

Abstract
Virtual screening of a library of commercially available compounds vs. the structure of Mycobacterium tuberculosis lumazine synthase identified 2-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)acetic acid (9) as a possible lead compound. Compound 9 proved to be an effective inhibitor of M. tuberculosis lumazine synthase with a Ki of 70 μM. Lead optimization through replacement of the carboxymethylsulfonamide sidechain with sulfonamides substituted with alkyl phosphates led to a four-carbon phosphate 38 that displayed a moderate increase in enzyme inhibitory activity (Ki 38 μM). Molecular modeling based on known lumazine synthase/inhibitor crystal structures suggests that the main forces stabilizing the present benzindolone/enzyme complexes involve π–π stacking interactions with Trp27 and hydrogen bonding of the phosphates with Arg128, the backbone nitrogens of Gly85 and Gln86, and the side chain hydroxyl of Thr87.
Keywords: Mycobacterium tuberculosis, lumazine synthase, inhibitor, virtual screening
1. Introduction
Riboflavin (vitamin B2) plays a crucial role in many biological processes, including photosynthesis and mitochondrial electron transport. Riboflavin is biosynthesized by plants and numerous microorganisms but not by animals. Whereas animals obtain riboflavin from dietary sources, certain microorganisms such as Gram-negative pathogenic bacteria and yeasts lack efficient riboflavin uptake systems and are therefore absolutely dependent on endogenous riboflavin biosynthesis.1–4 Riboflavin biosynthesis is therefore an attractive target for the design and synthesis of new antibiotics, which are urgently needed because pathogens are becoming drug resistant at an alarming rate. Lumazine synthase and riboflavin synthase catalyze the last two steps in the biosynthesis of riboflavin (4) (Scheme 1). The biosynthetic pathway starts off from one molecule of GTP,5–7 which is converted to 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione (1) by a sequence of ring opening, deamination, reduction, and dephosphorylation.8–10 Lumazine synthase catalyzes the condensation of 3,4-dihydroxy-2-butanone 4-phosphate (2) with 5-amino-6-ribitylamino-2,4(1H,3H)pyrimidinedione (1) yielding 6,7-dimethyl-8-D-ribityllumazine (3).11–25 The final process in the biosynthesis of riboflavin (4) involves a mechanistically unusual dismutation of two molecules of 6,7-dimethyl-8-D-ribityllumazine (3) to result in the formation of one molecule of riboflavin and one molecule of the pyrimidinedione derivative 1. 26–30
Scheme 1.

Although the precise details remain to be established, the lumazine synthase-catalyzed reaction most likely proceeds along a mechanistic pathway involving formation of the Schiff base 5, phosphate elimination affording 6, tautomerization to 7, ring closure, and dehydration to yield the lumazine 3 (Scheme 2).31 Variations of this mechanism are possible depending on the Schiff base geometry and possible isomerization, conformational changes, and the timing of phosphate elimination.
Scheme 2.

Screening drug-like compounds by computational docking against the structure of a known target has become a promising avenue in lead discovery.32–34 This approach is arguably much cheaper than experimental high-throughput screening and has been validated in other lead discovery projects.35,36 The successful history of lumazine synthase structure determination and inhibitor development provides a sound basis for the selection of lead compounds from commercial chemical libraries and possible structural modification in order to improve the binding properties. The design of most of the known inhibitors of lumazine synthase has been based on the structures of both substrates of lumazine synthase and the putative Schiff base intermediate in the enzymatic reaction.37,38
The ZINC database,39 maintained by the Shoichet group at UCSF, was subjected to virtual screening for the discovery of new lumazine synthase inhibitors. Receptor-based filtering followed by receptor-based docking of the structures into the lumazine synthase structure led to an interesting new scaffold, compound 9 (Figure 1), which contains “drug-like” features, such as a sulfonamide moiety, and therefore has more potential for drug development than mechanism-based probes containing polar ribityl side chains. The lead compound 9 displayed a Ki of 70 μM vs. M. tuberculosis lumazine synthase. It consists of an aromatic oxobenzindole ring system and a carboxymethylsulfonamide side chain. The inhibitor 9 lacks any close analogy to the pyrimidinedione ring or the ribityl side chain of the substrate 1, which explains its relatively low affinity for the enzyme. However, it has been recently demonstrated that the ribityl chain present in the substrate, which was thought to be necessary for binding to the enzyme, can be replaced by a simple hydroxybenzylidene moiety (compound 12) with retention of lumazine synthase inhibitory activity.40 Moreover, it has recently been shown that the attachment of aliphatic chains bearing phosphate or phosphonate groups on the heterocyclic core dramatically increases binding affinity of the inhibitors.41–45 An alkylphosphate chain consisting of 4–5 carbon atoms produces of the strongest inhibitory effect. It therefore seemed logical to replace the carboxymethyl group of the lead compound 9 with alkyl phosphate groups and test the influence of the length of aliphatic chains with 2 to 6 carbon atoms. The rationale behind this design is that the phosphate moiety in compound 13 is expected to bind in the same phosphate-binding pocket as the inhibitors 10 and 11.
Figure 1.

Representative M. tuberculosis lumazine synthase Inhibitors.
2. Results and Discussion
2.1. Virtual Screening
Virtual screening aims to discover inhibitors with novel scaffolds. In the present case, more than one million compounds were screened from the ZINC database,39 which has been prepared for docking screening. The M. tuberculosis lumazine synthase crystal structure in complex with the inhibitor 3-(1,3,7-trihydro-9-D-ribityl-2,6,8-purinetrione-7-yl) propane 1-phosphate (PDB: 1w19) was prepared using Protein Preparation tools in Maestro 7.5 (Schrödinger LLC, Portland, Oregon), removing all crystal water molecules. The compounds were first docked with Glide 4.0 (Schrödinger LLC, Portland, Oregon) High Throughput Virtual Screening mode without any constraints.46 A multi-stage screening workflow was applied, increasing the ligand conformational search and applying different scoring functions in order to rapidly reduce the number of possible hits. During the latter two stages of the screening, the following pharmacophore constraints were applied: three backbone hydrogen bonds to Ile83, Val81, Ala59 and the phosphate interaction to Arg128. At least three of these constraints had to be satisfied. The backbone hydrogen bonds are desirable because they could prevent future drug resistance issues. Based on solubility criteria posed by the ITC measurement, a solubility filter was also applied in the workflow. The score cut off of −10 was chosen based on docking of several known active compounds41 under the same conditions. The resulting 44 ligands were manually inspected and based on the interactions and their chemical scaffold, 12 compounds with different chemical scaffolds in total were selected for assay. Of these compounds, compound 9 demonstrated inhibition in the subsequent kinetic assay (Table 1). The principle of docking/pharmacophore based filtering is that it selects compounds complementary to the targeted site. Therefore, statistically, there is a much higher probability that the compounds selected by such a screening protocol will bind at the targeted site.
TABLE 1.
Inhibition of recombinant riboflavin synthase (RS) and/or lumazine synthase (LS) of M. tuberculosis by selected compounds.a
| Comp | Enzyme | Ks,b μM | Kcat,c m−1 | Ki,d μM | Kis,e μM | Mechanism |
|---|---|---|---|---|---|---|
| 9 | LS | 70 | 58 | |||
| 26 | LS | 9.8 ± 0.5 | 0.41 ± 0.01 | 260 ± 109 | 125 ± 57 | Partial |
| RS | 5.7 ± 0.04 | 0.30 ± 0.01 | > 1000 | |||
| 27 | LS | 15 ± 2 | 0.45 ± 0.01 | 99 ± 30 | 434 ± 115 | Mixed |
| RS | 5.6 ± 0.04 | 0.28 ± 0.01 | > 1000 | |||
| 38 | LS | 11 ± 1 | 0.44 ± 0.01 | 38 ± 8 | 572 ± 210 | Mixed |
| RS | 6.5 ± 0.3 | 0.29 ± 0.01 | > 1000 | |||
| 39 | LS | 19 ± 1 | 0.45 ± 0.01 | 563 ± 56 | Uncompetitive | |
| RS | 9.8 ± 0.6 | 0.56 ± 0.01 | 971 ± 179 | Uncompetitive | ||
| 40 | LS | 18 ± 1 | 0.45 ± 0.01 | 723 ± 91 | Uncompetitive | |
| RS | 9.8 ± 0.6 | 0.56 ± 0.01 | 898 ± 187 | Uncompetitive | ||
| 72 | LS | 16 ± 1 | 0.57 ± 0.01 | 832 ± 418 | 518 ± 94 | Mixed |
| RS | 4.1 ± 0.3 | 0.15 ± 0.01 | > 1000 | |||
| 77 | LS | 21 ± 2 | 0.61 ± 0.02 | 832 ± 418 | > 1000 | Mixed |
| RS | 7.8 ± 0.3 | 0.34 ± 0.01 | 897 ± 104 | Uncompetitive |
The assays with lumazine synthase were performed with dihydroxybutanone phosphate substrate held constant, while the concentration of the pyrimidinedione substrate was varied.
Ks is the substrate dissociation constant for the equilibrium E + S ⇌ ES.
Kcat is the rate constant for the process ES ⇌ E + P.
Ki is the inhibitor dissociation constant for the process E + I ⇌ EI.
Kis is the inhibitor dissociation constant process ES + I ⇌ ESI. Compounds 31–33, 54–55, 71, 73, 79 are not listed in this Table since they did not significantly inhibit either lumazine synthase or riboflavin synthase.
A straightforward synthesis (Scheme 3) was proposed for compounds having general structure 13. The synthesis started from commercially available benz[cd]indol-2(1H)-one (14), which on treatment with chlorosulfonic acid yielded compound 15.47
Scheme 3. Reagents and Conditions.

(a) ClSO3H, 0 °C.
Various procedures were tried unsuccessfully to produce the final compound 13. The solvent system, base and temperature were varied without any improvement in results. The phosphate group was therefore protected before performing the coupling reaction. The syntheses of compounds 26 and 27 were both carried out the same way starting with the appropriate amino alcohol. Accordingly, the synthesis was started from ethanolamine (16), which on Cbz protection provided compound 18 (Scheme 4). The phosphate functionality in compound 20 was introduced by treatment of 18 with di-tert-butyl diisopropyl phosphoramidite under 1-H tetrazole catalysis, followed by in situ oxidation with hydrogen peroxide.48,49 The Cbz group of compound 20 was removed via hydrogenolysis with Pd/C to afford the free amine compound 22. Reaction of compound 15 with the free amine 22 in the presence of triethylamine provided the sulfonamide 24. Subsequent deprotection of the phosphate with trifluoroacetic acid provided the desired product 26.50
Scheme 4. Reagents and Conditions.

(a) Benzylchloroformate, Et3N, CH2Cl2, r.t.; (b) iPr2NP(OtBu)2, H2O2, tetrazole, r.t.; (c) H2, Pd/C, MeOH; (d) 15, Et3N, THF, r.t.; (e) TFA, CH2Cl2, r.t.
Scheme 4 was shortened by removing the Cbz protection step and the subsequent deprotection by hydrogenation. The improved synthesis of oxobenzindole derivatives is outlined in Scheme 5. Simply mixing compound 15 with the respective amino alcohols in THF at room temperature provided the required intermediates 31–34 (Scheme 5). The phosphate group was introduced by treatment with di-tert-butyl diisopropyl phosphoramidite under tetrazole catalysis, followed by in situ oxidation with hydrogen peroxide to provide compounds 24 and 35–37. The final deprotection of the tert-butyl groups with trifluoroacetic acid produced the required substances 26, 38, 39 and 40 with different linker chain lengths between the phosphate group and the sulfonamide moiety.
Scheme 5. Reagents and Conditions.

(a) 15, Et3N, THF, r.t.; (b) iPr2NP(OtBu)2, H2O2, tetrazole, r.t.; (c) TFA, CH2Cl2, r.t.
Enzyme assays were performed with recombinant lumazine synthase and riboflavin synthase from M. tuberculosis. Assays with serial dilutions of each respective inhibitor were conducted in microtiter plates that were monitored photometrically over a period of 15 min. Data analysis was performed using Dynafit software.51 Compound 38, with a C-4 chain length, showed the best inhibitory activity (Ki of 38 μM) against M. tuberculosis lumazine synthase (Table 1). The inhibitory activity increases significantly as the connector chain length between the sulfonamide and phosphate groups is increased from two to four carbons.
The compounds studied had been designed to fit the active site of lumazine synthase. Since the product of lumazine synthase is the substrate of riboflavin synthase and there may be some similarity of the ligand recognition pattern of lumazine synthase and riboflavin synthase, the compounds under study were assayed versus the riboflavin synthase of M. tuberculosis. In fact, compounds 39, 40, 77 were found to be weak, uncompetitive inhibitors of the enzyme with Ki values in the upper μM range (Table 1).
2.2. Molecular Modeling of Ligand Binding to Lumazine Synthase
The X-ray crystal structure of the complex of 4-(6-chloro-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)butyl dihydrogen phosphate (11)42 with lumazine synthase [PDB code: 2C97] with resolution 2.00 Å allowed the rational docking and energy minimization of newly synthesized compounds in the active site of M. tuberculosis lumazine synthase. Docking of the lead and synthesized compounds in the active site of M. tuberculosis lumazine synthase was performed with GOLD (BST, version 3.0, 2005). Energy minimization was performed with the MMFF94s force field in order to reduce steric clashes between the GOLD-docked compounds and the protein.
Figure 2 was constructed by displaying the amino acid residues of the enzyme involved in hydrogen bonding with the inhibitors 27 and 38 in the hypothetical model derived from docking and energy minimization. The maximum distance between the heavy atoms participating in the hydrogen bonds shown in Figure 2 was set at 3.8 Å. The binding patterns of both compounds are rather similar to each other and are comparable to that observed in the crystal structure of 11 in M. tuberculosis lumazine synthase. In general, the binding most likely reflects the contributions of the charged residue Arg128 and several polar residues that interact with the ligands directly via hydrogen bonding. The phosphate moiety of the ligands is extensively hydrogen bonded to the side chain nitrogens of Arg128, as well as the backbone nitrogens of Gly85 and Gln86 and the side chain hydroxyl of Thr87. Besides hydrogen bonding, the interaction of M. tuberculosis lumazine synthase and the inhibitor is predominantly hydrophobic. As expected, the calculated structural model positions the benzindolone aromatic rings of the ligands stacked with the indole ring of Trp27, consistent with the experimental structures of different lumazine synthase inhibitor complexes.7,42,43,51–53 The carbonyl oxygen of the benzindolone moiety of both compounds hydrogen bonds with the backbone nitrogen of Ala59. Because of the longer aliphatic chain of 38, the hydrophobic moiety of this compound is slightly shifted to the protein compared to the position of this moiety in the model with compound 27. This shift is resulted in formation of two additional hydrogen bonds by carbonyl oxygen of the benzindolone moiety with main chain nitrogen of Ile60 and with side chain oxygen of Glu61. Apparently, the formation of the additional hydrogen bonds is reflected in the better binding constant found for compound 38.
Figure 2.

