

Abstract
We present a comparative study of two-photon excited fluorescence (TPEF) and second harmonic generation (SHG) imaging in turbid media at 800- and 1300-nm excitation. The depth-dependent decay of TPEF and SHG signals in turbid tissue phantoms is used to estimate the impact of light scattering on excitation intensity at each wavelength. A 50 to 80% increase in scattering length is observed using 1300-nm excitation, while peak TPEF emission intensity is obtained 10 to 20 μm beneath the surface for both sources. The increased penetration depth at 1300 nm is confirmed by TPEF and SHG microscopy of tissue phantoms composed of gelatin/microspheres and 3-D organotypic collagen-fibroblast cultures, respectively. Our results establish the feasibility of 1.3-μm excitation in nonlinear optical microscopy.
Keywords: two-photon excited fluorescence, second harmonic generation, nonlinear optical microscopy
The use of NIR ultrafast sources facilitates deep tissue penetration and enables simultaneous generation of contrast from multiple nonlinear processes such as two-photon excited fluorescence (TPEF), harmonic generation, and coherent anti-Stokes Raman scattering (CARS).
Improving multiphoton microscopy (MPM) imaging depth is a major technical challenge. The depth limits of MPM are a strong function of tissue light scattering. Each scattering event potentially results in the loss of coherent or ballistic excitation intensity in the focal plane and a commensurate quadratic reduction of MPM signal.1
It has been shown that the maximum depth (zmax) in TPEF microscopy scales as:
| (1) |
where is the scattering length for excitation light in a sample. Pave, f, and τ, are the laser average power, repetition rate, and pulse width, respectively.2 φ is the collected fraction of the generated fluorescence, and α is a setup parameter.
The most common strategies used to increase penetration depth in TPEF microscopy are increasing the laser average power and optimizing the collection efficiency of the microscope. Conversely, increasing Pave can lead to excitation saturation, photobleaching, and photodamage.
A second parameter that can be adjusted to increase penetration depth is the laser pulse width. Employing smaller τ yields larger zmax, as shown in Eq. (1). We have previously demonstrated improvement in penetration depth when using shorter laser pulse widths.3 However, using shorter pulses may decrease the efficiency of signal generation due to the fact that the spectrum associated with a short pulse might become broader than the absorption band of a fluorophore.
Theer et al. have demonstrated that the decrease of the repetition rate of the laser leads to increased penetration depth in TPEF microscopy [Eq. (1)].4 They were able to reach a depth of 1000 μm in mouse brain, although the use of low repetition rate lasers can result in a slower imaging rate.
From Eq. (1) it is evident that another approach to increase penetration depth in biological tissues is to increase the scattering length by using longer excitation wavelengths.
Because of the wavelength tuning limitation of Ti:sapphire lasers, there are only a few reports that describe the effect of longer excitation wavelengths on multiphoton microscopy.2,5
These studies confirm that NIR wavelengths substantially above ~1000 nm can be used as excitation sources in nonlinear microscopy. However, no rigorous wavelength-dependent comparisons have been performed to assess MPM performance in turbid tissues. Consequently, in this work, we present a quantitative analysis of the effect of excitation wavelength on penetration depth at 800 and 1300 nm using the depth-dependent decay of TPEF and SHG signals as a standardized performance metric.
A direct quantitative comparison was achieved by measuring the depth-dependent TPEF in turbid tissue phantoms with embedded fluorescent microspheres. Since only ballistic photons contribute to signal generation, fluorescence generated at the focal volume decreases exponentially with increasing depth (z):
| (2) |
where ls is the scattering length defined as the average distance between scattering events. In addition, we imaged labeled human fibroblast cells in collagen matrices using TPEF and SHG signals, respectively.
The experimental setup is shown in Fig. 1. The laser source was a Mai Tai® Ti:sapphire oscillator [Spectra-Physics, Mountain View, California, sub-100-fs pulses, 80 MHz, tunning wavelength 690 to 1020 nm]. The output of the Mai Tai® was used either directly for imaging or as a pump for an OPO (OPAL® Spectra-Physics) delivering tunable femtosecond pulses from 1.1 to 2.25 μm. The laser light was directed toward our home-built microscope and focused into the sample by a Zeiss Plan-Neofluar 40×0.75 NA objective. Images were acquired by a computer running MPScope software.6
Fig. 1.
