

Abstract
The product of the fushi tarazu (ftz) gene is shown to be a site-dependent activator of transcription. In vitro-defined binding sites act as ftz-dependent enhancers in cultured cells. Another homoeodomain-containing protein, the engrailed gene product, competes for homoeodomain-binding sites and counteracts ftz activation.
GENETIC analysis in Drosophila melanogaster has identified a group of regulatory genes that participate in embryonic pattern formation1,2. Many of these genes encode a 60 amino-acid sequence, the homoeodomain, that is highly conserved through evolution3–5. This domain contains a putative helix-turn-helix motif6 such as that found in a number of bacterial regulators7, and possesses a sequence-specific DNA-binding activity8,9. Numerous regulatory interactions occur among homoeodomain-encoding genes during development. For example, a combination of segmentation and homoeotic gene products, many of which contain homoeodomains1, specify the developmental fates of cells in striped patterns10,11. Furthermore, homoeodomain-containing proteins (homoeodomain proteins) regulate both each other12 and downstream (`cytodifferentiation'13) genes. Consequently, it has been attractive to think of homoeodomain proteins as direct regulators of transcription. Due to the complexity of cross-regulatory interactions however, mutations altering or eliminating one homoeodomain protein generally change the expression patterns of many other regulators, confounding efforts to distinguish direct from indirect regulation. Additionally, suspected target genes contain large cis-acting control regions that respond, directly or indirectly, to complex combinations of regulators14–17. As a result, identification of target genes directly regulated by homoeodomain proteins has been problematic.
Previous studies have documented instances in which homoeodomain proteins control transcription. The enhancer activity of a DNA fragment from the ftz gene, a homoeodomain-containing member of the pair-rule class of segmentation genes, suggested that the ftz gene product (ftz) activates its own expression in the embryo18. In a different approach a Drosophila tissue culture system was used to show that the Ultrabithorax (Ubx) gene product induces expression of its own promoter, and inhibits the Antennapedia (Antp) (P1) promoter (M. Krasnow and D. S. Hogness, personal communication, see ref. 19 for summary). In these cases however, the complexity of the regulatory circuitry and of the responding control regions leaves open the possibility that the homoeodomain proteins produce these transcriptional effects through intermediaries. To circumvent the complexities of natural target genes, we chose to use binding sites identified in vitro to build homoeodomain-responsive promoters. We find that ftz is a potent activator of transcription from these promoters in cultured cells.
During sequential subdivision of the Drosophila embryo to produce segmentally repeating patterns, combinations of regulatory gene products direct patterned expression of other regulators. How might these multiple inputs be integrated? At least where regulatory interactions between homoeodomain proteins are involved, hints at one important consideration have already been obtained. In vitro binding studies have shown that several distinct homoeodomains that have diverged considerably in their amino-acid sequences bind to the same DNA elements (homoeodomain-binding sites), albeit with somewhat different affinities9,20. This suggests that homoeodomain proteins with similar binding specificities may function by interacting with a family of related sites. If these proteins compete for binding sites and have distinct regulatory consequences once bound, target genes could respond differently to different combinations and/or different levels of the regulators.
Simple target promoters built from synthetic binding sites allow us to show that homoeodomain proteins can compete functionally in cultured cells. In contrast to ftz, expression of the engrailed (en) gene product (en) alone has no apparent effect on these promoters. However, co-expression of en with ftz counteracts ftz activation. Thus the regulatory effect of a homoeodomain protein can be influenced by a second homoeodomain protein.
A ftz-dependent enhancer
We inserted homoeodomain-binding sites9 adjacent to characterized promoters (Fig. 1) in an attempt to make these promoters responsive to homoeodomain proteins. These constructs (responders) were co-transfected into cultured Drosophila cells (Schneider line 2)21 either alone or with constructs producing homoeodomain protein (producers). The activity of a responder was assessed by the level of an enzyme activity, the expression of which was driven by the responding promoter.
Fig. 1.

Plasmids. a, Homoeodomain protein producer plasmids (Ac-ftz and pAc-en) contain complete cDNA coding sequences inserted into Marc Krasnow's actin 5C promoter/polyadenylation signal vector, pPAc. Homoeodomains are the solid black regions. Ac-ftz (pPAcG1100) was provided by Gary Winslow and Matthew Scott. Stop codons were introduced into the coding sequences to produce pAc-ftzSTOP and pAc-enSTOP (see below). b, Responder plasmids 1 and 2 containing truncated distal ADH promoters46 (from −33 and −86 nucleotides, respectively, to +53 nt; −86dADH includes a binding site for a factor that appears to be important in regulating ADH expression in the embryo47) driving chloramphenicol acetyltransferase (CAT) gene expression (pD –33CAT, pD –86CAT) were provided by Bruce England and Robert Tjian. Six tandem copies of a consensus homoeodomain-binding site9 (NP6, see c below) were inserted immediately upstream of these promoters. Responders 3 and 4 contain the hsp70 promoter48 driving β-galactosidase (β-gal) expression; the latter includes cis-acting heat-shock transcription factor binding sites49,50. Various homoeodomain-binding sites were inserted just upstream by Jean-Paul Vincent (J.-P. Vincent and P.H.O'F., unpublished results). Responder 5 (pAF0) contains a proximal ADH promoter and structural gene51. Again, NP6 was inserted at the 5'-end of the promoter. c, Sequences of homoeodomain-binding sites9 used in the responder constructs are shown. Tandem arrows show the relative orientations of the individual NP sites within the multimers. No orientation can be assigned to the palindromic RP and LP sites. d, Reference plasmids. pCOPiCAT contains a copia transposable element long terminal repeat (LTR) (including the copia promoter)21 fused to a CAT structural gene. hsp82LacZ (pLac82SU) was provided by Dale Dorsett (Sloan-Kettering Cancer Center, New York) and contains the hsp82 promoter driving a β-gal structural gene.
