

Abstract
The role of the Alzheimer’s Disease Neuroimaging Initiative Genetics Core is to facilitate the investigation of genetic influences on disease onset and trajectory as reflected in structural, functional, and molecular imaging changes; fluid biomarkers; and cognitive status. Major goals include (1) blood sample processing, genotyping, and dissemination, (2) genome-wide association studies (GWAS) of longitudinal phenotypic data, and (3) providing a central resource, point of contact and planning group for genetics within Alzheimer’s Disease Neuroimaging Initiative. Genome-wide array data have been publicly released and updated, and several neuroimaging GWAS have recently been reported examining baseline magnetic resonance imaging measures as quantitative phenotypes. Other preliminary investigations include copy number variation in mild cognitive impairment and Alzheimer’s disease and GWAS of baseline cerebrospinal fluid biomarkers and longitudinal changes on magnetic resonance imaging. Blood collection for RNA studies is a new direction. Genetic studies of longitudinal phenotypes hold promise for elucidating disease mechanisms and risk, development of therapeutic strategies, and refining selection criteria for clinical trials.
Keywords: Alzheimer’s Disease Neuroimaging Initiative (ADNI), Alzheimer’s disease, Mild cognitive impairment (MCI), Genome-wide association studies (GWAS), Copy number variation (CNV), Magnetic resonance imaging (MRI), Cerebrospinal fluid (CSF)
1. Introduction
1.1. Genetic factors in late onset Alzheimer’s disease and mild cognitive impairment
Genetic factors play an important role in late-onset Alzheimer’s disease (LOAD) as demonstrated by twin data indicating heritability in the range of 58%–79% [1]. The epsilon 4 allele of APOE is the strongest known genetic risk factor for AD with a two- to three-fold increased risk for AD in people with one epsilon 4 allele rising to about 12-fold in those with two alleles. Many other genes have also been identified, but until very recently only APOE had been consistently replicated. The APOE ε4 allele is associated with an earlier age of onset of AD [2], and may account for up to 50% of AD heritability [3]. If AD is ~60%–80% heritable [1], then approximately 30% of the genetic variance is presently unexplained after accounting for APOE. Other genes associated with LOAD have been difficult to find. An up-to-date online encyclopedia of all genetic association studies in LOAD, including systematic meta-analyses across datasets investigating overlapping polymorphisms, can be found at http://www.alzgene.org/ [4]. Genetic studies have also been performed in patients with amnestic mild cognitive impairment (MCI) [5,6]. Many approaches are now being applied to identify genes that play a role in the development and progression of AD and MCI, as briefly discussed in the following sections.
1.2. Genome-wide association studies
Genome-wide association studies (GWAS) employ tests of association between markers, called single nucleotide polymorphisms (SNPs), distributed across the genome and a phenotype of interest, which could be dichotomous (affected, unaffected) or quantitative (fluid biomarker levels, rate of longitudinal change on imaging metrics, etc.). This approach has identified susceptibility loci in several diseases (see http://www.genome.gov/26525384). GWAS of AD, listed on http://www.alzgene.org/ [4], have confirmed the strong influence of APOE, but evidence implicating other genes has been less convincing, despite many biologically plausible and interesting candidates. Other susceptibility genes appear to have modest effects and require very large samples to detect them using a case-control design. Two large GWAS recently implicated several new genes (CLU, PICALM, and CR1) [7,8]. The National Institute on Aging (NIA) sponsored Alzheimer’s Disease Genetics Consortium (G. Schellenberg, Principal Investigator; http://alois.med.upenn.edu/adgc/) is attempting to replicate and extend these findings. The AlzGene database provides a continuously updated summary of these findings [4]. By design, its content and meta-analysis results are dynamically changing and reflect the continuing evolution of leading candidate genes for AD and the biological pathways they may represent, regardless whether they emerged from genome-wide or candidate-gene designs [9]. The most robust findings from case-control GWAS and other types of genetic association studies can provide targets for examining quantitative pheno-types derived from Alzheimer’s Disease Neuroimaging Initiative (ADNI) imaging and other biomarker data sets.
1.3. Imaging genetics
ADNI provides a unique opportunity to combine imaging and genetics. Advances in brain imaging and high throughput genotyping enable new approaches to study the influence of genetic variation on brain structure and function [10–12]. As a result, imaging genetics has become an emergent transdisciplinary research field, where genetic variation is evaluated using imaging measures as quantitative traits (QTs) or continuous phenotypes. Imaging genetics studies have advantages over traditional case-control designs. An important consideration is that QT association studies have increased statistical power and thus decreased sample size requirements [13]. Additionally, imaging phenotypes may be closer to the underlying biological etiology of AD, making it easier to identify underlying genes.
SNPs and other polymorphisms in several genes, including APOE, have been related to neuroimaging measures in brain disorders such as MCI and AD and also in nondemented carriers (eg, [14,15]). However, analytic tools that relate a single gene to a few imaging measures are insufficient to provide comprehensive insight into the multiple mechanisms and imaging manifestations of these complex diseases. Although GWAS are increasingly performed, effectively relating high density SNP data to large scale image data remains a challenging task. Prior studies typically make significant reductions in one or both data types [16]. Whole brain studies usually focus on a small number of genetic variables, whereas genome-wide studies typically examine a limited number of imaging variables [11,17,18]. This restriction of target genotypes and/or phenotypes greatly limits the capacity to identify important relationships. ADNI is contributing new methods that begin to address the high dimensionality of both the imaging and genomic data [6,19,20]. Important challenges in imaging genomics include identifying optimal statistical modeling approaches and addressing power limitations and multiple comparison issues. Given the nature of these challenges, multidisciplinary teams and methods are needed, and ADNI is an ideal test bed for development of new analytic methods.