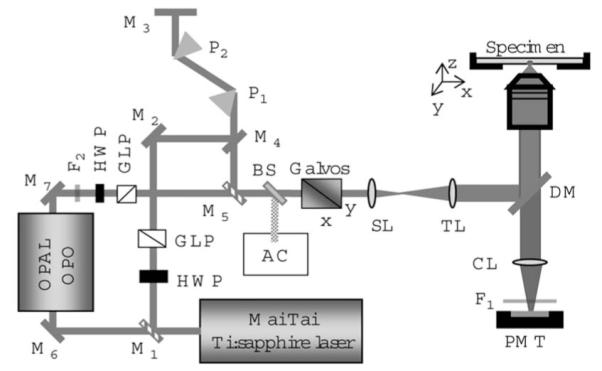
Schematic of the experimental setup. HWP: half-wave plate; GLP: Glan-laser polarizer; M: mirror (M1 and M5 mirrors held in flip mounts); BS: beam sampler; P: prism; AC: autocorrelator/spectrometer; SL: scanning lens; TL: tube lens; DM: dichroic mirror; CL: collection lens; F: filter; and PMT: photomultiplier tube.
Quantitative measurements of the effect of excitation wavelengths on the depth-dependent signal decay in TPEF microscopy were performed in phantoms with identical tissue-like scattering properties. The 800-nm phantom consisted of agarose gel containing as scatterers 1.8-μm nonfluorescent beads (Bangs Laboratories, Fishers, Indiana) at a concentration of 5.7×108 beads/mL, and as tracers 0.5-μm yellow-green (505/515) fluorescent beads (Molecular Probes, Eugene, Oregon) at a concentration of 2.5×109 beads/mL. The scattering length of the sample was estimated to be 200 μm by the Mie scattering calculator.7 The scattering of the sample was considered to be given mainly by the nonfluorescent beads. For the phantom used for 1300-nm excitation, the yellow-green fluorescent beads were replaced with 1-μm carboxylate microspheres (Molecular Probes) at a concentration of 4.5×108 beads/mL and labeled with a red-shifted dye (625/645) that can be efficiently excited by 1300 nm.
For the TPEF and SHG imaging studies, we used stained organotypic tissue models (RAFTs).8 They consisted of human dermal fibroblast cells dispersed in type-1 rat-tail collagen. The cells were stained with chloroaluminum sulfonated phtalocyanine (CASPc), a widely used photodynamic therapy agent and an efficient two-photon fluorophore at both 800 and 1300 nm.
TPEF signals measured in phantoms are plotted versus depth in Fig. 2(a) for 800- and 1300-nm excitation. The signal recorded at each depth was the average of the fluorescence peak intensities of five tracer beads at different x-y locations in the same z plane. The standard deviation of the measurements ranges between 5 and 10%. The laser power used for 800 (30 mW) and 1300 nm (70 mW) was adjusted such that the average intensity of spheres imaged at z=0 μm was the same and close to saturation for both wavelengths. All other experimental conditions such as pixel rate (0.67 MHz), frame dimensions (512×512 pixels), and PMT settings were kept identical.
Fig. 2.
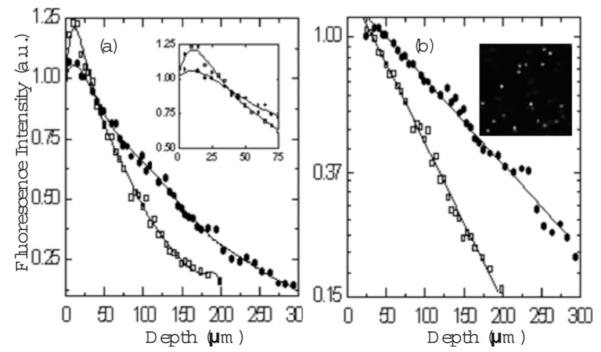
TPEF of fluorescent polystyrene beads in scattering agarose gels. (a) Fluorescent signal versus penetration depth for 800-nm (◻) and 1300-nm ([●) excitation wavelengths. The solid curves are a guide to the eye. (b) TPEF intensities (logarithmic scale) versus penetration depth for data starting from 25 μm. Inset: image of the phantom at 15 μm depth, using 1300-nm excitation wavelength.
For both excitation wavelengths, TPEF intensities reached a maximum value at a depth of 15 μm. The signal generated by using 800-nm excitation wavelength exhibits a slightly higher peak than the signal generated by using 1300-nm excitation wavelength, as shown in the inset of Fig. 2(a). At larger depths, TPEF signals dropped rapidly, becoming comparable to noise at depths of around 175 and 300 μm for 800-and 1300-nm excitation wavelengths, respectively.
