

Abstract
This paper describes a technique for rapidly exchanging the solution environment near a surface by displacing laminar flow fluid streams using sudden changes in applied pressure. The method employs off-chip solenoid valves to induce pressure changes, which is important in keeping the microfluidic design simple and the operation of the system robust. The performance of this technique is characterized using simulation and validated with experiments. This technique adds to the microfluidic tool box that is currently available for manipulating the solution environment around biological particles and molecules.
Introduction
Probing adherent neuronal cells with a stimulus, such as an exogenous neurotransmitter or drug, is a well-established neurobiological technique.1 Such an experiment typically takes the form of applying external pressure to a capillary, ejecting a volume of stimulant at the end of the capillary, which is positioned in the vicinity of the target cells. Improvements to this single-capillary “puffing” technique have included ejection systems with multiple outlets with independently controlled pressures to facilitate simultaneous experiments with different stimulants.2
Further advancements to these capillary-based methods were made possible with microfluidics and microfabrication. For non-adherent cells, one very successful approach has been to attach the cell on a micro-manipulator so the cell can be translated across the different fluid environments provided by a series of laminar fluid streams that are flowing side-by-side.3 In this format, each fluid stream essentially forms a distinct virtual container each with a different contained solution or dissolved molecules. The cell can be moved rapidly between the fluid streams and the switching time between each stream can be very short, on the order of a few to tens of milliseconds depending on the velocity by which the cell is being moved and the flow rate of the fluid streams. Besides using a micro-manipulator to move the cell, other micro-manipulation techniques have also been used, such as optical trapping.4 In particular, optical trapping has been used to shift a cell between two environments.5
In addition to translating a cell across laminar flow streams, other microfluidic approaches have been demonstrated to exchange solution around cells.6,7 Using a microfluidic weir, Li et al.6 trapped cells within a flow stream to exchange its environment. In another example, Cheong et al.8 created a system in which a number of cells could be incubated with a dye or other exogenous agent for a predetermined time, then the cell media could be completely exchanged to other experimental conditions. This microfluidic technique has advantages over washing within a well plate, particularly for observing rapid responses.9 For highly localized (within hundreds of nanometres) release of stimulant and activation of cells, two-photon uncaging of caged compounds10 or nanocapsules11,12 have proven to be highly effective. While it would not be possible with the present technique to selectively stimulate one cell or an island of cells, we can stimulate a subset of cells (i.e. a strip along the length of the channel).
The above methods, while highly successful for their respective applications, may not be appropriate in other situations. For example, translating a cell across laminar flow streams cannot be employed for adherent cells, and the use of integrated valves and chambers increases both the complexity of the device and the switching time. Here, we describe and characterize a simple method for switching solutions around adherent objects. Rather than translating a cell in solution across laminar flows, we used rapid pressure changes to displace streams of laminar flows across objects immobilized on the surface. Our method uses a simple microfluidic design and external solenoid valves to control flow.13
While our approach is simple to implement and robust in operation, it did require special design considerations and performance characterizations. For example, the use of off-chip valving has issues of backflow, because unlike on-chip valves, it is not possible to stop flow at a particular location on the device. Additionally, the parabolic flow profile that is characteristic of pressure-driven flow complicates the kinetics of laminar flow switching. Diffusional broadening is more severe near the surface than in the middle of the channel because the slower flow near the surface allows for more time for diffusion. Furthermore, there is a delay between the arrival of the laminar boundary in the middle of the channel versus near the surface. In the situation of a static laminar boundary, the slower flow near surfaces (e.g. the floor and roof of the microchannel) leads to greater diffusional broadening and an hourglass-like shape (not shown). In contrast, this dynamic shift of the laminar boundary happens more quickly near the center of the channel and thus leads to a bulged profile (see below). As a result, the switching time is dependent on a complex relationship between applied pressure and diffusion.14,15 Here, we characterize the effects of the fluid dynamics on the switching time of this system using both simulation and experiment.