Methods. pAc-en was made by inserting a blunt-ended EcoRI fragment, containing en cDNA leader, complete coding, and 180 nucleotides of 3′ non-translated sequences52, into the unique BamHI site of pPAc. An XbaI nonsense codon linker (NEB) was inserted into the unique MluI site following en amino-acid 406 (out of 552) to yield pAc-enSTOP. A second nonsense insertion after en amino-acid 81 (NotI site) was also constructed and tested as a negative control (see text). The en homoeodomain (amino acids 453–512) should not be produced by these `STOP' plasmids. pAc-ftzSTOP was similarly constructed from Ac-ftz by insertion of a nonsense codon linker into the unique XhoI site after the 16th amino acid of the homoeodomain6. A second nonsense insertion after the 33rd amino acid of the homoeodomain (EcoRV site) was also made and tested for activity (see text). Both of these `STOP' constructs are expected to produce a ftz protein truncated before the putative helix-turn-helix DNA-binding motif. To make the `+ sites' versions of responders 1 and 2, a SmaI-PstI fragment from an M13mp18 clone containing NP6 in the BamHI site9 was inserted into pD −33/−86CAT cut with XbaI (blunted) and PstI. Construction of responders 3 and 4 will be described in detail elsewhere. Briefly, they consist of a P-element vector (pSXhLac-7, obtained from Pieter Wensink, Brandeis University) with hsp70 promoter and leader sequences48 (−194 to +1,250 nucleotides) fused to the Escherichia coli LacZ structural gene, followed by hsp70 3′ non-coding sequences. Homoeodomain-binding sites were inserted into unique restriction sites immediately upstream of the promoter. Responder 5 contains the Drosophila melanogaster proximal ADH promoter and structural gene starting at −386 nucleotides51. A PstI-EcoRI (blunted) fragment from the M13mp18 clone containing NP6 was inserted into the unique PstI site immediately upstream of the proximal ADH promoter. pCOPiCAT was constructed by fusing a bluescript vector (Stratagene; cut with EcoRI and BamHI) with both the copia LTR (EcoRI-HindIII fragment) from pCOPneo21 and the CAT gene/poly(A)-site region (HindIII-BamHI fragment) from pSV2CAT53.
A responder, consisting of six homoeodomain-binding sites (NP6) upstream of a truncated promoter (the distal alcohol dehydrogenase promoter with 33 base pairs (bp) of upstream sequence: −33dADH), showed a 300-fold increase in activity when co-transfected with the ftz producer (Fig. 2). This increase in expression was entirely dependent on the ftz open reading frame in the producer, as shown by the insertion of stop codons at two different positions in the coding region (see legends of Figs 1 and 2). In addition, induction of transcription was strongly dependent on the homoeodomain-binding sites in the responder (Fig. 2). We conclude that activation by ftz is mediated via the inserted sites. As the inserted sequences are sites for in vitro binding by ftz, we suggest that the activation involves direct binding of ftz to the cis-acting sites in the responding promoter.
Fig. 2.

Ftz induces transcription in cultured cells. Drosophila cells (Schneider line 2, cultured as previously described54) were co-transfected with (1) either (+ftz) `producer' plasmid expressing ftz cDNA, or (–ftz) a control plasmid (either pAc-ftzSTOP or pPAC), and (2) the indicated `responder' plasmid (# 1–# 5, see Fig. 1), either with (upper panel) or without (lower panel) the NP6 homoeodomain-binding sites. Responder activity was ascertained by quantitating the appropriate enzymatic activity in whole-cell extracts. Activity was determined relative to the activity of a co-transfected reference gene plasmid; the results were essentially the same when expressed per μg of total protein in the extract (determined by the method described55). Activity is normalized to the basal level of each responder, without binding sites and `–ftz' (set at 1.0). Error bars indicate the range of values in duplicate transfections within the same experiment. Equivalent results were obtained in at least two separate experiments for each responder.