1.4. Copy number variation
Copy number variations (CNVs) are segments of DNA, ranging from 1 kilobase (kb) to several megabases (Mb), for which differences in regional copy number have been revealed by comparing two or more genomes. These differences can be gains (duplications), losses (deletions), or other more complex rearrangements. CNVs have been implicated in autism, schizophrenia, bipolar disorder, and cancer [21,22]. In addition, duplications of the APP gene on chromosome 21 have been shown to cause rare, early-onset familial forms of AD that follow Mendelian transmission. Only one study [23] has performed a genome-wide CNV scan in non-Mendelian LOAD, with 331 cases and 368 controls, but no new SNP CNVs were significant at a genome-wide threshold. However, a duplication in CHRNA7 was found, which warrants further investigation. Preliminary analysis of CNVs in the ADNI sample has suggested several genomic loci worthy of follow-up exploration and molecular verification [24,25]. CNV analysis may reveal variations in genomic microarchitecture associated with AD risk. Because CNVs may lead to increased or decreased expression of genes involved in AD, they might be useful in predicting response to particular treatments.
2. Genotyping the ADNI cohort
2.1. ADNI background, sample sources, and informed consent
Data discussed in this report were uploaded to and/or obtained from the ADNI database (www.loni.ucla.edu/ADNI). Detailed information about ADNI can be found at www.adni-info.org, and throughout the other papers in this Special Issue on ADNI. Clinical characteristics and methods are presented in References [26,27], magnetic resonance imaging (MRI) methods are in Reference [28], and those for cerebrospinal fluid (CSF) are in Reference [29]. The ADNI studies described later in the text were approved by institutional review boards of all participating institutions, and written informed consent was obtained from all participants or authorized representatives.
2.2. Brief history of the core and GWAS planning
Genetic assessment beyond APOE genotyping was not included in the original ADNI grant. Therefore, a Genetics Working Group (Members of the working group included: Andrew Saykin [Chair], Bryan DeChairo [Pfizer, industry representative, replaced by Elyse Katz in 2009], Lindsay Farrer, Tatiana Foroud, Robert Green, Steven Potkin, Eric Reiman, Gerard Schellenberg, Rudolph Tanzi, John Trojanowski, Christopher van Dyck, Michael Weiner and Kirk Wilhelmsen) was tasked in 2005 to develop plans for genetic studies. The working group considered potential data collection and analysis options, and recommended completing a GWAS using an Illumina Human BeadChip panel (Illumina, Inc, San Diego, CA). A proposal was generated and funds were obtained through the Foundation for National Institute of Health from Merck and Gene Network Sciences, and Pfizer contributed DNA extraction (by Cogenics) and other assistance to the project. A supplement from the National Institute of Biomedical Imaging and Bioengineering provided initial support for data analysis. The Illumina arrays were processed by TGen (Phoenix, AZ), a National Institute of Health Neuroscience Microarray Consortium site. Sample processing, storage, and distribution were provided by the National Institute on Aging-sponsored National Cell Repository for Alzheimer’s Disease (NCRAD). Quality control bioinformatics and sample identity confirmation were performed at Indiana University. This group worked with the ADNI Informatics Core to format, document, and post the final genotyping results on the ADNI Laboratory of Neuro Imaging (LONI) web site. With the ADNI Grand Opportunities (GO) grant in 2009, genetics efforts in ADNI were organized as a core to facilitate genetics research related to ADNI biomarkers in an integrated manner.
2.3. Genotyping methods
Genotyping using the Human610-Quad BeadChip (Illumina, Inc., San Diego, CA) included 620,901 SNP and CNV markers, and was completed on all ADNI-1 participants using the following protocol. A 7 mL sample of blood was taken in ethylenediaminetetraacetic acid (EDTA)-containing vacutainer tubes from all participants, and genomic DNA was extracted at Cogenics (now Beckman Coulter Genomics) using the QIAamp DNA Blood Maxi Kit (Qiagen, Inc, Valencia, CA) following the manufacturer’s protocol. Lymphoblastoid cell lines (LCLs) were established by transforming B lymphocytes with Epstein-Barr virus as described in Reference [30]. Genomic DNA samples were analyzed on the Human610-Quad BeadChip according to the manufacturer’s protocols (Infinium HD Assay; Super Protocol Guide; Rev. A, May 2008). Before initiation of the assay, 50ng of genomic DNA from each sample was examined qualitatively on a 1% Tris-acetate-EDTA agarose gel to check for degradation. Degraded DNA samples were excluded from further analysis. Of the initial 818 ADNI-1 samples, 731 were analyzed using DNA from peripheral blood and 87 were genotyped using DNA extracted from LCLs at NCRAD. Samples were quantitated in triplicate with PicoGreen® reagent (Invitrogen, Carlsbad, CA) and diluted to 50 ng/μL in Tris-EDTA buffer (10mM Tris, 1mM EDTA, pH 8.0). 200ng of DNA was then denatured, neutralized, and amplified for 22 hours at 37°C (this is termed the MSA1 plate). The MSA1 plate was fragmented with FMS re-agent (Illumina) at 37°C for 1 hour, precipitated with 2-prop-anol, and incubated at 4°C for 30 minutes. The resulting blue precipitate was resuspended in a resuspension, hybridization, and wash reagent (Illumina) at 48°C for 1 hour. Samples were then denatured (95°C for 20 minutes) and immediately hybridized onto the BeadChips at 48°C for 20 hours. The BeadChips were washed and subjected to single base extension and staining. Finally, the BeadChips were coated with XStain BeadChip solution 4 (Illumina), desiccated, and imaged on the BeadArray Reader (Illumina).