Results and discussion
Design of the apparatus
We used inexpensive, off-the-shelf components to construct a computer controlled solenoid valve manifold. The manifold passes pressure to polyethylene tubes and from there into integrated fluid reservoirs on the chip. Fig. 1 shows the design of our microfluidic chip, which mitigates backflow. This design used a 2-layer SU-8 on a silicon master, and consisted of small, shallow channels as flow restrictions (55 μm wide × 37 μm deep) placed between the inlet channels (200 μm wide × 125 μm deep) and the outlet channels (600 μm wide × 125 μm deep). We chose a 2-layer design for the flow restrictor due to ease of micro-fabrication, because very high aspect ratio structures can be difficult to fabricate reliably in SU-8. If an experiment can tolerate additional back flow, or if compensatory flow resistance is added by increasing the length of the inlet channels or severely restricting the width of the restriction, other designs could be used. Pressure applied at the reservoir pushes fluid through the flow restriction. Past the restriction, it can flow out through the tall, wide outlet channel or flow up into another inlet channel through another restriction. Without on-chip valves, some flow towards other inlets is unavoidable. The restrictions we used create a condition where forward flow towards the outlet is strongly preferred. Using fluorescent particle tracking through a digital image stack, we found that our design incorporating the restrictions cuts the backflow through any of the inlets to less than 2% of the influx through the pressurized inlet, thus suggesting that the restricted paths have a flow resistance approximately 40× that of the main outlet. Axial (i.e. along the channel) diffusion takes a long time to reach the flow restriction, if at all, because of the continuous steady-state flow towards the exit. If axial diffusion is an issue, it can be overcome by periodically pushing buffer through one inlet; the back-flow is sufficient to restore clear buffer to all inlets. Of course, care must be taken not to maintain clear buffer flow so long that it dilutes alternate inlets.
Fig. 1.
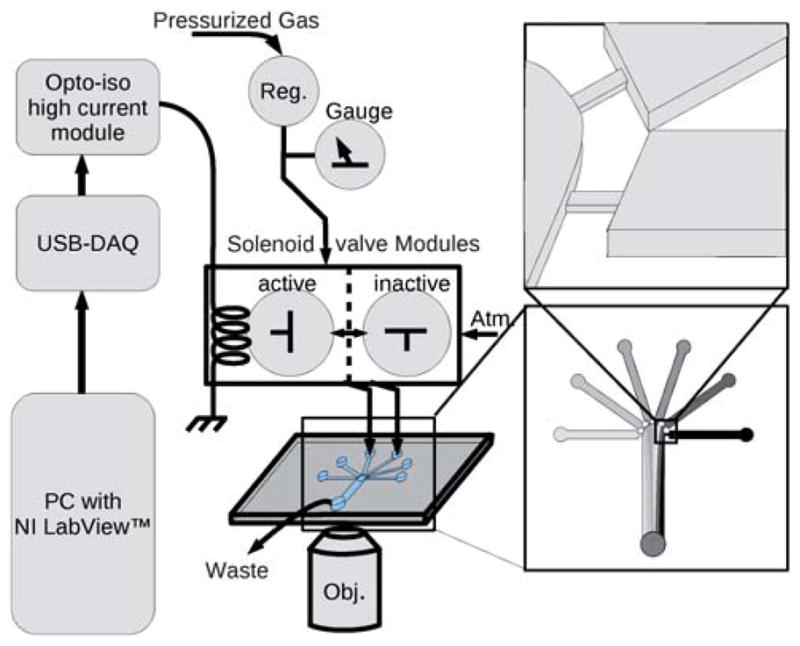
A schematic of the apparatus shows the computer (PC) at left connected to the solenoid valves through the USB-6008 DAQ device and the high power optical isolator board (Opto-iso high current module). The solenoid gas valves are 3-way normally closed type. Without current (shown as ‘inactive’) the valve is open to atmospheric pressure (Atm.). With applied current (‘active’) the valve connects the input pressure (Pressurized Gas) to the fluid in the on-chip reservoir. The input pressure is controlled with a micro regulator (Reg.). The chip is shown with expanded diagrams at right to show the narrow and shallow flow restrictions, which reduce backflow to unpressurized channels.
Fig. 2A shows a top-down view of the displacement of the laminar flow boundary. What cannot be seen in the top-down view is that the arrival of the stimulant at the surface is driven at least in part by diffusion: the no-slip boundary condition means that the surface had to wait longer for flow to deliver solute. As a consequence, the high concentration of solute delivered quickly to the center of the channel diffuses radially outward to the surface.
Fig. 2.

(A) From top to bottom, a micrograph shows the 8 inlets and the wide outlet, followed by fluorescence micrographs of green fluorescent beads (100 nm diameter) flowing into the wide outlet channel from the far left inlet channel. The flow exits at bottom, but the fluorescent front can be seen to move from left to right as laminar flow is established between flow from the leftmost and rightmost channels, illustrating the lateral front movement that would move dissolved stimulant across the surface. (B) Simulations of the fluid flow reveal the disparity in arrival time between fluid in the bulk and that near the surface. Although the dissolved solutes from the left (red shades) seem to have arrived at a location within the channel as seen from above, the cross section view at a position halfway down the channel reveals that it takes additional time for the solutes to actually reach the surface. (C) A schematic and micrographs show laminar flow along the left side of a microchannel as visualized by looking down the axis of the channel; in the micrographs on the right, a suspension of green fluorescent beads (20 nm diameter) can be seen to arrive in the center of the channel first and then spread toward the left side of the channel, qualitatively agreeing with the simulation shown in (B).