Methods. Cells were grown in Schneider's Drosophila medium54 supplemented with 12% fetal bovine serum. Transfection was by the calcium phosphate precipitation method, done essentially as described54. Cells were collected two days after transfection by scraping, rinsed once with phosphate-buffered saline and lysed by three cycles of freeze-thaw (−70 °C to room temperature). Extracts were prepared by centrifugation (15,000g, 5 min), and supernatants were either stored at −70 °C or assayed immediately. CAT activity (from responders 1 and 2, and pCOPiCAT reference gene used with responders 3 and 4) was determined as described56. Relative activities were the same whether or not extracts were heated (68 °C, 5 min) to destroy potential de-acetylase activity57. High activity extracts were diluted to keep determinations within the linear range of the assay, as shown by standard curves. CAT reaction time courses and extract mixing experiments indicated that little or no inhibitory activity was present in low activity extracts (data not shown). β-galactosidase (β-gal) activity (from responders 3 and 4 and hsp82LacZ reference gene used with responders 1, 2 and 5) was determined by fluorimetric assay58. Drosophila ADH activity was determined as described59. There was a detectable background activity in cells from control transfections (without responder plasmid) for β-gal and ADH (but not CAT). The β-gal background was at most 10% of the smallest plasmid-dependent activity, and so did not affect the results presented. The ADH background was relatively higher, about equal to the uninduced activities shown (responder 5), so the induction value for the −386pADH promoter (60-fold) is a minimum estimate. Amounts of plasmid DNA used per transfection (60-mm Petri dish) were as follows: 5 μg responder 1 (+/− sites) with 1 μg Ac-ftz (+/− STOP codon), 1 μg hsp82LacZ (reference gene), and 3 μg pAc-ftzSTOP; 1 μg responder 2 (+/− sites) with either 1 μg Ac-ftz and 8 μg pPAc, or 9 μg pPAc; 0.1 μg responder 3 (+/− sites) with 3 μg Ac-ftz (+/− STOP codon), 1 μg pCOPiCAT (reference gene), and 3 μg pPAc; 0.05 μg responder 4 (+/− sites) with 3 μg Ac-ftz (+/− STOP codon), 1 μg pCOPiCAT (reference gene), and 6 μg pAc-ftzSTOP; or, 7 μg responder 5 (+/− sites) with 3 μg Ac-ftz (+/− STOP codon), and 0.01 μg hsp82LacZ (reference gene). Amounts of responder DNA were chosen to give a readily quantifiable basal (uninduced) activity level, except responder 5 (see above). Small amounts of responders 3 and 4 were used because much larger amounts (1−2 μg) gave somewhat reduced induction ratios, consistent with either a titration of activating factor(s) or a `ceiling' on stable β-gal accumulation in the cells.
To test the generality of this effect, we constructed responsive promoters by placing the same DNA fragment containing the homoeodomain-binding sites (NP6) in other positions and adjacent to other promoters (Fig. 1b). In addition to the −33dADH promoter, we used a longer version, −86dADH. The −86dADH promoter had a higher basal level of expression than −33dADH (about 10–fold), but when homoeodomain-binding sites were included immediately upstream, it was also induced by ftz several hundredfold above its basal level (responder 2, Fig. 2). In addition, two configurations of the heat-shock 70 (hsp70) promoter were tested, one with sequences to −50 nucleotides, and one to −194 nucleotides. When homoeodomain-binding sites were added (at −50 or −194), ftz caused a 30- to 60-fold induction (responders 3 and 4, Fig. 2). In the −50 nucleotide configuration, the binding-site array was in the opposite orientation to that in the other responders. The fifth responder, the proximal ADH promoter to −386 nucleotides, gave a 60-fold binding-site-dependent ftz induction (Fig. 2). The basal activity levels of the responders (without ftz) were not significantly affected by the addition of homoeodomain-binding sites.
Although a strong ftz activation (30- to 500-fold) always depended on the presence of homoeodomain-binding sites, much smaller effects of ftz were sometimes seen in the absence of sites. In particular the −86dADH promoter (responder 2, Fig. 2) gave a ftz-dependent stimulation of about seven-fold. Some other promoters also exhibited small but significant stimulation when co-transfected with higher amounts of the ftz producer ducer (not shown). These effects could be due to adventitious ftz-binding sites in these promoters, or there might be small site-independent effects on some promoters at relatively high ftz concentrations. Nonetheless, the major effects of ftz were clearly site dependent.
We conclude that the homoeodomain-binding sites act as ftz-dependent enhancers22. That is, they act on all promoters tested, in either orientation, and at distances up to at least 386 nucleotides, to activate transcription.
Enhancer strength
Enhancer activity generally depends on the presence of multiple discrete elements which act synergistically23–27. The ftz-dependent enhancer used in the previous experiments consists of six tandem copies of a 12-nucleotide sequence. To test the importance of multiple copies we examined ftz activation of the −50hsp70 promoter with either six, four or two consensus copies (NP6, NP4, NP2, Fig. 3a). The reduction from six copies to four resulted in a large decrease in ftz-dependent activation, from about 60-fold to about 10-fold. With only two consensus copies, activation was reduced further to about two-fold. A similar response to the number of NP copies was seen with the −194hsp70 promoter (data not shown).
Fig. 3.