Structural representation of the hypothetical structure of compounds 27 and 38 bound in the active site of M. tuberculosis lumazine synthase The amino acid residues involved in the active site are presented in ball and sticks and colored differently for two different subunits. The carbonyl atoms are shown in yellow and green, and nitrogen and oxygen atoms are shown in blue and red, respectively. The respective inhibitor molecules are colored in magenta (C3-alkylphosphate 27) and cyan (C4-alkylphosphate 38). The black dashed lines represent the putative interactions between compounds 38 and enzyme molecules. This figure was generated by PyMol [DeLano, W. L. (2002), the PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA, USA] and programmed for wall-eyed (relaxed) viewing.
Comparison of these models with the known structures of M. tuberculosis lumazine synthase in complex with different substrate 1 analogues showed the lack of five hydrogen bonds involving the ribityl chains of the substrate 1 analogues,43,54 and at least three hydrogen bonds formed between oxygen/nitrogen atoms of heterocyclic rings of the substrate 1 analogues and main/side chain atoms of Val81, Ile83 and Trp27 (Figure 3). Trp27 is involved in binding through π-π stacking interactions. The hydrogen bonds involving the heterocyclic rings of the substrate 1 analogues seem to be particularly important for the affinities of the substrate analogues. For instance, the earlier discovered inhibitor 11,42,44 which also lacks the ribityl chain but contains oxygen/nitrogen substitutions in the heterocyclic ring, demonstrated very high inhibitory activity with an inhibition constant in the nanomolar range. The lumazine synthase inhibitory activities displayed by the benzindolone derivatives, which lack both a ribityl side chain and a pyrimidinedione system, are therefore surprising.
Figure 3.

Crystal structure of 10 [PDB Code: 1W29] shown in green and 11 [PDB Code: 2C97] shown in pink bound to M. tuberculosis Lumazine synthase. Hydrogen bonds formed by protein with the ribityl group, heterocyclic ring and phosphate group are shown for the compound 10. The diagram is programmed for wall-eyed (relaxed) viewing.
2.3. Thermodynamic Characterization of Binding
The apparent thermodynamic binding characteristics of compounds 26, 27, 38, 39 and 40 were obtained by Isothermal Titration Calorimetry (ITC). Analysis of the changes in heat accompanying the binding reaction allowed the binding enthalpy of the processes (ΔH) to be derived, the stoichiometry (n) and association constants (Ka) to be estimated, and the entropy (ΔS) and free energy (ΔG) of the binding reactions to be calculated. Figure 4 shows a typical calorimetric titration of M. tuberculosis lumazine synthase in 100 mM potassium phosphate buffer at pH 7.0 and 30 °C with the compound containing the benzindolone group and a C3- (27) or C4-alkylphosphate chain (38). Earlier crystallographic studies of lumazine synthase from various organisms (B. subtilis, S. pombe and A. aeolicus)21,51,55 suggested that orthophosphate ions are sufficient for the stability of the pentameric assemblies and consequently for the activity of the protein. Moreover, all attempts to crystallize any lumazine synthase in a phosphate-free environment and to obtain the thermodynamic characteristics have ended unsuccessfully. Thus, we have been forced to run our experiments in phosphate buffer and would like to emphasize that we are dealing with a tertiary binding reaction involving a phosphate ion, an inhibitor molecule, and free enzyme. As a result, the association constants and the binding free energy derived from our ITC experiments should be considered as “apparent” thermodynamic parameters. Fitting of the binding isotherm was achieved with a model using a “single set of identical sites” assuming that each of the active sites in the pentameric assembly is occupied by one inhibitor molecule. Binding of both compounds is exothermic with negative changes in binding enthalpy, although the titration of compounds to the reference phosphate buffer showed endothermic changes in the system. In order to correct for compound dilution, the compound was injected into phosphate buffer and the dilution heat was subtracted from the reaction heat. The measured negative enthalpy changes of −7.6 ± 1.3 kcal·mol−1 for C4-alkylphosphate afforded an apparent association constant of 7.7·103 ± 0.5·103 M−1 with stoichiometry 1 for the binding reaction, in agreement with the expected binding of one inhibitor molecule in each of the five active sites of the pentameric protein. The calculated entropy ΔS and free energy of binding ΔG were −7.2 cal·mol−1·deg−1 and −5.4 kcal·mol−1, respectively, which showed that the binding reaction is favored enthalpically. The measured enthalpy change for C3-alkylphosphate compound 27 was −5.2 ± 0.7 kcal·mol−1, which is less favorable than for C4-alkylphosphate compound 38, but the loss in favorable enthalpy was compensated by a favorable positive entropy change ΔS of 0.98 cal·mol−1·deg−1. The resulting free energy of binding ΔG and apparent association constant Ka were −5.5 kcal·mol−1 and 8.9·103 ± 3.5·103 M−1, respectively. A similar enthalpy-entropy compensation effect was observed for the binding reaction of M. tuberculosis lumazine synthase 1043 and for the reaction of C. albicans LS with 11.44
Figure 4.

Calorimetric titration profiles of M. tuberculosis lumazine synthase with C4-alkylphosphate (38) (a) and C3-alkylphosphate (27) (b) at 30 °C. Top panels. Raw data obtained for 35 injections 8 μL each. The differential power signal recorded in the control titration of compound to the potassium phosphate buffer is presented on the insertions of top panels (a). Bottom panels. Integrated heat of binding reaction after subtraction of integrated heat of compound dilution plotted against molar ratio of total ligand concentration to total pentameric protein concentration. The solid line shows the best fit to the data, according to the model that assumes a single set of identical sites. For comparison, the titration calorimetry curves for lumazine synthase from Bacillus anthracis and Schizosaccharomyces pombe, respectively, titrated with C4-alkylphosphate compound 38 presented on panels (c) and (d) do not indicate a binding reaction.
Notably, the titration calorimetry experiments with 38 vs. icosahedral lumazine synthase from Bacillus anthracis and the pentameric enzyme from Schizosaccharomyces pombe did not detect binding within the same experimental limits (Figure 4). A structural comparison of the active sites of the enzymes showed a single mutation in M. tuberculosis lumazine synthase, G136E, which could be responsible for a different affinity to the ligands. Thus, the compounds produced in this study can be considered as specific ligands for M. tuberculosis enzyme and leads for further development.
To determine the structural motif required for the activity, the intermediates 31, 32 and 33 (Figure 5) without the phosphate group were tested. They were found to be totally inactive. Although in the molecular modeling study the oxybenzindolone moieties were involved in π–π stacking interactions with Trp27, the compounds do not have any binding affinity in the active site. The evidence clearly indicates that the phosphate group is necessary for the inhibitory activity.
Figure 5.

To find metabolically stable inhibitors of M. tuberculosis lumazine synthase, compounds with phosphonate 41 and fluorophosphonate 42 moieties (Figure 6) were synthesized with C-3, C-4 and C-5 chain length. Phosphonate and fluorophosphonate groups are stable to hydrolysis by phosphatases. The fluoro derivatives were synthesized because they more closely resemble the phosphate group electronically than the phosphonates do. The pKa values of α,α-difluorophosphonates are closer to those of phosphates, while phosphonates are less acidic than phosphates. On the other hand, fluorophosphonates resemble phosphonates sterically (the van der Waals radii for fluorine and hydrogen are 1.35 and 1.20 Å, respectively).56
Figure 6.
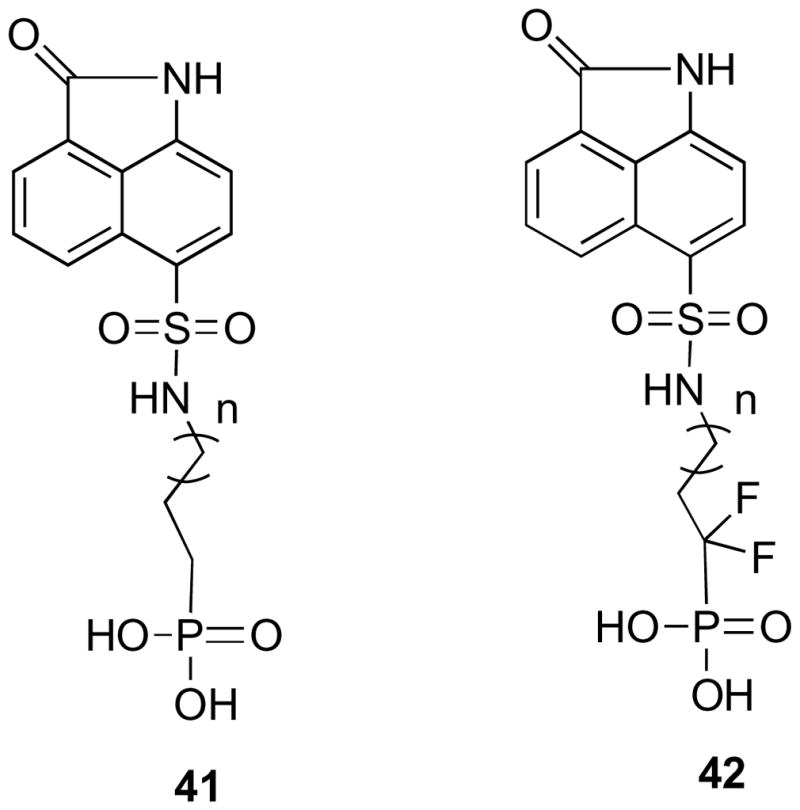
Compounds with phosphonate and fluorophosphonate moieties.
The synthesis (Scheme 6) started from diethylphosphite (45), which on treatment with 1,4-diiodobutane (43) and 1,5-diiodopentane (44) provided compounds 46 and 47.57,58 The iodo derivatives were converted to amines 50 and 51 via phthalimide derivatives 48 and 49.59,60 Treatment of benzindole sulfonyl chloride (Scheme 6) derivative 15 with amines 50 and 51 in THF provided the phosphonate derivatives 52 and 53. The deprotection of the ethyl groups with TMSBr furnished the desired phosphonate derivatives 54 and 55.
Scheme 6. Reagents and Conditions.

(a) NaH, THF, −20 °C 1h then −10 °C 18h; (b) DMF, 100 °C; (c) NH2NH2.H2O, EtOH, reflux; (d) 15, Et3N, THF, r.t.; (e) TMSBr, CH2Cl2, r.t.
The phosphonate derivatives 54 and 55 showed absolutely no inhibitory activity. At physiological pH, the difference between the phosphate and phosphonate groups is that a phosphate group is mainly diionic and a phosphonate group is mainly monoionic.
The synthesis of the difluorophosphonates 64–66 is outlined in Scheme 7. After much effort, reagent 56 was converted to the required compound 57.61–64 Reaction of 56 with activated zinc dust65 gave the stable [(diethoxyphosphinyl)difluoromethyl]zinc bromide, which on hydrolysis provided compound 57.
Scheme 7. Reagents and Conditions.

(a) i. Zn, Et2O, reflux, ii. H2O; (b) LDA, HMPA, −78 °C; (c) DMF, 100 °C.
Compound 57 when reacted at −78 °C with LDA in THF in the presence of hexamethylphosphoramide (HMPA) generated a lithium anion, which was treated with diiodo compounds to provide the desired products 61, 62 and 63 in approximately 20% yield. The poor yields observed in these reactions are probably due to the poor nucleophilicity of the highly stabilized lithium anion and its poor stability above −25 °C. Without HMPA, no alkylation was observed. An attempt was made to convert the iodo derivatives 61, 62 and 63 to amines via the standard Gabriel procedure by making the pthalimide derivatives 64, 65 and 66, but mixtures of products were obtained that were very hard to separate and characterize. Thus, the reverse strategy was considered, and accordingly, the sulfonyl chloride compound 15 was converted to the sulfonamide derivative 67 with ammonia in THF (Scheme 8).
Scheme 8. Reagents and Conditions.

(a) NH3, THF, rt; (b) K2CO3, THF, r.t.; (c) TMSBr, CH2Cl2, r.t.
Treatment of the sulfonamide derivative 67 with the iodo compounds 61, 62 and 63 in the presence of K2CO3 (Scheme 8) provided the protected benzindolone fluorophosphonate derivatives 68, 69 and 70. Finally, the removal of the ethyl groups with TMSBr furnished the desired products 71, 72 and 73 with fluorophosphonate groups. Surprisingly, the fluorophosphonate derivatives 71, 72 and 73 turned out to be inactive against M. tuberculosis lumazine synthase and M. tuberculosis riboflavin synthase, except for compound 72, which showed poor inhibitory activity against M. tuberculosis lumazine synthase (Table 1). Compound 72 corresponds to compound 38, bearing an alkylphosphate chain consisting of 4 carbon atoms, which exhibited the strongest enzyme inhibitory effect. Evidently, in this particular case, phosphonates and fluorophosphonates proved to be very poor mimics of the phosphate group of the inhibitors, although in other cases involving lumazine synthase, they were effective.41,66–67
The mechanism outlined in Scheme 2 is certainly reasonable, but questions still remain about the timing of phosphate elimination relative to the conformational reorganization of the side chain. Nevertheless, it is clear that the inorganic phosphate must eventually be released at some point to make way for new substrate. As mentioned above, the benzindolone moiety by itself does not have significant binding affinity considering that compound 31, 32 and 33 are totally inactive. On the other hand, the active phosphates must be able to displace inorganic phosphate from the phosphate binding pocket for any inhibitory activity and therefore the non-phosphate part of their structures must contribute positively in this regard. Compound 11, with a C-4 chain length, showed strong inhibitory activity (Ki 0.88 μM) against M. tuberculosis lumazine synthase. Compounds 77 and 79, which lack the phosphate group, were synthesized (Scheme 9). As expected, compounds 77 and 79 were inactive. This substantiates the point that although the pyrimidinedione ring alone does not have any binding affinity; it contributes positively to the binding of compound 11, since its phosphate group displaces inorganic phosphate from the phosphate binding pocket. The highlights of the synthesis of compounds 77 and 79 were the selective reduction of the nitro group to an amino group with sodium dithionate, and the removal of benzyl protecting groups under controlled hydrogenolysis using 1,4-cyclohexediene as a source of hydrogen.
Scheme 9. Reagents and Conditions.

