

Abstract
CEACAM1-4S (carcinoembryonic antigen-related cell adhesion molecule 1) is a type I membrane protein with a short (12 amino acid) cytoplasmic tail. Wild type CEACAM1-4S transfected MCF7 cells form glands with lumena when grown in 3D culture, while null mutations of two putative phosphorylation sites (T457A and S459A) in the cytoplasmic domain fail to undergo lumen formation. When gene chip analysis was performed on mRNA isolated from both wild type and T457A,S459A mutated CEACAM1-4S transfected MCF7 cells grown in 3D culture, calpain-9 (CAPN9) was identified out of over 400 genes with a >2 log 2 difference as a potential inducer of lumen formation. Inhibition of CAPN9 expression in MCF7/CEACAM1-4S cells by RNAi or by calpeptin or PD150606 inhibited lumen formation. Transfection of CAPN9 into wild type MCF7 cells restores lumen formation demonstrating that calpain-9 may plays a critical role in lumen formation. Additionally, we demonstrate that the apoptosis related kinase, PKC-δ is activated by proteolytic cleavage during lumen formation exclusively in wild type CEACAM1-4S transfected MCF7 cells grown in 3D culture, and that lumen formation is inhibited by either RNAi to PKC-δ or by the PKC-δ inhibitor rottlerin.
Keywords: CEACAM1, breast cancer, lumen formation, calpain, PKC-delta
Introduction
Epithelial cell polarization is a hallmark of glandular morphogenesis. In the case of mammary morphogenesis, many of the steps in this process can be observed in an in vitro model using mammary epithelial cells embedded in Matrigel, a convenient source of extracellular matrix (ECM). The interactions between these cells and the specific requirements for intergin expression that define their interaction with ECM have been extensively studied and reviewed [1-5]. In addition, cell-cell adhesion must play a role in the formation of acini, which will then undergo a polarization step that includes the formation of a lumen. Lumen formation is achieved by the induction of apoptosis in the central cells of the acini, leaving a single layer of polarized epithelia cells surrounding the newly formed space or lumen [6, 7].
CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule 1) is a type I transmembrane glycoprotein involved in cell-cell adhesion [8, 9] and highly expressed on the epithelia of mammary glands [10]. Through extensive alternative splicing, CEACAM1 has 11 different splice variants with isoforms consisting of 1-4 extracellular Ig-like domains, a transmembrane domain and either a long (L) or a short (S) cytoplasmic domain [11]. The short isoforms comprise 12 amino acids in the cytoplasmic tail, of which several residues have been shown to play critical roles in the ability of this isoform to induce lumen formation of MCF7 cells grown in a 3D model of mammary morphogenesis [7, 12]. In this regard the null mutation F454A has been shown to block the binding of CEACAM1-4S to actin and to inhibit lumen formation [12]. Furthermore, the double null mutant T457A, S459A also inhibits lumen formation [12]. Previously, these residues have been shown to be phosphorylated during lumen formation and the double mutant T457D,S459D, that mimics phosphorylation at these residues, supports lumen formation [12]. In general, these interactions define a role for the short cytoplasmic domain’s interaction with the cytoskeleton, while the ectodomains, specifically the N-terminal Ig-like domain, confer the function of CEACAM1 as a homotypic cell adhesion molecule [8, 13].
Previously, we have shown that CEACAM1 plays an essential role in reverting tumor cells to a normal lumen formation morphology in both the 3D Matrigel model and the in vivo humanized mouse fat pad model of mammary morphogenesis. First, anti-CEACAM1 antibody and CEACAM1 anti-sense blocked lumen formation of the nonmalignant human breast cell line MCF10F that expresses CEACAM1 when grown in Matrigel [6]. Second, transfection of the CEACAM1-4S isoform into MCF7 mammary carcinoma cells that do not express CEACAM1 or form lumena when grown in Matrigel, reverted these cells to a normal morphogenic program [7, 12]. We have also shown that CEACAM1-4S promoted lumen formation by mediating apoptosis of the central acinar cells that are not in contact with extracellular matrix [7, 14]. Although CEACAM1-4S transfected MCF7 cells did not form lumena in the humanized mouse mammary fat pad, the phosphorylation mimic mutations of Thr-457 (T457D) or Ser-459 (S459D) were able to induce lumen formation [14], suggesting that phosphorylation of Thr-457 or Ser-459 are essential for lumen formation.
Given these results, we reasoned that a comparison of the double null mutant T457A, S459A to the wild type gene transfected into MCF7 cells and grown in Matrigel would provide a unique opportunity to compare the mRNAs of cells that could make lumena versus those that could not, while otherwise maintaining the cell-cell adhesion properties of CEACAM1. When these comparisons were performed using the Affi U133 plus 2 gene chip, we found over 400 genes differed in expression >2 log 2. Among the genes identified, calpain-9 (CAPN9), immediately attracted our attention because there was no protein expression in the mutated CEACAM1-4S transfected cells and de novo expression in the wild type CEACAM1-4S transfected cells upon transfer to 3D culture. Furthermore, calpain had previously been identified by us as a potential regulator of apoptosis in CEACAM1-4S transfected MCF cells [7]. To further explore the possibility that a specific isoform of calpain, namely calpain-9 was involved, first we treated CEACAM1-4S transfected MCF7 cells grown in 3D culture with RNAi to CAPN9 and found 70% inhibition of lumen formation. Second, two pan-calpain inihibitors, calpectin and PD150606 are able to inhibit lumen formation by greater than 80%. Third, stable expression of CAPN9 in wild type MCF7 cells was able to retore lumen formation in the absence of CEACAM1.