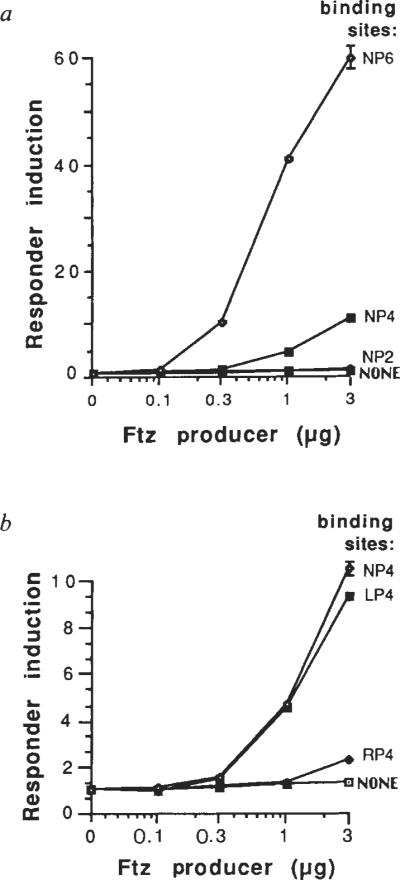
Alterations in binding sites change responsiveness to ftz. Different amounts of ftz producer (see below) were co-transfected with the −50hsp70 responder containing, a, either zero, two, four, or six NP binding sites (see Fig. 1), and, in b, four tandem copies of either the consensus (NP), or single-nucleotide alterations (LP, RP; see Fig. 1), or no homoeodomain-binding sites. Activity of each responder was determined as in Fig. 2, and was normalized to the basal level without ftz, set at 1.0, to give `Responder induction'. Error bars indicate the range of values of duplicate transfections. Similar results were obtained with the −194hsp70 responder. Amounts of plasmid DNA used per transfection were: 0.1 μg each responder with 3 μg pPAC, 1 μg pCOPiCAT (reference gene), and the amounts of ftz producer (Ac-ftz) shown. Total DNA was made constant by adding pAc-ftzSTOP. Control experiments using a plasmid with an actin 5C promoter driving CAT showed that expression levels were proportional to the amount of added DNA from 0.1 to 10 μg (data not shown).
The magnitude of the effect of reducing binding-site copy number from six to four was somewhat unexpected. Although in vitro studies suggested cooperative binding, the major effect was seen between one and two copies. An increase from four to six copies only marginally improved in vitro binding9. Perhaps site occupancy in vivo has a different dependence on the number of copies. Alternatively, the dramatic difference between transcriptional activation by four versus six copies might not represent a correspondingly large difference in site occupancy. Rather, an increase in the number of ftz-occupied sites might produce a disproportionately large increase in transcription. In either case, the copy number effect seen here is reminiscent of the cooperative or synergistic action of multiple enhancer elements in other systems.
If ftz activation requires interaction with specific binding sites in a responding gene, then site alterations that change binding affinity should also produce an altered ftz response. We therefore tested the effectiveness of two homoeodomain-binding sites, each altered from the consensus by a single nucleotide. These changes create a palindromic sequence related either to the left half (LP) or the right half (RP) of the original consensus (NP, see Fig. 1). Tandem repeats of four copies of each of these altered sequences (LP4, RP4) were compared to four copies of the original consensus (NP4). In vitro studies had shown that the change from NP to LP had no detectable effect on ftz-binding affinity, whereas the NP to RP change caused about a five-fold reduction in affinity9. Consistent with the in vitro results, LP4 and NP4 showed about the same ability to confer ftz activation to the −50hsp70 promoter, whereas RP4 had significantly reduced activity (Fig. 3b). Similar results were obtained with the same binding sites upstream of the −194hsp70 promoter (for example, Fig. 4b, data in the absence of en, 0 en). Thus these binding site alterations have parallel effects on in vitro binding affinity and transcriptional activation. This parallel behaviour supports our proposal that ftz binding is a prerequisite for activation.
Fig. 4.

Repression by en. Error bars indicate the range of values from duplicate transfections in the same experiment. Equivalent results were obtained in at least two separate experiments. a, Cells were co-transfected (see Fig. 2 legend) with (1) producer plasmids Acftz (`with ftz'), pAc-en (+en), pAc-ftzSTOP (`without ftz'), and/or pAc-enSTOP (-en), and (2) the indicated responder (see Fig. 1) with the NP6 homoeodomain-binding sites. Without homoeodomain-binding sites, there was no response to either ftz (Fig. 2) or en (data not shown). Amounts of plasmid DNA used per transfection were as follows: 5 μg responder 1 with 1 μg hsp82LacZ (reference gene) and either (1) (−ftz, −en) 4 μg pAc-ftzSTOP, (2) (+ftz, −en) 1 μg Ac-ftz and 3 μg pAc-enSTOP, or (3) (+ftz, +en) 1 μg Ac-ftz, 1 μg pAc-en, and 2 μg pAc-enSTOP; 0.1 μg responder 3 with 1 μg pCOPiCAT (reference gene) and either (1) (−ftz, −en) 3 μg pAc-ftzSTOP and 3 μg pPAc, (2) (+ftz, −en) 3 μg Ac-ftz and 3 μg pAc-enSTOP, or (3) (+ftz, +en) 3 μg Ac-ftz and 3 μg pAc-en; 0.05 μg responder 4 with 0.2 μg pCOPiCAT (reference gene), 3 μg Ac-ftz (+/−STOP codon, = `without/with ftz'), and 6 μg pAc-en (+/−STOP codon, = `−/+en'); 7 μg responder 5 with 0.01 μg hsp82LacZ (reference gene) and either (1) (−ftz, −en) 3 μg pAc-ftzSTOP, (2) (−ftz, +en) 2 μg pAc-ftzSTOP and 1 μg pAc-en, (3) (+ftz, −en) 1 μg Ac-ftz, 1 μg pAc-ftzSTOP, and 1 μg pAc-enSTOP, or (4) (+ftz, +en) 1 μg Ac-ftz, 1 μg pAc-ftzSTOP, and 1 μg pAc-en. `ND', response with en alone not determined in this experiment; several other experiments with responder 1 showed that en had a small negative effect with binding sites (see text). b, Different amounts of en producer were co-transfected with a constant amount of ftz producer and the −194hsp70 responder containing either no homoeodomain-binding sites, or the NP4, LP4, or RP4 binding site array (see Fig. 1). Amounts of plasmid DNA per transfection were: 0.05 μg each responder with 3 μg Ac-ftz, 1 μg pCOPiCAT (reference gene), and the amounts of en producer (pAc-en) shown. Total DNA was made constant with pAc-enSTOP. Responder activities for both a and b were determined as described in the Fig. 2 legend.