(a) Na2S2O4, 1,4-dioxane, MeOH, buffer, reflux; (b) ClCOCO2Et, Et3N, THF, rt; (c) C2H5COCl, Et3N, THF, rt; (d) Lindlar catalyst, 1,4-cyclohexadine, ethanol, r.t.
Compounds 27 and 38, with C-3 and C-4 alkyl phosphate chains, showed better inhibitory activity against M. tuberculosis lumazine synthase. The inhibitory activity increases significantly with an increase in the alkyl phosphate chain length connector between sulfonamide and phosphate groups from two carbons to four carbons, but then the inhibitory activity decreases as the chain length increases. The expected pKa of the phosphonate derivatives 54 and 55 suggest that these derivatives are mainly monoionic, thus, they are unable to displace the inorganic phosphate. Although the fluorophosphonate derivatives 71, 72 and 73 have favorable pKa values, it seems they are also unable to displace the inorganic phosphate from the phosphate binding pocket. Fluorophosphonate derivative 72, which corresponds to compound 38 bearing an alkyl phosphate chain consisting of 4 carbon atoms, showed some inhibitory activity against M. tuberculosis lumazine synthase, with Ki values of 832 μM and Kis 518 μM. Molecular modeling of fluorophosphonate-lumazine synthase complex, did not reveal a structural basis for these unexpected findings.
To delve deeper into the nature of the phosphate binding site, information obtained by REDOR NMR spectroscopy of complexes of lumazine synthase from S. cerevisiae with phosphonate reaction intermediate analogues 80 was used.45 The 15N{31P} REDOR NMR spectra of those complexes indicated that mobility of the Lys92 side chain could facilitate the required active site release of phosphate during the enzyme catalysis. Although the exact nature of the phosphate binding pocket still remains elusive, it is clear that the inorganic phosphate must be displaced by organic phosphate from the phosphate binding pocket for any enzyme inhibitory activity. These compounds have a pyrimidinedione ring and a ribityl side chain, which contribute to their affinity for lumazine synthase. The binding of oxybenzindolone compounds in the active site of lumazine synthase depends on their abilities to bind at the phosphate binding site. The metabolically stable fluorophosphonate derivative 72, which showed moderate activity, could possibly be used as a 19F NMR mechanistic probe to study the structure of the occupied phosphate binding sites of lumazine synthase.
In virtual screening, finding novel active scaffolds is the most important success criterion. The oxybenzindolone derivatives provide unique structural probes of the active site of lumazine synthase. The lead compound contains “drug-like” features and therefore has more potential for drug development than the mechanism probes containing polar ribityl side chains. Although the oxybenzindolone moiety has poor affinity to the active site, the enzyme inhibitory activity could possibly be optimized by improving the binding affinity of the oxybenzindolone moiety through incorporation of oxygen- or nitrogen-containing functional groups. Optimization of the enzyme inhibitory activity through logical structure manipulation of the lead compound 9 already provided compound 38 with moderately improved inhibitory activity (Ki 38 μM) against M. tuberculosis lumazine synthase.
3. Experimental
3.1.1. 2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonyl Chloride (15)
Chlorosulfonic acid (3.2 mL) was added slowly to compound 14 (1.0 g, 5.9 mmol). The reaction mixture was stirred at 0 °C for 1 h and at room temperature for 2 h. The mixture was then poured into ice water (20 mL). The precipitate was washed with water (2 × 10 mL) and dried to give compound 15 as yellow solid (0.76 mg, 44%): mp 143–146 °C. 1H NMR (300 MHz, MeOH-d4) δ 10.34 (brs, 1 H), 8.68 (d, J = 8.3 Hz, 1 H), 8.38 (d, J = 7.9 Hz, 1 H), 8.20 (d, J = 7.0 Hz, 1 H), 8.12 (dd, J = 7.0 Hz, 8.3 Hz, 1 H), 7.22 (d, J = 7.9 Hz, 1 H); 13C NMR (75 MHz, MeOH-d4) δ 169.0, 147.3, 135.5, 133.0, 132.5, 129.1, 128.0, 127.6, 126.5, 124.7, 105.2; EIMS m/z (rel intensity) 267 (M+, 4), 232 (5), 155 (38), 91 (C5H7+, 100); CIMS m/z (rel intensity) 268 (MH+, 76), 232 (53), 197 (100). Anal. Calcd for C11H6ClNO3S: C 49.36, H 2.26, N 5.23, S 11.98. Found C 49.53, H 2.08, N 5.47, S 12.21.
3.1.2. Benzyl-2-(di-tert-butoxyphosphoryloxy)ethylcarbamate (20)
Tetrazole (3 wt% solution in CH3CN, 32 mL, 11.0 mmol) and di-tert-butyl-N,N-diisopropylphosphoramidite (95%, 2.73 mL, 8.2 mmol) were added to a solution of benzyl-2-hydroxyethylcarbamate (18) (1.07 g, 5.48 mmol) in THF (20 mL) and the mixture was stirred at room temperature for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 1.0 mL, 24 mmol) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aqueous Na2SO3 (10%, 50 mL) was added with water bath cooling. After 25 min, the organic solvents were removed under reduced pressure and the aqueous residue was extracted with EtOAc (3 × 30 mL). The combined extracts were washed with brine, dried, and evaporated. The residue was purified by silica gel column chromatography, eluting with EtOAc-petroleum ether (1:1), to afford the product 20 (1.36 g, 64%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.37-7.25 (m, 5 H), 5.64 (br s, 1 H), 5.03 (s, 2 H), 3.96 (m, 2 H), 3.38 (q, J = 5.1 Hz, 2 H), 1.41 (s, 18 H); 13C NMR (75 MHz, CDCl3) δ 156.3, 136.5, 128.1, 127.7, 82.2, 82.1, 66.1, 64.1, 37.3, 30.1, 29.9, 29.8, 29.5; EIMS m/z (rel intensity) 796 (2MNa+, 100), 774 (2M+, 94), 410 (MNa+, 48), 388 (MH+, 80); HRMS m/z calcd for C18H30NO6P (MH+) 388.1889, found 388.1880.
3.1.3. Benzyl-2-(di-tert-butoxyphosphoryloxy)propylcarbamate (21)
Tetrazole (3 wt% solution in CH3CN, 4.5 mL, 1.91 mmol) and di-tert-butyl-N,N-diisopropylphosphoramidite (95%, 0.45 mL, 1.43 mmol) were added to a solution of benzyl-2-hydroxypropylcarbamate (19) (200 mg, 0.96 mmol) in THF (5 mL) and the mixture was stirred at room temperature for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 0.4 mL, 24 mmol) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aqueous Na2SO3 (10%, 50 mL) was added with water bath cooling. After 25 min, the organic solvents were removed under reduced pressure and the aqueous residue was extracted with EtOAc (3 × 10 mL). The combined extracts were washed with brine, dried, and evaporated. The residue was purified by silica gel column chromatography, eluting with EtOAc-petroleum ether (1:1), to provide the product 21 (250 mg, 65%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.24 (m, 5 H), 5.76 (br s, 1 H), 5.00 (s, 2 H), 3.92 (m, 2 H), 3.23 (m, 2 H), 1.76 (m, 2 H), 1.38 (s, 18H); 13C NMR (75 MHz, CDCl3) δ 156.2, 136.3, 128.2, 127.7, 82.5, 82.4, 66.3, 65.5, 41.1, 30.1, 29.51, 29.47; EIMS m/z (rel intensity) 825 (2MNa+, 2), 424 (MNa+, 100); HRMS m/z calcd for C19H32NO6P (MNa+) 424.1865, found 424.1864.
3.1.4. Di-tert-butyl-2-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)ethyl Phosphate (24)
Method 1
A solution of 20 (1.17 g, 3.02 mmol) in MeOH (30 mL) with Pd/C (5%, 0.21 g) was hydrogenated at 50 psi for 4 h. The mixture was filtered through Celite, washed with MeOH (2 × 10 mL), and the filtrate was evaporated to give 2-aminoethyl di-tert-butyl phosphate (22) (604 mg, 79%) as colorless oil, which was used directly for the next reaction. A solution of amine 22 (100 mg, 0.39 mmol) and Et3N (0.11 mL, 0.79 mmol) in THF (2 mL) was added to a solution of compound 15 (106 mg, 0.39 mmol) in THF (3 mL) at 0 °C. After 5 min, the cooling bath was removed and the reaction mixture was stirred overnight. The mixture was diluted with water and extracted with CH2Cl2 (3 × 15 mL). The combined extracts were dried and evaporated and the residue was purified by silica gel column chromatography, eluting with ethyl acetate-petroleum ether (9:1), to provide compound 24 (135 mg, 71%) as yellow solid: mp 200–203 °C.
Method 2
Tetrazole (3 wt% solution in CH3CN, 1.6 mL, 0.68 mmol) and di-tert-butyl-N,N-diisopropylphosphoramidite (95%, 0.16 mL, 0.51 mmol) were added to a solution of 31 (100 mg, 0.34 mmol) in THF (4 mL) and the mixture was stirred at room temperature for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 0.3 mL, 1.71 mmol) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aqueous Na2SO3 (10%, 5 mL) was added with water bath cooling. After 25 min, the organic solvents were removed under reduced pressure and the aqueous residue was extracted with EtOAc (3 × 10 mL). The combined extracts were washed with brine, dried, and evaporated. The residue was purified by silica gel column chromatography, eluting with EtOAc-petroleum ether (9:1), to give 24 (110 mg, 67%) as a as yellow solid: mp 200–203 °C. 1H NMR (300 MHz, CDCl3) δ 9.55 (brs, 1 H), 8.68 (d, J = 8.4 Hz, 1 H), 8.09 (d, J = 7.5 Hz, 1 H), 8.03 (d, J = 7.0 Hz, 1 H), 7.79 (m, 1 H), 6.96 (d, J = 7.5 Hz, 1 H), 6.34 (brs, 1 H), 3.99 (m, 2 H), 3.12 (m, 2 H), 1.38 (s, 18 H); 13C NMR (75 MHz, CDCl3) δ 169.8, 142.2, 132.7, 130.5, 130.0, 128.6, 126.8, 125.3, 124.9, 104.9, 83.4, 83.3, 65.6, 43.4, 29.7; negative ion EIMS m/z (rel intensity) 483 [(M − H+)−, 78], 464 (20), 276 (50); HRMS m/z calcd for C21H29N2O7PS (M − H+)− 483.1355, found 483.1348.
3.1.5. Di-tert-butyl-3-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)propyl Phosphate (25)
A solution of 21 (0.3 g, 0.75 mmol) in MeOH (30 mL) with Pd/C (5%, 20 mg) was hydrogenated at 50 psi for 4 h. The mixture was filtered through Celite, washed with MeOH (2 × 10 mL), and the filtrate was evaporated to give 2-aminopropyl di-tert-butyl phosphate (23) (140 mg, 70%) as colorless oil, which was used directly for the next reaction. A solution of amine 23 (100 mg, 0.37 mmol) and Et3N (0.1 mL, 0.75 mmol) in THF (2 mL) was added to a solution of compound 15 (100 mg, 0.37 mmol) in THF (3 mL) at 0 °C. After 5 min, the cooling bath was removed and the reaction mixture was stirred overnight. The mixture was diluted with water and extracted with CH2Cl2 (3 × 15 mL). The combined extracts were dried and evaporated and the residue was purified by silica gel column chromatography, eluting with ethyl acetate-petroleum ether (9:1), to give compound 25 (112 mg, 60%) as yellow solid: mp 196–198 °C. 1H NMR (300 MHz, acetone-d6) δ 8.98 (d, J = 8.4 Hz, 1 H), 8.39 (d, J = 7.5 Hz, 1 H), 8.34 (d, J = 7.0 Hz, 1 H), 8.18 (m, 1 H), 7.35 (d, J = 7.5 Hz, 1 H), 4.16 (dd, J = 6.3, 12.8 Hz, 2 H), 3.26 (t, J = 6.9 Hz, 2 H), 2.03 (m, 2 H), 1.62 (s, 18 H); 13C NMR (75 MHz, acetone-d6) δ 169.2, 142.4, 132.4, 130.1, 129.4, 128.6, 126.8, 126.5, 124.7, 124.4, 104.2, 81.8, 81.7, 63.8, 63.7, 39.1, 29.9; EIMS m/z (rel intensity) 521 (MNa+, 92), 499 (MH+, 100), 387 (62); negative ion EIMS m/z (rel intensity) 995 [(2M − H+)−, 34], 533 (100), 497 [(M − H+)−, 91]; HRMS m/z calcd for C22H31N2O7PS 521.1487 (MNa+), found 521.1492.
3.1.6. 2-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)ethyl Dihydrogen Phosphate (26)
TFA (0.06 mL, 0.8 mmol) was added to a solution of 24 (40 mg, 0.083 mmol) in CH2Cl2 (5 mL) and the solution was allowed to stand at room temperature for 16 h. The mixture was evaporated, and the residue was washed with CH2Cl2 (3 ×10 mL) and dried to yield 26 (23 mg, 74%) as a light-yellow solid: mp 185–188 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.72 (d, J = 8.4 Hz, 1 H), 8.14 (d, J = 7.5 Hz, 1 H), 8.10 (d, J = 7.0 Hz, 1 H), 7.92 (m, 1 H), 7.05 (d, J = 7.5 Hz, 1 H), 3.89 (dd, J = 6.0, 13.0 Hz, 2 H), 3.13 (t, J = 5.8 Hz, 2 H); 13C NMR (75 MHz, MeOH-d4 and DMSO-d6) δ 169.1, 142.7, 132.7, 130.8, 129.9, 128.6, 127.1, 126.5, 125.1, 124.7, 104.9, 64.0, 42.9; EIMS m/z (rel intensity) 395 (MNa+, 25), 373 (MH+, 100), 338 (37), 293 (26), 274 (21); negative ion EIMS m/z (rel intensity) 743 [(2M − H+)−, 100], 371 [(M − H+)−, 51], 236 (10); HRMS m/z calcd for C13H13N2O7PS (MH+) 373.0259, found 373.0252. Anal. Calcd for C13H13N2O7PS·1.5 H2O: C, 39.10; H, 4.04; N, 7.02; P, 7.76. Found: C, 39.15; H, 3.73; N, 7.02; P, 7.89.