Simulation and observation of the cross-section of flow during laminar flow switching
To illustrate the overall flow pattern that we will be exploring in the context of surface exposure to dissolved solutes we simulated using COMSOL the initiation of a flow down one side of a microchannel. We used simplified geometry for the simulation, but the essential down-stream dynamic is the same. From a top-down perspective, the solute flow seems to have reached a downstream region in the main channel (see Fig. 2B, image 2), but in fact the surface at that location has not yet been exposed to significant concentrations of dissolved solute. Only later do the surface flow and diffusion actually bring the stimulant to the surface. The cross-section through Z is shown on the right; it takes until frame 4 for significant concentrations of solutes to reach the surface.
Visualization of the lateral motion of the dye front (as opposed to the downstream motion of the dye itself) is rather difficult in experiment. Ideally, it would be visualized by looking “down the barrel” of the channel. We approximated this view with experiment to qualitatively confirm the simulation. We made a microchip to similar specifications as the one in the simulation and initiated a flow from one side. We placed the chip on its side and imaged through the side of the chip using an inverted microscope with a 20× objective. The roughness of the PDMS channel side-walls necessitated using a fluid that was matched in refractive index to PDMS. We made one inlet fluorescent by adding fluorescent microspheres to observe the two flows. We imaged at a 90° bend in the channel; the flow around the bend blurs the images somewhat. The results can be seen in the insets in Fig. 2C. Qualitatively, the fluorescent fluid is brightest near the center of the channel first, then it spreads to the sides and bottom, as predicted by the simulation. A haze can be seen due to imaging at a corner. When designing future experiments, it would be wise to avoid stimulating surfaces near any corners, which might introduce unintended fluid behaviours (e.g. local eddies). We suspect that in this case, however, the haze is caused by the fluorescence originating in front of the focal plane: in order to image the area of interest, we had to focus through a space that was subsequently filled with analyte.
Using TIRF and epi-fluorescence to quantify the difference in arrival times between flow near the surface and in bulk
In the following section, we explore the relationship of flow profile to surface concentration. The understanding of this relationship is important in designing any system that will be used to perfuse rapidly surfaces with dissolved solutes. To characterize the delay between the bulk and surface arrival of the dissolved solutes during laminar flow switching, we used TIRF (total-internal-reflection fluorescence) and epi-fluorescence to measure the switching times near the surface and in bulk. Epi-fluorescence has a relatively deep depth of field of several microns, whereas TIRF excites only the bottom ~100 nm near the surface. This fact allows us to image simultaneously both the nanometre-thin layer of fluid near the surface and the micrometre-thick layer above the surface. Fig. 3A and 3B schematically depicts our simultaneous two-color TIRF (at 488 nm) and epi-fluorescence (at 633 nm) experiment.
Fig. 3.

(A) Schematic showing optical strategy for two-color visualization of flow switching near the surface and in bulk. Red, 633 nm laser light is aligned for epi-fluorescence, while blue, 488 nm light is aligned for TIRF (total-internal-reflection fluorescence). (B) Diagram showing the progression of the dye front as seen in plan view and in cross-section with red fluorescence originating throughout the channel and green fluorescence originating only at the surface. (C) Graph showing the position of the dye front at the surface (green) and averaged across the focal region (red). The red, which represents the bulk, proceeds across the channel faster than the green, which represents the surface. Error bars represent the standard error among the data taken at several locations along the channel.
For this two-color experiment, we used the following two dyes: fluorescein for 488 nm excitation using TIRF and DDAO for 633 nm excitation using epi-fluorescence. Thus, the green fluorescence channel from fluorescein reports the arrival of fluids within an ~100 nm thick layer near the surface and the red fluorescence from DDAO reports the arrival of fluid that is a few micrometres above the surface. We use this micrometre-thick layer of fluid above the surface to estimate and represent the behaviour of the bulk fluid.
Fig. 3C shows the experimental result, where the front positions of the surface (monitored with TIRF) and bulk (monitored with epi-fluorescence) fluid flows are plotted as a function of time. The front positions are defined as the 50% maximal intensity of the red and green fluorescence, respectively, and were evaluated for each frame of a video sequence taken as the flow is displaced from left to right (see Fig. 3B) after the pressures applied to the inlet reservoirs have been changed. Therefore, the front positions represent the lateral positions of the laminar flow boundary as the boundary is swept from the left to the right of the channel. From the plot, we estimated that the dye front (laminar boundary) micrometres above the glass surface (monitored by epi-fluorescence) proceeds by as much as 100 ms ahead of the surface dye front (monitored by TIRF). This implies that, under some conditions, the bulk flow could be switched back and forth above a surface without any significant exposure of the surface to the dissolved solutes.