APOE genotyping was performed at the time of participant enrollment and included in the ADNI database. The two SNPs (rs429358, rs7412) that define the epsilon 2, 3, and 4 alleles, are not on the Human610-Quad BeadChip, and therefore were genotyped using DNA extracted by Cogenics from a 3 mL aliquot of EDTA blood, as described earlier in the text. Polymerase chain reaction amplification was followed by HhaI restriction enzyme digestion, resolution on 4% Metaphor Gel, and visualization by ethidium bromide staining [11].
2.4. Data availability, formats and updated release
BeadStudio 3.2 software was (Illumina) used to generate SNP genotypes from bead intensity data. After performing sample verification and quality control bioinformatics, the genotype data for 818 ADNI participants was uploaded to the ADNI website (http://www.loni.ucla.edu/ADNI) and publicly released in complete form on April 16, 2009. In the first 6 months, there were 42,270 downloads (counted as one per sample) by 94 different users.
GenomeStudio v2009.1 (Illumina), an updated version of BeadStudio, was recently used to reprocess the array data for all 818 samples. GenomeStudio project files containing the genotype information for all samples, as well as new Final Reports will be released in April 2010 on the ADNI LONI web site. In addition, the widely used PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/) data format will be provided to facilitate analysis by other groups. A comparison of calls for all 620,901 markers from the two versions of Final Reports for each sample indicated that only nine samples had mismatches in genotypes successfully called in the two versions, and those mismatches comprised <0.01% of all markers. Although the new files are highly similar to the initial release, these data may be helpful to investigators given the new formats and additional information provided. Finally, a new DNA source file is included to identify for each genotyped sample whether peripheral blood (n = 731) or immortalized LCL (n = 87) derived DNA was analyzed.
3. Initial progress in genetic investigations within ADNI
3.1. Genetic analyses of baseline MRI scans
Several baseline MRI GWAS have recently been published on line and are briefly summarized here along with a candidate gene study. Potkin et al. [11] performed the first GWAS of AD cases (n = 172) and controls (n = 209). A case-control analysis identified APOE and the adjacent risk gene, TOMM40 (translo-case of outer mitochondrial membrane 40), at the significance threshold of P < 10−6. A QT analysis using hippocampal atrophy as the phenotype identified 21 genes or chromosomal regions with at least one SNP with a P-value reaching 10−6, including EFNA5, CAND1, MAGI2, ARSB, and PRUNE2. These genes are involved in regulation of protein degradation, apoptosis, neuronal loss, and neurodevelopment. The TOMM40 finding is consistent with other recent GWAS results, and a recent sequencing study has generated considerable interest in possible independent influences of this gene [31].
Shen et al. [6] completed a GWAS of 729 ADNI participants on 142 brain-wide MRI phenotypic measures of grey matter density (GMD), volume, and cortical thickness derived using FreeSurfer automated parcellation and voxel-based morphometry methods, as previously described [32]. Data analyzed included genotypes based on 530,992 quality controlled SNPs from 175 AD, 351 MCI, and 203 controls. Cluster analysis of GWAS results (P < 10−7) confirmed SNPs in APOE and TOMM40 as strongly associated with multiple brain regions and revealed other novel SNPs proximal to the EPHA4, TP63 and NXPH1 genes. Detailed analyses of rs6463843 (flanking NXPH1) indicated reduced global and regional GMD in TT homozygotes and increased vulnerability to right hippocampal GMD loss in AD patients with the TT genotype. NXPH1 codes for neurexophilin-1, a protein implicated in synaptogenesis, that forms a tight complex with alpha neurexins, a group of proteins that promote adhesion between dendrites and axons. This adhesion is a key factor in synaptic integrity, the loss of which is a hallmark of AD.
Stein et al. [19] explored the relation between 448,293 quality-controlled SNPs and each of 31,622 voxels of the entire brain across 740 ADNI participants, where GWAS was performed on a tensor-based morphometry metric at each voxel location. A novel method was developed to address the multiple comparison problem and computational burden associated with this unprecedented amount of data. No SNP survived the strict significance threshold, but several genes including CSMD2 and CADPS2 were identified. These genes have high relevance to brain structure and are good candidates for further exploration.