3.1.7. 3-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)propyl Dihydrogen Phosphate (27)
TFA (0.07 mL, 0.8 mmol) was added to a solution of 25 (40 mg, 0.081 mmol) in CH2Cl2 (5 mL) and the solution was allowed to stand at room temperature for 16 h. The mixture was evaporated, the residue was washed with CH2Cl2 (3 × 10 mL), and the product was dried to provide 27 (21 mg, 68%) as a light-yellow solid: mp 192–195 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.65 (d, J = 8.4 Hz, 1 H), 8.07 (d, J = 7.6 Hz, 1 H), 8.01 (d, J = 7.0 Hz, 1 H), 7.84 (m, 1 H), 7.00 (d, J = 7.6 Hz, 1 H), 3.91 (dd, J = 6.3, 12.8 Hz, 2 H), 2.97 (t, J = 6.8 Hz, 2 H), 1.75 (m, 2 H); 13C NMR (75 MHz, MeOH-d4) δ 171.4, 143.8, 133.8, 131.5, 131.0, 130.1, 128.1, 127.8, 126.1, 106.1, 64.8, 40.4, 31.7; EIMS m/z (rel intensity) 387 (MH+, 100), 338 (3), 311 (3); negative ion EIMS m/z (rel intensity) 771 [(2M − H+)−, 100], 385 [(M − H+)−, 36]; HRMS m/z calcd for C14H15N2O7PS (MH+) 387.0416, found 387.0419. Anal. Calcd for C14H15N2O7PS·H2O: C, 41.59; H, 4.24; N, 6.93. Found: C, 41.35; H, 4.31; N, 6.99.
3.1.8. N-(2-Hydroxyethyl)-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide (31)
Ethanolamine (0.05 mL, 0.84 mmol) was added to solution of compound 15 (150 mg, 0.56 mmol) in THF (5 mL) and the reaction mixture was stirred at room temperature for 12 h. THF was removed under vacuum and the residue was purified by silica gel column chromatography, eluting with EtOAc-petroleum ether (4:6), to give compound 31 (115 mg, 70%) as yellow solid: mp 192–195 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.72 (d, J = 8.4 Hz, 1 H), 8.12 (d, J = 7.5 Hz, 1 H), 8.10 (d, J = 6.8 Hz, 1 H), 7.90 (m, 1 H), 7.04 (d, J = 7.5 Hz, 1 H), 3.48 (t, J = 6.0 Hz, 1 H), 2.95 (d, J = 6.0 Hz, 1 H); 13C NMR (125 MHz, MeOH-d4) δ 171.1, 144.2, 134.1, 132.3, 132.2, 131.0, 128.1, 128.0, 126.5, 106.4, 61.8, 45.9; EIMS m/z (rel intensity) 315 (MNa+, 85), 293 (MH+, 42); negative ion EIMS m/z (rel intensity) 291 [(M − H+)−, 10], 248 (100); HRMS m/z calcd for C13H12N2O4S (MNa+) 315.0416, found 315.0420.
3.1.9. N-(2-Hydroxybutyl)-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide (32)
4-Amino-1-butanol (28) (0.08 mL, 0.84 mmol) was added to solution of compound 15 (150 mg, 0.56 mmol) in THF (5 mL) and the reaction mixture was stirred at room temperature for 12 h. THF was removed under vacuum and the residue was purified by silica gel column chromatography, eluting with EtOAc-petroleum ether (4:6), to give compound 32 (120 mg, 67%) as yellow solid: mp 238–240 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.73 (d, J = 8.2 Hz, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 8.11 (d, J = 6.9 Hz, 1 H), 7.90 (dd, J = 7.1, 8.4 Hz, 1 H), 7.04 (d, J = 7.6 Hz, 1 H), 3.39 (t, J = 6.0 Hz, 1 H), 2.87 (d, J = 6.6 Hz, 1 H), 1.42 (m, 4 H); 13C NMR (75 MHz, MeOH-d4) δ 171.5, 143.9, 133.8, 131.5, 131.1, 130.7, 128.1, 126.3, 126.2, 106.0, 62.2, 43.7, 30.6, 27.1; EIMS m/z (rel intensity) 321 (MH+, 83), 283 (34); negative ion EIMS m/z (rel intensity) 639 [(2M − H+)−, 66], 319 [(M − H+)−, 100], 248 (79); HRMS m/z calcd for C15H16N2O4S (MH+) 321.0909, found 321.0911.
3.1.10. N-(5-Hydroxypentyl)-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide (33)
5-Amino-1-pentanol (29) (0.3 g, 2.9 mmol) was added to a solution of compound 15 (200 mg, 0.75 mmol) in THF (5 mL) and the reaction mixture was stirred at room temperature for 24 h under argon gas. THF was removed under vacuum and yellow solid was purified using silica gel column chromatography, eluting with MeOH-EtOAc (8:92) to provide compound 33 (180 mg, 70%) as a yellow solid: mp 186–189 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.71 (d, J = 8.4 Hz, 1 H), 8.10 (d, J = 7.5 Hz, 1 H), 8.09 (d, J = 6.9 Hz, 1 H), 7.89 (t, J = 8.3 Hz, 1 H), 7.04 (d, J = 7.6 Hz, 1 H), 3.36 (t, J = 6.4 Hz, 2 H), 2.85 (t, J = 6.8 Hz, 2 H), 1.35 (m, 4 H), 1.22 (m, 2 H); 13C NMR (75 MHz, MeOH-d4) δ 171.3, 143.9, 133.8, 131.4, 131.1, 130.7, 128.3, 128.0, 126.1, 105.9, 62.6, 43.7, 32.9, 30.3, 23.8; negative ion ESIMS m/z (rel intensity) 667 ([2M − H+]−, 100), 333 ([M − H+]−, 45); HRMS m/z calcd for C16H18N2O4S (MH+) 335.1066, found 335.1063.
3.1.11. N-(6-Hydroxyhexyl)-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide (34)
6-Amino-1-hexanol (30) (315 mg, 2.69 mmol) was added to a solution of compound 15 (180 mg, 0.672 mmol) in THF (7 mL) and the mixture was allowed to stir overnight. THF was removed under vacuum and the residue was purified using silica gel column chromatography, eluting with EtOAc-hexane (8:2) and then MeOH-EtOAc (5:95), to result in starting material 15 (72 mg) and pure compound 34 as a yellow solid (100 mg, 45%): mp 190–192 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.73 (d, J = 8.4 Hz, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 8.11 (d, J = 6.9 Hz, 1 H), 7.91 (dd, J = 7.2, 8.3 Hz, 1 H), 7.05 (d, J = 7.5 Hz, 1 H), 3.39 (t, J = 6.5 Hz, 2 H), 3.34 (s, 1 H), 2.85 (t, J = 6.9 Hz, 2 H), 1.33 (m, 4 H), 1.13 (m, 4 H); 13C NMR (125 MHz, MeOH-d4) δ 171.2, 143.5, 133.8, 131.2, 131.1, 130.0, 128.0, 127.9, 126.0, 105.5, 62.3, 43.7, 33.1, 30.5, 27.0, 26.5; ESIMS m/z (rel intensity) 371 (MNa+, 51), 349 (MH+, 100); negative ion ESIMS m/z (rel intensity) 695 ([2M − H+]−, 100), 347 ([M − H+]−, 47); HRMS m/z calcd for C17H20N2O4S (MH+) 349.1222, found 349.1227.
3.1.12. Di-tert-butyl-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butyl Phosphate (35)
Tetrazole (3 wt% solution in CH3CN, 0.4 mL, 0.16 mmol) and di-tert-butyl-N,N-diisopropylphosphoramidite (95%, 0.24 mL, 0.12 mmol) were added to a solution of 32 (0.25 mg, 0.078 mmol) in THF (3 mL) and the reaction mixture was stirred at room temperature under argon for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 0.1 mL) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aquous Na2SO3 (10%, 2 mL) was added with water bath cooling. After 25 min, the organic solvents were evaporated and the aqueous residue was extracted with EtOAc (3 ×10 mL). The extracts were washed with brine, dried with Na2SO4, and evaporated. The resulting residue was purified by silica gel column chromatography, eluting with 90:10 EtOH-hexane, to yield pure compound 35 (24 mg, 60%) as a yellow residue. 1H NMR (300 MHz, MeOH-d4) δ 8.72 (d, J = 8.4 Hz, 1 H), 8.11 (d, J = 7.6 Hz, 1 H), 8.09 (d, J = 7.0 Hz, 1 H), 7.90 (dd, J = 7.1, 8.3 Hz, 1 H), 7.04 (d, J = 7.6 Hz, 1 H), 3.79 (dd, J = 5.9, 12.0 Hz, 2 H), 2.89 (t, J = 6.3 Hz, 2 H), 1.55-1.40 (22 H); 13C NMR (75 MHz, MeOH-d4) δ 171.4, 143.9, 133.8, 131.5, 131.1, 130.6, 128.3, 128.0, 126.2, 106.0, 84.1, 84.0, 67.7, 43.2, 30.1, 30.08, 28.3, 26.9; EIMS m/z (rel intensity) 1024 (2MH+, 45), 513 (MH+, 100), 457 (8); negative ion EIMS m/z (rel intensity) 556 (M − HCOO−, 38], 511 [(M − H+)−, 100]; HRMS m/z calcd for C23H33N2O7PS 513.1824 (MH+), found 513.1822.
3.1.13. Di-tert-butyl-5-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentyl Phosphate (36)
Tetrazole (3 wt% solution in CH3CN, 3.0 mL, 1.3 mmol) and di-tert-butyl-N,N-diisopropylphosphoramidite (95%, 0.26 mL, 0.75 mmol) were added to a solution of 33 (180 mg, 0.54 mmol) in THF (5 mL) and the reaction mixture was stirred at room temperature under argon for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 1.5 mL) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aqueous Na2SO3 (10%, 5 mL) was added with water bath cooling. After 25 min, the organic solvents were evaporated and the aqueous residue was extracted with EtOAc (3 × 10 mL). The extracts were washed with brine, dried with Na2SO4, and evaporated. The resulting yellow residue was purified on a silica gel column made with EtOAc-hexane (7:3), eluting with EtOAc-hexane (9:1), to result in pure compound 36 (125 mg, 44%). 1H NMR (300 MHz, acetone-d6) δ 10.22 (s, 1 H), 8.66 (d, J = 8.1 Hz, 1 H), 8.04 (d, J = 7.5 Hz, 1 H), 7.98 (d, J = 6.9 Hz, 1 H), 7.82 (t, J = 6.9 Hz, 1 H), 7.01 (d, J = 7.6 Hz, 1 H), 6.78 (t, J = 4.8 Hz, 1 H), 3.73 (dd, J = 5.1, 6.3 Hz, 2 H), 2.82 (q, J = 5.7 Hz, 2 H), 1.36 (m, 22 H), 1.22 (m, 2 H); 13C NMR (75 MHz, acetone-d6) δ 206.1, 169.1, 143.2, 133.0, 130.7, 129.7, 127.6, 127.3, 125.51, 125.0, 104.858, 82.0, 66.8, 66.7, 43.1. 29.6, 22.9; ESIMS m/z (rel intensity) 549 (MNa+, 100); negative ion ESIMS m/z (rel intensity) 531 (92), 525 ([M − H+]−, 100); HRMS m/z calcd for C24H35N2O7PS (MNa+) 549.1800, found 549.1802.
3.1.14. Di-tert-butyl-5-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)hexyl Phosphate (37)
Tetrazole (3 wt% solution in CH3CN, 2.1 mL, 1.0 mmol) and di-tert-butyl-N,N-diisopropyl-phosphoramidite (95%, 0.20 mL, 0.8 mmol) were added to a solution of 34 (172 mg, 0.5 mmol) in THF (5 mL) and the reaction mixture was stirred at room temperature under argon for 16 h. The mixture was cooled to 0 °C and H2O2 (70% aqueous, 1.2 mL) was added. After 15 min, the cooling bath was removed and the mixture was stirred for a further 6 h, and then aqueous Na2SO3 (10%, 5 mL) was added with water bath cooling. After 25 min, the organic solvents were evaporated and the aqueous residue was extracted with EtOAc (3 × 10 mL). The extracts were washed with brine, dried with Na2SO4, and evaporated. The resulting yellow residue was purified by silica gel column chromatography, eluting with EtOAc-hexane (8:1), to result in pure compound 37 (95 mg, 18%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 9.91 (s, 1 H), 8.66 (d, J = 8.4 Hz, 1 H), 8.06 (d, J = 7.6 Hz, 1 H), 7.98 (d, J = 7.0 Hz, 1 H), 7.73 (t, J = 7.3 Hz, 1 H), 6.99 (d, J = 7.6 Hz, 1 H), 6.90 (s, 1 H), 3.84 (q, J = 6.5 Hz, 2 H), 2.87 (q, J = 4.2 Hz, 2 H), 1.47 (m, 22 H), 1.16 (m, 4 H); 13C NMR (75 MHz, CDCl3) δ 169.8, 142.9, 142.2, 132.5, 130.3, 129.7, 128.6, 126.6, 125.1, 124.8, 105.1, 82.9, 82.8, 66.9, 66.8, 42.7. 29.6, 29.3, 25.8, 24.8; ESIMS m/z (rel intensity) 541 (M+, 9), 429 (100); negative ion ESIMS m/z (rel intensity) 539 ([M − H+]−, 3), 483 ([M-tBu+]−, 100); HRMS m/z calcd for C25H37N2O7PS (MH+) 541.2137, found 541.2136.
3.1.15. 4-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butyl Dihydrogen Phosphate (38)
TFA (0.03 mL, 0.35 mmol) was added to a solution of 35 (18 mg, 0.035 mmol) in CH2Cl2 (2 mL) and the solution was stirred at room temperature for 16 h. The mixture was evaporated, and the residue was washed with CH2Cl2 (3 ×10 mL) and dried under vacuum to yield compound 38 (35 mg, 69%) as a light yellow solid: mp 194–196 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.72 (d, J = 8.1 Hz, 1 H), 8.12 (d, J = 6.3 Hz, 1 H), 8.09 (d, J = 5.4 Hz, 1 H), 7.90 (dd, J = 7.2, 8.4 Hz, 1 H), 7.05 (d, J = 7.5 Hz, 1 H), 3.83 (dd, J = 6.0, 12.3 Hz, 2 H), 2.88 (t, J = 6.6 Hz, 2 H), 1.56 (t, J = 6.3 Hz, 2 H), 1.52 (t, J = 5.1 Hz, 2 H); 13C NMR (75 MHz, MeOH-d4) δ 171.5, 143.9, 133.8, 131.5, 130.6, 128.3, 128.0, 126.2, 106.0, 67.0, 43.3, 28.5, 26.9; EIMS m/z (rel intensity) 801 (2MH+, 55), 401 (MH+, 100), 303 (19); negative ion EIMS m/z (rel intensity) 799 [(2M − H+)−, 100], 399 [(M − H+)−, 21]; HRMS m/z calcd for C15H17N2O7PS (MH+) 401.0572, found 401.0580. Anal. Calcd for C15H17N2O7PS·1 H2O: C, 43.06; H, 4.58; N, 6.70. Found: C, 43.35; H, 4.25; N, 6.56.
