

Grk1, or rhodopsin kinase, has been known for its critical function in visual pigment deactivation in photoreceptors, with its deficiency leading to photoreceptor dysfunction and light-induced photoreceptor cell death. This study was an investigation of whether and how enhancing pigment phosphorylation might affect photoreceptor viability.
Abstract
Purpose.
Photoreceptor rhodopsin kinase (Rk, G protein–dependent receptor kinase 1 [Grk1]) phosphorylates light-activated opsins and channels them into an inactive complex with visual arrestins. Grk1 deficiency leads to human retinopathy and heightened susceptibility to light-induced photoreceptor cell death in the mouse. The goal of this study was to determine whether excess Grk1 activity is protective against photoreceptor cell death.
Methods.
Grk1-overexpressing transgenic mice (Grk1+) were generated by using a bacterial artificial chromosome (BAC) construct containing mouse Grk1, along with its flanking sequences. Quantitative reverse transcription-PCR, immunoblot analysis, immunostaining, and activity assays were combined with electrophysiology and morphometric analysis, to evaluate Grk1 overexpression and its effect on physiologic and morphologic retinal integrity. Morphometry and nucleosome release assays measured differences in resistance to photoreceptor cell loss between control and transgenic mice exposed to intense light.
Results.
Compared with control animals, the Grk1+ transgenic line had approximately a threefold increase in Grk1 transcript and immunoreactive protein. Phosphorylated opsin immunochemical staining and in vitro phosphorylation assays confirmed proportionately higher Grk1 enzyme activity. Grk1+ mice retained normal rod function, normal retinal appearance, and lacked evidence of spontaneous apoptosis when reared in cyclic light. In intense light, Grk1+ mice showed photoreceptor damage, and their susceptibility was more pronounced than that of control mice with prolonged exposure times.
Conclusions.
Enhancing visual pigment deactivation does not appear to protect against apoptosis; however, excess flow of opsin into the deactivation pathway may actually increase susceptibility to stress-induced cell death similar to some forms of retinal degeneration.
Retinal degenerations are among the major causes of severe visual impairment in industrialized countries.1,2 Age-related macular degeneration affecting the geriatric population is the most prevalent, but other inherited retinal degenerations, including retinitis pigmentosa, also contribute to the visual toll in the remaining segments of population from newborns to middle-aged individuals.3,4 Despite extensive heterogeneity, these disorders have photoreceptor loss as a final common denominator leading to irreversible blindness.5–9 Insight into cell death and protection pathways provides an opportunity to mitigate or reverse visual decline in large groups of patients, regardless of the cause.4,10–12
Over the past several decades, genetic and light-induced photoreceptor cell death models have emerged as the key means of studying and defining the mechanisms of degeneration.13–15 Rod photoreceptors have been the major focus of these studies, as they are lost early in most retinal degenerations (i.e., retinitis pigmentosa and macular degeneration), followed by a secondary decline in cones.3,8 Two major categories of cell death pathways have been described in rod dominant rodent models5—transducin-dependent or transducin-independent pathways—based on whether cell death in a particular paradigm is suppressed by null mutation in rod α-transducin.16,17 Normally, in rods, transducin is the G protein mediator of visual signaling initiated by light-induced excitation of the visual pigment rhodopsin. Photoreceptor degeneration resulting from the rhodopsin mutations studied to date appears to occur independent of transducin and cannot be blocked by transducin's absence. Furthermore, bright light-induced damage, modeling potential light-aggravated cell death in human retinal degeneration including macular degeneration,18–20 appears to be unaffected by null mutations in transducin mouse models,16 suggesting that even light-induced retinal degeneration can occur in the absence of photosignaling. However, transducin mutations do seem to suppress dim light-mediated cell damage in a strain of mice lacking the visual pigment deactivation pathway.16 In summary, these findings suggest at least two alternative photoreceptor cell death pathways initiated by rhodopsin.
Besides the transducin-mediated visual signaling pathway, another physiologic pathway initiated by rhodopsin bleach is the deactivation pathway. Limiting the lifetime of photobleached rhodopsin activity by this pathway has been regarded largely as beneficial and cytoprotective, but other evidence suggests that it may serve as a potential cell death pathway independent of transducin. The key component responsible for initiating the rhodopsin deactivation pathway is rhodopsin kinase (Rk) or G protein–dependent receptor kinase 1 (Grk1). Grk1 catalyzes the light-dependent phosphorylation of opsin in both rods and cones, thus diverting the active intermediate into an inactive complex with visual arrestins.21,22 Absence of either Rk or arrestin prolongs the lifetime of photoactivated visual pigment intermediates and renders rods insensitive to repeated stimulation, as seen in patients with Oguchi disease and, in some cases, retinal degeneration.23–26 Grk1−/− mice are susceptible to intense light-induced and -independent photoreceptor degeneration16,27–29 highlighting a protective role of this pathway. Other biochemical studies, however, suggest a more complex role, with overphosphorylation and arrestin binding of constitutively active opsin mutants playing a potential role in retinal degenerations ranging from retinitis pigmentosa to retinal degenerations in Drosophila.17,30,31
To further investigate whether a deactivation bias would have a beneficial or detrimental effect on photoreceptor survival, we overexpressed Grk1 in mice, by using bacterial artificial chromosome (BAC) transgenic technology,32,33 and examined photoreceptor susceptibility in our established Grk1+ overexpressing line. The Grk1+ overexpressing mice retained normal retinal morphology, light sensitivity, and peak signal amplitude when reared in normal cyclic room light. In an intense light environment, excess Grk1 afforded no added protection against light-induced apoptosis, based on morphologic studies. Instead, light-induced photoreceptor cell loss appeared aggravated by Grk1 overexpression, but only with longer durations of exposure. Our findings point to a possible novel role for visual pigment inactivation in photoreceptor cell death supported by other studies that correlate excessive rhodopsin phosphorylation with vertebrate and invertebrate photoreceptor cell death.31,34,35
Materials and Methods
Transgenesis and Genotype Analysis
The BAC clone RP23–89F9 in the pBACe3.6 vector was purchased (BAC-PAC Services; Invitrogen, Carlsbad, CA), and the episome was purified by two sequential CsCl gradients. The DNA integrity was verified by NotI restriction endonuclease digestion. Transgenic mice were produced by pronuclear microinjection of the constructs according to methodologies established by the Roswell Park Cancer Institute (RPCI) Gene Targeting and Transgenic Core Facility.32,33,36 Briefly, fertilized F2 oocytes were derived from an F1 x F1 cross of (C57BL/10Ros-pd x C3h/HeRos) mice after superovulation of the females (3–4 weeks of age). Eggs were retrieved, microinjected, and reimplanted in SW (CD-1) pseudopregnant female mice. Incorporation of the BAC into the progeny genome was verified by PCR amplification of BAC ends from tail genomic DNA using primers designed specifically for the 5′ (forward: 5′-GCGGCCGCTAATACGACTCAC-3′; reverse: 5′-CTGACCTCTCTCTGCCTGAG-3′) and 3′ (forward: 5′-CAGGATGGACCATGGGTCACTG-3′; reverse: 5′-CCCGAATTGACTAGTGGGTAG-3′) BAC ends.