Engrailed represses ftz activation
The homoeodomain-binding site used to demonstrate ftz-dependent activation is also bound by the en protein in vitro8,9. Nonetheless, when en producer plasmid was co-transfected with the responder constructs that are inducible by ftz, no activation occurred. In some cases, a small but reproducible reduction in expression (two to threefold) was observed with, but not without, binding sites in the responders (data not shown).
To test whether en could bind to the same sites as ftz in vivo, we produced ftz and en together, reasoning that if en bound, it might affect ftz activation. Indeed, with each of the binding-site-containing responders, ftz activation was reduced between 30 and 100-fold by the presence of en (Fig. 4a). This effect of the en producer is entirely dependent on the en open reading frame, as insertion of stop codons at either of two different positions in the coding region abolished effectiveness (see Fig. 1 legend and Fig. 4). Thus, the combination of en with ftz can have an effect on transcription that is qualitatively distinct from that of ftz alone.
We examined the degree of en antagonism of ftz induction using different amounts of transfected en producer. Whether responders contained NP6 (data not shown), NP4, or LP4 binding sites, ftz induction was similarly titrated by increasing amounts of en producer (Fig. 4b). This is consistent with the fact that en, like ftz (see above), binds equally well to these sites in vitro9. As mentioned above, the in vitro affinity of ftz for RP4 is less than for NP4 (about fivefold), and ftz activation of RP4-containing responders in transfected cells is less than activation of NP4 responders. The in vitro affinity of en for RP4 is also lower than for NP4 (about 25-fold), and although the RP4 responder is induced less than three-fold by ftz, en is still able to significantly antagonize this activation (Fig. 4b). Consistent with the fact that, in vitro, the NP to RP site alteration reduces en affinity more than it reduces ftz affinity (25-compared to fivefold), the en repression with RP4 seems to be less effective than with NP4 (that is, Fig. 4b, the RP4 and NP4 curves cross). Although this distinction is reproducible (data not shown), its significance is, nonetheless, uncertain because it relies on comparison of small differences in the activities of the responders when repressed by en.
Discussion
The results presented here have shown that ftz is a powerful site-dependent activator of transcription. Sequences identified as in vitro binding sites for bacterially produced homoeodomain proteins9, when inserted upstream of promoters, act as ftz-dependent enhancers in cultured cells. This suggests that ftz acts by binding to the inserted sites to activate the linked promoter. However, we cannot formally eliminate the possibility that ftz is acting indirectly by altering the activity of another regulator that functions at the sites.
Another homoeodomain protein, en, is also able to bind in vitro to the sites that confer ftz responsiveness in our assay9. Although we do not observe transcriptional activation by en in cultured cells, we find that en negates activation by ftz. The parallel of in vitro binding and functional antagonism in vivo suggests the simple hypothesis that binding competition might be responsible for repression by en. That is, en, by binding to the sites, may prevent ftz binding and produce a complex that is ineffective at activating transcription (or possibly a complex that actively represses transcription). There are however, numerous alternative explanations for the observed repression. Perhaps en interacts with and inactivates ftz, or perhaps en interferes with accessory or intermediary factors necessary for ftz action.
Although we cannot yet specify mechanisms for the transcriptional activation by ftz or for the functional antagonism by en, we favour the simple model that both proteins act directly at the homoeodomain-binding sites. If such a model is correct, we might expect that other homoeodomain proteins having related DNA-binding specificities in vitro would affect transcription from the same promoters that are activated by ftz. Indeed, in preliminary experiments the Ubx gene product activated expression in a binding site (NP6)-dependent manner (albeit less effectively than ftz). Outside the homoeodomain, which specifies DNA-binding activity, Ubx and ftz share no significant homology. As Ubx can bind to homoeodomain-binding sites also bound by ftz (Gavis and D. S. Hogness, personal communication), this result is compatible with direct action of these proteins on the target sequences. In addition, we have found that transfection with a combination of ftz and Ubx producers activated the responder to levels intermediate to those obtained with ftz or Ubx alone. Such an averaging effect could occur if the proteins were similar in that they compete for the cis-acting sites, but different in that they produce complexes activating transcription to different degrees. A fourth homoeodomain protein, the even-skipped (eve) gene product, which has a binding specificity related to that of ftz, was also found to alter transcription in a binding site (NP6)-dependent fashion. Eve, like en, was inactive on its own but strongly antagonized induction by ftz and Ubx. As several members of the homoeodomain family of regulators seem to act on the same sites in our transfection assay, we propose that they act by directly binding to the sites, and that when present together they compete for binding sites and influence transcription according to the activity of the resulting complex.