3.1.16. 5-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentyl Dihydrogen Phosphate (39)
TFA (0.1 mL, 1.27 mmol) was added to a solution of 36 (120 mg, 0.23 mmol) in CH2Cl2 (3 mL) and the solution was allowed to stir at room temperature for 16 h overnight. The mixture was evaporated, and the residue was washed with CH2Cl2 (3 × 10 mL) and dried to provide compound 39 (65 mg, 62%): mp 209–212 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.71 (d, J = 8.4 Hz, 1 H), 8.11 (d, J = 7.5 Hz, 1 H), 8.09 (d, J = 6.9 Hz, 1 H), 7.91 (t, J = 7.3 Hz, 1 H), 7.04 (d, J = 7.6 Hz, 1 H), 3.73 (q, J = 6.6 Hz, 2 H), 2.82 (t, J = 6.6 Hz, 2 H), 1.40 (m, 6 H); 13C NMR (75 MHz, MeOH-d4) δ 171.5, 143.9, 133.8, 131.5, 131.1, 130.6, 128.3, 128.0, 126.2, 106.0, 67.4, 43.6, 30.9, 30.8, 30.1, 23.6; ESIMS m/z (rel intensity) 829, ([2M + H]+, 7), 415 (MH+, 100), 317 (10); negative ion ESIMS m/z (rel intensity) 827 ([2M − H+]−,100), 413 ([M − H+]−,19); HRMS m/z calcd for C16H19N2O7PS (MH+) 415.0729, found 415.0725. Anal. Calcd for C16H19N2O7PS: C 46.38, H 4.62, N 6.67, S 7.74. Found C 45.99, H 4.67, N 6.63, S 7.78.
3.1.17. 5-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)hexyl Dihydrogen Phosphate (40)
TFA (0.12 mL, 1.7 mmol) was added to a solution of 37 (90 mg, 0.17 mmol) in CH2Cl2 (5 mL) and the solution was allowed to stir at room temperature for 16 h overnight. The mixture was evaporated, and the residue was washed with CH2Cl2 (3 × 10 mL) and dried to give compound 40 (22 mg, 31%): mp 224–228 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.73 (d, J = 8.4 Hz, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 8.11 (d, J = 6.9 Hz, 1 H), 7.91 (dd, J = 7.2, 8.3 Hz, 1 H), 7.05 (d, J = 7.6 Hz, 1 H), 3.83 (q, J = 6.6 Hz, 2 H), 2.82 (t, J = 6.9 Hz, 2 H), 1.45 (m, 2 H), 1.35 (m, 2 H), 1.18 (m, 4 H); 13C NMR (75 MHz, MeOH-d4) δ 171.5, 143.9, 133.8, 131.5, 131.2, 130.8, 128.3, 128.1, 126.3, 106.0, 67.6, 67.5, 43.7, 31.3, 31.2, 30.4, 27.1, 26.0; ESIMS m/z (rel intensity) 451, (MNa+, 10), 429 (MH+, 100); HRMS m/z calcd for C17H21N2O7PS (MH+) 429.0885, found 429.0890. Anal. Calcd for C17H21N2O7PS: C 47.66, H 4.94, N 6.54, S 7.45. Found C 47.31, H 4.70, N 6.47, S 7.45.
3.1.18. Diethyl 4-Iodobutylphosphonate (46)
Sodium hydride (60%) in mineral oil (0.75 g, 18.8 mmol) was suspended in anhydrous THF (70 mL) and the reaction mixture cooled to −20 °C. A solution of diethyl phosphate (2.0 g, 14.5 mmol) in THF (10 mL) was introduced under argon. After stirring at −20 °C for 1 h, 1,4-diiodobutane (43) (3.8 mL, 29.0 mol) was added and the mixture was stirred at −10 °C for 18 h. The mixture was then concentrated under vacuum, taken up in EtOAc (30 mL) and water (30 mL), and the pH adjusted to 8 with concentrated HCl. The EtOAc was washed with brine, dried over Na2SO4, and evaporated to a colorless oil, which was purified by silica gel flash column chromatography with CH2C12-EtOAc (3:7) as eluant to afford 46 (2.6 g, 57%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.09-4.01 (m, 4 H), 3.20 (t, J = 6.6 Hz, 2 H), 1.86 (m, 2 H), 1.70 (m, 4 H), 1.27 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 61.5, 61.41, 33.8, 33.6, 25.4, 23.5, 23.4, 16.4, 16.4, 5.5; EIMS m/z (rel intensity) 321 (MH+, 15), 343 (MNa+, 100); HRMS m/z calcd for C8H18IO3P (MNa+) 342.9936, found 342.9944.
3.1.19. Diethyl 4-Iodopentylphosphonate (47)
Sodium hydride (60%) in mineral oil (0.75 g, 18.8 mmol) was suspended in anhydrous THF (70 mL) and the reaction mixture cooled to −20 °C. A solution of diethyl phosphate (2.0 g, 14.5 mmol) in THF (10 mL) was introduced under argon. After stirring at −20 °C for 1 h, 1,5-diiodopantane (44) (2.6 mL, 17.4 mol) was added and the mixture was stirred at −10 °C for 18 h. The mixture was then concentrated in vacuo, taken up in EtOAc (30 mL) and water (30 mL), and the pH adjusted to 8 with concentrated HCl. The EtOAc was washed with brine, dried over Na2SO4, and evaporated to a colorless oil, which was purified by flash silica gel column chromatography using CH2C12 as eluant to yield 47 (2.5 g, 52%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 3.78-3.70 (m, 4 H), 2.84 (t, J = 6.9 Hz, 2 H), 1.53-1.20 (m, 6 H), 1.16 (m, 2 H), 0.96 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 60.8, 60.7, 32.3, 30.8, 30.5, 25.8, 23.9, 20.9, 20.8, 15.9, 15.86, 5.9; EIMS m/z (rel intensity) 335 (MH+, 100), 207 (14); HRMS m/z calcd for C9H20IO3P (MH+) 335.0273, found 335.0283.
3.1.20. Diethyl 4-(1,3-Dioxoisoindolin-2-yl)butylphosphonate (48)
A DMF solution (5 mL) of iodobutylylphosphonate 46 (1.0 g, 3.13 mmol) and of potassium phthalimide (1.16 g, 6.26 mmol) was stirred at 100 °C for 8 h. The mixture was cooled to 25 °C and the precipitated material filtered off. The filtrate was concentrated in vacuo and the residue taken up in water (50 mL). After extraction with diethyl ether (3 × 30 mL), the organic phase was dried (Na2SO4) and evaporated, and the residue purified by flash silica gel column chromatography using CH2C12-EtOAc (1:9) as eluant to afford 48 (0.9 g, 85%) as white solid: mp 52–54 °C. 1H NMR (300 MHz, CDCl3) δ 7.75 (m, 4 H), 4.13-4.02 (m, 4 H), 3.64 (t, J = 6.7 Hz, 2 H), 1.91-1.72 (m, 4 H), 1.63-1.57 (m, 2 H), 1.34 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 169.4, 135.2, 133.0, 123.9, 62.94, 62.86, 37.9, 30.1, 29.9, 26.1, 24.2, 20.7, 20.6, 16.8, 16.7; EIMS m/z (rel intensity) 340 (MH+, 2), 362 (MNa+, 100); HRMS m/z calcd for C16H22NO5P (MNa+) 362.1133, found 362.1139.
3.1.21. Diethyl 4-(1,3-Dioxoisoindolin-2-yl)pentylphosphonate (49)
A DMF (5 mL) solution of iodopentylphosphonate 47 (1.0 g, 3.0 mmol) and potassium phthalimide (1.11 g, 6.0 mmol) was stirred at 100 °C for 8 h. The mixture was cooled to 25 °C and the precipitated material filtered off. The filtrate was concentrated in vacuo and the residue taken up in water (50 mL). After extraction with diethyl ether (3 × 30 mL), the organic phase was dried (Na2SO4), evaporated and the residue purified by flash silica gel column chromatography using silica gel and CH2C12- EtOAc (1:9) as eluant to afford 49 (0.91 g, 86%) as yellowish semisolid. 1H NMR (300 MHz, MeOH-d4) δ 7.76 (m, 4 H), 4.09-4.03 (m, 4 H), 3.62 (t, J = 7.0 Hz, 2 H), 1.85-1.60 (m, 6 H), 1.42 (m, 2 H), 1.30 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, MeOH-d4) δ 169.6, 135.2, 133.2, 124.0, 63.04, 62.95, 38.4, 28.9, 28.6, 28.4, 26.5, 24.6, 23.0, 22.9, 16.8, 16.7; EIMS m/z (rel intensity) 354 (MH+, 87), 376 (MNa+, 100); HRMS m/z calcd for C17H24NO5P (MNa+) 376.1290, found 376.1296.
3.1.22. Diethyl 4-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butylphosphonate (52)
Compound 48 (0.4 g, 1.3 mmol) was dissolved in absolute ethanol (8 mL) and 98% hydrazine hydrate (0.18 mL, 3.9 mmol) was added. The reaction mixture was stirred at reflux for 3 h. The ethanol was removed in vacuo and the residue purified by flash column chromatography with silica gel, eluting with 5% MeOH in ethyl acetate, to afford compound 50 (0.2 g, 81%) as a colorless oil. Amine 50 (117 mg, 0.56 mmol) and derivative 15 (100 mg, 0.37 mmol) were dissolved in THF (5 mL). The reaction mixture was stirred at room temperature for 12 h. The THF was removed in vacuo and the residue purified by flash silica gel column chromatography, eluting with 1% MeOH in ethyl acetate, to afford compound 52 (0.1 g, 61%) as yellow oil. 1H NMR (300 MHz, MeOH-d4) δ 8.68 (d, J = 8.3 Hz, 1 H), 8.08 (d, J = 7.6 Hz, 1 H), 8.04 (d, J = 7.2 Hz, 1 H), 7.93 (dd, J = 7.2, 8.3 Hz, 1 H), 7.02 (d, J = 7.5 Hz, 1 H), 3.98 (m, 4 H), 2.86 (m, 2 H), 1.61 (m, 2 H), 1.46 (m, 4 H), 1.24 (t, J = 7.1 Hz, 6 H); 13C NMR (75 MHz, MeOH-d4) δ 171.3, 143.8, 133.9, 133.7, 131.5, 131.0, 130.5, 128.2, 127.9, 126.1, 106.0, 63.1, 63.0, 43.1, 31.1, 30.9, 26.0, 24.1, 20.4, 20.3, 16.73, 16.65; EIMS m/z (rel intensity) 441 (MH+, 100), 463 (MNa+, 69); negative ion EIMS m/z (rel intensity) 439 [(M − H+)−, 100]; HRMS m/z calcd for C19H25N2O6PS (MH+) 441.1249, found 441.1251.
3.1.23. Diethyl 4-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentylphosphonate (53)
Compound 49 (0.25 g, 0.7 mmol) was dissolved in absolute ethanol (5 mL) and 98% hydrazine hydrate (0.1 mL, 2.1 mmol) was added. The reaction mixture was stirred at reflux for 3 h. The ethanol was removed in vacuo and the residue purified by silica gel flash column chromatography, eluting with 5% MeOH in ethyl acetate, to afford compound 51 (0.13 g, 82%) as a colorless oil. Amine 51 (125 mg, 0.56 mmol) and derivative 15 (100 mg, 0.37 mmol) were dissolved in THF (5 mL). The reaction mixture was stirred at room temperature for 12 h. The THF was removed in vacuo and the residue purified by silica gel flash column chromatography with silica gel, eluting with 1% MeOH in ethyl acetate, to afford compound 53 (110 mg, 67%) as a yellow oil. 1H NMR (300 MHz, MeOH-d4) δ8.67 (d, J = 8.4 Hz, 1 H), 8.07 (d, J = 7.6 Hz, 1 H), 8.04 (d, J = 7.0 Hz, 1 H), 7.86 (dd, J = 7.0, 8.4 Hz, 1 H), 7.01 (d, J = 7.6 Hz, 1 H), 4.01 (m, 4 H), 2.84 (t, J = 6.5 Hz, 2 H), 1.49 (m, 2 H), 1.46 (m, 6 H), 1.25 (t, J = 7.1 Hz, 6 H); 13C NMR (75 MHz, MeOH-d4) δ 171.3, 143.8, 133.9, 133.7, 131.4, 131.1, 130.6, 128.2, 127.9, 126.5, 126.1, 106.0, 63.1, 63.0, 43.4, 29.8, 28.3, 28.1, 26.3, 24.5, 22.7, 22.6, 16.74; EIMS m/z (rel intensity) 455 (MH+, 100), 477 (MNa+, 44); negative ion EIMS m/z (rel intensity) 453 [(M − H+)−, 100]; HRMS m/z calcd for C20H27N2O6PS (MH+) 455.1406, found 455.1411.
3.1.24. 4-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butylphosphonic Acid (54)
Bromotrimethylsilane (0.6 mL, 0.45 mmol) was added to a solution of compound 52 (50 mg, 0.12 mmol) in CH2Cl2 (4 mL). After stirring for 36 h at 25 °C, the volatiles were removed in vacuo, and the residue was purified by passing it through lipophilic Sephadex (LH20), using CH2Cl2-MeOH (8:2) as eluant, to produce 54 (28 mg, 64%) as a yellowish solid: mp 259–261 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 11.13 (s, 1 H), 8.65 (d, J = 8.1 Hz, 1 H), 8.09 (d, J = 6.9 Hz, 1 H), 8.00 (d, J = 7.5 Hz, 1 H), 7.93 (dd, J = 7.2, 8.1 Hz, 1 H), 7.78 (t, J = 7.2 Hz, 1 H), 7.04 (d, J = 7.5 Hz, 1 H), 2.70 (m, 2 H), 1.39 (m, 6 H); 13C NMR (125 MHz, MeOH-d4 and DMSO-d6) δ 169.4, 135.2, 133.0, 123.9, 62.94, 62.86, 37.9, 30.1, 29.9, 26.1, 24.2, 20.7, 20.6, 16.8, 16.7; EIMS m/z (rel intensity) 791 (2MNa+, 100), 384 (MNa+, 27), 179 (38); negative ion EIMS m/z (rel intensity) 767 [(2M − H+)−, 100], 383 [(M − H+)−, 75]; HRMS m/z calcd for C15H17N2O6PS (MNa+) 407.0443, found 407.0441. Anal. Calcd for C15H17N2O6PS.CH2Cl2: C, 40.95; H, 4.08; N, 5.97. Found: C, 41.18; H, 4.16, N, 6.30.
3.1.25. 4-(2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentylphosphonic Acid (55)
Bromotrimethylsilane (0.6 mL, 0.44 mmol) was added to the solution of compound 53 (50 mg, 0.11 mmol) in CH2Cl2 (4 mL). After stirring for 36 h at 25 °C, the volatiles were removed in vacuo, and the residue was purified by lipophilic Sephadex column (LH20), using CH2Cl2-MeOH (8:2) as eluant, to result in 55 (34 mg, 61%) as a yellowish solid: mp 294–296 °C (dec). 1H NMR (500 MHz, DMSO-d6) δ 11.13 (s, 1 H), 8.66 (d, J = 8.3 Hz, 1 H), 8.09 (d, J = 6.9 Hz, 1 H), 8.01 (d, J = 7.5 Hz, 1 H), 7.94 (dd, J = 7.2, 8.1 Hz, 1 H), 7.76 (t, J = 7.2 Hz, 1 H), 7.05 (d, J = 7.5 Hz, 1 H), 2.72 (m, 2 H), 1.30 (m, 6 H), 1.18 (m, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 169.0, 142.6, 132.5, 130.4, 129.8, 128.9, 127.0, 126.2, 125.0, 124.5, 104.9, 42.2, 28.7, 27.9, 27.2, 27.0, 26.9, 22.4; negative ion EIMS m/z (rel intensity) 795 [(2M − H+)−, 100], 398 [(M − H+)−, 55]. Anal. Calcd for C16H19N2O6PS·H2O: C, 46.15; H, 5.08; N, 6.73. Found: C, 46.22; H, 4.93, N, 6.8.