Grk1-overexpressing transgenic mice were maintained on a C57BL/6 background and crossed with albino BALB/c mice, to generate pigment-matched Leu450Met heterozygotes at the Rpe65 locus. These mice show intermediate light susceptibility.37 Genotypes at the Rpe65 locus were confirmed by differential digestion of the amplified allelic bands with MwoI.38 All procedures complied with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Breeding protocols were approved by the Roswell Park Cancer Institute's Institutional Animal Care and Use Committee. Experimental procedures, light treatment protocols, and the light exposure chamber designs were approved by the State University of New York at Buffalo Institutional Animal Care and Use Committee and by the Washington University Animal Studies Committee. The Grk1−/− mice were kindly provided by Jason Chen (Virginia Commonwealth University, Richmond, VA) and housed in an approved facility at the University of Southern California. Whenever necessary, globes or the retinas were dissected or fixed in absolute darkness after euthanatization, with the aid of a pair of night vision goggles or night vision microscope adapters under an infrared source.
Evaluation of Transgene Overexpression
Total RNA and protein fractions were obtained from Grk1+ and control wild-type (WT) mouse eyes or retinas (TRIzol; Invitrogen) and stored under ethanol. Pellets were resuspended just before their use in real-time RT-PCR and quantitative immunoblot analysis.
Real-time PCR was performed on transgene-positive and -negative RNA fractions, as previously described.39 Reverse transcription reactions contained 5 μg of total RNA annealed to random hexamers in the presence of reverse transcription buffer (1×), excess deoxynucleotide triphosphates, and MMLV reverse transcriptase in a total volume of 25 μL. Optimized gene-specific primer pairs were used in the subsequent amplification reaction. The primer sequences have been published.40,41 The mouse rod arrestin (Arr1) primers were 5′-CACATCCAGTGAAGTGGCTAC-3′ (forward) and 5′-TCTGTGTTCTCTCCAGTATC-3′ (reverse). cDNA amplification was monitored at 60°C in the presence of SYBR Green, as previously detailed, in a thermocycler (MyiQ; Bio-Rad, Hercules, CA).39 Cycles to threshold (Ct) values were compared among Grk1+ and WT samples, either directly or after correction for actin or β2-microglobulin by subtraction (ΔCt). Relative levels of Grk1 were estimated from differences in ΔCt (ΔΔCt) for Grk1+ and control samples based on the formula R = 2−ΔΔCt.
Immunoblot analysis was performed on protein pellets (TRIzol; Invitrogen) resuspended in 250 μL/eye solubilization buffer consisting of 7 M urea, 2 M thiourea, 4% CHAPS, 1% ampholytes pH 3–10, 0.5% Triton X-100, and 10 mg/mL dithiothreitol. Total protein was measured by the Bradford method (Bio-Rad, Hercules, CA) before addition of the gel loading dye, denaturation, and electrophoresis. After addition of the loading buffer, various protein aliquots from transgene-positive (Grk1+) and their transgene-negative (WT) eyes were loaded onto a 6% polyacrylamide gel, fractionated, and transferred to PVDF membranes. The membranes were blocked with 2% nonfat dried milk in Tris-buffered saline containing 0.02% Tween-20 and incubated sequentially with one or both G8 or D11 monoclonal antibodies specific for C- and N-terminal Grk1 epitopes (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). Some blots were incubated with rabbit anti-arrestin 1 antibody C10C10.42,43 After washes with Tris-buffered saline, horseradish peroxidase-coupled rabbit anti-mouse IgG (1:3000) was added to the blots, and incubation was continued for another hour. The blots were developed by exposure to a chemiluminescent substrate consisting of 1.25 mM luminol and 200 μM p-coumaric acid with hydrogen peroxide added to 0.003% concentration. The signal was recorded by using a gel image documentation platform (Storm; GE Health Care, Piscataway, NJ) equipped with a digital chemiluminescence camera. After image acquisition, the blots were generally reprobed with anti-actin (Santa Cruz Biotechnology) monoclonal antibody. The signal strength for each band was estimated in arbitrary pixel units after background subtraction (Quantity-One software; Bio-Rad). The net signal volume for the Grk1 band in each sample was plotted against the total volume, protein volume, or actin signal volume of that sample in the linear region. The slope ratios yield relative estimates of Grk1 levels in Grk1+ and control samples.
Immunostaining
Grk1+ and WT eyes were obtained from room light- or dark-adapted mice after euthanatization, fixed with 4% paraformaldehyde in 0.1 M Na-phosphate buffer (pH 7.4), and frozen-sectioned onto positively charged slides, as previously described.44 After they were blocked, the 10-μm cryosections were incubated with one or more of the following primary antibodies raised against: phosphorylated rhodopsin (monoclonal A11–82P, 1:10),45 arrestin 1 (polyclonal C10C10, 1:100),42 or Grk1 (polyclonal 8585,1:400; gift of Robert Lefkowitz, Duke University, Durham, NC). The sections were then secondarily labeled with Alexa-568-labeled anti-rabbit and Alexa-488 anti-mouse secondary IgG (Invitrogen, Molecular Probes; Eugene, OR), either in combination or alone, before viewing. Twelve-bit images were acquired with a laser scanning confocal microscope (LSM; Carl Zeiss Meditec, Inc., Dublin, CA) and accompanying software. Single optical slices, as well as the integrated signals from projection were compared across samples.
Rhodopsin Phosphorylation Assays
Rhodopsin phosphorylation was quantified in retinal samples according to previously established methods with minor modification.46,47 Endogenous substrate activities were measured in the light and dark with 6 μg of mouse retinal homogenate as the source of both the Grk1 enzyme and the rhodopsin substrate, using the same reaction mixture as previously described,43,48 except for the addition of 5 mM NaF and 3 mM EDTA and the exclusion of exogenous Grk1 substrate. After a 2.5-minute incubation, the reactions were terminated with the addition of SDS sample buffer. SDS gels were run to separate other retinal proteins from the rhodopsin bands, which were cut out and counted in a scintillation counter. Counts per minute (cpm) from corresponding dark samples were subtracted, so that only light-stimulated phosphate incorporation was used to determine GRK1 activity, which was calculated by dividing cpm by the specific activity of ATP (in cpm per picomole), the incubation time (2.5 minutes), and the amount of enzyme protein (6 μg). Exogenous substrate activities were assayed in the same way, except that 50 μg of dark-adapted Grk1−/− retinal homogenate was added as a source of exogenous substrate. Grk1−/− mice are homozygous null for the Grk1 allele and lack functional Grk1, but have normal rhodopsin in their retinas.