Characterization of front velocity versus channel depth and pressure
The simultaneous two-color TIRF and epi-fluorescence experiment described above (Fig. 3) allowed us to monitor only up to a few micrometres from the surface. In order to understand the fluidic behaviour and predict limitations for surface exposure at different flow rates, we felt we needed to explore deeper into the channel. The difference in the switching time of the laminar flow boundary between the flow near the surface and that near the middle of the channel was even greater. To measure more quantitatively the rate at which this front moves across the channel at different depths, we used line-confocal detection.16 Here, the 488 nm laser was focused to a line, and the fluorescence from this line was passed through a slit placed at the image plane and then focused onto a single-photon detector. As the dye front moves across the channel, it occupies progressively more of the line, and the fluorescence intensity increases. We measured the slope of the linear region of this fluorescence increase to determine the velocity at which the dye front propagates across the channel. We focused the line at different depths in the channel and used different pressure differentials to move the dye front.
Closer to the middle of the channel, the dye front moves faster across the channel than near the surface, as would be expected from a parabolic, pressure driven flow profile. Fig. 4A shows this effect, where the transit velocity of the dye front is plotted against the channel depth for a pressure differential between the two inlet reservoirs of 1 PSI. At this low pressure differential of 1 PSI, the transit velocity is 63 μm s−1 at the surface and 111 μm s−1 deep within the channel. A full duty cycle at the surface for one field-of-view of ~50 μm is approximately 1.6 s at 1 PSI.
Fig. 4.
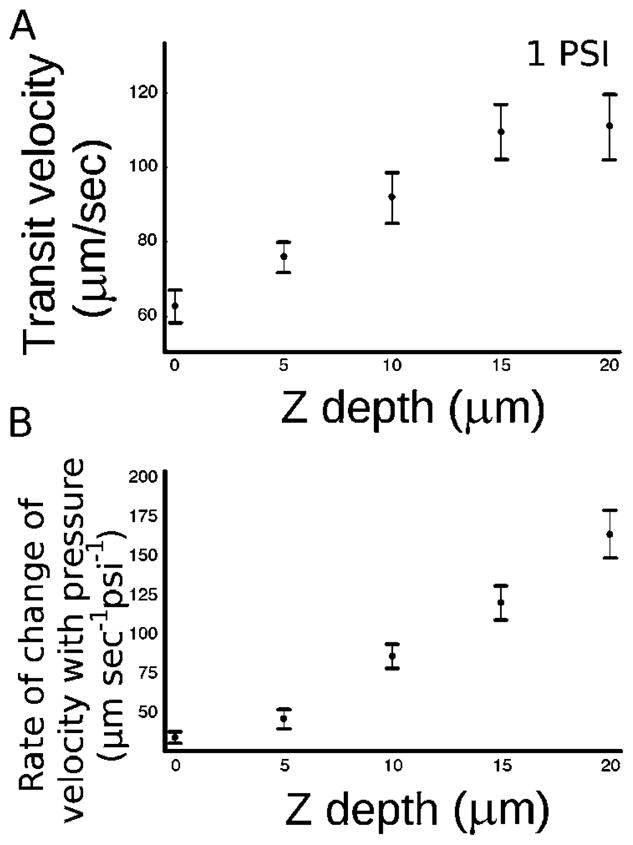
(A) A plot of the transit velocity of the dye front versus Z depth of the channel during laminar flow switching. The surface of the coverslip (floor of the channel) is at 0 Z depth. 1 PSI is the pressure differential between the inlets. (B) A plot of the rate of velocity increase with increasing pressure versus Z depth of the channel. This plot shows that most of the effect on velocity increase by increasing the applied pressure is seen in the center of the channel rather than near the surface. Error bars represent the standard error of the measurements.
More significantly, Fig. 4B shows the response to an increased pressure differential is greater at locations closer to the center of the channel. Fig. 4B plots the slope of the transit velocity with respect to pressure (ΔV/ΔP) against the channel depth (Z). From this experiment, we see that if the pressure differential between the inlet reservoirs is increased in order to cause faster movement of the dye front (i.e. the laminar flow boundary), the lateral front velocity increase is seen mostly at the center of the channel and not at the surface. This fact has important implications for surface stimulation. For example, extrapolating from our data, at 10 PSI pressure differential, our measurements indicate a limitation of 260 ms (rather than the 160 ms duty cycle that might be predicted as a result of a ten-fold higher pressure) to sweep the laminar boundary across a 50 μm field-of-view near the surface. Thus, to reach 2000 μm s−1 (in order to stimulate a 50 μm field of adherent biological particles on the order of our solenoid valves’ switching speed, 50 ms) would require pressures of about 60 PSI. Visualizing the dye front at the center of the channel would be quite deceptive; it can make a complete transit of the channel in less than 50 ms at 13 PSI, but this would leave much of the surface unexposed. Our system, as built, with a maximum pressure of 25 PSI, is capable of a full duty cycle (and thus minimum stimulation time for a 50 μm field of view) of 110 ms. Of course, the minimum time for stimulation of a single cell or single organelle would be much faster because that would correspond to the time it takes for the laminar boundary to traverse the small object rather than the 50 μm field of view of our microscope. At present, the minimum time for stimulating a biological particle using our current set-up is constrained by the limit of our solenoid valves, or 50 ms. This is undoubtedly a conservative limitation as with more advanced solenoid valves, this could be significantly improved.