We also searched for possible targets of calpain cleavage that were also involved in apoptosis. In this regard, we analyzed the cleavage of PKC-δ, a highly studied mediator of apoptosis [15-17] that is cleaved by the action of both caspases and calpain [18, 19] and has previously been shown to be associated with CEACAM1 [20]. We show here that PKC-δ is cleaved during the time course of lumen formation for CEACAM1-4S transfected MCF7 cells, and that the cleavage is blocked by the use of CAPN-9 RNAi. In addition, both PKC-δ RNAi and rottlerein, a specific inhibitor of PKC-δ, are able to inhibit lumen formation by greater than 50%. Taken together, these data provide strong evidence that calpain-9 and PKC-δ play major roles in lumen formation in the 3D culture system utilizing CEACAM1-4S transfected MCF7 cells.
Materials and Methods
Materials and cell lines
Monoclonal antibodies anti-Calpain-9 and anti-PKC-δ were from Abnova (Taipei, Taiwan); polyclonal ant-PKC-δ was from Santa Cruz Biotechnology (Santa Cruz, CA); anti-β-actin was from Abcam (Cambridge, MA). RNAi oligos (Suppl. Table 1, bottom), RNAi negative control-low GC duplex, Lipofectamine 2000 transfecting reagent and Opti-MEM® I reduced serum medium were from Invitrogen Corporation (Carlsbad, CA). Calpectin, rottlerin and PD-150606 were from Calbiochem (San Diego, CA). The MCF7 cell line was obtained from ATCC (HTB-22™) and cultured in MEM with 10% Fetal Bovine Serum. CEACAM1-4S in the pHβ-actin vector and the double null mutants (T457A, S459A), as well as the Matrigel sandwich assay, were previously described [7, 12]. Lumen formation was observed over a 1-6 day period using an inverted light microscope (phase contrast) with maximal lumen formation usually seen at 6 days. In each case two hundred acini were scored for lumen formation and statistical analysis was performed using Fisher’s exact test. In cases where RNA was required, day 4 acini were treated with 10 mL of Matrisperse for 3 hrs at 4°C, cells harvested and RNA isolated for Gene chip analysis.
Microarray analysis
Microarray analysis of RNA isolated from MCF7 cells transfected with CEACAM1-S wild-type (SW) or CEACAM1-S double-A mutant T457A,S459A (DA) grown in Matrigel for 4 days was performed by the Functional Genomics Core - Microarray Service at Beckman Research Institute (Duarte, CA) using the GeneChip® Human Genome U133 Plus 2.0 (including 47,000 gene transcripts) arrays manufactured by Affymetrix.
RT-PCR
Total RNA was extracted from cells using the Tri-Reagent (Molecular Research Center) according to the manufacturer’s instruction. From 1 μg of each total RNA, 20 μl of cDNA was synthesized with a Ommiscript® RT kit (Qiagen, Valencia, CA). PCR was performed with the iCycler Thermal Cycler (Bio-Rad) for 35 cycles at 55°C annealing temperature. The FideliTaq™ PCR Master Mix (USB, Cleveland, Ohio) was used in each experiment to ensure tube to tube consistency in PCR reactions. Primers (Suppl. Table 1, top) were designed for amplification of specific genes. A glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) control amplimer set was used to assess RNA integrity (data not shown). Reaction products (15 μl) were visualized after electrophoresis in 1.4% agarose gel containing SYBR® Safe DNA gel stain Invitrogen Corporation (Carlsbad, CA).
qPCR
mRNA expression level of ADD3, SASH1, SCIN, SDC2, and GAPDH was quantitated with primers (Suppl. Table 1, middle) using the iQ™5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). Briefly, one μl cDNA from the reverse transcription reaction with 20 pmol of each primer in a total volume of 25 μl using the Sense Mix Plus SYBR® (Quantace Inc, Norwood, MA) and the following conditions: initial denaturation step at 94°C for 3 min; followed by 35-40 cycles of 94°C for 10 sec, 55°C for 30 sec, 72°C for 40 sec. Fluorescence was measured at the end of the extension step at 72°C. Subsequently, a melting curve was recorded between 55°C to 95°C every 0.5°C with a hold every 1 second. Levels of mRNA (triplicate) were compared after correction by use of concurrent GAPDH message amplification.
RNAi treatment
Cells were transfected with RNAi oligos (Suppl. Table 1, bottom) using the Lipofectamine 2000 transfection agent (Invitrogen Corporation). Cells were split, seeded at 50% confluence in T25 flasks overnight, washed twice with 1X PBS, and treated with RNAi oligos (100nM) and transfection agent (1:100 dilution) in Opti-MEM® I reduced serum medium for 6 hours without removing the transfection solution, after which, cells were added back to MEM medium with 10% fetal bovine serum overnight, harvested and transferred to Matrigel.
Transfection of MCF7 cells with CAPN9
MCF7 cells were stably transfected with pCMV-XL5-CAPN9 (Origene, Rockville, MD) using the Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA), selected with 1000 μg/ml Gentamincin (Invitrogen Corporation), and single clones isolated by limited dilution. CAPN9 transfected MCF7 cells were grown in Matrigel as described above and scored for % lumen formation at day 6 using an inverted light microscope. Statistical analysis was performed using Fischer’s exact test.
Western blotting
Cells were lysed with lysis buffer (10 mM Tris-HCl, pH7.4, 100mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 0.05% sodium deoxycholate, 10% glycerol, 1% Triton X-100, and proteinase inhibitor cocktail (Roche Applied Science, Indianapolis, IN)) on ice for thirty minutes, and the protein concentration was determined using the Bio-Rad protein assay. Fifty micrograms of protein from each sample were resolved by SDS gel electrophoresis, Western blotted with appropriate antibodies and detected using the Odyssey® Infrared Imaging System (LI-COR Biotechnology, Lincoln, Nebraska).