Electrophysiology
The mice were dark-adapted overnight before the experiments. After euthanatization, the eyes were removed under dim red light. All subsequent manipulations were performed under infrared light. The eyes were hemisected, and each retina was isolated and stored in Locke solution at 4°C. A quarter of the isolated retina was mounted on filter paper, photoreceptor side up, and placed on the recording chamber with an electrode connected to the bottom. A reference electrode was placed above the retina. The perfusion Locke solution (112 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2, 1.2 mM CaCl2, 10 mM HEPES, 20 mM NaHCO3, 3 mM Na2-succinate, 0.5 mM Na-glutamate, and 10 mM glucose) was equilibrated with 95% O2/5% CO2, heated to 34–37°C, and contained, in addition, 2 mM l-glutamic acid to block the higher order components of photoresponse.49 The electrode solution (140 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2, 1.2 mM CaCl2, 3 mM HEPES, and 10 mM glucose [pH 7.4]) under the retina contained, in addition, 10 mM BaCl2 to suppress glial components.50,51 Responses were amplified by a differential amplifier (DP-311; Warner Instruments, Hamden, CT). Intensity–response data were fit by the equation
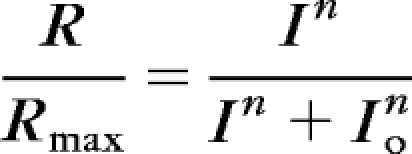 |
where R is the transient peak amplitude of response, Rmax is the maximum response amplitude, I is flash intensity, and Io is the flash intensity necessary to generate a half-maximum response. Monochromatic 20-ms test flashes (500 nm) were delivered from a calibrated light source via computer-controlled shutters. Flash intensity was set by a combination of calibrated neutral-density filters.
Light Exposure
Age-, sex- and, when possible, littermate-matched, F1 hybrid Grk1 overexpressors (Grk1+) and control animals lacking the BAC transgene (WT) were used in all light-exposure experiments at 8 to 12 weeks of age. For cyclic room light exposure, the mice were maintained in a conventional facility at 30% to 70% humidity, at 22 ± 1.5°C and 60 lux in 12-hour cyclic light. For intense light-exposure experiments, the mice were first placed in light-tight ventilated boxes in absolute darkness in their home cages for 16 hours before exposure to light. Their pupils were dilated by instillation of a drop of 1% atropine (Alcon, Fort Worth, TX) in each eye, and they were returned to dark home cages for 30 minutes before exposure to intense light. The mice were exposed to 10,000 lux of cool, bright, white fluorescent light for 3 or 12 hours in individual acrylic cages within light-tight air-conditioned chambers with reflective interiors approved by the IACUC. Light intensity was measured with the spectroradiometer (ILT900; International Light Technologies, Peabody, MA). The temperature was maintained at 25°C to 27°C during the light exposure episodes. The exposure was initiated at 8 to 9 AM and terminated 3 or 12 hours thereafter. Exposure of the matched groups of Grk1+ and WT control animals were performed in parallel and terminated simultaneously during the afternoon hours. After a recovery period in the dark, the mice were euthanatized, and the eyes or retinas evaluated with morphometric measures or apoptosis assays.
Retinal Morphometry
Grk1+ and WT eyes were embedded in glycol methacrylate for morphometry, according to standard protocols.31 After exposure to light and recovery, the mice were deeply anesthetized and subjected to cardiac perfusion with fresh 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (pH 7.4). Before enucleation, the lateral canthus of each eye was marked with a 5-0 suture (Ethilon; Ethicon, Somerville, NJ) to preserve orientation. A small amount of soft tissue was preserved around the eye so that a flat surface could be cut at the medial canthus on which the globe could maintain position within the plastic embedding mold. After fixation in 4% PFA for 24 hours at 4°C, the eyes were dehydrated in an increasing series of ethanol. They were embedded in resin (JB-4 Plus; Polysciences, Inc., Warrington, PA), according to the manufacturer's recommendations. Sections (3 μm) were cut along the vertical meridian at the level of the optic nerve and stained with hematoxylin and eosin. Mosaic photographs were taken at 10× with a fluorescence microscope (Axioimager and Axiovision, ver. 4.6 software; Carl Zeiss Meditec, Inc.). The outer nuclear layer (ONL) thickness was measured at nine equidistant positions, 250 μm apart, starting from the optic nerve along the superior and inferior hemiretina (−9 to +9; Image Tool for Windows ver 3.00 software; provided in the public domain by the University of Texas Health Science Center, San Antonio). Central retinal ONL thickness was based on the average of measurements from positions −3 to +3. P was determined from t-test statistics based on both parametric and nonparametric models (Prism; GraphPad, San Diego, CA, or Excel, Microsoft, Redmond, WA).
Apoptosis Assays
Apoptosis in Grk1+ and WT retinal tissues was measured by ELISA (Cell Death ELISA; Roche, Indianapolis, IN). For ELISA-based nucleosome release assays, individual retinas retrieved from light-treated animals were homogenized in 400 μL of lysis buffer. Free nucleosomes were quantified in the cleared supernatants after a 1:200 to 1:400 dilution, according to the manufacturer's instructions.
TUNEL staining of frozen globe sections was performed (In Situ Cell Death Detection kit; Roche) according to the manufacturer's instructions, with some modifications. Light-treated and control eyes were fixed in situ by cardiac perfusion with 4% paraformaldehyde in mice under deep anesthesia to minimize postmortem autolysis artifacts. The globes were retrieved after euthanatization and sectioned on a cryostat (5 μm) after embedding (Precision Cryoembedding System; Pathology Innovations LLC, Wyckoff, NJ) at −22°C in embedding medium for frozen sections (TBS Tissue Freezing Medium; Triangle Biomedical Sciences, Durham, NC). After a 15-minute permeabilization period, terminal deoxynucleotidyl transferase reactions were performed on the eye sections in the presence of the biotinylated dUTP substrate as specified by the manufacturer. Fluorescein-tagged streptavidin was used to visualize apoptotic nuclei. The parallel negative controls lacked transferase. Fluorescence images were recorded with a confocal microscope (LSM 510 Meta mounted on an Axiovert 200M; Carl Zeiss Meditec, Inc.).