Several precedents exist for competition among regulators for cis-acting sites. For example, in the lysis/lysogeny switch of bacteriophage λ, cro and repressor compete for related sites in the rightward operator28. In eukaryotes, a number of regulatory proteins have been shown to be members of protein families with related DNA-binding specificities29–37. In fact, a member of the nuclear hormone receptor family, thyroid hormone receptor, was recently shown to mediate negative effects on transcription from oestrogen-responsive DNA elements, apparently by competing with oestrogen receptor for binding to the elements38. It is possible that regulatory gene families in general evolve by duplication and divergence to produce members with overlapping functions. The modular nature of protein structure (for example, see ref. 39) would make it likely that some domains retain their original function whereas others diverge by losing one interaction and possibly gaining a new functional interaction. The members of such protein families would interdependently regulate target genes because of an overlapping function (such as DNA binding to the same family of sites), but would have differing effects based on a diverged function (such as activation versus repression of transcription).
Competition for binding sites could dramatically influence the action of homoeodomain proteins during embryonic pattern formation. Each of the many known homoeodomain proteins is expressed in a complex pattern during embryogenesis. As a result of overlap of these patterns, binding sites are exposed to a mixture of homoeodomain proteins that varies in composition in a complex spatio-temporal fashion. As the outcome of binding competition will depend on the relative concentrations of the binding proteins, the enhancer activity of a site might also vary in a detailed spatio-temporal pattern. Additionally, the outcome of competition depends on the relative affinity of each particular site for the different binding proteins. As a result, each of the related binding sites might undergo transitions in occupancy at distinct relative concentrations of homoeodomain proteins and would consequently have a unique saptio-temporal programme of activity (Fig. 5).
Fig. 5.
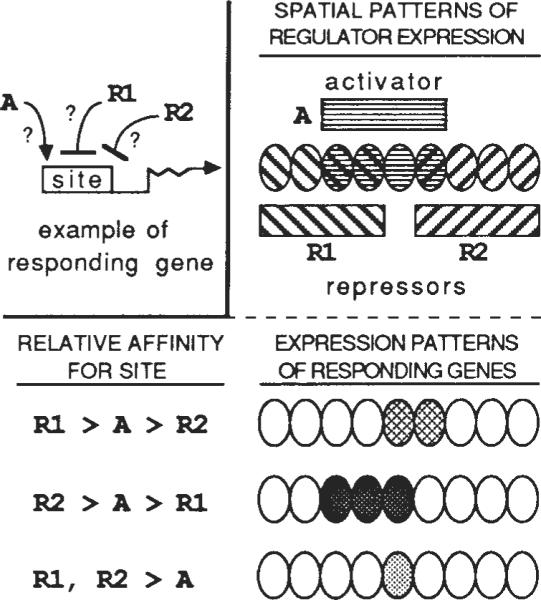
Multiple patterns of gene expression can be generated by competition among regulators for binding to related but different cis-acting sites. Beginning with overlapping patterns of expression of three regulatory proteins with related DNA-binding specificities, three novel patterns are produced by differential competition. A is an activator, whereas R1 and R2 are repressors. Related cis-acting sites in the responding genes have different relative affinities for the regulators. The first responding gene has a binding site with relative affinities R1 > A > R2, so that it is activated by A and repressed by R1, but not by R2. The second responding gene has a site with relative affinities R2 > A > R1, whereas the site in the third gene has a higher affinity for both R1 and R2 than for A. Such a scheme could operate in the embryo to refine spatial patterns of regulatory gene expression and to subdivide domains of expression. This simple example assumes that regulator concentrations are equal and constant in space and time. Similar competitive interactions could also generate multiple domains of responder expression from a gradient of regulator concentration.
Although the potential exists in the embryo for competition to occur among homoeodomain proteins for binding sites, the situation is likely to be more complicated than that in cultured cells. For example, other work performed in our laboratory has shown that promoters responsive to ftz in culture do not produce a ftz-dependent pattern of expression in the embryo (J.-P. Vincent, personal communication). A number of interactions or modifications might occur during embryogenesis to change the behaviour of regulators. For example, phosphorylation can potentially produce40 or unmask41,42 acidic domains that activate transcription43. Accordingly, ftz and en, both of which are known to be phosphorylated44,45, might not always have the same transcriptional effects that we have observed in these experiments.
As yet, we have no direct evidence that competitive interactions among transcription factors are important in embryonic development. Nonetheless, the prevalence of a highly conserved DNA-binding domain among developmental regulators leads us to propose that competitive interactions will have a general role in control mechanisms governing spatial-and tissue-specific transcription.