Results
Gene chip analysis of wild type vs null mutant T457A,S459A CEACAM1-4S in Matrigel
Our previous studies have demonstrated that CEACAM1-4S transfected MCF7 cells were able to form glandular lumena in a 3D model of mammary morphogenesis, while the cytoplasmic domain null mutant T457A,S459A of CEACAM1-4S transfected cells were not able to form lumena [12]. In order to determine which genes were regulated by the action of CEACAM1, these two cell lines were selected for a comparative gene chip analysis. In this way, the comparative analysis is not affected by the absence of the cell adhesive domains of CEACAM1-4S, but lacks only the signal transduction capability of CEACAM1-4S in its cytoplasmic domain. mRNA was isolated from 4d acini, the time point at which nascent lumena are visible, and subjected to a comparative analysis on the Affi U133 plus 2 gene chip. Over 400 transcripts with > 2 log 2 ratio were identified (supplementary Table 2). These results suggested that the process of lumen formation, including the secretory function of these glands, involves a relatively large number of genes. The top 13 hits (~6 log 2 ratio) are shown in Table 1. Based on a preliminary examination of the top thirteen hits for their possible connection to ECM, apoptosis and lumen formation, we selected 4 genes (SDC2, SASH1, ADD3 and SCIN) for validation by Q-RT-PCR.
Table 1. Gene chip analysis of CEACAM1-4S wild type vs null mutants grown in Matrigel1.
| Symbol | Description | GenBank | SW mean intensity |
DA mean intensity |
Log2 ratio |
|---|---|---|---|---|---|
| TCN1 | transcobalamin I | NM_001062 | 12.5249 | 2.07485 | 10.45 |
| LOC283352 | hypothetical protein | BG475827 | 11.9552 | 5.10348 | 6.85 |
| CA2 | carbonic anhydrase II | M36532 | 9.47973 | 2.65061 | 6.83 |
| SDC2 | syndecan 2 | AL577322 | 9.62272 | 2.82015 | 6.80 |
| ID4 | inhibitor of DNA binding 4 |
AW157094 | 8.69142 | 1.98875 | 6.70 |
| SDC2 | syndecan 2 | AI380298 | 8.8886 | 2.42465 | 6.46 |
| HS6ST2 | heparan sulfate 6-O-sulfotransferase 2 |
AI767756 | 9.28247 | 2.8628 | 6.42 |
| SLITRK6 | SLIT and NTRK-like family, member 6 |
AL137517 | 9.21892 | 2.85992 | 6.36 |
| SASH1 | SAM and SH3 domain containing 1 |
AU144882 | 7.66824 | 1.32889 | 6.34 |
| LOC283352 | hypothetical protein LOC283352 |
BC035922 | 8.3298 | 2.19897 | 6.13 |
| ADD3 | adducin 3 (gamma) | NM_019903 | 11.5247 | 5.40862 | 6.12 |
| IMMP2L | IMP2 inner mitochondrial membrane protease-like |
AI784580 | 9.04834 | 3.03058 | 6.02 |
| SCIN | scinderin | AF276507 | 9.69706 | 3.77289 | 5.92 |
SW= wild type sequence; DA= T457A,S45A double mutant; log 2 ratio= SW/DA.
Validation was performed by isolating mRNA from acini over the time course of lumen formation (Fig 1A) and performing Q-RT-PCR. In the cases of SDC2, SASH1, and ADD3, negligible transcript levels were seen for the vector control and mutant CEACAM1-4S transfected cells, while the wild type CEACAM1 transfected cells exhibited a 4-10 fold increase in transcript expression. Even in the case of SCIN, a 4-fold increase in transcript expression was seen over the time-course of the analysis. These results suggest that the gene chip analysis reported transcript levels reliably. Further validation studies by RNAi inhibition of lumen formation are underway.
Figure 1. Quantitative PCR analysis of expression levels of SDC2, SASH1, ADD3, and SCIN in CEACAM1-4S transfected cells grown in matrigel and expression of calpain genes by PCR.

A: MCF7 cells transfected with vector only (blue), wild type (red), or double mutant T457A,S459A CEACAM1-4S (magenta) were grown in Matrigel for 0, 2, 4, or 6d, cells harvested, RNA isolated, and Q-PCR analysis performed using primers for ADD3, SASH1, SCIN, SDC2 and GAPDH. Results were normalized to GAPDH. B: MCF7 cells transfected with vector only (V), wild type (S), or double mutant T457A,S459A CEACAM1-4S (DA) were grown in Matrigel for 0, 2, 4, or 6d, cells harvested, RNA isolated, and RT-PCR analysis performed using primers for CAPN1, CAPN2, CAPN9, and CAPNS1 (M= DNA markers).
Identification of a role for calpain-9 in lumen formation
Since a role for calpain was previously deduced from our studies on the mechanism of apoptosis in lumen formation [7], we examined our gene chip results for a possible up-regulation of any of the 11 known calpain genes. Strikingly, we noted that a single calpain gene, Calpain-9 (CAPN9), was significantly up-regulated (log 2 = 2.47) compared to other calpains (log 2 <1) (Suppl. Table 3) found in our gene chip analysis (Suppl. Table 2). For comparative purposes we determined the expression of CAPN1, 2, 9 and their common subunit CAPNS1 by RT-PCR over the time course of lumen formation for vector transfected, wild type CEACAM1 transfected, and double mutant CEACAM1 transfecetd MCF7 cells (Fig 1B). As expected, CAPN1 and 2, the ubiquitously expressed isoforms of calpain and the common subunit CAPNS1 do not vary for any of the cells tested over the time period; however, CAPN9 shows little or no expression in the vector control cells at day 0 and low level expression for wild type transfected cells at day 0, with increasing amounts over time. Since mRNA expression does not always correlate with protein expression, protein expression levels of CAPN9 over time was analyzed by western blot analysis (Fig 2A). The results demonstrate low expression of CAPN9 at day 0, increasing by 4-fold to 8-fold at days 4 and 6 in Matrigel for the wild type CEACAM1 transfected cells with negligible expression in vector control and double mutant transfected cells. These data indicate that, as judged by protein levels, CAPN9 expression is directly related to lumen formation. Going back to the RT-PCR results, it appears that there is minimal expression of CAPN9 in the vector control cells, but some expression in the double mutant transfected cells. These data may indicate CAPN9 regulation at the level of both transcription and translation. Future studies to investigate the mechanism of CAPN9 regulation are planned.