There are two limitations that determine how fast fluid can flow and thus the speed at which the laminar flow boundary can be switched. The first, as mentioned above, is a hardware constraint: valves and regulators are only rated to particular maximal pressures. The second limitation is imposed by Stokes drag. If flow becomes too fast, the particle of interest can no longer be maintained stably in the flow or adhered to the surface. For other situations where the solution environment around the non-adherent particle is changed by moving the particle (e.g. which is attached to a micropipette3 or held by an optical trap5) across laminar flow streams, this stokes drag imposes the limit on the fluid switching speed. Fortunately, for particles near the surface, although the laminar flow switching speed is slower compared with that in the middle of the channel, the flow velocity and stokes drag near the surface also is correspondingly less. Therefore, while the laminar flow switching speed is slower for fluid near the surface than at the channel center for a given average flow rate, it is possible to employ a greatly increased average flow rate to arrive at fast switching speeds at the surface.
Disruption of surface adsorbed biological particles
Once we developed an understanding of the constraints of this system, we applied it to change the solution around surface immobilized synaptic vesicles. To demonstrate that our system could detect immediate and dramatic effects, we applied strong base and SDS to surface-immobilized vesicles, because both conditions illicit large changes from the vesicles. For these studies, we used vesicles isolated from transgenic mice expressing synaptopHluorin, a pH sensitive green fluorescent protein (GFP) fused to the lumenal domain of the synaptic vesicle protein VAMP/synaptobrevin.17 We suspect that the distribution of fluorescence intensity we observed is due to differential percentages of native VAMP and VAMP-pHluorin construct.
In this experiment, synaptopHluorin vesicles were loaded into the chip through the waste well and allowed to settle and adhere onto the glass surface, after which buffer was flushed through all input reservoirs. One inlet reservoir was then exchanged with the SDS/NaOH solution. The laminar boundary was then established and video acquisition was started with the camera focused on the buffer side of the laminar boundary. Pressure to the buffer well was then released by opening the valve to atmosphere, causing the SDS/NaOH front to propagate rapidly across the channel and across the field of view. Fig. 5 shows the results. The vesicles brighten briefly as their internal pHluorin molecules are exposed to the strong base (pHluorin is pH sensitive and becomes brighter in a basic solution), then the vesicles dim immediately as they are disrupted by the SDS detergent and/or removed from the surface. This all occurs within one frame such that the initial burst of fluorescence overwhelms the average intensity of that frame, and the vesicle is likely disrupted before the frame is completed. When we attempted to change solutions around vesicles in bulk, the pH induced intensity shift seems to be limited by diffusion. If a pH change were induced in a different manner (e.g. in a well-plate, without stirring) the change induced in the vesicle could be deceptively slow as the reagent takes time to reach the vesicle. This experiment demonstrates that the solution near surface can be switched with hundreds of millisecond time resolution, which should be sufficient for many biological studies. Synaptic vesicles are on the order of 50 nm in diameter. Thus, the flow sampled by the synaptic vesicles represents the flow near the no-slip boundary condition of the walls, and therefore shows the most stringent conditions for fast switching.
Fig. 5.

SynaptopHluorin vesicles (synaptic vesicles labeled with a pH sensitive green fluorescent protein) settled on the surface of a microchannel. At time 0 in the graph, we shifted a laminar boundary across the adsorbed vesicles. The vesicles were rapidly exchanged into a solution of SDS and NaOH. The intensity trace from the vesicles is shown on the left, and representative frames from the image sequence are shown on the right. As the pHluorin molecules come into contact with the alkaline solution they de-quench and brighten, followed shortly by the vesicle being disrupted and removed, resulting in a loss of fluorescence.