Stein et al. [20] in a second GWAS focusing on regional structural brain degeneration, mapped the 3D profile of temporal lobe volume differences using tensor-based morphometry in 742 ADNI participants. This study identified rs10845840, a SNP located in GRIN2B, as associated with greater temporal lobe atrophy and with AD. It is particularly notable that GRIN2B encodes the N-methyl-D-aspartate glutamate receptor NR2B subunit, which is a target for memantine therapy to decrease excitotoxic damage.
Ho et al. [33] investigated the relationship between an obesity-associated candidate gene (FTO) and regional brain volume differences in 206 ADNI control participants. Systematic brain volume deficits were detected in cognitively normal obesity-associated risk allele carriers, as well as in subjects with increased body mass index. The results of this study indicate that a very common susceptibility gene for obesity is associated with detectable deficits in brain structure, which may indirectly influence future risk for neurodegenerative disease.
3.2. Preliminary candidate gene and GWAS of MRI changes at 12-month follow-up
Saykin et al. [34] examined genetic predictors of 12-month change in hippocampal volume and GMD using a candidate and GWAS approach. Scan data included baseline and 12-month 1.5T MRI scans from 627 non-Hispanic Caucasian ADNI participants (141 AD, 60 MCI to AD converters, 241 MCI-stable, 185 healthy controls) that were analyzed as in [6,32] with the additional step of registration of follow-up to baseline scans.
For the candidate gene analysis, common SNPs with high minor allele frequency (≥20%) in the top 30 genes of the Alz-Gene database [4] were examined. Regression models were performed using SAS/STAT 9.3 (SAS Institute Inc, Cary, NC) to test the ability of SNPs to predict hippocampal volume and GMD changes. Models including diagnostic group (AD, MCI to AD converters, MCI-stable, healthy controls), SNP (0,1,2), parental history of dementia (0,1,2), APOE ε4 carrier status (0,1), and interactions were computed. Covariates included baseline age, sex, education, handedness, and total intracranial volume. A total of 732 SNPs were available with minor allele frequency ≥20%. A corrected significance threshold of P < .01 was determined using Bonferroni adjustment (.01/732 = 1.37 × 10−5). Using this model and criterion, five AD genes (NEDD9, SORL1, DAPK1, IL1B, and SORCS1) showed significant SNPs associated with hippocampal volume or GMD changes after accounting for APOE, diagnosis, and other factors. In addition, SNPs from several other candidate genes (MYH13, TNK1, ACE, PRNP, MAPT, PCK1, GAPDHS, and APP) showed less robust indications of possible association that nonetheless encourage further investigation in relation to imaging phenotypes.
The preliminary GWAS was performed using PLINK software release v1.06 (http://pngu.mgh.harvard.edu/~purcell/plink/), which permits modeling of additive main effects for each SNP on a genome-wide basis but not group × genotype interaction effects as described for candidate gene analyses. Results of main effects for SNPs identified associations with hippocampal change measures. Manhattan plots show chromosomal locations for association peaks for hippocampal volume (Fig. 1) and GMD (Fig. 2). As expected, APOE (rs429358, the epsilon 4 marker) and TOMM40 (rs2075650) showed significant genome-wide association with both change measures. In addition, a SNP not previously linked to AD was significantly associated with rate of hippocampal volume loss. This SNP, rs12449237 located at 16q22.1, is intergenic between CDH8 (cadherin 8, type 2) and LOC390735. CDH8 codes for a calcium-dependent cell adhesion protein implicated in synaptic adhesion and axonal growth and guidance. Neuronal cadherin interacts with presenilin-1 [35], and cell adhesion molecules may be decreased in MCI and AD [36], suggesting the possibility of a mechanistic relationship to AD that warrants investigation. Several other loci or genes labeled in Figs. 1 and 2, while not reaching genome-wide significance, showed association signals worthy of follow-up, including SLC6A13 with volume change, and MAD2L2, LOC728574, QPCT and GRB2 with change in GMD.
Fig. 1.

Manhattan plot for percent change in hippocampal volume over 1 year.
Fig. 2.

Manhattan plot for percent change in hippocampal grey matter density over 1 year.
In sum, initial analyses indicate that common variations in major AD candidate genes, as well as other novel loci, are related to rate of longitudinal hippocampal structural change over 12 months in the ADNI cohort. Replication in independent samples and further characterization of these relationships over time will be important.
3.3. Copy number variation
CNVs are alterations in DNA sequence resulting in gains (duplications) and losses (deletions) of genomic segments. Because CNVs often encompass or overlap genes, they may have important roles in disease. Swaminathan et al. [25] analyzed CNVs in data from 193 AD, 116 MCI, and 131 control participants with blood-derived DNA samples that remained after CNV-specific quality control. Although no excess CNV burden was observed in AD and MCI cases relative to controls, gene-based analysis revealed CNVs in cases but not controls in regions of TP53TG3, CSMD1, SLC35F2, HNRNPCL1, and UBTFL2, as well as the candidate gene, CHRFAM7A. Using a different approach and larger sample, Macciardi et al. [24] observed large deletions that could disrupt pathways of biological significance including axonal development.