Figure 2. Expression of Calpain-9 protein in MCF7 cells transfected with vector, CEACAM1-4S wild type or double mutant T457A,S459A grown in Matrigel in the presence or absence of RNAi.

A: MCF7 cells transfecetd with vector only (V), wild type (S), or double mutant T457A,S459A CEACAM1-4S (DA) were grown in Matrigel for 2, 4, or 6d, cells harvested, protein separated by SDS gel electrohoresis, and western blotted with anti-calpain-9 or anti-β-actin antibodies. B: Same as above (day 4 only), cells treated with lipofectamine only, RNAi control oligo, PKC-δ RNAi oligo 1, or CAPN9 RNAi oligo 1 and western blotted with anti-calpain-9 or anti-β-actin antibodies.
To test for a functional role for Calpain-9, we performed three types of experiments. First, the effect of calpain specific inhibitors on lumen formation was also tested. Treatment of wild type CEACAM1 transfected MCF7 cells with 10 μM calpectin or 20 μM PD150606 reduced lumen formation from 95% in DMSO treated controls to 15% or 5%, respectively (Fig 3, Table 2). Second, we compared the ability of a Calpain-9 specific RNAi to reduce protein expression levels to lipofectamine, negative control low GC siRNA and 1 other RNAi used in other experiments. As shown in Fig 2B, only the Calpain-9 specific siRNA was able to knock down the expression of Calpain-9 in wild type CEACAM1-4S transfected cells. When these cells were examined for lumen formation, lumen formation was significantly reduced in Calpain-9 specific siRNA treated cells compared to low GC siRNA treated controls (Fig 4, Table 2). Third, we stably transfected wild type MCF7 cells with CAPN-9 to determine if the expression of this gene alone could induce lumen formation in the absence of CEACAM1-4S. While MCF7 cells transfected with vector gave a background of 17% lumen formation, MCF7 cells stably transfected with CAPN-9 gave 67% lumen formation (Fig 5, Table 2). Although the lumen formation is not as high as seen for CEACAM1-4S trabsfected cells, it is remarkable that transfection with CAPN-9 alone is sufficient to give rise to lumen formation. These data further establish a functional role for calpain in lumen formation and suggest that one specific isoform, namely Calpain-9, may be a critically important gene in the lumen formation program.
Figure 3. Inhibition of lumen formation of MCF7/CEACAM1-4S cells by calpain inhibitors calpectin and PD150606.

A-B: MCF7 cells transfected with wild type CEACAM1 were treated with 0.05% DMSO for 6d in Matrigel and examined at 50x (A) or 400x (B) by phase contrast microscopy. After examination of 200 acini, the percent lumen formation was 95% (Table 2), essentially identical to untreated controls (data not shown). C-D: Same as above treated with 10 μM calpectin in 0.05% DMSO and examined at 50x (C) or 400x (D) by phase contrast microscopy (15% lumen formation, Table 2). E-F: same as above treated with 20 μM PD150606 in 0.05% DMSO and examined at 50X (E) or 400X (F) by phase contrast microscopy (5% lumen formation, Table 2).
Table 2. Lumen formation for transfected MCF7 cells treated with RNAi oligos or small molecule inhibitors1.
| Cells | Treatment | Percent Lumen |
|---|---|---|
| MCF7/CEACAM1-4S | None | 93 |
| MCF7/CEACAM1-4S | Lipofectamine | 81 |
| MCF7/CEACAM1-4S | Low GC RNAi | 67* |
| MCF7/CEACAM1-4S | CAPN-9 RNAi oligo 1 | 30*** |
| MCF7/CEACAM1-4S | CAPN-9 RNAi oligo 2 | 27*** |
|
| ||
| MCF7/CEACAM1-4S | 0.05% DMSO | 95 |
| MCF7/CEACAM1-4S | Calpeptin (10 μM) | 15*** |
| MCF7/CEACAM1-4S | PD150606 (20 μM) | 5*** |
|
| ||
| MCF7 | None | 17 |
| MCF7/CAPN9 | ------- | 67*** |
|
| ||
| MCF7/CEACAM1-4S | PKC-δ oligo 1 | 45*** |
| MCF7/CEACAM1-4S | PKC-δ oligo 2 | 33*** |
| MCF7/CEACAM1-4S | PKC-δ oligo 3 | 17*** |
| MCF7/CEACAM1-4S | Rottlerin (100 nM) | 45*** |
Lumen formation was scored as described in Methods. For the experiments with MCF7/CEACAM1-4S cells this occurred at day 4, while for MCF7 cells that over-expressed CAPN9, this occurred at day 6. Two hundred acini counted for each experiment
p<0.035
p<0.0001
compared to No treatment or to treatment with 0.05% DMSO.
Figure 4. Inhibition of lumen formation of MCF7/CEACAM1-4S cells by RNAi to calpain-9.