3.1.26. Diethyl 1,1-Difluoro-4-iodobutylphosphonate (61)
A solution of LDA (2.9 mL, 5.85 mmol, 2M solution) in dry THF (10 mL) was added at −78 °C to a solution of diethyl (difluoromethy1)phosphonate (57) (1.0 g, 5.32 mmol) in HMPA (2 mL). After stirring for 1 h at −78 °C, a solution of diiodopropane (58) (1.35 mL, 11.69 mmol) in THF (10 mL) was added and the mixture was allowed to stir for 6 h at −78 °C. The reaction mixture was then poured into ether (50 mL)-20% H3PO4 (10 mL) and the aqueous layer was extracted with ether (20 mL × 2). The combined ether extracts were washed with brine, dried over Na2SO4, and concentrated under vacuum. The residual oil was purified by silica gel column chromatography using CH2Cl2-30% EtOAc as eluent to give 61 (0.35 g, 19%) as a yellow oil: 1H NMR (300 MHz, CDC13) δ 4.23 (dt, J = 7.2, 14.9 Hz, 4 H), 3.18 (t, J = 6.6 Hz, 2 H), 2.09 (m, 4 H), 1.34 (t, J = 6.9 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 121.5 (dt), 64.4, 64.3, 34.6 (m), 24.8, 24.8, 16.3, 16.2, 4.9; EIMS m/z (rel intensity) 357 (MH+, 100), 379 (MNa+, 78); HRMS m/z calcd for C8H16F2IO3P (MH+) 356.9928, found 356.9927.
3.1.27. Diethyl 1,1-Difluoro-5-iodopentylphosphonate (62)
A solution of LDA (2.5 mL, 4.97 mmol, 2M solution) in dry THF (10 mL) was added at −78 °C to a solution of diethyl (difluoromethy1)phosphonate (57) (0.85 g, 4.52 mmol) in HMPA (2 mL). After stirring for 1 h at −78 °C, a solution of diiodobutane (59) (1.3 mL, 9.94 mmol) in THF (10 mL) was added and the mixture was allowed to stir for 6 h at −78 °C. The reaction mixture was then poured into ether (50 mL)-20% H3PO4 (10 mL) and the aqueous layer was extracted with ether (20 mL × 2). The combined ether extracts were washed with brine, dried over Na2SO4, and concentrated under vacuum. The residual oil was purified by silica gel column chromatography using CH2Cl2-30% EtOAc as eluent to give 62 (0.28 g, 17%) as a yellow oil: 1H NMR (300 MHz, CDC13) δ 4.22 (dt, J = 7.2, 14.9 Hz, 4 H), 3.13 (t, J = 6.8 Hz, 2 H), 2.03 (m, 2 H), 1.80 (m, 2 H), 1.66 (m, 2 H), 1.34 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 112.3 (dt), 63.8, 63.7, 32.2 (m), 21.3, 21.2, 15.8, 15.7, 5.2; EIMS m/z (rel intensity) 371 (MH+, 100), 343 (29); HRMS m/z calcd for C9H18F2IO3P (MH+) 371.0085, found 371.0082.
3.1.28. Diethyl 1,1-Difluoro-5-iodohexylphosphonate (63)
A solution of LDA (2.5 mL, 4.97 mmol, 2M solution) in dry THF (10 mL) was added at −78 °C to a solution of diethyl (difluoromethy1)phosphonate (57) (0.85 g, 4.52 mmol) in HMPA (2 mL). After stirring for 1 h at −78 °C, a solution of diiodopentane (60) (1.3 mL, 9.94 mmol) in THF (10 mL) was added and the mixture was allowed to stir for 6 h at −78 °C. The reaction mixture was then poured into ether (50 mL)-20% H3PO4 (10 mL) and the aqueous layer was extracted with ether (20 mL × 2). The combined ether extracts were washed with brine, dried over Na2SO4, and concentrated under vacuum. The residual oil was purified by silica gel column chromatography using CH2Cl2-30% EtOAc as eluent to give 63 (0.4 g, 23%) as a yellow oil: 1H NMR (300 MHz, CDC13) δ 4.13 (dt, J = 7.1, 15.0 Hz, 4 H), 3.06 (t, J = 6.9 Hz, 2 H), 1.90 (m, 2 H), 1.7.2 (m, 2 H), 1.49 (m, 2 H), 1.34 (m, 2 H), 1.24 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, CDCl3) δ 119.0 (dt), 64.1, 64.0, 33.5 (m), 32.8, 29.8, 19.4, 19.3, 16.2, 16.1, 6.2; EIMS m/z (rel intensity) 385 (MH+, 55), 329(MH+ – C4H8, 100); HRMS m/z calcd for C10H20F2IO3P (MH+) 385.0241, found 385.0243.
3.1.29. 2-Oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide (67)
Compound 15 (300 mg, 1.12 mmol) was dissolved in dry THF (10 mL). Ammonia gas was dissolved in the reaction mixture at −78 °C for 20 min. The reaction mixture was stirred at room temperature for 6 h. The yellow precipitate was filtered and dried to give compound 67 (0.22 g, 79%) as yellow solid: mp 272–275 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 8.62 (d, J = 8.3 Hz, 1 H), 8.09 (d, J = 7.0 Hz, 1 H), 8.03 (d, J = 7.5 Hz, 1 H), 7.92 (dd, J = 7.3 Hz, 8.0 Hz, 1 H), 7.05 (d, J = 7.5 Hz, 1 H); 13C NMR (75 MHz, DMSO-d6) δ 169.4, 142.2, 132.8, 130.7, 130.09, 130.08, 127.1, 126.3, 125.2, 124.5, 105.1; EIMS m/z (rel intensity) 531 [(2M+ + Cl− , 60], 496 (2M+, 100), 283 [(M+) + Cl−), 33], 248 (M+, 85). Anal. Calcd for C11H8N2O3S: C, 53.22; H, 3.25; N, 11.28. Found: C, 53.24; H, 3.33, N, 11.42.
3.1.30. Diethyl 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butyl-phosphonate (68)
K2CO3 (83 mg, 0.6 mmol) was added to the solution of iodo derivative 61 (79 mg, 0.22 mmol) and amine derivative 67 (50 mg, 0.20 mmol) in DMF (3 mL). The reaction mixture was stirred at room temperature for 12 h. Water (25 mL) was added to the reaction mixture and the mixture was extracted with diethyl ether (20 mL × 3). The combined organic extracts were washed with brine, dried (Na2SO4), and evaporated. The residue was purified by flash silica gel column chromatography, eluting with EtOAc-petroleum ether (7:3), to afford compound 68 (60 mg, 63%) as a yellow oil. 1H NMR (300 MHz, acetone-d6) δ 8.73 (d, J = 8.4 Hz, 1 H), 8.18 (d, J = 7.5 Hz, 1 H), 8.10 (d, J = 7.0 Hz, 1 H), 7.94 (t, J = 8.1 Hz, 1 H), 7.28 (d, J = 7.5 Hz, 1 H), 6.80 (s, 2 H), 4.24-4.14 (m, 4 H), 4.07 (t, J = 6.7 Hz, 2 H), 2.24-2.09 (m, 4 H), 1.27 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, acetone-d6) δ 168.2, 144.1, 134.0, 131.5, 131.0, 130.8, 127.3, 126.5, 125.6, 120.6, 104.4, 64.9, 64.8, 40.1, 32.0 (m), 21.2, 16.6, 16.5; EIMS m/z (rel intensity) 499 (MNa+, 100), 477 (MH+, 23), 444 (29); HRMS m/z calcd for C19H23F2N2O6PS (MNa+) 499.0880, found 499.0889.
3.1.31. Diethyl 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentyl-phosphonate (69)
K2CO3 (83 mg, 0.6 mmol) was added to a solution of iodo derivative 62 (82 mg, 0.22 mmol) and amine derivative 67 (50 mg, 0.20 mmol) in DMF (3 mL). The reaction mixture was stirred at room temperature for 12 h. Water (25 mL) was added to the reaction mixture and the mixture was extracted with diethyl ether (20 mL × 3). The combined organic extracts were washed with brine, dried (Na2SO4), and evaporated. The residue was purified by flash silica gel column chromatography, eluting with EtOAc-petroleum ether (7:3), to afford compound 69 (65 mg, 66%) as a yellow oil. 1H NMR (300 MHz, acetone-d6) δ 8.87 (d, J = 8.4 Hz, 1 H), 8.31 (d, J = 7.6 Hz, 1 H), 8.23 (d, J = 7.0 Hz, 1 H), 8.07 (t, J = 8.1 Hz, 1 H), 7.38 (d, J = 7.3 Hz, 1 H), 6.94 (s, 2 H), 4.38-4.28 (m, 4 H), 4.15 (t, J = 6.8 Hz, 2 H), 2.29-2.18 (m, 4 H), 1.83 (m, 2 H), 1.43 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, acetone-d6) δ 168.2, 144.2, 133.8, 131.5, 130.9, 130.7, 127.3, 126.4, 125.5, 125.4, 120.6 (dt), 104.4, 64.8, 64.7, 40.3, 34.2 (m), 19.02, 18.91, 16.7, 16.6; EIMS m/z (rel intensity) 491 (MH+, 100), 471 (10); negative ion EIMS m/z (rel intensity) 489 [(M − H+)−, 100], 457 (34); HRMS m/z calcd for C20H25F2N2O6PS (MH+) 491.1217, found 491.1216.
3.1.32. Diethyl 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)-hexyl- phosphonate (70)
K2CO3 (83 mg, 0.6 mmol) was added to the solution of iodo derivative 63 (84 mg, 0.22 mmol) and amine derivative 67 (50 mg, 0.20 mmol) in DMF (3 mL). The reaction mixture was stirred at room temperature for 12 h. Water (25 mL) was added to the reaction mixture and the mixture was extracted with diethyl ether (20 mL × 3). The combined organic extracts were washed with brine, dried (Na2SO4), and evaporated. The residue was purified by flash silica gel column chromatography, eluting with EtOAc-petroleum ether (7:3), to afford compound 70 (60 mg, 59%) as a yellow oil. 1H NMR (300 MHz, acetone-d6) δ 8.71 (d, J = 8.4 Hz, 1 H), 8.16 (d, J = 7.5 Hz, 1 H), 8.06 (d, J = 6.9 Hz, 1 H), 7.90 (t, J = 8.2 Hz, 1 H), 7.20 (d, J = 7.7 Hz, 1 H), 6.78 (s, 2 H), 4.21 (m, 4 H), 3.96 (t, J = 6.8 Hz, 2 H), 2.03 (m, 2 H), 1.86 (m, 2 H), 1.60 (m, 2 H), 1.48 (m, 2 H), 1.30 (t, J = 7.0 Hz, 6 H); 13C NMR (75 MHz, acetone-d6) δ 168.1, 144.2, 133.7, 131.5, 130.9, 130.6, 127.3, 126.3, 125.4, 125.3, 120.4 (dt), 104.3, 64.8, 64.7, 40.5, 34.3 (m), 27.1, 21.2, 21.1, 16.7, 16.6; EIMS m/z (rel intensity) 505 (MH+, 100), 447 (45); HRMS m/z calcd for C21H27F2N2O6PS (MH+) 505.1374, found 505.1375.
3.1.33. 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)butylphosphonic Acid (71)
Bromotrimethylsilane (0.6 mL, 0.42 mmol) was added to the solution of compound 68 (50 mg, 0.10 mmol) in CH2Cl2 (4 mL). After stirring for 36 h at 25 °C, the volatiles were removed in vacuo, and the residue was purified by passing it through lipophilic Sephadex (LH20) using CH2Cl2-MeOH (8:2) as eluant to give 71 (32 mg, 73%) as a yellowish solid: mp 127–130 °C. 1H NMR (500 MHz, MeOH-d4) δ 8.66 (d, J = 8.1 Hz, 1 H), 8.15 (d, J = 7.5 Hz, 1 H), 8.00 (d, J = 6.6 Hz, 1 H), 7.84 (t, J = 7.5 Hz, 1 H), 7.11 (d, J = 7.5 Hz, 1 H), 3.98 (t, J = 6.6 Hz, 2 H), 2.15 (m, 2 H), 2.07 (m, 2 H); 13C NMR (125 MHz, MeOH-d4) δ 169.7, 144.0, 134.3, 131.7, 131.3, 127.2, 126.6, 126.2, 125.7, 124.9, 122.2 (dt), 105.3, 40.8, 32.4 (m), 21.6; EIMS m/z (rel intensity) 841 (2MH+, 63), 421 (MH+, 100); negative ion ESIMS m/z (rel intensity) 839 ([2M − H+]−, 100), 419 ([M − H+]−, 33); HRMS m/z calcd for C15H15F2N2O6PS (MH+) 421.0435, found 421.0431. Anal. Calcd for C15H15F2N2O6PS·H2O: C, 41.10; H, 3.91; N, 6.39; P, 7.07. Found: C, 41.32; H, 4.09; N, 6.58; P, 6.97.
3.1.34. 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)pentylphosphonic Acid (72)
Bromotrimethylsilane (0.6 mL, 0.42 mmol) was added to the solution of compound 69 (50 mg, 0.10 mmol) in CH2Cl2 (4 mL). After stirring for 36 h at 25 °C, the volatiles were removed in vacuo, and the residue was purified by passing it through lipophilic Sephadex (LH20) using CH2Cl2-MeOH (8:2) as eluant to give 72 (27 mg, 61%) as a yellowish solid: mp 170–173 °C. 1H NMR (500 MHz, MeOH-d4) δ 8.70 (d, J = 8.1 Hz, 1 H), 8.17 (d, J = 7.5 Hz, 1 H), 8.07 (d, J = 6.6 Hz, 1 H), 7.87 (t, J = 7.5 Hz, 1 H), 7.14 (d, J = 7.5 Hz, 1 H), 3.97 (t, J = 6.6 Hz, 2 H), 2.12 (m, 2 H), 1.86 (m, 2 H), 1.68 (m, 2 H); 13C NMR (125 MHz, MeOH-d4) δ 169.7, 144.3, 134.4, 131.8, 131.3, 127.4, 126.8, 126.2, 121.0 (dt), 105.4, 40.9, 34.5 (m), 29.4, 19.6; EIMS m/z (rel intensity) 869 (2MH+, 100), 435 (MH+, 74); negative ion ESIMS m/z (rel intensity) 867 ([2M − H+]−, 100), 433 ([M − H+]−, 25); HRMS m/z calcd for C16H17F2N2O6PS (MH+) 435.0591, found 435.0593. Anal. Calcd for C16H17F2N2O6PS: C, 44.24; H, 3.94; N, 6.45; P, 7.13. Found: C, 44.39; H, 4.25; N, 6.62; P, 6.92.
3.1.35. 1,1-Difluoro-4-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)hexylphosphonic Acid (73)
Bromotrimethylsilane (0.5 mL, 0.42 mmol) was added to the solution of compound 70 (50 mg, 0.10 mmol) in CH2Cl2 (4 mL). After stirring for 36 h at 25 °C, the volatiles were removed in vacuo, and the residue was purified by passing it through lipophilic Sephadex (LH20) using CH2Cl2-MeOH (8:2) as eluant to give 73 (30 mg, 68%) as a yellowish solid: mp 90–93 °C. 1H NMR (300 MHz, MeOH-d4) δ 8.65 (d, J = 8.1 Hz, 1 H), 8.13 (d, J = 7.5 Hz, 1 H), 8.00 (d, J = 6.6 Hz, 1 H), 7.83 (t, J = 7.5 Hz, 1 H), 7.07 (d, J = 7.5 Hz, 1 H), 3.90 (t, J = 6.6 Hz, 2 H), 2.02 (m, 2 H), 1.84 (m, 2 H), 1.78 (m, 2 H), 1.42 (m, 2 H); 13C NMR (125 MHz, MeOH-d4) δ 169.7, 144.2, 134.3, 131.7, 131.23, 131.18, 127.3, 126.1, 125.7, 122.5 (dt), 105.3, 41.0, 34.8 (m), 29.3, 27.7, 21.7; EIMS m/z (rel intensity) 897 (2MH+, 100), 449 (MH+, 86); negative ion ESIMS m/z (rel intensity) 895 ([2M − H+]−, 100), 447 ([M − H+]−, 12); HRMS m/z calcd for C17H19F2N2O6PS (MH+) 449.0748, found 449.0758. Anal. Calcd for C17H19F2N2O6PS·H2O: C, 43.78; H, 4.54; N, 6.01; P, 6.64. Found: C, 43.72; H, 4.69; N, 5.78; P, 6.41.