Results
Generation of Grk1-Overexpressing Mice
We used the RP23-89F9 BAC clone (Fig. 1A) for transgenesis, because it contains the full-length mouse Grk1 gene along with the entire 5′ and 3′ regulatory flanking sequences needed for accurate expression of the gene in rods and cones. A total of 14 mice were generated from pronuclear injection of oocytes with the BAC construct. Screening of the tail genomic DNA from these mice by PCR revealed transgene-specific bands in three lines (Fig. 1B). A 273-bp band corresponding to the 5′ BAC vector junctional sequence was evident in these three lines, but was missing from the control and WT mice. All three lines also contained the 3′ BAC sequences consistent with the presence of intact transgene in one or more copies in the transgene-positive lines (not shown). The progeny of lines 1 and 2 were normal in appearance and behavior, and produced normal-sized litters. Transgene-positive animals from line 2 had the same Grk1 transcript levels within their retinas, as did the WT, by real-time RT-PCR. Line 3 descendents were variable in fertility, with a significant proportion of pseudohermaphrodites and produced smaller sized litters. Line 3 was therefore not investigated further in this study.
Figure 1.

Transgenic construct and resultant founders. (A) The BAC clone RP23–89F9 contained a fragment of mouse chromosome 8 (0.22 Mb) spanning the Grk1 gene with its exons (□), introns, and the full flanking sequences (solid line) inserted into the EcoRI site of pBACe3.6. The position of the 5′ PCR primer (filled arrow and arrowhead) and the 3′ primer (open arrow and arrowhead) sets are shown. The 5′ and 3′ primer sets amplified a 273- and 261-bp junctional insert–vector fragment, respectively. (B) PCR analysis of the genomic DNAs from mice generated by pronuclear injections of oocytes with a 5′ primer set. Bold arrow: the position of the band generated by amplification. The bottom band in each lane represents a possible primer dimer. Grk1+, Grk1 overexpressing mouse line 1, WT, transgene-negative wild-type mouse.
Transgene positive animals from line 1, designated Grk1+, expressed significantly higher mRNA levels of Grk1 based on measurements of transcript amplification by real-time reverse transcription PCR (Fig. 2A). The detection threshold was attained by an average of 1.5 cycles earlier among cDNA samples derived from Grk1+ mice compared with WT samples in real-time amplification, both when corrected and uncorrected for actin. This difference indicated Grk1 in Grk1+ eyes at 300% ± 50% of the WT levels. Mouse visual arrestin 1 (Arr1) or opsin (mOps) Ct and transcript contents were not affected in the Grk1+ or WT samples.
Figure 2.

Grk1 expression levels in Grk1+ transgenic eyes. (A) Real-time RT-PCR was used to estimate relative Grk1, Arr1 (visual arrestin 1), or rhodopsin (Ops) transcript levels in Grk1+ eyes as a percent of the WT levels. The percentages were based on the net number of cycles to threshold based on the formula R = 100(2−ΔΔCt). Results from at least 6 to 10 amplifications from various Grk1 and WT RNA pools were used to arrive at the average and SEM. (B) Immunoblot analysis from Grk1+ and WT eye proteins were sequentially stained with specific antibodies for Grk1, Arr1 (rod arrestin), and actin at varying loads. Band volume (pixel depth × area) quantification and background subtraction were performed after digital acquisition of chemiluminescent images. (C) Grk1 band volumes were plotted against the actin band volume for increasing volumes of Grk1+ and WT samples. The units on each axis are arbitrary and depend on the density. The experiments represent an average of at least three blots from three independent pools of Grk1+ and WT eyes. (D) Relative levels of Grk1 in Grk1+ as a percentage of WT littermates were based on the ratio of the slopes in (C).
The immunoblots also showed higher levels of Grk1 immunoreactive protein in Grk1+ eye samples compared with WT when stained with a highly specific monoclonal antibody (G8; Fig. 2B). This C-terminal epitope-specific antibody recognizes only a single band migrating at 67-kDa, corresponding to full-length Grk1 protein.52 Similarly, monoclonal antibody D11, specific for the N-terminal domain also showed higher levels of full-length Grk1 in Grk1+ eyes compared with control mice (data not shown). In contrast, in the same blot, the Arr1 or actin antibody immunoreactive bands appeared invariant across the Grk1+ and WT mice. Relative expression of Grk1 between Grk1+ and WT eyes was further quantified by plotting background-subtracted Grk1 band volumes (pixel depth × area) against increasing actin band volumes with increasing sample loads (Fig. 2C). Comparison of the slopes showed Grk1 protein in Grk1+ eyes at 280% ± 50% of the WT levels (Fig. 2D). The slope ratio was identical whether total protein, volume, or Arr1 load was used for normalization. Immunofluorescent staining of eye sections showed Grk1 expression confined to photoreceptor outer segments and to a minor extent to the photoreceptor synaptic terminals in Grk1+ animals in a pattern of distribution similar to that of WT retina (data not shown).
Increased Light-Dependent Phosphorylation of Rhodopsin in Grk1+ Mice
To determine whether Grk1 excess affects rhodopsin phosphorylation levels in vivo, we compared Grk1 and WT mouse eye sections stained with antibody against phosphorylated opsin. The antibody recognizes the C-terminally phosphorylated opsin generated exclusively in light-exposed retinas. As seen in comparing Figures 3A and 3B, higher levels of immunostaining were evident in outer segments of the Grk1+ than in those of the WT eyes, suggesting higher levels of rhodopsin in phosphorylation. In addition, higher specific labeling appeared in perinuclear regions in light-exposed Grk1+ eyes, suggesting possible higher levels of soluble forms of phosphorylated opsin in the Grk1+ mice. The immunostaining appeared entirely light-dependent, as the eyes adapted and retrieved in absolute dark show no specific immunostaining (Figs. 3C, 3D). The fully dark-adapted state in these sections was verified with Arr1 immunostaining, showing localization confined to inner segments, and perinuclear and synaptic terminals (Fig. 3D).
Figure 3.

Enhanced light-dependent phospho-rhodopsin staining in the Grk1+ outer retina. Frozen sections of Grk1+ and WT eyes were incubated with antibodies against phosphorylated rhodopsin (monoclonal, A, B) and Arr1 (polyclonal, C, D). The rhodopsin and Arr1 stainings were visualized with Alexa-488 (green) and -568 (red) secondary antibodies, respectively. (A, B) Sections from light-adapted eyes; (C, D) sections from fully dark-adapted eyes. OS, outer segment; IS, inner segment; ONL, outer nuclear layer, IR, inner retina. Bar, 10 μm.