A: MCF7 cells transfected with wild type CEACAM1-4S treated with RNAi control oligo in Matrigel for 6d and examined by phase contrast microscopy (50x). After examination of 200 acini, lumen formation was 67% compared to 81% for lipofectamine treated controls or 93% for untreated controls (Table 2). B: Same as above treated with RNAi oligo 1 for CAPN9 (30% lumen formation, Table 2).
Figure 5. Lumen formation of MCF7 cells transfected with CAPN-9.
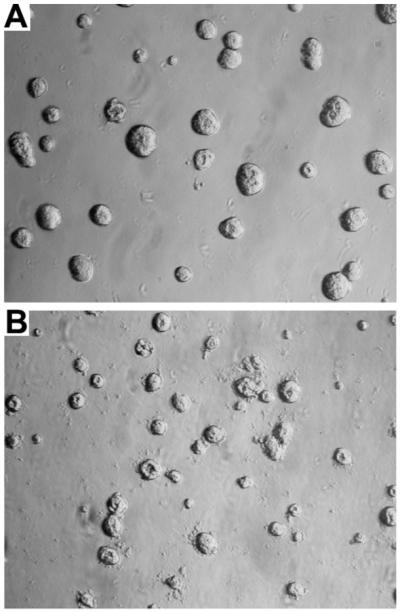
MCF7 cells stably transfected with CAPN-9 were grown in Matrigel for 6 days and scored for lumen formation. A: Wild type cells have a background of 17% lumen formation (Table 2). B: CAPN-9 transfected cells have 67% lumen formation (Table 2). Magnification 50X.
Identification of a role for PKC-δ in lumen formation
Since we have already shown that phosphorylation of Thr-457 and/or Ser-459 plays a critical role in the function of CEACAM1-4S in lumen formation [12] and it has been suggested that PKC isozymes can phosphorylate Thr/Ser residues in murine CEACAM1 [21], we examined the gene chip results for possible clues to the identification of the kinase involved. Two PKC isozymes exhibited modest upregulation in this gene screen, PKC-ε (log 2= 1.87) and PKC-δ (log 2= 1.24) (Suppl. Table 3). Since it is well known that endogenous kinases may be activated and/or relocalized by other cellular processes to access their intended targets, a modest or even no degree of up-regulation may not reveal their potential roles in lumen formation. In order to narrow down the potential leads, we surveyed the literature for possible connections among PKC-ε and/or PKC-δ and CEACAM1 and/or any of the other top hits in our gene screen list. In one recent publication PKC-δ was linked to CEACAM1 in a study on anti-CD98 stimulated F9 embryonic carcinoma cells [22]. This study was of interest because these cells underwent CEACAM1 mediated aggregation, along with nuclear translocation of PKC-δ when treated with anti-CD98, and aggregation was blocked by the PKC-δ inhibitor, rottlerin. In a study on osteoblasts, PKC-δwas linked to both Syndecan-2 and apoptosis [23]. Since Syndecan-2 was a major hit in our study and apoptosis is one of the critical steps in lumen formation, we decided to further investigate PKC-δ. Confocal analysis of PKC-δ using anti-PKC-δ antibodies revealed no significant changes in its levels or cellular localization between wild type or mutant transfected MCF7 cells grown in Matrigel (data not shown). However, when western blot analysis using anti-PKC-δ antibodies was performed on these cells, a significant amount of the cleaved form of PKC-δ was found in the wild type cells vs the vector control and mutant cells at day 4 (Fig 6A). This is of interest because the cleaved form of PKC-δ is known to be associated with cells undergoing apoptosis [15, 24]. Based on these data, we tested the ability of 3 different RNAi oligos to inhibit PKC-δ expression in the vector control, wild type, and mutant transfected MCF7 cells grown in 3D culture. One of the three RNAi oligos (PKC-δ #3) gave greater than a 90% reduction of PKC-δ levels as analyzed by western blots (Fig 6B, lane 3). When these cells treated with RNAi oligo #3 and grown in Matrigel were scored for percent lumen formation, >80% inhibition of lumen formation was found compared to untreated controls (Fig 7, Table 2). In contrast, cells treated with PKC-δ RNAi oligos #1 and #2 exhibited intermediate levels of inhibition of lumen formation (Table 2). Upon close examination, we note that RNAi inhibition of PKC-δ expression also reduced the number of acini per field (Fig 7), suggesting that PKC-δ is essential for cell growth. We also tested the effect of the PKC-δ inhibitor rottlerin on lumen formation. Since this inhibitor was toxic to the cells at the concentrations normally used (1- 10 μM), we performed a dose response study and found that 100 nM rottlerin was sufficient to inhibit >50% lumen formation in MCF7/CEACAM1-4S vs vector control transfected MCF7 cells (Fig 7, Table 2). Taken together, these data strongly indicate that PKC-δ is required for lumen formation. Finally, we tested the effect of CAPN9 RNAi on the cleavage of PKC-δ (Fig 6B, lane 4). The results demonstrate that lowering the levels of CAPN9 reduce the amount of PKC-δ cleavage, suggesting the possibility that PKC-δ is a direct target of CAPN9, thus linking the expression of CAPN9 to the activation of PKC-δ
Figure 6. Cleavage of PKC-δ and its inhibition by RNAi to PKCδ or calpain-9 in CEACAM1-4S transfected cells grown in Matrigel.