3.1.36. Ethyl 2-(2,4-Bis(benzyloxy)-6-chloropyrimidin-5-ylamino)-2-oxoacetate (76)
Triethylamine (0.3 mL, 2.2 mmol) was added to a solution of compound 75 (80 mg, 0.23 mmol) in THF (5 mL). The solution was cooled to 0 °C and ethyl 2-chloro-2-oxoacetate (31 μL, 0.28 mmol) was added dropwise. The reaction mixture was stirred at 0 °C for 12 h. The solvent was distilled off under reduced pressure and the residue was dissolved in dichloromethane (20 mL) and washed with water (2 × 15 mL). The organic layer was dried and solvent was distilled off. The residue was flash column chromatographed with silica gel, eluting with 35% ethyl acetate in hexane, to afford compound 76 (78 mg, 76%) as a white semisolid. 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 1 H), 7.44-7.30 (m, 10 H), 5.41 (s, 2 H), 5.36 (s, 2 H), 4.37 (q, J = 7.13, 2 H), 1.37 (t, J = 7.14, 3 H); 13C NMR (75 MHz, CDCl3) δ 166.4, 161.7, 159.7, 158.3, 154.5, 135.4, 135.0, 128.42, 128.37, 128.23, 128.18, 128.1, 127.8, 108.7, 70.1, 69.7, 63.6, 13.8; HRMS m/z calcd for C22H20ClN3O5 (MNa+) 464.0989, found 464.0991. Anal. Calcd for C22H20ClN3O5: C, 59.80; H, 4.56; N, 9.51. Found: C, 59.83; H, 4.31; N, 9.80.
3.1.37. Ethyl 2-(6-Chloro-2,4,dioxo-1,2,3,4-tetrahydropyrimidin-5-ylamino)-2-oxoethanoate (77)
Lindlar catalyst (5 mg) was added to a solution of compound 76 (50 mg, 0.113 mmol) in anhydrous ethanol (4 mL). 1,4-Cyclohexediene (90 μL, 1.13 mmol) was added and argon was bubbled through the reaction mixture for 10 min. The mixture was stirred at room temperature for 12 h. The reaction mixture was filtered through celite, which was then washed with ethanol (2 × 5 mL). The solution was concentrated and the residue was washed several times with CH2Cl2 and THF. Finally, compound 77 was precipitated out by dissolving it in MeOH and adding excess diethyl ether to furnish pure compound 77 (20 mg, 68%) as a white amorphous solid: mp 174–176 °C. 1H NMR (300 MHz, MeOH-d4) δ 4.37 (q, J = 6.9 Hz, 2 H), 1.37 (t, J = 6.9 Hz, 3 H); 13C NMR (75 MHz, MeOH-d4) δ 162.8, 160.8, 158.8, 149.6, 129,6, 107.9, 64.2, 14.2; negative ion EIMS m/z (rel intensity) 260 [(M − H+)−, 100], 224 (43); HRMS m/z calcd for C8H8ClN3O5 (MNa+) 284.0050, found 284.0053. Anal. Calcd for C8H8ClN3O5·0.65 H2O: C, 35.15; H, 3.43; N, 15.37. Found: C, 34.8; H, 3.18; N, 15.10.
3.1.38. N-(6-Chloro-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)propionamide (78)
Triethylamine (0.3 mL, 2.2 mmol) was added to a solution of compound 75 (80 mg, 0.23 mmol) in THF (5 mL). The solution was cooled to 0 °C and propionyl chloride (25 μL, 0.28 mmol) was added dropwise. The reaction mixture was stirred at 0 °C for 12 h. The solvent was distilled off under reduced pressure and the residue was dissolved in dichloromethane (20 mL) and washed with water (2 × 15 mL). The organic layer was dried and solvent was distilled off. The residue was flash column chromatographed with silica gel, eluting with 35% ethyl acetate in hexane, to afford compound 78 (65 mg, 70%) as white solid: mp 122–124 °C. 1H NMR (300 MHz, CDCl3) δ 7.42-7.32 (m, 10 H), 5.35 (s, 2 H), 5.33 (s, 2 H), 2.33 (q, J = 7.23, 2 H), 1.37 (t, J = 7.1, 3 H); 13C NMR (75 MHz, CDCl3) δ 172.8, 166.9, 161.2, 158.7, 135.5, 135.3, 128.4, 128.2, 127.7, 110.5, 70.0, 69.5, 29.3, 9.7; HRMS m/z calcd for C21H20ClN3O3 (MNa+) 420.1091, found 420.1088. Anal. Calcd for C21H20ClN3O3: C, 63.40; H, 5.07; N, 10.56. Found: C, 63.66; H, 4.82; N, 10.78.
3.1.39. N-(6-Chloro-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)propanamide (79)
Lindlar catalyst (5 mg) was added to a solution of compound 78 (50 mg, 0.13 mmol) in anhydrous ethanol (4 mL). 1,4-Cyclohexediene (0.1 mL, 1.3 mmol) was added and argon was bubbled through the reaction mixture for 10 min. The mixture was stirred at room temperature for 12 h. The reaction mixture was filtered through celite, which was then washed with ethanol (2 × 5 mL). The solution was concentrated and the residue was washed several times with CH2Cl2 and THF. Finally, compound 79 was precipitated out by dissolving it in MeOH and adding excess diethyl ether to furnish pure compound 79 (18 mg, 66%) as a white amorphous solid: mp 208–211 °C (dec). 1H NMR (300 MHz, MeOH-d4) δ 2.38 (q, J = 7.6 Hz, 2 H), 1.18 (t, J = 7.6 Hz, 3 H); 13C NMR (75 MHz, MeOH-d4) δ 177.1, 163.1, 152.0, 147.2, 109.8, 29.8, 10.1; EIMS m/z (rel intensity) 262 (M − H+ + 2Na+, 82), 240 (MNa+, 100), 130 (35); negative ion EIMS m/z (rel intensity) 216 [(M − H+)−, 100], 180 (25); HRMS m/z calcd for C7H8ClN3O3 (MNa+) 240.0152, found 240.0153. Anal. Calcd for C7H8ClN3O3·1.4 H2O: C, 34.62; H, 4.48; N, 17.30. Found: C, 34.80; H, 4.18; N, 17.10.
3.2. Computational Methods
3.2.1. Screening the Database
The ZINC database maintained by the Shoichet group at UCSF is a pre-filtered comprehensive database of available compounds.39 The ligands in this database have multiple protonation and tautomeric states. Approximately 1.03 million compounds were downloaded in SDF format with multiple tautomeric and protonation states (pH range 5.75–8.25) selected from following vendors: Asinex, Chembridge, Maybridge, Nanosyn and Sigma-Aldrich. The M. tuberculosis lumazine synthase crystal structure in complex with inhibitor 3-(1,3,7-trihydro-9-D-ribityl-2,6,8-purinetrione-7-yl) propane 1-phosphate (PDB: 1w19) was prepared using Protein Preparation tools in Maestro 7.5 (Schrödinger LLC, Portland, Oregon) removing all crystal water molecules. The compounds were first docked with Glide 4.0 (Schrödinger LLC, Portland, Oregon) High Throughput Virtual Screening mode without any constraints. The top 5% of hits were then screened using Glide Standard Precision (SP), applying the following pharmacophore constraints: 3 backbone hydrogen bonds to Ile83, Val81, Ala59 and the phosphate interaction to Arg128. At least 3 of these constraints had to be satisfied. For all hits with Glide Score -5 or better, logS and logP were calculated with QikProp 2.5 (Schrödinger LLC, Portland, Oregon). Compounds that were predicted with reasonable solubility (logP < 2.5 and logS > −3.6) were selected resulting in 6742 compounds. These selected compounds were docked again with Glide Extra Precision (XP). Glide XP not only offers a more extensive conformational search but also recognizes multiple protein-ligand motifs such as enclosure of lipophilic ligand substructures by hydrophobic protein residues and various hydrogen bonds in hydrophobic environments. In addition, the scoring function has additional penalty terms including desolvation of polar groups and solvent exposure of hydrophobic ligand group designed to remove potential false positives.46 The backbone hydrogen bond constraints were again applied. The final 44 compounds that had Glide Score of −10 or better were inspected visually and 12 of them were tested based on the chemical scaffolds and interaction with the receptor for inhibition properties by enzymatic assay and Isothermal Titration Calorimetry (ITC). The visual inspection is a common practice which is important to avoid testing false positives due to assumptions and shortcomings in docking methods and scoring functions.68 The score cutoff of −10 was chosen based on docking of several known active compounds41 under the same condition.
3.2.2. Molecular Modeling
Using Sybyl (Tripos, Inc., version 7.1), the crystal structure of the complex of compound 11 was clipped to include information within a 12 Å sphere of one of the 5 equivalent ligand molecules. Docking of the lead and synthesized compounds in the active site of M. tuberculosis lumazine synthase was performed with GOLD (BST, version 3.0, 2005). The programs were set to conduct 10,000 repeats for each ligand under identical conditions. The best fit result from GOLD docking was added to complex, MMFF94 charges were loaded, and the energy of the complex was minimized using the Powell method to a termination gradient of 0.05 kcal/mol employing the MMFF94s force field, which was used to minimize steric clashes between the GOLD-docked compounds and the protein.
3.3. Isothermal Titration Calorimetry
Calorimetric measurements for binding analysis were carried out by using a VP-ITC MicroCalorimeter (MicroCal, Inc., Northampton, MA, USA). The reference cell was filled with water and the instrument was calibrated using standard electrical pulses. Binding isotherms were measured by direct titration. A solution of M. tuberculosis lumazine synthase (0.05 mM) in 100 mM potassium-phosphate buffer at pH 7.1 in a total volume of 1.451 mL was titrated at 30 °C with 35 identical 8 μL injections at 3 min intervals. The syringe was filled with 5 mM compound dissolved in the same buffer. The heat evolved after each injection was obtained from the integral of the calorimetric signal. All data were evaluated with the Microcal Origin50 Software package (Microcal Software, INC., Northampton, MA, USA) supplied with the calorimeter. The heat of dilution was subtracted from the observed heat in the binding experiments. The association constants, Ka, binding enthalpy, ΔH, and stoichiometry, n, were obtained by fitting the data to standard equations for the binding using a model for one set of independent and identical binding sites as implemented in the MicroCal Origin50 package. The binding entropy ΔS and free energy ΔG of the binding were calculated from the basic thermodynamic equations, ΔG = −RTlnK and the Gibbs-Helmholtz equation ΔG = ΔH − TΔS.
3.4. Enzymes Used in Kinetic Assays
Recombinant lumazine synthase and riboflavin synthase of M. tuberculosis were prepared as described earlier.43,69 The purified enzymes had specific activity of 1.2 μmol mg−1 h−1 (lumazine synthase) and 2.6 μmol mg−1 h−1 (riboflavin synthase).
3.4.1. Kinetic Assay of Lumazine Synthase Inhibitors
Assay mixtures contained 100 mM Tris hydrochloride, pH 7.0, 100 mM NaCl, 2% (v/v) DMSO, 5 mM dithiothreitol, 100 μM 2, 2 μM lumazine synthase, variable concentrations of 1 (3 – 100 μM) and inhibitor (0 – 200 μM) in a volume of 0.2 mL. Assay mixtures were prepared as follows. A solution (170 μL) containing 105 mM NaCl, 5.3 mM dithiothreitol, 118 μM 2, 2.4 μM lumazine synthase in 105 mM Tris hydrochloride, pH 7.0, was added to 4 μL of inhibitor in 100% (v/v) DMSO in a well of a 96-well microtiter plate. The reaction was started by adding 20 μL of a solution containing 105 mM NaCl, 5.3 mM dithiothreitol, and substrate 1 (30 – 1000 μM) in 105 mM Tris hydrochloride, pH 7.0. The formation of 6,7-dimethyl-8-D-ribityllumazine (3) was measured online for a period of 30 min at 27 °C with a computer-controlled plate reader at 408 nm (εLumazine = 10,200 M−1cm−1).
3.4.2. Kinetic Assay of Riboflavin Synthase Inhibitors
Assay mixtures contained 100 mM Tris hydrochloride, pH 7.0, 100 mM NaCl, 2% (v/v) DMSO, 5 mM dithiothreitol, 1 μM riboflavin synthase, variable concentrations of 3 (3 – 60 μM) and inhibitor (0 – 200 μM) in a volume of 0.2 mL. Assay mixtures were prepared as follows. A solution (170 μL) containing 105 mM NaCl, 5.3 mM dithiothreitol, 1.2 μM riboflavin synthase in 105 mM Tris hydrochloride, pH 7.0, was added to 4 μL of inhibitor in 100% (v/v) DMSO in a well of a 96-well microtiter plate. The reaction was started by adding 20 μL of a solution containing 105 mM NaCl, 5.3 mM dithiothreitol, and substrate 3 (30 – 600 μM) in 105 mM Tris hydrochloride, pH 7.0. The formation of riboflavin (4) was measured online for a period of 30 min at 27 oC with a computer-controlled plate reader at 470 nm (εRiboflavin = 9,600 M−1cm−1).