Acknowledgments
We thank Jill Jongens, John Little, Delia Lakich, Rich Kostriken, Jean-Paul Vincent, Bruce Edgar, Bill Kalionis and Steve DiNardo for critical advice on the manuscript. Additionally, we thank Claude Desplan and all members of the O'Farrell laboratory for help and encouragement. We thank Jean-Paul Vincent, Gary Winslow, Mark Krasnow, Anne Ephrussi and Dale Dorsett for sharing plasmid constructs and information before publication. We would also like to acknowledge our debt to Mark Krasnow whose earlier work stimulated these studies. This work was funded by an NIH grant to P.H.O. and by an ACS post-doctoral grant to J.B.J.
References
- 1.Scott MP, O'Farrell PH. A. Rev. cell. Biol. 1986;2:49–80. doi: 10.1146/annurev.cb.02.110186.000405. [DOI] [PubMed] [Google Scholar]
- 2.Scott MP, Carroll SB. Cell. 1987;51:689–698. doi: 10.1016/0092-8674(87)90092-4. [DOI] [PubMed] [Google Scholar]
- 3.Scott MP, Weiner AJ. Proc. natn. Acad. Sci. U.S.A. 1984;81:4115–4119. doi: 10.1073/pnas.81.13.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGinnis W, Garber RL, Wirz J, Kuroiwa A, Gehring WJ. Cell. 1984;37:403–408. doi: 10.1016/0092-8674(84)90370-2. [DOI] [PubMed] [Google Scholar]
- 5.Gehring WJ, Hiromi Y. A. Rev. Genet. 1986;20:147–173. doi: 10.1146/annurev.ge.20.120186.001051. [DOI] [PubMed] [Google Scholar]
- 6.Laughon A, Scott MP. Nature. 1984;310:25–31. doi: 10.1038/310025a0. [DOI] [PubMed] [Google Scholar]
- 7.Pabo CO, Sauer RT. A. Rev. Biochem. 1984;53:293–321. doi: 10.1146/annurev.bi.53.070184.001453. [DOI] [PubMed] [Google Scholar]
- 8.Desplan C, Theis J, O'Farrell PH. Nature. 1985;318:630–635. doi: 10.1038/318630a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desplan C, Theis J, O'Farrell PH. Cell. 1988;54:1081–1090. doi: 10.1016/0092-8674(88)90123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiNardo S, O'Farrell PH. Genes Dev. 1987;1:1212–1225. doi: 10.1101/gad.1.10.1212. [DOI] [PubMed] [Google Scholar]
- 11.Howard K, Ingham P. Cell. 1986;44:949–957. doi: 10.1016/0092-8674(86)90018-8. [DOI] [PubMed] [Google Scholar]
- 12.Hafen E, Levine M, Gehring W. Nature. 1984;307:287–298. doi: 10.1038/307287a0. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Bellido A. Ciba Fdn Symp. 1975;29:161–182. doi: 10.1002/9780470720110.ch8. [DOI] [PubMed] [Google Scholar]
- 14.Bender W, et al. Science. 1983;221:23–29. doi: 10.1126/science.221.4605.23. [DOI] [PubMed] [Google Scholar]
- 15.Kuner JM, et al. Cell. 1985;42:309–316. doi: 10.1016/s0092-8674(85)80126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beachy PA, Helfand SL, Hogness DS. Nature. 1985;313:545–551. doi: 10.1038/313545a0. [DOI] [PubMed] [Google Scholar]
- 17.Kassis JA, Wong ML, O'Farrell PH. Molec. cell. Biol. 1985;5:3600–3609. doi: 10.1128/mcb.5.12.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiromi Y, Gehring WJ. Cell. 1987;50:963–974. doi: 10.1016/0092-8674(87)90523-x. [DOI] [PubMed] [Google Scholar]
- 19.Robertson M. Nature. 1987;327:556–557. [Google Scholar]
- 20.Hoey T, Levine M. Nature. 1988;332:858–861. doi: 10.1038/332858a0. [DOI] [PubMed] [Google Scholar]
- 21.Rio DC, Rubin GM. Molec. cell. Biol. 1985;5:1833–1838. doi: 10.1128/mcb.5.8.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serfling E, Jasin M, Schaffner W. Trends Genet. 1985;1:224–230. [Google Scholar]
- 23.DeFranco D, Yamamoto KR. Molec. cell. Biol. 1986;6:993–1001. doi: 10.1128/mcb.6.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herr W, Clarke J. Cell. 1986;45:461–470. doi: 10.1016/0092-8674(86)90332-6. [DOI] [PubMed] [Google Scholar]
- 25.Ptashne M. Nature. 1986;322:697–701. doi: 10.1038/322697a0. [DOI] [PubMed] [Google Scholar]
- 26.Schirm S, Jiricny J, Schaffner W. Genes Dev. 1987;1:65–74. doi: 10.1101/gad.1.1.65. [DOI] [PubMed] [Google Scholar]
- 27.Ondek B, Gloss L, Herr W. Nature. 1988;333:40–45. doi: 10.1038/333040a0. [DOI] [PubMed] [Google Scholar]