3.4. GWAS of cerebrospinal fluid biomarkers
Levels and ratios of CSF biomarkers (Aβ1–42, t-tau, p-tau181p, p-tau181p/ Aβ1–42, and t-tau/ Aβ1–42) have been shown to be promising biomarkers for AD diagnosis[27,29]. Kim et al. [37] conducted a GWAS of CSF bio-markers on 374 non-Hispanic Caucasian ADNI participants. This analysis identified SNPs in the regions of APOE, TOMM40, and EPC2 that reached genome-wide significance for one or more of the CSF biomarkers. In addition, association signals were noted in CCDC134, ABCG2, SREBF2 and NFATC4, although these did not reach genome-wide significance. Although APOE and TOMM40 are well-established candidate genes, EPC2 is a novel finding requiring further characterization and independent replication.
3.5. Imaging associations with PICALM
The recent large case/control GWAS noted earlier in the text [7,8] that identified new promising AD candidates did not include neuroimaging measures or MCI participants. We examined key brain regions implicated in early AD to determine whether variation in these new gene loci were predictive of structural changes. Entorhinal cortex (EC) neurodegeneration is one of the earliest known structural findings in MCI and AD. Fig. 3 shows the association of the previously reported PICALM (phosphatidylinositol-binding clathrin assembly protein) SNP and mean bilateral EC thickness at baseline [32]. Covariates included baseline age, sex, education, handedness, and intracranial volume. The number of G alleles of rs3851179 was linearly associated with reduction of EC thickness at baseline across groups (P < .002). This remained significant after adding APOE status as a covariate (P = .006), suggesting PICALM may have an independent contribution to neurodegenerative changes. Although there was a main effect of SNP across groups, the pattern of results as shown in Fig. 3 suggests that the combined AD and MCI-converter (probable AD within 12 months) group drive this effect. This finding illustrates the potential synergy between large case/control GWAS and an imaging genetics approach.
Fig. 3.
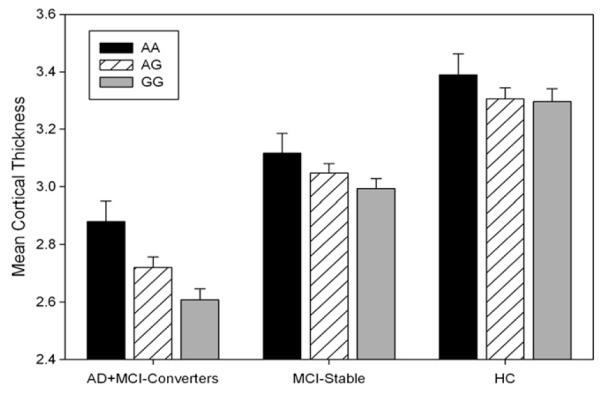
Mean (±SE) bilateral entorhinal cortex thickness on baseline MRI as a function of PICALM (rs3851179) genotype and diagnosis group. Participants who had a baseline diagnosis of MCI but converted to probable AD within a year are included with the AD at baseline group. The MCI-stable group included participants with a stable MCI diagnosis over 1 year. See text for details.
4. Future directions
Several rapidly emerging technologies have the potential to enhance the genetic information yield from ADNI-1 as well as to open new possibilities for investigating the influence of genetic variation in the ADNI-GO and planned ADNI-2 projects which will examine early amnestic MCI in addition to the current diagnostic groups. Here we briefly discuss future resequencing, RNA and replication studies.
4.1. Deep resequencing
Although GWAS have identified genes and chromosomal regions of interest, association does not permit inference of causation. As a result, targeted sequencing or deep resequencing of genes or regions associated with a phenotype is often employed to catalog all sequence variation (SNPs, insertion/deletions, etc), as a first step in determining which variation(s) directly contribute to disease risk. Deep resequencing has helped to determine how sequence variations in genes are associated with several psychiatric disorders [38–40]. These techniques hold promise for uncovering new variants that may be involved in the pathogenesis of LOAD, yet there has been limited application of this approach to date [41]. Promising genes and regions such as those described in this report can be selectively targeted for resequencing in the next few years. In 5–10 years, whole genome sequencing for all ADNI samples is likely to become cost effective.
4.2. Gene expression and RNA studies on peripheral blood samples
Analysis of aberrant gene expression profiles in AD is difficult because tissue samples for expression analysis cannot typically be acquired from living patients. Additionally, postmortem RNA degradation and protein modification may potentially confound the interpretation of data obtained from neuropathological samples. Expression profiling of peripheral blood mononuclear cells (PBMCs) may offer advantages in deciphering gene regulation patterns. Circulating blood is accessible and there is communication between the brain and immune system through multiple mechanisms that may be expressed in blood. In addition, abnormal APP expression, altered levels of antioxidant enzymes, oxidative damage to DNA, RNA, and protein, deregulated cytokine secretion, and augmented rates of apoptosis are features shared by AD brain and blood lymphocytes, and PBMCs have been successfully employed in investigating other neurological diseases [42]. Peripheral blood cells share more than 80% of the transcriptome with other tissues types including brain, suggesting that these cells can be used for molecular profiling in humans [43]. PBMCs have been used for gene expression profiling in AD with encouraging results [42,44,45].