3.4.3. Evaluation of kinetic data
The velocity-substrate data were fitted for all inhibitor concentrations with a non-linear regression method using the program DynaFit.69 Different inhibition models were considered for the calculation. Ki and Kis values ± standard deviations were obtained from the fit under consideration of the most likely inhibition model.
Supplementary Material
Figure 7.
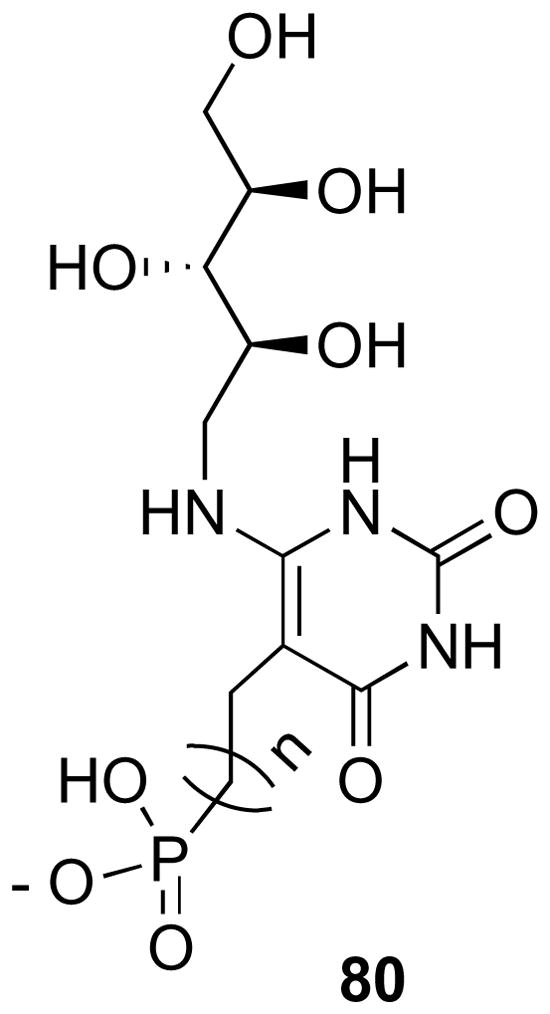
Phosphonate reaction intermediate analogues.
Footnotes
Supporting Information Available. 1H and 13C NMR spectra of compounds 20, 21, 24, 25, 26, 27, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 46, 47, 48, 49, 52, 53, 54, 55, 61, 62, 63, 67, 68, 69, 70, 71, 72, 73, 76, 77, 78 and 79.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- 1.Wang A. I Chuan Hsueh Pao. 1992;19:362–368. [Google Scholar]
- 2.Oltmanns O, Lingens F. Z Naturforsch. 1967;22(b):751–754. [PubMed] [Google Scholar]
- 3.Logvinenko EM, Shavlovsky GM. Mikrobiologiya. 1967;41:978–979. [Google Scholar]
- 4.Neuberger G, Bacher A. Biochem Biophys Res Commun. 1985;127:175–181. doi: 10.1016/s0006-291x(85)80141-8. [DOI] [PubMed] [Google Scholar]
- 5.Richter G, Ritz H, Katzenmeier G, Volk R, Kohnle A, Lottspeich F, Allendorf D, Bacher A. J Bacteriol. 1993;175:4045–4051. doi: 10.1128/jb.175.13.4045-4051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritz H, Schramek N, Bracher A, Herz S, Eisenreich W, Richter G, Bacher A. J Biol Chem. 2001;276:22273–22277. doi: 10.1074/jbc.M100752200. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser J, Schramek N, Eberhardt S, Püttmer S, Schuster M, Bacher A. Eur J Biochem. 2002;269:5264–5270. doi: 10.1046/j.1432-1033.2002.03239.x. [DOI] [PubMed] [Google Scholar]
- 8.Richter G, Fischer M, Krieger C, Eberhardt S, Lüttgen H, Gerstenschläger I, Bacher A. J Bacteriol. 1997;179:2022–2028. doi: 10.1128/jb.179.6.2022-2028.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacher A, Richter G, Ritz H, Eberhardt S, Fischer M, Krieger C. Methods Enzymol. 1997;280:382–389. doi: 10.1016/s0076-6879(97)80129-2. [DOI] [PubMed] [Google Scholar]
- 10.Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. Vol. 3. CRC Press; Boca Raton, FL: 1992. [Google Scholar]
- 11.Richter G, Volk R, Krieger C, Lahm HW, Röthlisberger U, Bacher AJ. Bacteriol. 1992;174:4050–4056. doi: 10.1128/jb.174.12.4050-4056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richter G, Krieger C, Volk R, Kis K, Ritz H, Götze E, Bacher A. Methods Enzymol. 1997;280:374–382. doi: 10.1016/s0076-6879(97)80128-0. [DOI] [PubMed] [Google Scholar]
- 13.Fischer M, Römisch W, Schiffmann S, Kelly M, Oschkinat H, Steinbacher S, Huber R, Eisenreich W, Richter G, Bacher A. J Biol Chem. 2002;277:41410–41416. doi: 10.1074/jbc.M206863200. [DOI] [PubMed] [Google Scholar]
- 14.Steinbacher S, Schiffmann S, Richter G, Huber R, Bacher A, Fischer M. J Biol Chem. 2003;278:42256–42265. doi: 10.1074/jbc.M307301200. [DOI] [PubMed] [Google Scholar]
- 15.Fischer M, Haase I, Kis K, Meining W, Ladenstein R, Cushman M, Schramek N, Huber R, Bacher A. J Mol Biol. 2003;326:783–793. doi: 10.1016/s0022-2836(02)01473-0. [DOI] [PubMed] [Google Scholar]
- 16.Fischer M, Haase I, Feicht R, Richter G, Gerhardt S, Changeux JP, Huber R, Bacher A. Eur J Biochem. 2002;269:519–526. doi: 10.1046/j.0014-2956.2001.02674.x. [DOI] [PubMed] [Google Scholar]
- 17.Bacher A, Eberhardt S, Fischer M, Mörtl S, Kis K, Kugelbrey K, Scheuring J, Schott K. Methods Enzymol. 1997;280:389–399. doi: 10.1016/s0076-6879(97)80130-9. [DOI] [PubMed] [Google Scholar]
- 18.Haase I, Mörtl S, Köhler P, Bacher A, Fischer M. Eur J Biochem. 2003;270:1025–1032. doi: 10.1046/j.1432-1033.2003.03478.x. [DOI] [PubMed] [Google Scholar]
- 19.Haase I, Fischer M, Bacher A, Schramek N. J Biol Chem. 2003;278:37909–37915. doi: 10.1074/jbc.M303090200. [DOI] [PubMed] [Google Scholar]
- 20.Schramek N, Haase I, Fischer M, Bacher A. J Am Chem Soc. 2003;125:4460–4466. doi: 10.1021/ja028226k. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Meining W, Fischer M, Bacher A, Ladenstein R. J Mol Biol. 2001;306:1099–1114. doi: 10.1006/jmbi.2000.4435. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Meining W, Cushman M, Haase I, Fischer M, Bacher A, Ladenstein R. J Mol Biol. 2003;328:167–182. doi: 10.1016/s0022-2836(03)00186-4. [DOI] [PubMed] [Google Scholar]
- 23.Scheuring J, Kugelbrey K, Weinkauf S, Cushman M, Bacher A, Fischer M. J Org Chem. 2001;66:3811–3819. doi: 10.1021/jo001739u. [DOI] [PubMed] [Google Scholar]
- 24.Mörtl S, Fischer M, Richter G, Tack J, Weinkauf S, Bacher A. J Biol Chem. 1996;271:33201–33207. doi: 10.1074/jbc.271.52.33201. [DOI] [PubMed] [Google Scholar]
- 25.Meining W, Mörtl S, Fischer M, Cushman M, Bacher A, Ladenstein RJ. Mol Biol. 2000;299:181–197. doi: 10.1006/jmbi.2000.3742. [DOI] [PubMed] [Google Scholar]
- 26.Illarionov B, Eisenreich W, Bacher A. Proc Natl Acad Sci U S A. 2001;98:7224–7229. doi: 10.1073/pnas.131610698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illarionov B, Kemter K, Eberhardt S, Richter G, Cushman M, Bacher A. J Biol Chem. 2001;276:11524–11530. doi: 10.1074/jbc.M008931200. [DOI] [PubMed] [Google Scholar]
- 28.Eberhardt S, Richter G, Gimbel W, Werner T, Bacher A. Eur J Biochem. 1996;242:712–719. doi: 10.1111/j.1432-1033.1996.0712r.x. [DOI] [PubMed] [Google Scholar]
- 29.Gerhardt S, Schott AK, Kairies N, Cushman M, Illarionov B, Eisenreich W, Bacher A, Huber R, Steinbacher S, Fischer M. Structure (Camb) 2002;10:1371–1381. doi: 10.1016/s0969-2126(02)00864-x. [DOI] [PubMed] [Google Scholar]
- 30.Illarionov B, Haase I, Bacher A, Fischer M, Schramek N. J Biol Chem. 2003;278:47700–47706. doi: 10.1074/jbc.M305050200. [DOI] [PubMed] [Google Scholar]
- 31.Volk R, Bacher A. J Am Chem Soc. 1988;110:3651–3653. [Google Scholar]
- 32.Lyne PD. Drug Discov Today. 2002;7:1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 33.Garg A, Tewari R, Raghava GPS. BMC Bioinformatics. 2010;11:S53. doi: 10.1186/1471-2105-11-S1-S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okimoto N, Futatsugi N, Fuji H, Suenaga A, Morimoto G, Yanai R, Ohno Y, Narumi T, Taiji M. PLoS Comput Biol. 2009;5:e1000528. doi: 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen IJ, Neamati N, Nicklaus MC, Orr A, Anderson L, Barchi JJ, Kelly JA, Pommier Y, MacKerell AD. Bioorg Med Chem. 2000;8:2385–2398. doi: 10.1016/s0968-0896(00)00180-2. [DOI] [PubMed] [Google Scholar]
- 36.Hopkins SC, Vale RD, Kuntz ID. Biochemistry. 2000;39:2805–2814. doi: 10.1021/bi992474k. [DOI] [PubMed] [Google Scholar]
- 37.Kis K, Volk R, Bacher A. Biochemistry. 1995;34:2883–2892. doi: 10.1021/bi00009a019. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Meining W, Cushman M, Haase I, Fischer M, Bacher A, Ladenstein R. Journal of Molecular Biology. 2003;328:167–82. doi: 10.1016/s0022-2836(03)00186-4. [DOI] [PubMed] [Google Scholar]
- 39.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talukdar A, Breen M, Bacher A, Illarionov B, Fischer M, Georg G, Ye QZ, Cushman M. J Org Chem. 2009;74:5123–5134. doi: 10.1021/jo900238q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cushman M, Sambaiah T, Jin G, Illarionov B, Fischer M, Bacher A. J Org Chem. 2004;69:601–612. doi: 10.1021/jo030278k. [DOI] [PubMed] [Google Scholar]
- 42.Morgunova E, Illarionov B, Sambaiah T, Haase I, Bacher A, Cushman M, Fischer M, Ladenstein R. FEBS J. 2006;273:4790–804. doi: 10.1111/j.1742-4658.2006.05481.x. [DOI] [PubMed] [Google Scholar]
- 43.Morgunova E, Meining W, Cushman M, Illarionov B, Haase I, Jin G, Bacher A, Cushman M, Fischer M, Ladenstein R. Biochemistry. 2005;44:2746–2758. doi: 10.1021/bi047848a. [DOI] [PubMed] [Google Scholar]
- 44.Morgunova E, Saller S, Haase I, Cushman M, Bacher A, Fischer M, Ladenstein R. J Biol Chem. 2007;282:17231–41. doi: 10.1074/jbc.M701724200. [DOI] [PubMed] [Google Scholar]
- 45.Cushman M, Yang D, Mihalic JT, Chen J, Gerhardt S, Huber R, Fischer M, Kis K, Bacher A. J Org Chem. 2002;67:6871–6877. doi: 10.1021/jo020144r. [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 47.Guan H, Laird AD, Blake RA, Tanga C, Liang C. Bioorg Med Chem Lett. 2004;14:187–190. doi: 10.1016/j.bmcl.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 48.Nurminen EJ, Mattinen JK, Lonnberg H. J Chem Soc, Perkin Trans. 1998;2:1621. [Google Scholar]
- 49.Heasley HH, Jarosz R, Lynch KR, Macdonald TL. Bioorg Med Chem Lett. 2004;14:2735–2740. doi: 10.1016/j.bmcl.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 50.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 51.Gerhardt S, Haase I, Steinbacher S, Kaiser JT, Cushman M, Bacher A, Huber R, Fischer M. J Mol Biol. 2002;318:1317–1329. doi: 10.1016/s0022-2836(02)00116-x. [DOI] [PubMed] [Google Scholar]
- 52.Klinke S, Zylberman V, Vega DR, Guimaraes BG, Braden BC, Goldbaum FA. J Mol Biol. 2005;353:124–37. doi: 10.1016/j.jmb.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 53.Ladenstein R, Schneider M, Huber R, Bartunik HD, Wilson K, Schott K, Bacher A. J Mol Biol. 1998;203:1045–1070. doi: 10.1016/0022-2836(88)90128-3. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Illarionov B, Morgunova E, Jin G, Bacher A, Fischer M, Ladenstein R, Cushman M. J Org Chem. 2008;73:2715–2724. doi: 10.1021/jo702631a. [DOI] [PubMed] [Google Scholar]
- 55.Ritsert K, Huber R, Turk D, Ladenstein R, Schmidt-Bäse K, Bacher A. J Mol Biol. 1995;253:151–167. doi: 10.1006/jmbi.1995.0542. [DOI] [PubMed] [Google Scholar]
- 56.Stirtan WG, Withers SG. Biochemistry. 1996;35:15057–15064. doi: 10.1021/bi9606004. [DOI] [PubMed] [Google Scholar]
- 57.Kim CU, Luh BY, Misco PF, Bronson JJ, Hitchcock MJM, Ghazzouli I, Martin JC. J Med Chem. 1990;33:1207–1213. doi: 10.1021/jm00166a019. [DOI] [PubMed] [Google Scholar]
- 58.Pevzner LM. Russ J Gen Chem. 2006;76:1354–1358. [Google Scholar]
- 59.Bako P, Novak T, Ludanyi K, Pete B, Toke L, Keglevich G. Tetrahedron: Asymmetry. 1999;10:2373–2380. [Google Scholar]
- 60.Chun YJ, Park JH, Oh GM, Hong SI, Kim YJ. Synthesis. 1994:909–910. [Google Scholar]
- 61.Billups WE, Arney BE, Jr, Lin L-J. J Org Chem. 1984;49:3437–3438. [Google Scholar]
- 62.Burton DJ, Takei R, Shina-Ya S. J Fluorine Chem. 1981;18:197–202. [Google Scholar]
- 63.Burton DJ, Flynn RM. J Fluorine Chem. 1980;15:263–266. [Google Scholar]
- 64.Burton DJ, Flynn RM. J Fluorine Chem. 1977;10:329–332. [Google Scholar]
- 65.Picotin G, Miginiac P. J Org Chem. 1997;52:4796–4798. [Google Scholar]
- 66.Cushman M, Mihalic JT, Kis K, Bacher A. J Org Chem. 1999;64:3838–3845. [Google Scholar]
- 67.Yu TY, O’Connor RD, Sivertsen AC, Chiauzzi C, Poliks B, Fischer M, Bacher A, Haase I, Cushman M, Schaefer J. Biochemistry. 2008;47:13942–13951. doi: 10.1021/bi8015789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klebe G. Virtual screening: Scope and limitation. In: Alvarez Juan, Shoichet Brian., editors. Virtual Screening in Drug Discovery. Chap 1. CRC Press; Boca Raton, FL: 2005. pp. 3–24. [Google Scholar]
- 69.Kaiser J, Illarionov B, Rohdich F, Eisenreich W, Saller S, den Brulle JV, Cushman M, Bacher A, Fischer M. Anal Biochem. 2007;365:52–61. doi: 10.1016/j.ab.2007.02.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.