMed] [Google Scholar]
- Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, Wang X, Zarnegar R, Michalopoulos GK. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Research. 2002;62:2064–2071. [PubMed] [Google Scholar]
- Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, Kim BJ, Bae JH, Hong YK, Lee KS, Lee SH, Yoo NJ, Jang JJ, Pack S, Zhuang Z, Schmidt L, Zbar B, Lee JY. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Research. 1999;59:307–310. [PubMed] [Google Scholar]
- Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nature Reviews Immunology. 2002;2:664–674. doi: 10.1038/nri887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, Liu Z, Murase N, Ezure T, Yokomuro S, Poli V, Demetris AJ. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology. 1999;29:403–411. doi: 10.1002/hep.510290244. [DOI] [PubMed] [Google Scholar]
- Sakata H, Takayama H, Sharp R, Rubin JS, Merlino G, LaRochelle WJ. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth & Differentiation. 1996;7:1513–1523. [PubMed] [Google Scholar]
- Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- Seol DW, Chen Q, Smith ML, Zarnegar R. Regulation of the c-met proto-oncogene promoter by p53. Journal of Biological Chemistry. 1999;274:3565–3572. doi: 10.1074/jbc.274.6.3565. [DOI] [PubMed] [Google Scholar]
- Seol DW, Chen Q, Zarnegar R. Transcriptional activation of the hepatocyte growth factor receptor (c-met) gene by its ligand (hepatocyte growth factor) is mediated through AP-1. Oncogene. 2000;19:1132–1137. doi: 10.1038/sj.onc.1203404. [DOI] [PubMed] [Google Scholar]
- Seol DW, Zarnegar R. Structural and functional characterization of the mouse c-met proto-oncogene (hepatocyte growth factor receptor) promoter. Biochimica et Biophysica Acta. 1998;1395:252–258. doi: 10.1016/s0167-4781(97)00202-9. [DOI] [PubMed] [Google Scholar]
- Stein B, Cogswell PC, Baldwin AS., Jr Functional and physical associations between NF-kappa B and C/EBP family members: a Rel domain-bZIP interaction. Mol Cell Biol. 1993;13:3964–3974. doi: 10.1128/mcb.13.7.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Hayashi N, Yamada Y, Yoshihara H, Miyamoto Y, Ito Y, Ito T, Katayama K, Sasaki Y, Ito A. Expression of the c-met protooncogene in human hepatocellular carcinoma. Hepatology. 1994;20:1231–1236. [PubMed] [Google Scholar]
- Tavian D, De Petro G, Benetti A, Portolani N, Giulini SM, Barlati S. u-PA and c-MET mRNA expression is co-ordinately enhanced while hepatocyte growth factor mRNA is down-regulated in human hepatocellular carcinoma. International Journal of Cancer. 2000;87:644–649. [PubMed] [Google Scholar]
- Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nature Reviews Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, Michalopoulos GK, Zarnegar R. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Molecular Cell. 2002;9:411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Webber EM, Kirillova I, Peschon JJ, Fausto N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatology. 1998;28:959–970. doi: 10.1002/hep.510280410. comment. [DOI] [PubMed] [Google Scholar]
- Zou C, Ma J, Wang X, Guo L, Zhu Z, Stoops J, Eaker AE, Johnson CJ, Strom S, Michalopoulos GK, DeFrances MC, Zarnegar R. Lack of Fas antagonism by Met in human fatty liver disease. Nat Med. 2007;13:1078–1085. doi: 10.1038/nm1625. [DOI] [PubMed] [Google Scholar]