A: MCF7 cells transfected with vector (V), wild type (S) or double mutant T457,S459 CEACAM1-4S (DA) were grown in Matrigel for 2, 4, or 6d, cells harvested, proteins separated by SDS gel electrophoresis, and western blotted with anti-PKC-δ or anti-β-actin antibodies (arrows show position of intact (FL) and cleaved fragment (KD) of PKC-δ). B: MCF7 cells transfected with wild type CEACAM1-4S and treated with lipofectamine only (lane 1), RNAi control oligo (lane 2), RNAi oligo 3 to PKC-δ (lane 3) or RNAi oligo 2 to CAPN9 (lane 4) were grown in Matrigel for 4d, cells harvested, proteins separated by SDS gel electrophoresis, and western blotted with anti-PKC-δ and anti-β-actin antibodies (arrows show position of intact and cleaved fragment of PKC-δ).
Figure 7. Inhibition of lumen formation of MCF7/CEACAM1-4S cells by PKC-δ RNAi or PKC-δ inhibitor rottlerin.

A: MCF7/CEACAM1-4S cells treated with RNAi oligo 3 for PKC-δ (17% lumen formation, Table 2). B: MCF7 cells transfected with vector only and treated with 100 nM rottlerin (< 5% lumen formation, data not shown). C: MCF7/CEACAM1-4S cells treated with 100 nM rottlerin (45% lumen formation, Table 2).
Discussion
MCF7 cells that lack CEACAM1 expression and fail to undergo lumen formation when grown in a 3D culture, but regain this function when transfected with wild-type but not the double mutant T457A,S459A version of CEACAM1-4S, present a model of mammary morphorgenesis that is amenable to a full gene expression survey. To begin to assess the role of ~400 genes that changed by >2 log 2 between the two cell lines, we chose the mechanism of lumen formation by apoptosis because this was the basis of our assay system and represents a fundamental biological phenomenon lacking mechanistic details. Among over 400 genes differentially expressed, we found the de novo expression of CAPN9, a member of the calpain gene family that was previously thought to have tissue specific expression in the GI tract [25, 26]. Since calpain had been previously identified by us to be expressed in the central luminal cells undergoing apoptosis [7], we thought that this “tissue-specific” isoform, rather than the ubiquitous calpain isoforms CAPN1 and CAPN2, may be responsible for the either the initiation or execution phase of apoptosis. Indeed, analysis of the protein expression levels of CAPN9 over the time course of lumen formation suggested a close relationship between CAPN9 expression and lumen formation. When this relationship was tested by inhibition of CAPN9 expression using RNAi to CAPN9 that was shown to block protein expression, lumen formation was inhibited by 70%. Similarly, treatment of MCF7/CEACAM1-4S cells with two different calpain inhibitors significantly inhibited lumen formation. Finally, stable transfection of CAPN9 into MCF7 cells (lacking CEACAM1) was sufficient to induce lumen formation. Thus, CAPN9 may be the key protease that is responsible for lumen formation in these cells. Although CAPN9 was thought to be restricted to the GI tract, its expression in the breast may have been missed because lumen formation is only expected during active phases of mammary morphogenesis. The identification of CAPN9 in this model of mammary morphogenesis may have implications in DCIS (ductal carcinoma in situ) where lumen formation is abnormal. In this regard, CAPN9 has been identified as a potential tumor suppressor [27], and thus one would predict that down-regulation/silencing of CAPN9 may reduce lumen formation leading directly to conditions such as DCIS. Based on the studies shown here, we predict that the regulation of CAPN9 expression is linked to CEACAM1 expression.
It should be noted that MCF7 cells lack caspase-3, a key regulator of apoptosis in the extrinsic pathway [28]. As a result, many researchers have used this cell line to study cells deficient in the extrinsic pathway of apoptosis. However, it was shown that anti-cancer drugs activate caspase-8 in MCF7 cells, indicating that caspase-8 may act as a substitute for caspase-3 as an executioner caspase [29]. Although in the case of lumen formation in MCF7 cells, we have implicated the intrinsic pathway in terms of activation of Bax and release of cytochrome c from the mitochondria [7], it remains a possibility that the intrinsic pathway is utilized in breast cells that have normal expression of caspase-3. If this is true, there may be several pathways to lumen formation. However, we have previously shown that while MCF10F cells, that express both CEACAM1 and caspase-3 form lumen in 3D culture, MCF10F cells transfected with a CEACAM1 antisense gene do not form lumena [6]. This study rules out the possibility that expression of caspase-3 alone (unlike CAPN-9) is sufficient for lumen formation.