Materials and Methods
Fabrication of the microfluidic chip and assembly of solenoid valves
We assembled the valve manifold using eight 3-way solenoid valves (Parker Hannifin Corp. Pneutronic brand, Cleveland, OH). These valves require 200 mA of current to operate, which is more than most USB devices can provide. Therefore, we added an optical isolator board (Futurlec, New York, NY) and generic 12 volt AC adapter to provide the higher current needed. The optical isolation board was controlled using a USB-6008 data acquisition and input/output device (National Instruments, Austin, TX).
House compressed air was connected to the solenoid valves using silicone tubing through a micro-regulator (Omega, Stamford, CT). This device stepped the ~20 PSI house gas to a more precise, low pressure. We measured this final pressure with a digital gauge (Cole-Palmer, Vernon Hills, IL). The regulator and gauge allowed adjustments from 0.1 PSI to full house pressure with 0.1 PSI precision.
Fabrication of microfluidic chips by replica molding from SU-8 on silicon master
The fabrication of microfluidic chips in PDMS has been described extensively elsewhere. Briefly, SU-8 masters were fabricated on silicon substrates by exposure of SU-8-2050 photoresist (Micro-chem, Newton, MA) to UV light through a photomask and development with propylene glycol methyl ether acetate. This master was then coated with trideca-fluoro-1,1,2,2,-tetrahydrooctyl trichlorosilane (Gelest, Morrisville, PA) by gas-phase deposition and used to cast polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI) per the manufacturer’s specifications. Once cast, PDMS replicas were punched with a 3 mm cork-borer and sealed by exposure to oxygen plasma followed by immediate contact to a glass coverslip.
The reservoirs were formed by sealing a second piece of PDMS (punched with a syringe needle corresponding to the OD of the polyethylene tubing) over the larger holes created with the cork borer. This created a condition where pressure could be applied to the enclosed reservoir. We filled these chambers through the narrow hole with a fine syringe needle.
Characterization of the laminar flow switching velocity
We directed a 488 nm laser (Coherent Sapphire, Santa Clara, CA) through a series of lenses, including a pair of cylindrical lenses, which produced a line focused in one dimension within the focal plane. A custom polychroic (Chroma filters, Rockingham, VT) was used to direct the 488 nm light into the back aperture of the objective. The fluorescence from this focal line was filtered with a bandpass filter (Chroma, Rockingham, VT) and aligned to a slit (400 μm × 50 μm) at the primary image plane of the objective lens. An aspheric lens was then used to collect this light and direct it onto the active area of an avalanche photodiode-based single photon counting module (Pacer components, North Palm Beach, FL). Data was collected using a MCS card (National Instruments, Austin, TX).
We established a laminar flow boundary by applying pressure simultaneously to two wells using the computer controlled solenoid valve manifold described above. One well contained deionized water, while the other contained a pH 8 buffered solution of 100 μM carboxyfluorescein (Sigma, St. Louis, MO). We positioned the illuminated line just outside of the fluorescein on the far side of the laminar boundary. The long axis of the illumination line was placed perpendicular to the fluid flow. We initiated data collection and then turned off the pressure to the clear buffer well. This caused the laminar boundary to move across the channel, and the fluorescence count to increase nearly linearly as the dye front enters and fills the illumination line. We chose a linear region of this fluorescence increase and correlated it to the velocity of the dye front.
We repeated this procedure for 5 depths within the channel (0, 5, 10, 15 and 20 μm). The channel was molded from a master with a height of 140–150 μm. We then repeated this entire procedure with 5 acquisitions at different depths for 3 different applied pressures (1, 2 and 4 PSI).
Side imaging of channel cross-section during flow
We made a master as above with a simple Y channel, but with a right angle bend in the outlet channel. We made a 42% sucrose solution with no dyes and a second solution spiked with 30 nm diameter green fluorescent beads (Polysciences, Warrington, PA). We used a 42% sucrose solution for two reasons. First, it is close to the index of refraction of PDMS so wall defects are optically hidden, which makes it easier to image through the side wall of the right angle bend. Second, diffusion is slowed in this high-sucrose solution so that the laminar flow boundary remained sharp along the length of the channel. The use of particles slows diffusion further, and they were chosen for this reason, in order to catch the dynamics of interest for demonstration purposes.
The sample reservoir with no beads was pressurized first, then the camera and image acquisition was initiated, and finally the reservoir with the fluorescent beads was pressurized. This sequence sent a wave of fluorescent sample down the channel as shown in the simulation in Fig. 2B. The images corresponding to the stages of fluorescence arrival are shown as insets in the sequence of cross-section images.