Phosphorylating activities of Grk1+ and WT retinal extracts were further quantified and compared by two independent in vitro assays. One method relied on measuring the incorporation of radioactive phosphate into ample endogenous rhodopsin substrate already present in excess in the retinal extracts from Grk1+ and WT mice. Alternatively, radioactive phosphate incorporation was measured by using excess exogenously added unphosphorylated rhodopsin from Grk1−/− mice, which lack light-dependent phosphorylation altogether. As seen in Figure 4, retinal extracts from Grk1+ mice incorporated 32P at 2.3 or 2.8 times the levels of WT mouse extracts using either endogenous (Fig. 4A) or exogenous rhodopsin (Fig. 4B) substrate. The relative levels of inorganic phosphate incorporation using both types of assays were consistent with the relative levels of proteins and RNA estimated from the studies described (Figs. 2A–D). The differences between Grk1+ and WT levels were highly significant; and, in every instance, Grk1+ and WT retinas could be differentiated by their enzymatic activity levels without knowledge of genotyping results. Grk1−/− retinal extracts lacking Grk1 failed to incorporate any phosphate into their rhodopsin bands (data not shown).
Figure 4.

Increased rhodopsin phosphorylating activity of Grk1+ retinal extract. Six micrograms of the homogenates of Grk1+ and WT retinas were incubated with 10 μM [γ-32P] ATP, 50 mM potassium phosphate (pH 7.0), 10 mM magnesium acetate, 5 mM sodium fluoride, and 3 mM EDTA in the absence (A) or presence of exogenous substrate (B), which was 50 μg of Grk1−/− retinal homogenate, containing normal rhodopsin but lacking Grk1 enzyme. Light-stimulated activities are calculated from counts per minute incorporated into rhodopsin and are the mean ± SEM of results in at least six mice. ***P < 0.001.
Effects of Cyclic Light on the Retina of Grk1+ Mice
To evaluate photoreceptor physiology, we characterized the function of rods in ERG recordings from isolated retinas. This method allowed us to pharmacologically isolate the photoreceptor component (a-wave) from the retina response. As rods represent 97% of photoreceptors in mouse retina, the photoresponses recorded by this method were generated almost exclusively by rods. We found that the saturating retina response, which is a function of the maximum response per rod and the number of rods per unit of retinal area, were similar in WT and Grk1+ retinas (Figs. 5A, 5B). This result confirms the lack of degeneration in the Grk1+ retina and indicates that the saturating response per rod was also not altered by the overexpression of Grk1. Even though ERG recordings do not allow precise analysis of photoresponse kinetics, we observed no dramatic differences in the dim-flash kinetics between the rod responses of WT and Grk1+ retinas. In addition, the two genotypes of retina had identical intensity-response functions (Fig. 5A). Rod sensitivity, as measured by the light intensity necessary to produce a half-saturating response (Io), was identical in WT and Grk1+ littermates (Fig. 5B). Together, these results indicate that rod photosignaling was not significantly affected by the overexpression of Grk1.
Figure 5.

Physiology and morphology of Grk1+ retina. (A) Fractional a-wave amplitude of transretinal ERG recordings as a function of flash intensity from WT (n = 8) and Grk1+ (n = 6) retinas. Data were fit with the equation in the text. (B) Physiological parameters obtained from WT and Grk1+ retinas. Io is the flash intensity that gives the half-maximum response. Rmax is the maximum amplitude of a-wave. Data are expressed as the mean ± SEM. (C) Structure of Grk1+ and WT retinas were viewed by light microscopy. Sections (3 μm) of plastic-embedded retina were cut along the vertical meridian of the globe at the level of the optic nerve and stained with hematoxylin and eosin. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; ON, optic nerve; O, ora serrata. (D) Comparison of Grk1+ and WT ONL thicknesses in micrometers at nine equidistant positions along the superior and inferior hemiretina (+0–9 superior retina, −9–0 inferior retina). Data are expressed as the mean ± SD (n = 8 WT and 8 Grk1+). Scale bar, 50 μm.
Consistent with these findings, histologic sections of the Grk1+ retina appeared qualitatively indistinguishable from those of their WT littermates (Fig. 5C.). There was also no significant difference in outer nuclear layer (ONL) measurements based on quantitative morphometry between the background strain and the Grk1+ transgenic mice, even at ages beyond 18 months (Fig. 5D).
Effects of Intense White Light on the Retina of Grk1+ Mice
We used an intense rhodopsin bleach paradigm similar to the one used by Grimm et al.,40 to test the sensitivity of photoreceptors to stress-induced death in the Grk1+ line. The majority of mice used throughout this study were heterozygous for the Leu450met polymorphism, making their retinas moderately susceptible to bright-light–induced damage. Mice with these backgrounds generally show little photoreceptor loss when reared under cyclic light, but undergo consistent photoreceptor damage at light intensities between 5 and 15 klux. We did not find any significant differences in the thickness of the ONL in the dark-adapted mice (data not shown). We compared photoreceptor loss in Grk1+ and WT mice under uniform 10 klux of white-light illumination using ONL thickness measurements. We initially focused on the central region flanking the optic disc from positions –3 to +3 (−750 to +750 μm; Fig. 6), given its direct exposure to focused incident light and hence greatest propensity to cell loss. Progressive reduction in thickness was evident with increasing duration of intense light from light microscopic comparison of glycol methacrylate embedded sections in both Grk1+ and WT mice. Central outer retinal thicknesses for Grk1+ and WT retinas were not significantly different at 0 and 3 hours after exposure (Figs. 6A, 6B). At the 12-hour time point, however, a reproducible, statistically significant 80% (P < 0.0004) reduction in ONL thickness was noted in the Grk1+ line compared with that in the WT (Fig. 6A). This strongly suggests a different rate of cell loss at 12 hours. The decrease in thickness extended into the retinal periphery beyond the –3 to +3 positions, but was confined to the photoreceptor layer (Figs. 6B, 6C). The far peripheral retina was spared photoreceptor damage in both Grk1+ and WT retinas, presumably as a result of its position near the poles of the lens, with little exposure to intense light (Fig. 6D). These findings support the notion that excess Grk1 does not provide added protection against photoreceptor damage and may instead aggravate photoreceptor cell loss in intense light.
Figure 6.