Having demonstrated that CAPN9 is involved in lumen formation, we searched for possible targets of this protease. A comparison of the crystal structure of mini-calpain-9 to mini-calpain-1 [24] predicted that calpain-9 activation and its substrate targets were different from the ubiquitously expressed calpain-1 [27]. We hypothesized that calpain-9 would be an initiator protease in apoptosis because it was newly expressed and that it should be connected in some way to CEACAM1 because it was mainly expressed in the wild type CEACAM1 transfected cells. In a search of the literature for early apoptotic proteins that are both cleaved for activation and related to CEACAM1 expression, we found a literature reference to PKC-δ. In that study BAF-3 cells stimulated with anti-CD98 antibodies underwent CEACAM1 mediated cell-cell adhesion with concomitant translocation of PKC-δ to the cell-cell adhesion sites [20]. Importantly, the PKC-δ inhibitor rottlerin was able to inhibit the cell-cell adhesion step, indicating that it was connected in some way to CEACAM1, the cell adhesion protein responsible for the cell-cell adhesion activity. PKC-δ is an especially attractive candidate as a possible target for calpain-9 because it is only activated after proteolytic cleavage and has a major role in the apoptotic mechanism of many cells [15, 24]. Significantly, PKC-δ was linked to syndecan-2 mediated apoptosis in osteoblasts and its inhibition by rottlerin reduced apoptosis in these cells [23]. Since syndecan-2 was one of the top hits in our gene screen, this connection may link it to both CEACAM1 and CAPN9. At first glance, this connection suggests PKC-δ may act to prevent apoptosis, a finding in keeping with studies that suggests it may act as either a pro- or anti-apoptotic factor [15], but apparently unrelated to the apoptotic mechanism of lumen formation. However, in our analysis, we are unable to separate the living from the apoptotic cells. As depicted in Fig 8, the acinar cells contacting ECM are protected from apoptosis while the central cells with no contact to ECM are apoptotic. This fact alone qualifies the central cells for apoptosis according to the mechanism called anoikis [30, 31]. As currently envisioned, anoikis only requires engagement of integrins to ECM to promote an anti-apoptotic signal. In this respect proper expression and engagement of β1-intergin has been shown to be critical for lumen formation [2, 3]. However, we know that the central acinar cells do not apoptose unless they (and the surrounding ECM contacting cells) also express the cell-cell adhesion molecule CEACAM1. Furthermore, the gene screen results implicate a second ECM related cell surface molecule in the process, namely syndecan-2. Thus, the ECM requirement for anoikis is not limited to integrins but requires additional ECM-related factors plus cell-cell adhesion factors to initiate the apoptotic program. Two possibilities exist, in one scenario the central acinar cells may die by neglect by virtue of not receiving anti-apoptotic signals, or in another scenario, the peripheral epithelial cells that contact ECM and express CEACAM1 may deliver an apoptotic signal to the central acinar cells (Fig 8). At this point we cannot distinguish the two possible scenarios because all the cells are analyzed together (they are part of the same acinus and must be isolated as a functional unit). Nonetheless, activation of PKC-δ may generate an anti-apoptotic signal in the peripheral cells, while generating a pro-apoptotic signal in the central acinar cells, in keeping with its dual activities. In the case of the peripheral cells expressing syndecan-2, activation of PKC-δ and/or ECM engagement may prevent syndecan-2 from delivering an apoptotic signal as described for osteoblasts. In the case of the central acinar cells, the apoptotic signal of syndecan-2 may be fully engaged. These possibilities will be explored in future studies.
Figure 8. Model of CEACAM1 mediated lumen formation.

The peripheral acinar cells (beige) receive a life signal (plus sign) from ECM (blue mesh) via their interaction with cell surface integrins (green), while the central acinar cells (grey) do not receive this life signal. Instead the central acinar cells receive an apoptotic signal from CEACAM1 (red, minus sign). The apoptotic signal induces CAPN9 (brown) that cleaves PKC-δ (blue), that in turn, initiates apoptosis. Neither CEACAM1 nor CAPN9 are able to overcome the life signal produced by the ECM-integrin interaction in the peripheral acinar cells. Note: In the absence of CEACAM1, the central acinar cells do not die, CAPN9 is not induced, and PKC-δ is not cleaved.
Returning to PKC-δ, our data demonstrate that inhibition of CAPN9 by RNAi prevents PKC-δ activation as measured by inhibition of its cleavage. However these data do not tell us if PKC-δ is a direct target of CAPN9, nor do they reveal possible downstream targets of PKC-δ. A tempting possibility is that PKC-δ may phosphorylate the critical T457 or S459 residues in CEACAM1-4S. However, in in vitro studies, we have found that the cytoplasmic domain peptide of CEACAM1-4S is not a good substrate for PKC-δ (data not shown), thus, we believe that PKC-δ may be involved down-stream from CEACAM1 and its phosphorylation targets still need to be identified.
In summary, we have identified a number of genes involved in lumen formation of mammary epithelial cells that are down-stream from CEACAM1, including a novel isoform of calpain, namely CAPN9, that may initiate the process of apoptosis by proteolytic cleavage of various target proteins. We have identified PKC-δ as one possible target of CAPN9.
Supplementary Material
Acknowledgements
We thank the Functional Genomics Core/Microarray Service at the Beckman Research Institute of City of Hope for the gene screen analysis and Dr. Terry D. Lee for Figure 8. This work was supported by NIH grant CA84202.
References
- [1].Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009;28:167–176. doi: 10.1007/s10555-008-9178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weigelt B, Bissell MJ. Unraveling the microenvironmental influences on the normal mammary gland and breast cancer. Semin Cancer Biol. 2008;18:311–321. doi: 10.1016/j.semcancer.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mailleux AA, Overholtzer M, Brugge JS. Lumen formation during mammary epithelial morphogenesis: insights from in vitro and in vivo models. Cell Cycle. 2008;7:57–62. doi: 10.4161/cc.7.1.5150. [DOI] [PubMed] [Google Scholar]