4.3. MicroRNAs and gene regulation
MicroRNAs (miRNAs) are a large family of short (21–25 nucleotide), noncoding regulatory RNAs involved in post-transcriptional gene regulation and silencing. They bind to complementary sites typically located in the 3′ untranslated regions (3′-UTR) of target messenger RNA (mRNA) molecules. They repress mRNA expression, which results in gene silencing by interfering with translation or by targeting mRNA for degradation [46]. The miRNA database miRBase (http://www.mirbase.org/; version 13.0) identified 706 validated miRNAs in the human genome, although there may be a thousand or more. There is evidence that miRNAs play a role in synaptic development, plasticity, and long-term memory [47]. Recent studies have shown that miRNAs can regulate expression of genes such as APP and BACE1, suggesting that miRNAs may play a role in AD pathophysiology [48–50].
4.4. Replication, meta-analyses and collaboration with other cohort studies
It is essential for all genetics findings to undergo replication in independent samples given the heightened risk of false discovery. A challenge for replication studies of findings from ADNI, particularly for imaging genetics analyses, is the lack of comparable data sets. Fortunately, World Wide ADNI efforts will generate excellent replication opportunities in the future. Similarly, collaborative analyses of large pooled data sets with substantial power will become possible. In the interim, possibilities exist for replication and joint analysis studies with existing cohorts that acquired relatively similar neuroimaging and genetics data sets.
5. Conclusion
There is considerable momentum in genetic studies of AD and MCI. Although it is beyond the scope of ADNI to support longitudinal gene expression and mRNA studies and targeted deep resequencing at this time, the relevant data will be collected and banked and the core will provide a venue to foster new proposals and collaborations so that the most scientifically promising leads and directions are pursued.
The availability of advanced brain imaging techniques such as MRI and positron emission tomography paired with the ability to capture over a million genetic markers by current array technology makes this an exciting time for imaging genetics research, where there is great potential for discovery. Many promising preliminary results have emerged to be followed-up by replication and detailed characterization studies. As the ADNI-1 cohort is followed beyond the present 3 years and augmented by new participants and updated methods (e.g., amyloid positron emission tomography and CSF biomarkers), the prospects for generation of new knowledge regarding disease mechanisms that can inform treatment development and clinical trials design are compelling. Analyses addressing the role of genetic variation can be expected to enhance the value of this important initiative.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904 and RC2 AG036535-01). ADNI is funded by the NIA, the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as nonprofit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles.
Data analysis was supported in part by NIA grant R01 AG19771 and Indiana Economic Development Corporation (number 87884) to Dr Saykin; NIA P30 AG10133-18S1 to Drs B. Ghetti and Saykin; NIBIB R03 EB008674 and an Indiana CTSI award (NCRR RR025761) to Dr Shen, U01 AG032984 to Dr G. Schellenberg, P20 RR020837 to Dr Potkin, R01 N5059873 to Dr Huentelman, as well as U24 RR021992, P30 AG010129 and K01 AG030514 and a Dana Foundation grant. Samples from the NCRAD, support by U24 AG21886 to Dr Foroud, were used in this study.
Appendix
Appendix.
Expanded Gene Names and Chromosomal Locations
| Gene Abbreviation | Gene Name | Chromosomal Location |
|---|---|---|
| ABCG2 | ATP-binding cassette, sub-family G, member 2 | 4q22.1 |
| ACE | angiotensin I converting enzyme 1 | 17q23.3 |
| APOE | apolipoprotein E | 19q13.2 |
| APP | amyloid beta (A4) precursor protein | 21q21.3 |
| ARSB | arylsulfatase B | 5q14.1 |
| BACE1 | beta-site APP-cleaving enzyme 1 | 11q23.3 |
| CADPS2 | Calcium-dependent secretion activator 2 | 7q31.32 |
| CAND1 | cullin-associated and neddylation-dissociated 1; TBP-interacting protein TIP120A | 12q14.3 |
| CCDC134 | coiled-coil domain containing 134; | 22q13.2 |
| CDH8 | cadherin 8, type 2 | 16q21.1 |
| CHRFAM7A | cholinergic nicotinic receptor, alpha 7 subunit, exons 5-10 and family with sequence similarity 7A, exons A-E fusion |
15q13.2 |
| CHRNA7 | cholinergic nicotinic receptor, alpha 7 subunit | 15q13.3 |
| CLU | clusterin, apolipoprotein J | 8p21.1 |
| CR1 | complement component (3b/4b) receptor 1 | 1q32.2 |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 |
| CSMD2 | CUB and Sushi multiple domains 1 | 1p34.3 |
| DAPK1 | death-associated protein kinase 1 | 9q34.1 |
| EFNA5 | ephrin-A5 | 5q21.3 |
| EPC2 | enhancer of polycomb homolog 2 | 2q23.1 |
| EPHA4 | ephrin type-A receptor 4 | 2q36.1 |
| GAPDHS | glyceraldehyde-3-phosphate dehydrogenase, spermatogenic | 19q13.12 |
| GRB2 | growth factor receptor-bound protein 2 | 17q25.1 |
| GRIN2B | glutamate N-methyl D-aspartate receptor, subunit 2B | 12p13.1 |
| HNRNPCL1 | heterogeneous nuclear ribonucleoprotein C-like 1 | 1p36.21 |
| IL1B | interleukin 1, beta | 2q14 |