Evidence of differential photoreceptor cell loss in Grk1+ and WT retinas. (A) Average ONL thickness for Grk1+ central retina positions −3 to 3 is shown as a percent of thickness of WT ONL in the corresponding region after equivalent treatments for 0, 3, and 12 hours in intense light (10,000 lux). Rpe65 Leu450Met heterozygous, sex-matched mice underwent dark adaptation before exposure to intense light and remained 10-days in the dark after exposure before euthanatization and retrieval of the globes (n = 6–12 per time point). **Statistical significance, P < 0.0004. (B, C) Comparison of ONL thickness across the entire superior (+0–9) and inferior (−9–0) retina between untreated and treated mice. (D) Representative central and far peripheral retinas of Grk1+ and WT mice after 12-hour exposure to intense light. Because of the severe light-induced damage in the central retina of the Grk1+ animals, the outer and inner photoreceptor segments were obliterated, with the ONL reduced to a single row of nuclei against a markedly thinned OPL. OS, outer segment; ON, optic nerve; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Apoptosis in the Retina of Grk1+ Mice
To determine whether increased apoptosis is the mechanism of the observed differences in photostress-related death, we used ELISA to quantify and compare the levels of nucleosome released from Grk1+ and WT mouse retinas after exposure to an intense light environment. As seen in Figure 7A, after 12 hours of exposure to intense light, the immunoreactive nucleosomes were present at substantial levels that were easily measurable in both Grk1+ and WT extracts, even at the 1:400 dilutions of the supernatants. However, there was no significant difference between the Grk1+ and WT samples, suggesting a similar apoptotic response to intense light, despite higher photoreceptor loss in Grk1+ mice. Soluble nucleosomes were not detectible in supernatants prepared from Grk1+ retinas in the absence of intense light.
Figure 7.

Apoptosis in Grk1+ and WT retinas. (A) Nucleosome release was measured in Grk1+ and WT retinas using immunoassay. Litter, sex, Rpe65 genotype (Leu450Met), and pigment-matched mice were exposed to either cyclic light (CL, only GRK+) or intense light (10,000 lux) for 12 hours. Diluted retinal lysate from individual eyes (20 μL) was examined for the presence of immunoreactive nucleosomes by using sandwich ELISA in microtiter well plates. Absorbance at λ405 nm (OD) was measured on a plate reader (n = 8 for both Grk1+ and WT). (B) TUNEL staining of Grk1+ and WT eyes was performed after exposure of the mice to ambient cyclic light (CL, 60 lux) or intense light (IL,10,000 lux) after 12 to 24 hours of dark adaptation. Scale bar: solid, 25 μm; dashed, 7 μm.
To localize site of apoptosis responsible for the observed nucleosome release, we performed TUNEL staining. TUNEL staining was evident only in the photoreceptor nuclei after 12 to 24 hours of exposure to intense white light. TUNEL-positive nuclei were confined to the outer nuclei and were not seen in any of the eye sections from mice reared in room light (Fig. 7B).
Discussion
Prior studies suggested that excess bleach or protracted lifetime of constitutively active rhodopsin may increase the susceptibility of photoreceptors to cell death, even in the absence of phototransduction signaling. Phosphorylation of light-activated opsin by Grk1 and subsequent capping by visual arrestins provides a mechanism of channeling the activated intermediate away from visual signal transduction pathway and may serve as a potential mechanism for mitigating cell death. Shifting the balance in favor of this inactivation pathway by overexpression of Grk1, however, did not confer the expected resistance based on our findings and increased retinal susceptibility to photoreceptor cell loss under prolonged exposure to bright light. This paradoxical effect, while somewhat unexpected, underscores the crucial role of balance between inactivation and activation for optimal survival of photoreceptors exposed to light-induced stress. This effect also points to a potential detrimental role for excess inactivation in photoreceptor cell death, consistent with some of the findings that have been reported in the literature.31,34
The Grk1+ transgenic retinas retained morphology and rod amplitude indistinguishable from their WT counterparts, at least to 18 months of age, when reared in subsaturating light or even when exposed to relatively brief periods of intense light. The relative resilience of the Grk1+-overexpressing retinas has further been corroborated by Krispel et al.,53 who showed normal morphology and physiology of photoreceptors in a separate transgenic mouse line overexpressing bovine GRK1 in rod photoreceptors under the control of a rhodopsin promoter. At this point, the effect of intense light on Grk1 levels is unknown. Although such changes could affect the expression or stability of protein during the course of treatment, the relative overexpression levels should theoretically be maintained, as the Grk protein and gene in both Grk1+ and WT mice are identical in every respect and hence are expected to respond similarly to light and other genetic modifiers during the course of treatment.
Prolonged photostress in our experimental paradigm unmasked a higher susceptibility of Grk1+ photoreceptors to cell loss. With nearly the entire rhodopsin population expected to be activated within 1 minute at 10,000 lux in both Grk1+ and WT retina, a toxic threshold level of hyperphosphorylated opsin adducts may be reached faster in the presence of higher levels of Grk1, explaining the observed differences in susceptibility. Prior studies have suggested that the aberrantly active rhodopsin with K296E opsin mutation,34 responsible for human retinitis pigmentosa, is hyperphosphorylated in transgenic mice and that this hyperphosphorylation, rather than the persistent activity, may be the cause of cell death in mice carrying this human mutation. In addition, mice deficient in Rpe65 and the 11-cis retinal opsin chromophore accumulate apo-opsin in the phosphorylated state in the retina before the ensuing degeneration. Opsin hyperphosphorylation54 and the resulting increased affinity for Arr1 has been associated with internalization of the complex and photoreceptor degeneration in Drosophila as well.55–57 Phospho-opsin–Arr1 interactions have been further found to affect the nuclear localizations of cell death mediators including JNK3 and Mdm2.48 In the context of light-dependent cell death, two pathways have been described in the mouse, both triggered by rhodopsin activation.16,58 A transducin-dependent path parallels the visual phototransduction signaling path and mediates damage from low light. A second poorly characterized transducin-independent pathway mediates damage by intense light. Our findings suggest that excess flow of rhodopsin through the Grk1-visual arrestin pathway may be responsible for the toxic effects of intense light, as illustrated in the model in Figure 8. The model highlights that, while favoring deactivation by Grk1 overexpression may be inconsequential or even protective in dim light, an increased flux of activated rhodopsin, driven preferentially en mass into a phosphorylated arrestin-bound state in Grk1+ animals, could equate with increased susceptibility to cell death.
Figure 8.

A model for Grk1-mediated photoreceptor cell death. Phototransduction pathways in dark, low, and intense light in model WT and Grk1+ photoreceptors are illustrated. In the absence of light, the light-dependent pathways including transducin-dependent visual signaling and Grk1-mediated deactivation are both quiescent, regardless of the levels of Grk1. In low light, both light-dependent pathways become stimulated with increased rhodopsin flow through both activation and deactivation channels. In this state, biased flow toward the deactivation pathway has little impact on photoreceptor integrity and may actually be protective. In intense light, the differential flow of signal through the Grk1-mediated deactivation pathway becomes a liability acting as a transducin-independent cell death pathway. Dashed arrows: subnormal flux through a pathway below that expected of the WT levels. Progressively bolder solid arrows: increased flux over those in the WT state.