Simultaneous imaging of fluorescence from bulk solution using epi-illumination and surface using TIRF imaging
We carried out simultaneous two-color TIRF imaging in green (488 nm excitation) and epi-fluorescence imaging in red (633 nm excitation). To achieve TIRF, we directed the output of a 488 nm laser off axis into the side of the back aperture of a 60× objective lens (NA= 1.45); the reflection from the glass–water interface produces an evanescent wave field on the glass surface to excite fluorescence. To achieve epi-illumination, we focused the output of a 633 nm laser onto the back aperture of the same 60× objective lens to produce a wide-field homogeneous illumination in the object plane. The fluorescence in both colors was filtered with a dual-color bandpass filter (Semrock, Rochester, NY) and directed onto a color CCD camera (Cohu, Poway, CA).
We established a laminar flow boundary as described above. We positioned the boundary just outside the illuminated area. We then released pressure applied the buffer-only reservoir and observed the dye front move across the visible area. The video sequence was digitized and processed with in-house developed software written in Mathematica. The flow was directed along the y-axis of the video sequence, and the front moved laterally across the x-axis. The average x-axis distance at which the intensity was 50% of the maximum was measured for each frame and then this value was plotted against the frame number for the video sequence.
Disruption of surface adsorbed biological particles
To test our ability to observe the effects of a chemical on biological structures, we “stimulated” synaptic vesicles on the surface of the glass with an aggressive mixture of SDS and sodium hydroxide. We isolated synaptic vesicles from the brains of transgenic mice. These mice were engineered to express synaptopHluorin, a modified synaptic vesicle integral membrane protein (VAMP) that has a pH sensitive GFP molecule attached to its terminus within the synaptic vesicle lumen.17 The strain was developed in the Murthy laboratory17 and samples were kindly provided by the Bajjalieh laboratory. The mouse brains were dissected and immediately frozen at −80 °C for later use in the isolation and purification of synaptopHluorin vesicles.
The vesicle preparation was adapted from protocols described elsewhere,18 but briefly, frozen brain tissue was reduced to fine particles in a Waring blender then homogenized using a rotary homogenizer in a HEPES buffered, 0.3 M sucrose homogenization buffer. The resulting homogenate was then centrifuged at 47 000 g to pellet cell debris. The supernatant was then centrifuged at 137 000 g on a sucrose step gradient where the vesicles settled on a cushion of HEPES buffered 1.5 M sucrose, from where the vesicles were pipetted out and frozen at −80 °C until used.
To start the experiment, the purified vesicle solution was diluted by 100 times into a HEPES buffered 0.15 M potassium chloride solution, which was then introduced into the channel system through the waste well. Buffer was then added to all of the inlets wells except one, which was exchanged with 0.1 % SDS and 100 mM sodium hydroxide. The vesicles were observed in 488 nm illumination TIRF as described above. The computer controlled solenoid valve manifold was programmed to apply pressure to a buffer well and the alkaline/surfactant well simultaneously and a laminar boundary was established. The location could be seen with the TIRF microscope as a line past which all vesicles were absent.
We focused the microscope just to the far side of this boundary, then initiated detection and the computer controlled stimulus. At a pre-determined time, the computer actuated the valve to release pressure from the buffer inlet (by opening it to atmospheric pressure) and initiated the sweep of the laminar boundary across the channel. This caused the vesicles to brighten briefly as the alkaline pH fully de-quenched the pH sensitive fluorophores, then go dark to background levels as the vesicle was solubilized by SDS.
Conclusions
The ability to switch rapidly the solution around surface adhered molecules/particles will find broad utility. Although it is possible to change solutions in microchannels using integrated valves and pumps, these devices tend to be complex to fabricate and operate. The technique presented here utilizes a simple channel that is easy to fabricate, and the fluid switching speed achieved is faster than can be attained using integrated valves and pumps. There are two key considerations in implementing this laminar flow switching technique: (1) the need to use flow restrictions to prevent back flow from one inlet into another, and (2) the need to account for the slower fluid movement near the surface than in bulk, owing to the presence of the parabolic flow profile characteristic of pressure-driven flow. This paper describes these two design considerations, and has demonstrated a laminar flow-boundary switching velocity at the surface of 63 μm s−1 at a low pressure differential of 1 PSI and 171 μm s−1 at 4 PSI. For a 10 μm diameter adherent cell and at our maximal pressure of 25 PSI differential (limited by the regulators and solenoid valves we currently use), this would correspond to exchanging the surrounding solution within 11 ms. This switching speed may be further increased using external valves and regulators which can withstand the higher applied pressure. Because the Stokes drag experienced by a particle near the surface is much reduced as compared to bulk solution, this can be done without risk of detaching the cell due to shear stress.
Peterman et al.19 suggested that a microfluidic device for stimulating adherent neuronal cells could contribute to a chemical interface for a retinal prosthetic and developed a prototype cell stimulator. Spatially and temporally controlled chemical stimulation are critical for such an application. Characterizing the environment at the surface has direct implications for the design of such experiments. A device similar to that shown here may be useful to optimize the proper conditions for future clinical applications. Careful consideration must be given to the conditions near the cells when stimulation (and flow) are fast. The surface may experience significantly lower concentrations than would be suggested by the average concentrations across the depth of the channel.