| MAD2L2 | mitotic arrest deficient homolog-like 2 | 1p36.22 |
| MAGI2 | membrane associated guanylate kinase, WW and PDZ domain containing 2; atrophin-1-interacting protein A | 7q21.11 |
| MAPT | microtubule-associated protein tau | 17q21.31 |
| MYH13 | myosin, heavy chain 13 | 17p13.1 |
| NEDD9 | neural precursor cell expressed, developmentally down-regulated 9 | 6p24.2 |
| NFATC4 | nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 4 | 14q11.2 |
| NXPH1 | neurexophilin 1 | 7p22 |
| PCK1 | phosphoenolpyruvate carboxykinase 1 | 20q13.31 |
| PICALM | phosphatidylinositol binding clathrin assembly protein | 11q14.2 |
| PRNP | prion protein | 20p13 |
| PRUNE2 | prune homolog 2 | 9q21.2 |
| QPCT | glutaminyl-peptide cyclotransferase | 2p22.2 |
| SLC35F2 | solute carrier family 35, member F2 | 11q22.3 |
| SLC6A13 | solute carrier family 6 (neurotransmitter transporter, GABA), member 13 | 12p13.33 |
| SORCS1 | sortilin-related VPS10 domain containing receptor 1 | 10q25.1 |
| SORL1 | sortilin-related receptor containing LDLR class A repeats | 11q24.1 |
| SREBF2 | sterol regulatory element binding transcription factor 2 | 22q13.2 |
| TNK1 | tyrosine kinase, non-receptor, 1 | 17p13.1 |
| TOMM40 | translocase of outer mitochondrial membrane 40 homolog | 19q13.32 |
| TP53TG3 | tumor protein p53 target 3 | 16p13 |
| TP63 | tumor protein p63 | 3q28 |
| UBTFL2 | upstream binding transcription factor, RNA polymerase I-like 2 | 11q14.3 |
Footnotes
Data used in the preparation of this article were obtained from the ADNI database (www.loni.ucla.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. For a complete list of investigators involved in ADNI see: http://www.loni.ucla.edu/ADNI/Collaboration/ADNI_Authorship_list.pdf.
References
- [1].Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- [2].Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ashford JW, Mortimer JA. Non-familial Alzheimer’s disease is mainly due to genetic factors. J Alzheimer’s Dis. 2002;4:169–77. doi: 10.3233/jad-2002-4307. [DOI] [PubMed] [Google Scholar]
- [4].Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- [5].Barabash A, Marcos A, Ancin I, Vazquez-Alvarez B, de Ugarte C, Gil P, et al. APOE, ACT and CHRNA7 genes in the conversion from amnestic mild cognitive impairment to Alzheimer’s disease. Neurobiol Aging. 2009;30:1254–64. doi: 10.1016/j.neurobiolaging.2007.11.003. [DOI] [PubMed] [Google Scholar]
- [6].Shen L, Kim S, Risacher SL, Nho K, Swaminathan S, West JD, et al. Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: a study of the ADNI cohort. Neuroimage. doi: 10.1016/j.neuroimage.2010.01.042. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- [9].Sleegers K, Lambert JC, Bertram L, Cruts M, Amouyel P, Van Broeckhoven C. The pursuit of susceptibility genes for Alzheimer’s disease: progress and prospects. Trends Genet. 2010;26:84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- [10].Glahn DC, Paus T, Thompson PM. Imaging genomics: mapping the influence of genetics on brain structure and function. Hum Brain Mapp. 2007;28:461–3. doi: 10.1002/hbm.20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PLoS One. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7:818–27. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- [13].Potkin SG, Turner JA, Guffanti G, Lakatos A, Torri F, Keator DB, et al. Genome-wide strategies for discovering genetic influences on cognition and cognitive disorders: methodological considerations. Cogn Neuropsychiatry. 2009;14:391–418. doi: 10.1080/13546800903059829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cuenco KT, Lunetta K, Baldwin CT, McKee AC, Guo J, Cupples LA, et al. Distinct variants in SORL1 are associated with cerebrovascular and neurodegenerative changes related to Alzheimer disease. Arch Neurol. 2008;65:1640–8. doi: 10.1001/archneur.65.12.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wishart HA, Saykin AJ, Rabin LA, Santulli RB, Flashman LA, Guerin S, et al. Increased prefrontal activation during working memory in cognitively intact APOE e4 carriers. Am J Psychiatry. 2006;163:1603–10. doi: 10.1176/ajp.2006.163.9.1603. [DOI] [PubMed] [Google Scholar]
- [16].Glahn DC, Thompson PM, Blangero J. Neuroimaging endopheno-types: strategies for finding genes influencing brain structure and function. Hum Brain Mapp. 2007;28:488–501. doi: 10.1002/hbm.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Seshadri S, DeStefano A, Au R, Massaro J, Beiser A, Kelly-Hayes M, et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham study. BMC Med Genet. 2007;8:S15. doi: 10.1186/1471-2350-8-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baranzini SE, Wang J, Gibson RA, Galwey N, Naegelin Y, Barkhof F, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet. 2009;18:767–78. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stein JL, Hua X, Lee S, Ho AJ, Leow AD, Toga AW, et al. Voxelwise genome-wide association study (vGWAS) Neuroimage. 2010 Feb 17; doi: 10.1016/j.neuroimage.2010.02.032. [Epublication ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stein JL, Hua X, Morra JH, Lee S, Hibar DP, Ho AJ, et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuro-image. 2010;51:542–54. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–23. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- [22].Wain LV, Armour JA, Tobin MD. Genomic copy number variation, human health, and disease. Lancet. 2009;374:340–50. doi: 10.1016/S0140-6736(09)60249-X. [DOI] [PubMed] [Google Scholar]