Apoptosis has been regarded as the mechanism responsible for photoreceptor degeneration under light-induced stress6,12,59; however, it is increasingly apparent that other extra-apoptotic mechanisms could play a significant role in photoreceptor cell death. Although we did not find any dramatic increase in apoptosis in Grk1+ mice, the possibility of a small difference cannot be ruled out, given the scatter inherent in the nucleosome release and TUNEL staining. We also used real-time reverse transcription-PCR to compare the baseline transcript levels of several apoptosis mediators in Grk1+ and WT retinas previously associated with light-induced damage,40 including the Ap1 components c-Fos and c-Jun, as well as Bad, Bax, Bcl2, and the caspases, and did not find any differences in the levels (data not shown). Apoptosis-independent cell death pathways, such as necrosis and autophagy could also contribute to photoreceptor loss mediated by Grk1 in intense light.60 Of note, we found increasing levels of inner segment rhodopsin and Grk1 staining in Grk1+ photoreceptors, especially after loss of outer segments, suggesting involvement of internalized outer segment proteins or vacuoles in cell death (data not shown). Studies in Drosophila have shown internalization of the opsin–arrestin complex to be a key trigger of cell death, but evidence of such mechanism in vertebrate models has been sparse.54 It is unlikely that light-induced apoptotic DNA fragmentation elsewhere in the retina overwhelmed and masked apoptotic differences in photoreceptor layer, given that virtually all apoptotic nuclei observed in TUNEL assays were confined to the photoreceptor layer.
Our findings suggest that Grk1 expression levels could act as modifiers of photoreceptor susceptibility in the face of environmental light-induced stress. Diseases ranging from age-related macular degeneration to retinitis pigmentosa are known for their heterogeneity, and part of this vast heterogeneity is likely to be dictated by variable response to environmental changes and stress throughout a lifetime. Study of the precise role of the photoreceptor deactivation pathway components as disease modifiers and mechanisms remains an important future area of investigation. Furthermore, since Grk1+ mice express excess Grk1 in both rods and cones, the availability of these mice provides key opportunities to study, by single-cell recordings, the impact of the Grk1 pathway on the deactivation and recovery of photoreceptors.
Acknowledgments
The authors thank Kenneth Gross for helpful discussions on BAC transgenesis, and Mary Kay Ellsworth for excellent help in propagation and maintenance of the mouse lines.
Footnotes
Supported by a challenge grant from Research to Prevent Blindness and by National Institutes of Health Grants EY016662 to the Department of Ophthalmology at the State University of New York at Buffalo, EY02687 to the Department of Ophthalmology and Visual Sciences at Washington University, EY03040 to the Doheny Eye Institute Department of Ophthalmology and Cell and Neurobiology at the University of Southern California, R01EY015851 (CMC) and R01EY19312 (VJK). VJK is the recipient of a Career Development Award from Research to Prevent Blindness and the Karl Kirchgessner Foundation. CMC is the Mary D. Allen Chair in Vision Research at Doheny Eye Institute and a recipient of a Senior Scientific Investigator Award from Research to Prevent Blindness.
Disclosure: T. Whitcomb, None; K. Sakurai, None; B.M. Brown, None; J.E. Young, None; L. Sheflin, None; C. Dlugos, None; C.M. Craft, None; V.J. Kefalov, None; S.C. Khani, None
References
- 1.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy: the Beaver Dam eye study. Ophthalmology 1992;99:933–943 [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology 2007;114:253–262 [DOI] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368:1795–1809 [DOI] [PubMed] [Google Scholar]
- 4.Rattner A, Nathans J. Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci 2006;7:860–872 [DOI] [PubMed] [Google Scholar]
- 5.Stone J, Maslim J, Valter-Kocsi K, et al. Mechanisms of photoreceptor death and survival in mammalian retina. Prog Retin Eye Res 1999;18:689–735 [DOI] [PubMed] [Google Scholar]
- 6.Adler R, Curcio C, Hicks D, Price D, Wong F. Cell death in age-related macular degeneration. Mol Vis 1999;5:31 [PubMed] [Google Scholar]
- 7.Travis GH. Mechanisms of cell death in the inherited retinal degenerations. Am J Hum Genet 1998;62:503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson GR, Owsley C, Curcio CA. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res Rev 2002;1:381–396 [DOI] [PubMed] [Google Scholar]
- 9.Mendes HF, van der SJ, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med 2005;11:177–185 [DOI] [PubMed] [Google Scholar]
- 10.Marigo V. Programmed cell death in retinal degeneration: targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle 2007;6:652–655 [DOI] [PubMed] [Google Scholar]
- 11.Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, et al. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol 2008;38:253–269 [DOI] [PubMed] [Google Scholar]
- 12.Chang GQ, Hao Y, Wong F. Apoptosis. final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 1993;11:595–605 [DOI] [PubMed] [Google Scholar]
- 13.Reme CE, Grimm C, Hafezi F, Iseli HP, Wenzel A. Why study rod cell death in retinal degenerations and how? Doc Ophthalmol 2003;106:25–29 [DOI] [PubMed] [Google Scholar]
- 14.Organisciak DT, Darrow RM, Barsalou L, et al. Light history and age-related changes in retinal light damage. Invest Ophthalmol Vis Sci 1998;39:1107–1116 [PubMed] [Google Scholar]
- 15.Elizabeth RP, Yu MJ, Nusinowitz S, Chang B, Heckenlively JR. Mouse models of age-related macular degeneration. Exp Eye Res 2006;82:741–752 [DOI] [PubMed] [Google Scholar]
- 16.Hao W, Wenzel A, Obin MS, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet 2002;32:254–260 [DOI] [PubMed] [Google Scholar]
- 17.Brill E, Malanson KM, Radu RA, et al. A novel form of transducin-dependent retinal degeneration: accelerated retinal degeneration in the absence of rod transducin. Invest Ophthalmol Vis Sci 2007;48:5445–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol 2006;90:1060–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomany SC, Cruickshanks KJ, Klein R, Klein BE, Knudtson MD. Sunlight and the 10-year incidence of age-related maculopathy: the Beaver Dam Eye Study. Arch Ophthalmol 2004;122:750–757 [DOI] [PubMed] [Google Scholar]
- 20.Algvere PV, Marshall J, Seregard S. Age-related maculopathy and the impact of blue light hazard. Acta Ophthalmol Scand 2006;84:4–15 [DOI] [PubMed] [Google Scholar]