Demonstrating the disruption of surface adsorbed vesicles serves to illustrate the technique’s ability to generically apply a stimulant to surface-bound particles. As examples of possible future applications, we suggest that this could be integrated with microfluidic patch clamps20 and used to apply neurotransmitter selectively to a stripe of neuronal cells. It could also be used to strip cells or cellular connections from a stripe of a channel in order to observe their re-growth for the study of contact inhibition.
Acknowledgments
The authors gratefully acknowledge the Murthy laboratory from which the synaptopHluorin mouse strain originated and the Bajjalieh laboratory in which the mice were raised. This work was supported by NIH (NS 052637 & 062725).
Notes and references
- 1.Palmer M, Wuerthele S, Hoffer B. Neuropharmacology. 1980;19:931–938. doi: 10.1016/0028-3908(80)90001-5. [DOI] [PubMed] [Google Scholar]
- 2.Marchand AR, Pearlstein E. J Neurosci Methods. 1995;60:99–105. doi: 10.1016/0165-0270(94)00225-6. [DOI] [PubMed] [Google Scholar]
- 3.Sinclair J, Pihl J, Olofsson J, Karlsson M, Jardemark K, Chiu DT, Orwar O. Anal Chem. 2002;74:6133–6138. doi: 10.1021/ac026133f. [DOI] [PubMed] [Google Scholar]
- 4.Shelby JP, Mutch SA, Chiu DT. Anal Chem. 2004;76:2492–2497. doi: 10.1021/ac035531v. [DOI] [PubMed] [Google Scholar]
- 5.Eriksson E, Enger J, Nordlander B, Erjavec N, Ramser K, Goksor M, Hohmann S, Nystrom T, Hanstorp D. Lab Chip. 2007;7:71–76. doi: 10.1039/b613650h. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Huang J, Tibbits GF, Li PC. Electrophoresis. 2007;28:4723–4733. doi: 10.1002/elps.200700312. [DOI] [PubMed] [Google Scholar]
- 7.Di Carlo D, Lee LP. Anal Chem. 2006;78:7918–7925. doi: 10.1021/ac069490p. [DOI] [PubMed] [Google Scholar]
- 8.Cheong R, Wang CJ, Levchenko A. Mol Cell Proteomics. 2009;8:433–442. doi: 10.1074/mcp.M800291-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warrick J, Meyvantsson I, Ju J, Beebe DJ. Lab Chip. 2007;7:316–21. doi: 10.1039/b613350a. [DOI] [PubMed] [Google Scholar]
- 10.Pettit DL, Wang SSH, Gee KR, Augustine GJ. Neuron. 1997;19:465–471. doi: 10.1016/s0896-6273(00)80361-x. [DOI] [PubMed] [Google Scholar]
- 11.Sun B, Chiu DT. J Am Chem Soc. 2003;125:3702–3703. doi: 10.1021/ja029942p. [DOI] [PubMed] [Google Scholar]
- 12.Dendramis KA, Allen PB, Reid PJ, Chiu DT. Chem Commun. 2008:4795–4797. doi: 10.1039/b806685j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen PB, Chiu DT. BBA - Mol Basis Dis. 2008;1782:326–334. doi: 10.1016/j.bbadis.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beard DA. J Appl Phys. 2001;89:4667–4669. [Google Scholar]
- 15.Kamholz AE, Yager P. Biophys J. 2001;80:155–160. doi: 10.1016/S0006-3495(01)76003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiro PG, Kuyper CL, Chiu DT. Electrophoresis. 2007;28:2430–2438. doi: 10.1002/elps.200600730. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Burrone J, Tyler WJ, Hartman KN, Albeanu DF, Murthy VN. Proc Natl Acad Sci U S A. 2005;102:6131–6136. doi: 10.1073/pnas.0501145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hell JW, Maycox PR, Stadler H, Jahn R. EMBO J. 1988;7:3023–3029. doi: 10.1002/j.1460-2075.1988.tb03166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterman MC, Bloom DM, Lee C, Bent SF, Marmor MF, Blumenkranz MS, Fishman HA. Invest Ophthalmol Visual Sci. 2003;44:3144–3149. doi: 10.1167/iovs.02-1097. [DOI] [PubMed] [Google Scholar]
- 20.Pantoja R, Nagarah JM, Starace DM, Melosh NA, Blunck R, Bezanilla F, Heath JR. Biosens Bioelectron. 2004;20:509–517. doi: 10.1016/j.bios.2004.02.020. [DOI] [PubMed] [Google Scholar]