- [5].Shaw KR, Wrobel CN, Brugge JS. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia. 2004;9:297–310. doi: 10.1007/s10911-004-1402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang J, Hardy JD, Sun Y, Shively JE. Essential role of biliary glycoprotein (CD66a) in morphogeneesis of the human mammary epithelial cell line MCF10F. J. Cell Science. 1999;112:4193–4205. doi: 10.1242/jcs.112.23.4193. [DOI] [PubMed] [Google Scholar]
- [7].Kirshner J, Chen CJ, Liu P, Huang J, Shively JE. CEACAM1-4S, a cell-cell adhesion molecule, mediates apoptosis and reverts mammary carcinoma cells to a normal morphogenic phenotype in a 3D culture. Proc Natl Acad Sci U S A. 2003;100:521–526. doi: 10.1073/pnas.232711199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- [9].Kuespert K, Pils S, Hauck CR. CEACAMs: their role in physiology and pathophysiology. Curr Opin Cell Biol. 2006;18:565–571. doi: 10.1016/j.ceb.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Obrink B. CEA adhesion molecules: multifunctional proteins with signal-regulatory properties. Curr Opin Cell Biol. 1997;9:616–626. doi: 10.1016/S0955-0674(97)80114-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barnett TR, Drake L, Pickle W., 2nd Human biliary glycoprotein gene: characterization of a family of novel alternatively spliced RNAs and their expressed proteins. Mol Cell Biol. 1993;13:1273–1282. doi: 10.1128/mcb.13.2.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen CJ, Kirshner J, Sherman MA, Hu W, Nguyen T, Shively JE. Mutation analysis of the short cytoplasmic domain of the cell-cell adhesion molecule CEACAM1 identifies residues that orchestrate actin binding and lumen formation. J Biol Chem. 2007;282:5749–5760. doi: 10.1074/jbc.M610903200. [DOI] [PubMed] [Google Scholar]
- [13].Teixeira AM, Fawcett J, Simmons DL, Watt SM. The N-domain of the biliary glycoprotein (BGP) adhesion molecule mediates homotypic binding: domain interactions and epitope analysis of BGPc. Blood. 1994;84:211–219. [PubMed] [Google Scholar]
- [14].Yokoyama S, Chen CJ, Nguyen T, Shively JE. Role of CEACAM1 isoforms in an in vivo model of mammary morphogenesis: mutational analysis of the cytoplasmic domain of CEACAM1-4S reveals key residues involved in lumen formation. Oncogene. 2007;26:7637–7646. doi: 10.1038/sj.onc.1210577. [DOI] [PubMed] [Google Scholar]
- [15].Reyland ME. Protein kinase Cdelta and apoptosis. Biochem Soc Trans. 2007;35:1001–1004. doi: 10.1042/BST0351001. [DOI] [PubMed] [Google Scholar]
- [16].Sitailo LA, Tibudan SS, Denning MF. The protein kinase C delta catalytic fragment targets Mcl-1 for degradation to trigger apoptosis. J Biol Chem. 2006;281:29703–29710. doi: 10.1074/jbc.M607351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cerda SR, Mustafi R, Little H, Cohen G, Khare S, Moore C, Majumder P, Bissonnette M. Protein kinase C delta inhibits Caco-2 cell proliferation by selective changes in cell cycle and cell death regulators. Oncogene. 2006;25:3123–3138. doi: 10.1038/sj.onc.1209360. [DOI] [PubMed] [Google Scholar]
- [18].Basu A, Adkins B, Basu C. Down-regulation of caspase-2 by rottlerin via protein kinase C-delta-independent pathway. Cancer Res. 2008;68:2795–2802. doi: 10.1158/0008-5472.CAN-07-6244. [DOI] [PubMed] [Google Scholar]
- [19].van Raam BJ, Drewniak A, Groenewold V, van den Berg TK, Kuijpers TW. Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood. 2008;112:2046–2054. doi: 10.1182/blood-2008-04-149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kakugawa K, Hattori M, Beauchemin N, Minato N. Activation of CEA-CAM-1-mediated cell adhesion via CD98: involvement of PKCdelta. FEBS Lett. 2003;552:184–188. doi: 10.1016/s0014-5793(03)00924-4. [DOI] [PubMed] [Google Scholar]
- [21].Edlund M, Wikstrom K, Toomik R, Ek P, Obrink B. Characterization of protein kinase C-mediated phosphorylation of the short cytoplasmic domain isoform of C-CAM. FEBS Lett. 1998;425:166–170. doi: 10.1016/s0014-5793(98)00222-1. [DOI] [PubMed] [Google Scholar]
- [22].Kakugawa K, Hattori M, Beauchemin N, Minato N. Activation of CEA-CAM-1-mediated cell adhesion via CD98: involvement of PKCdelta. FEBS Lett. 2003;552:184–188. doi: 10.1016/s0014-5793(03)00924-4. [DOI] [PubMed] [Google Scholar]
- [23].Orosco A, Fromigue O, Hay E, Marie PJ, Modrowski D. Dual involvement of protein kinase C delta in apoptosis induced by syndecan-2 in osteoblasts. J Cell Biochem. 2006;98:838–850. doi: 10.1002/jcb.20826. [DOI] [PubMed] [Google Scholar]
- [24].Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- [25].Sorimachi H, Ishiura S, Suzuki K. A novel tissue-specific calpain species expressed predominantly in the stomach comprises two alternative splicing products with and without Ca(2+)-binding domain. J Biol Chem. 1993;268:19476–19482. [PubMed] [Google Scholar]
- [26].Sorimachi H, Kimura S, Kinbara K, Kazama J, Takahashi M, Yajima H, Ishiura S, Sasagawa N, Nonaka I, Sugita H, Maruyama K, Suzuki K. Structure and physiological functions of ubiquitous and tissue-specific calpain species. Muscle-specific calpain, p94, interacts with connectin/titin. Adv Biophys. 1996;33:101–122. doi: 10.1016/s0065-227x(96)90026-x. [DOI] [PubMed] [Google Scholar]
- [27].Davis TL, Walker JR, Finerty PJ, Jr., Mackenzie F, Newman EM, Dhe-Paganon S. The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J Mol Biol. 2007;366:216–229. doi: 10.1016/j.jmb.2006.11.037. [DOI] [PubMed] [Google Scholar]
- [28].Kagawa S, Gu J, Honda T, McDonnell TJ, Swisher SG, Roth JA, Fang B. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin Cancer Res. 2001;7:1474–1480. [PubMed] [Google Scholar]
- [29].Engels IH, Stepczynska A, Stroh C, Lauber K, Berg C, Schwenzer R, Wajant H, Janicke RU, Porter AG, Belka C, Gregor M, Schulze-Osthoff K, Wesselborg S. Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene. 2000;19:4563–4573. doi: 10.1038/sj.onc.1203824. [DOI] [PubMed] [Google Scholar]
- [30].Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- [31].Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76:1352–1364. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.