- 21.Palczewski K, Saari JC. Activation and inactivation steps in the visual transduction pathway. Curr Opin Neurobiol 1997;7:500–504 [DOI] [PubMed] [Google Scholar]
- 22.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem 1998;67:653–692 [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto S, Sippel KC, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997;15:175–178 [DOI] [PubMed] [Google Scholar]
- 24.Khani SC, Nielsen L, Vogt TM. Biochemical evidence for pathogenicity of rhodopsin kinase mutations correlated with the Oguchi form of stationary night blindness. Proc Natl Acad Sci USA 1998;95:2824–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 1995;10:360–362 [DOI] [PubMed] [Google Scholar]
- 26.Nakazawa M, Wada Y, Tamai M. Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmol 1998;116:498–501 [DOI] [PubMed] [Google Scholar]
- 27.Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA 1999;96:3718–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Brown B, Rife L, Craft CM. Slowed photoresponse recovery and age-related degeneration in cones lacking G protein-coupled receptor kinase 1. Adv Exp Med Biol 2006;572:133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi S, Hao W, Chen CK, Simon MI. Gene expression profiles of light-induced apoptosis in arrestin/rhodopsin kinase-deficient mouse retinas. Proc Natl Acad Sci USA 2001;98:13096–13101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li T, Franson WK, Gordon JW, Berson EL, Dryja TP. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci USA 1995;92:3551–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Shi G, Concepcion FA, Xie G, Oprian D, Chen J. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci 2006;26:11929–11937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen LB, McCormick SP, Young SG. A new approach for studying gene regulation by distant DNA elements in transgenic mice. Scand J Clin Lab Invest Suppl 1999;229:33–39 [DOI] [PubMed] [Google Scholar]
- 33.Heintz N. BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci 2001;2:861–870 [DOI] [PubMed] [Google Scholar]
- 34.Rim J, Oprian DD. Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry 1995;34:11938–11945 [DOI] [PubMed] [Google Scholar]
- 35.Rim J, Faurobert E, Hurley JB, Oprian DD. In vitro assay for trans-phosphorylation of rhodopsin by rhodopsin kinase. Biochemistry 1997;36:7064–7070 [DOI] [PubMed] [Google Scholar]
- 36.Heintz N. Analysis of mammalian central nervous system gene expression and function using bacterial artificial chromosome mediated transgenesis. Hum Mol Genet 2000;12:937–943 [DOI] [PubMed] [Google Scholar]
- 37.Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci 2001;21:53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SR, Fishkin N, Kong J, Nakanishi K, Allikmets R, Sparrow JR. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci USA 2004;101:11668–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young JE, Gross KW, Khani SC. Conserved structure and spatiotemporal function of the compact rhodopsin kinase (GRK1) enhancer/promoter. Mol Vis 2005;11:1041–1051 [PubMed] [Google Scholar]
- 40.Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat Genet 2000;25:63–66 [DOI] [PubMed] [Google Scholar]
- 41.Young JE, Kasperek EM, Vogt TM, Lis A, Khani SC. Conserved interactions of a compact highly active enhancer/promoter upstream of the rhodopsin kinase (GRK1) gene. Genomics 2007;90:236–248 [DOI] [PubMed] [Google Scholar]
- 42.Knospe V, Donoso LA, Banga JP, Yue S, Kasp E, Gregerson DS. Epitope mapping of bovine retinal S-antigen with monoclonal antibodies. Curr Eye Res 1988;7:1137–1147 [DOI] [PubMed] [Google Scholar]
- 43.Zhu X, Brown B, Li A, Mears AJ, Swaroop A, Craft CM. GRK1-dependent phosphorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Neurosci 2003;23:6152–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young JE, Vogt T, Gross KW, Khani SC. A short, highly active photoreceptor-specific enhancer/promoter region upstream of the human rhodopsin kinase gene. Invest Ophthalmol Vis Sci 2003;44:4076–4085 [DOI] [PubMed] [Google Scholar]
- 45.Adamus G, Zam ZS, McDowell JH, Shaw GP, Hargrave PA. A monoclonal antibody specific for the phosphorylated epitope of rhodopsin: comparison with other anti-phosphoprotein antibodies. Hybridoma 1988;7:237–247 [DOI] [PubMed] [Google Scholar]
- 46.Lee RH, Brown BM, Lolley RN. Autophosphorylation of rhodopsin kinase from retinal rod outer segments. Biochemistry 1982;21:3303–3307 [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis 2002;8:462–471 [PubMed] [Google Scholar]
- 48.Song X, Gurevich EV, Gurevich VV. Cone arrestin binding to JNK3 and Mdm2: conformational preference and localization of interaction sites. J Neurochem 2007;103:1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sillman AJ, Ito H, Tomita T. Studies on the mass receptor potential of the isolated frog retina. I. General properties of the response. Vision Res 1969;9:1435–1442 [DOI] [PubMed] [Google Scholar]
- 50.Bolnick DA, Walter AE, Sillman AJ. Barium suppresses slow PIII in perfused bullfrog retina. Vision Res 1979;19:1117–1119 [DOI] [PubMed] [Google Scholar]
- 51.Nymark S, Heikkinen H, Haldin C, Donner K, Koskelainen A. Light responses and light adaptation in rat retinal rods at different temperatures. J Physiol 2005;567:923–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao X, Huang J, Khani SC, Palczewski K. Molecular forms of human rhodopsin kinase (GRK1). J Biol Chem 1998;273:5124–5131 [DOI] [PubMed] [Google Scholar]
- 53.Krispel CM, Chen D, Melling N, et al. RGS expression rate-limits recovery of rod photoresponses. Neuron 2006;51:409–416 [DOI] [PubMed] [Google Scholar]
- 54.Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem 2007;282:32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iakhine R, Chorna-Ornan I, Zars T, et al. Novel dominant rhodopsin mutation triggers two mechanisms of retinal degeneration and photoreceptor desensitization. J Neurosci 2004;24:2516–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron 2000;28:129–138 [DOI] [PubMed] [Google Scholar]
- 57.Satoh AK, Ready DF. Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol 2005;15:1722–1733 [DOI] [PubMed] [Google Scholar]
- 58.Chen L, Wu W, Dentchev T, et al. Light damage induced changes in mouse retinal gene expression. Exp Eye Res 2004;79:239–247 [DOI] [PubMed] [Google Scholar]
- 59.Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res 2005;24:275–306 [DOI] [PubMed] [Google Scholar]
- 60.Reme CE, Wolfrum U, Imsand C, Hafezi F, Williams TP. Photoreceptor autophagy: effects of light history on number and opsin content of degradative vacuoles. Invest Ophthalmol Vis Sci 1999;40:2398–2404 [PubMed] [Google Scholar]