The presence of fluorescence maxima at 15-μm depths [Fig. 2(a)] for both excitation wavelengths is a consequence of subsurface scattering, as predicted in Ref. 9. Although TPEF is expected to decrease monotonically with increasing depth [Eq. (2)], collection efficiency close to the source in an epifluorescent geometry can be enhanced by scattering. When the laser is focused on the surface of the phantom, only the photons emitted inside the cone angle defined by the numerical aperture (θNA) of the objective can be collected. When the laser is focused ~15 μm inside the phantom, additional fluorescent photons outside θNA are collected through backscattering. Furthermore, the larger collection enhancement observed when using 800- versus 1300-nm excitation wavelengths is attributed to increased scattering of the emitted photons at shorter wavelengths. In our experiment, the emission sources consisted of fluorescent photons corresponding to a maximum of 645 nm for 1300-nm excitation and to a maximum of 515 nm for 800-nm excitation. Further studies are necessary for quantifying the signal enhancement attributed to different emission sources.
Considering only TPEF intensities generated at penetration depths larger than 15 μm, logarithmic plots of signal versus depth display excellent linear fits to Eq. (2) [Fig. 2(b)]. Figure 2(b) also shows a representative image of the phantom at 15 μm depth and 1300-nm excitation. The decay constant is quantified by the total attenuation coefficient μt, defined as a linear combination of the absorption μa and scattering μs coefficients for both excitation and emission photons. In our samples, most of the absorption is due to water. Since the absorption coefficients of water at 800 and 1300 nm are several orders of magnitude smaller than the scattering coefficients, the attenuation coefficient can be attributed to scattering only. Fluorescent photons generated in the laser focal point are emitted in all directions, while ballistic photons are required for signal production. Consequently, the scattering coefficient of the excitation photons carries a larger weight than the scattering coefficient of the emission photons.1 Therefore, the attenuation coefficient can be approximated by the scattering coefficient of the excitation photons μs=1/ls. The scattering lengths corresponding to the excitation wavelengths can be retrieved from the slopes of the two curves. For 800-nm excitation, ls=181 μm, while for 1300-nm excitation, ls=333 μm. These values are comparable with ls=200 and 370 μm at 800 and 1300 nm, respectively, predicted by the Mie scattering calculator.7
For the RAFT sample, the total signal from TPEF and SHG was simultaneously recorded. Figure 3(a) shows the 3-D reconstruction of the sample corresponding to the 800-nm excitation. The 3-D reconstruction of the sample corresponding to 1300 nm is presented in Fig. 3(b). By placing long-pass filters at 400 and 650 nm in front of the PMT, we confirmed that only TPEF from stained cells was observable and the SHG signal originated exclusively from collagen fibers within the sample. From the 3-D representations of the entire raster-scanned volume with 800- and 1300-nm excitation wavelengths, it is clear that more signal can be obtained at greater depths when using 1300-nm excitation. The depth dependence of collagen fiber SHG signals are presented in Fig. 4 for 800- and 1300-nm excitation wavelengths. Following the same procedure as for the gelatin/microsphere phantom samples, we calculated scattering lengths from the SHG intensity decays. For 800-nm excitation, ls=235 μm, while for 1300-nm excitation, ls=357 μm, an increase of approximately 50%. We noticed that the signal obtained with the 1300-nm excitation could be increased even more by increasing the excitation average power without photobleaching. This was not the case when 800 nm was used as excitation.
Fig. 3.
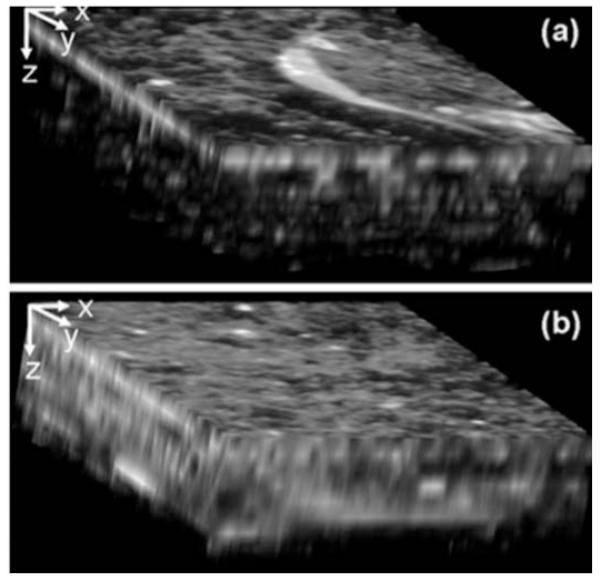
TPEF and SHG images of the RAFT model. 3-D reconstruction of the volume scanned within the sample excited with (a) 800 nm and (b) 1300 nm. The dimensions are 100×100×160 μm in the xyz directions (in the figure, x:z=1:5).
Fig. 4.