- 28.Ptashne M. A Genetic Switch—Gene Control and Phage λ. Cell; Cambridge, Massachusetts: 1986. [Google Scholar]
- 29.Dorn A, Bollekens J, Staub A, Benoist C, Mathis D. Cell. 1987;50:863–872. doi: 10.1016/0092-8674(87)90513-7. [DOI] [PubMed] [Google Scholar]
- 30.Jones NC, Rigby PWJ, Ziff EB. Genes Dev. 1988;2:267–281. doi: 10.1101/gad.2.3.267. [DOI] [PubMed] [Google Scholar]
- 31.Santoro C, Mermod N, Andrews PC, Tjian R. Nature. 1988;334:218–224. doi: 10.1038/334218a0. [DOI] [PubMed] [Google Scholar]
- 32.Von der Ahe D, et al. Nature. 1985;313:706–709. doi: 10.1038/313706a0. [DOI] [PubMed] [Google Scholar]
- 33.Chowdhury K, Deutsch U, Gruss P. Cell. 1987;48:771–778. doi: 10.1016/0092-8674(87)90074-2. [DOI] [PubMed] [Google Scholar]
- 34.Struhl K. Cell. 1987;50:841–846. doi: 10.1016/0092-8674(87)90511-3. [DOI] [PubMed] [Google Scholar]
- 35.Lambert PF, Spalholz BA, Howley PM. Cell. 1987;50:69–78. doi: 10.1016/0092-8674(87)90663-5. [DOI] [PubMed] [Google Scholar]
- 36.Chalepakis G, et al. Cell. 1988;53:371–382. doi: 10.1016/0092-8674(88)90157-2. [DOI] [PubMed] [Google Scholar]
- 37.Franza BR, Rauscher FJ, III, Josephs SF, Curran T. Science. 1988;239:1150–1153. doi: 10.1126/science.2964084. [DOI] [PubMed] [Google Scholar]
- 38.Glass CK, Holloway JM, Devary OV, Rosenfeld MG. Cell. 1988;54:313–323. doi: 10.1016/0092-8674(88)90194-8. [DOI] [PubMed] [Google Scholar]
- 39.Rusconi S, Yamamoto KR. EMBO J. 1987;6:1309–1315. doi: 10.1002/j.1460-2075.1987.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sorger PK, Lewis MJ, Pelham HRB. Nature. 1987;329:81–84. doi: 10.1038/329081a0. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto KR. A. Rev. Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 42.Sen R, Baltimore D. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 43.Giniger E, Ptashne M. Nature. 1987;330:670–672. doi: 10.1038/330670a0. [DOI] [PubMed] [Google Scholar]
- 44.Krause HM, Klemenz R, Gehring WJ. Genes Dev. 1988;2:1021–1036. doi: 10.1101/gad.2.8.1021. [DOI] [PubMed] [Google Scholar]
- 45.Gay NJ, Poole SJ, Kornberg TB. Nucleic Acids Res. 1988;16:6637–6647. doi: 10.1093/nar/16.14.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heberlein U, England B, Tjian R. Cell. 1985;41:965–977. doi: 10.1016/s0092-8674(85)80077-5. [DOI] [PubMed] [Google Scholar]
- 47.Heberlein U, Tjian R. Nature. 1988;331:410–415. doi: 10.1038/331410a0. [DOI] [PubMed] [Google Scholar]
- 48.Lis JT, Simon JA, Sutton CA. Cell. 1983;35:403–410. doi: 10.1016/0092-8674(83)90173-3. [DOI] [PubMed] [Google Scholar]
- 49.Pelham HRB. Cell. 1982;30:517–528. doi: 10.1016/0092-8674(82)90249-5. [DOI] [PubMed] [Google Scholar]
- 50.Parker CS, Topol J. Cell. 1984;37:273–283. doi: 10.1016/0092-8674(84)90323-4. [DOI] [PubMed] [Google Scholar]
- 51.Posakony JW, Fischer JA, Maniatis T. Cold Spring Harb. Symp. quant. Biol. 1985;50:515–520. doi: 10.1101/sqb.1985.050.01.063. [DOI] [PubMed] [Google Scholar]
- 52.Poole SJ, Kauvar LM, Drees B, Kornberg T. Cell. 1985;40:37–43. doi: 10.1016/0092-8674(85)90306-x. [DOI] [PubMed] [Google Scholar]
- 53.Gorman CM, Moffat LF, Howard BH. Molec. cell. Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Nocera PP, Dawid IB. Proc. natn. Acad. Sci. U.S.A. 1983;80:7095–7098. doi: 10.1073/pnas.80.23.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradford MM. Analyt. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 56.Kimura A, Israel A, Le Bail O, Kourilsky P. Cell. 1986;44:261–272. doi: 10.1016/0092-8674(86)90760-9. [DOI] [PubMed] [Google Scholar]
- 57.Crabb DW, Dixon JE. Analyt. Biochem. 1987;163:88–92. doi: 10.1016/0003-2697(87)90096-0. [DOI] [PubMed] [Google Scholar]
- 58.Stuart GW, Searle PF, Chen HY, Brinster RL, Palmiter RD. Proc. natn. Acad. Sci. U.S.A. 1984;81:7318–7322. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maroni G. Biochem. Genet. 1978;16:509–523. doi: 10.1007/BF00484215. [DOI] [PubMed] [Google Scholar]