- [23].Heinzen EL, Need AC, Hayden KM, Chiba-Falek O, Roses AD, Strittmatter WJ, et al. Genome-wide scan of copy number variation in late-onset Alzheimer’s disease. J Alzheimer’s Dis. 2010;19:69–77. doi: 10.3233/JAD-2010-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Macciardi F, Guffanti G, Torri F, Rasmussen J, Lakatos A, Turner JA, et al. Increased CNV-Region Deletions in MCI and AD Subjects in the ADNI Sample. International Conference on Alzheimer’s Disease (ICAD 2010); Honolulu, HI. 2010. [Google Scholar]
- [25].Swaminathan S, Kim S, Shen L, Risacher SL, Foroud T, Pankratz N, et al. Preliminary Analysis of Copy Number Variation in the ADNI Cohort. International Conference on Alzheimer’s Disease (ICAD 2010); Honolulu, HI. 2010. [Google Scholar]
- [26].Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack C, Jagust W, et al. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin N Am. 2005;15:869–77. doi: 10.1016/j.nic.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–6. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- [31].Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. doi: 10.1038/tpj.2009.69. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009;6:347–61. doi: 10.2174/156720509788929273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, et al. Commonly carried allele within FTO, an obesity-associated gene, relates to accelerated brain degeneration in the elderly. Proc Natl Acad Sci U S A. 2010 Apr 19; [Epublication ahead of print] [Google Scholar]
- [34].Saykin AJ, Shen L, Risacher SL, Kim S, Nho K, West JD, et al. Genetic predictors of 12 month change in MRI hippocampal volume in the Alzheimer’s Disease Neuroimaging Initiative cohort: analysis of leading candidates from the AlzGene database. Imaging Consortium, International Conference on Alzheimer’s Disease; Vienna, Austria. 2009. [Google Scholar]
- [35].Uemura K, Kuzuya A, Aoyagi N, Ando K, Shimozono Y, Ninomiya H, et al. Amyloid beta inhibits ectodomain shedding of N-cadherin via down-regulation of cell-surface NMDA receptor. Neuroscience. 2007;145:5–10. doi: 10.1016/j.neuroscience.2006.12.022. [DOI] [PubMed] [Google Scholar]
- [36].Hochstrasser T, Weiss E, Marksteiner J, Humpel C. Soluble cell adhesion molecules in monocytes of Alzheimer’s disease and mild cognitive impairment. Exp Gerontol. 2010;45:70–4. doi: 10.1016/j.exger.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, Foroud T, et al. Genome-wide association study of CSF biomarkers amyloid beta 1-42, tau and tau phosphorylated at threonine 181 in the ADNI cohort. International Conference on Alzheimer’s Disease (ICAD); (in press) [Google Scholar]
- [38].Meyer-Lindenberg A, Straub RE, Lipska BK, Verchinski BA, Goldberg T, Callicott JH, et al. Genetic evidence implicating DARPP-32 in human frontostriatal structure, function, and cognition. J Clin Invest. 2007;117:672–82. doi: 10.1172/JCI30413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Licinio J, Dong C, Wong ML. Novel sequence variations in the brain-derived neurotrophic factor gene and association with major depression and antidepressant treatment response. Arch Gen Psychiatry. 2009;66:488–97. doi: 10.1001/archgenpsychiatry.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A. 2004;101:12604–9. doi: 10.1073/pnas.0405077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Coon KD, Valla J, Szelinger S, Schneider LE, Niedzielko TL, Brown KM, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6:194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- [42].Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM. Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging. 2007;28:1795–809. doi: 10.1016/j.neurobiolaging.2006.08.004. [DOI] [PubMed] [Google Scholar]
- [43].Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–32. doi: 10.1016/j.lab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- [44].Kalman J, Kitajka K, Pakaski M, Zvara A, Juhasz A, Vincze G, et al. Gene expression profile analysis of lymphocytes from Alzheimer’s patients. Psychiatr Genet. 2005;15:1–6. doi: 10.1097/00041444-200503000-00001. [DOI] [PubMed] [Google Scholar]
- [45].Cosentino M, Colombo C, Mauri M, Ferrari M, Corbetta S, Marino F, et al. Expression of apoptosis-related proteins and of mRNA for dopaminergic receptors in peripheral blood mononuclear cells from patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2009;23:88–90. doi: 10.1097/wad.0b013e318184807d. [DOI] [PubMed] [Google Scholar]
- [46].Liu Z, Sall A, Yang D. MicroRNA: an emerging therapeutic target and intervention tool. Int J Mol Sci. 2008;9:978–99. doi: 10.3390/ijms9060978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Corbin R, Olsson-Carter K, Slack F. The role of microRNAs in synaptic development and function. BMB Rep. 2009;42:131–5. doi: 10.5483/bmbrep.2009.42.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, et al. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–23. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–20. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimer’s Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]