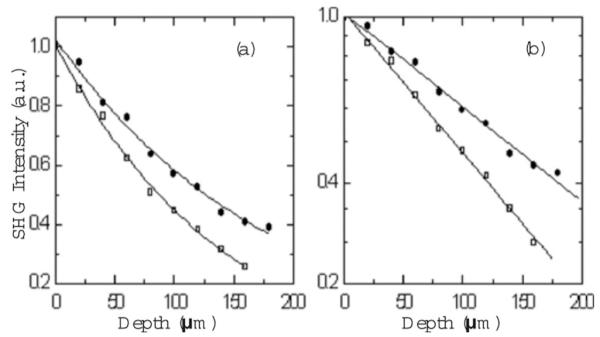
SHG signal of collagen. (a) SHG signal versus penetration depth for 800- (◻) and 1300-nm (●) excitation wavelengths. The solid curves are a guide to the eye. (b) SHG intensities (logarithmic scale) versus penetration depth.
In summary, we studied the effect of excitation wavelength on nonlinear optical microscopy in tissues by measuring the depth dependence of 800- and 1300-nm TPEF in tissue phantoms. Our results show a 50 and 80% increase in scattering lengths by using the longer excitation wavelength in tissue phantoms made of fibrillar collagen and gelatin, respectively. Practical advantages of using 1300-nm excitation are demonstrated by collecting TPEF and SHG images of stained cells and collagen at different depths within a collagen matrix. Although our system is not optimized for image quality and depth, it is expected that the use of sources in the 1300-nm region combined with properly designed microscopes, as described by Oheim et al. in Ref. 2, could result in 2- to 3-fold improvements in imaging depth compared to conventional 800-nm sources. This would significantly enhance MPM performance, particularly for in vivo imaging applications where there is considerable interest in accessing structures in the 500- to 1000-μm region.10
Acknowledgments
We thank Dr. Chung-Ho Sun for helpful discussions and for preparing the RAFTS, and Michael Herrick for his assistance with the OPAL. This research was supported partially by the NIH Laser Microbeam and Medical Program (P41-RR01192), the U.S. Air Force Office of Scientific Research, Medical Free-Electron Laser Program (F49620-00-2-0371 and FA9550-04-1-0101), and the Beckman Foundation.
References
- 1.Dunn AK, Wallace VP, Coleno M, Berns MW, Tromberg BJ. Influence of optical properties on two-photon fluorescence imaging in turbid samples. Appl. Opt. 2000;39:1194–1201. doi: 10.1364/ao.39.001194. [DOI] [PubMed] [Google Scholar]
- 2.Oheim M, Beaurepaire E, Chaigbneau E, Mertz J, Charpak S. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J. Neurosci. Methods. 2001;111:29–37. doi: 10.1016/s0165-0270(01)00438-1. also see erratum at Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J. Neurosci. Methods. 2001;112:205. doi: 10.1016/s0165-0270(01)00438-1.
- 3.Tang S, Krasieva TB, Chen Z, Tempea G, Tromberg BJ. Effect of pulse duration on two-photon excited fluorescence and second harmonic generation in nonlinear optical microscopy. J. Biomed. Opt. 2006;11:020501–020503. doi: 10.1117/1.2177676. [DOI] [PubMed] [Google Scholar]
- 4.Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000-μm in living brains by use of a Ti:Al2O3 rgenerative amplifier. Opt. Lett. 2003;28:1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- 5.Chu SW, Chen IH, Liu TM, Chen PC, Sun CK. Multimodal nonlinear spectral microscopy based on a femtosecond Cr-:forsterite laser. Opt. Lett. 2001;26:1909–1911. doi: 10.1364/ol.26.001909. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen QT, Tsai PS, Kleinfeld D. MPScope: a versatile software suite for multiphoton microscopy. J. Neurosci. Methods. 2006;156:351–359. doi: 10.1016/j.jneumeth.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 7. See http://omlc.ogi.edu/calc/mie_calc.html.
- 8.Chiu LL, Sun CH, Yeh AT, Torkian B, Karamzadeh A, Tromberg BJ, Wong BJF. Photodynamic therapy on keloid fibroblast in tissue-engineered keratinocyte-fibroblast co-culture. Lasers Surg. Med. 2005;37:231–244. doi: 10.1002/lsm.20213. [DOI] [PubMed] [Google Scholar]
- 9.Beaurepaire E, Mertz J. Epifluorescence collection in two-photon microscopy. Appl. Opt. 2002;41:5376–5382. doi: 10.1364/ao.41.005376. [DOI] [PubMed] [Google Scholar]
- 10.Tromberg BJ. “Current and emerging applications of multiphoton microscopy,” Introduction to Part IV. In: Masters BR, So PTC, editors. Handbook of Biomedical Nonlinear Optical Microscopy. Oxford Univ. Press; New York: 2008. pp. 707–715. [Google Scholar]