

Abstract
To determine if different racial groups shared common types of vaginal microbiota we characterized the composition and structure of vaginal bacterial communities in asymptomatic and apparently healthy Japanese women in Tokyo, Japan and compared them with those of White and Black women from North America. The composition of vaginal communities was compared based on community profiles of terminal restriction fragments of 16S rRNA genes and phylogenetic analysis of cloned 16S rRNA gene sequences of the numerically dominant bacterial populations. The types of vaginal communities found in Japanese women were similar to those of Black and White women. As with White and Black women, most vaginal communities were dominated by lactobacilli, and only four species of Lactobacillus (L. iners, L. crispatus, L. jensenii and L. gasseri) were commonly found. Communities dominated by multiple species of lactobacilli were common in Japanese and White women, but rare in Black women. The incidence in Japanese women of vaginal communities with several non-Lactobacillus species at moderately high frequencies was intermediate between Black women and White women. The limited number of community types found among women in different ethnic groups suggests that host genetic factors, including the innate and adaptive immune systems, may be more important in determining the species composition of vaginal bacterial communities than are cultural and behavioral differences.
Keywords: Vaginal microbiota, 16S rRNA genes, bacterial communities, Japanese women
Introduction
Each area of the human body has a unique collection of microorganisms. Though previous studies have tended to focus on understanding etiological agents of infectious disease, there has recently been an increased emphasis on the role of indigenous microbiota in the maintenance of human health. The notion of a mutualistic relationship between the human host and its microbiome is being increasingly appreciated. (Backhed et al., 2005; Dethlefsen et al., 2007). As a partner in a mutualism, the hosts exerts strong selective pressures on determining the composition of microbiota found at different anatomical sites (Backhed et al., 2005), and in turn the microbiota of humans are essential to many aspects of normal human physiology, ranging from nutrition, metabolic activities, homeostasis of the immune system (Backhed et al., 2004; Kelly et al., 2004; Rakoff-Nahoum et al., 2004; Cash et al., 2006) and competitive exclusion of pathogens. Moreover, the microbiota in the human body may reduce the risk of cancer, diabetes, obesity, arthritic diseases, cardiovascular disease, and other serious ailments (Naruszewicz et al., 2002; Ohashi et al., 2002; Rayes et al., 2002; Rozanova et al., 2002; Tannock, 2002). Hence, the benefits of accurately defining the human microbiota and understanding their ecological function and role in human health may be crucially important for understanding differential risk to disease, and ultimately in disease prevention and treatment.
The human vagina is a dynamic and complex microbial ecosystem in which the symbiotic vaginal microbiota play an important protective role in maintaining the health of women. Disrupting these communities may increase susceptibility to various urogenital infections including sexually transmitted diseases and HIV in women (Schwebke, 2005; McClelland et al., 2008). Recently, several studies have characterized the vaginal microbial communities using cultivation-independent methods, and tested whether differences in the species composition of vaginal bacterial communities may predispose certain individuals to bacterial vaginosis and various infectious diseases (Hyman et al., 2005; Fredricks et al., 2005; Verhelst et al., 2004; Vitali et al., 2007; Shi et al., 2009). Previously, we have shown that there were several kinds of vaginal microbial communities found in healthy North American White and Black women. The community types in White women were similar to those of Black women, but the relative frequencies of the types differed (Zhou et al., 2004; Zhou et al., 2007). In this study, we extended our previous work by determining the bacterial community types in vaginas of apparently healthy Asian women from Tokyo, Japan and compared them to those of White and Black women. The aim of this study was to determine whether the vaginal communities were similar types in all three racial groups.
Materials and Methods
Clinical study
The samples used in this study were collected as part of a study previously described by Parsonnet et al. (Parsonnet et al. 2008). Vaginal samples were obtained from 73 Japanese women from the Tokyo area of Japan. The subjects were evenly distributed among three age groups: 18–25, 26–34, and 35–45 years old. For inclusion in this study the women enrolled must self declare that they were born in Japan and be of Japanese ancestry. The criteria for enrollment of healthy and asymptomatic Japanese women were essentially the same as those employed for a previously reported study of White and Black women (Zhou et al., 2007). Women of Japanese decent were eligible for enrollment if they had a history of regular menstrual cycles for the past two years and agreed to refrain from the use of mouth wash, mouth rinse or medicated drop or sprays, douching substances, vaginal medications, suppositories, feminine sprays, genital wipes, or contraceptive spermicides, and from sexual intercourse for 48 hours prior to sample collection. Subjects were also required to refrain from bathing, showering, or swimming within 2 hours prior to sample collection. Women were excluded if they worked in healthcare settings; had been hospitalized in the past six weeks; had experienced a genital or sexually-transmitted infection within the past six weeks; were pregnant, actively trying to become pregnant, or suspected they were pregnant; had been diagnosed as having diabetes, kidney failure, hepatitis, HIV infection or TSS; were currently suffering from sinus infection or pharyngitis (self-declared); or had taken immunosuppressive drugs, chemotherapy, or systemic or topical antimicrobial drugs within the prior 30 days. The subjects enrolled did not have vaginal symptoms associated with BV during the 6 weeks prior to enrollment. When the vaginal samples were collected the attending health care practitioner noted any signs of possible genital infections (e.g., abnormal discharge, cervicitis, or foul odor). None of the subjects in this study had signs of vaginal infection. These were samples of convenience and Nugent scores (Nugent et al., 1991) were not available.
Written informed consent was obtained from the subjects prior to the collection of any information or clinical samples. Subjects were removed from the study if they failed to meet the inclusion criteria or satisfied any of exclusion criteria any time during the study. The study protocol and informed consent document were reviewed and approved by the Ethics Committee-Sogo Clinical Pharmacology Co., Ltd.
Each vaginal sample was collected by a physician by inserting a swab into the vagina (without using a speculum) and swabbing the mid-upper vaginal walls approximately 5 cm past the introitus. The labia were spread during this procedure to minimize the potential for contamination by perineal flora (Parsonnet et al., 2008). The swab was placed in a sterile cryovial and stored at −70° until analysis. When collecting of the vaginal sample the attending health care practitioner noted any signs of possible genital infections (e.g., abnormal discharge, cervicitis, or foul odor); none of the subjects in this study had signs of vaginal infection.
Extraction of genomic DNA
The bacterial cells retrieved on swabs were resuspended in 2ml cell lysis solution from Wizard DNA purification kit (Promega, Madison, WI, USA). Genomic DNA was isolated from 0.5 ml aliquots of the cell suspensions using a two-step cell lysis procedure as previously described (Zhou et al., 2007). Briefly, bacterial cell walls were disrupted enzymatically by mixing with mutanolysin (50 μg) and lysozyme (500 μg) followed by incubation for 1 hour at 37°C. The cells were then mechanically disrupted by 6 freeze-thaw cycles. Each cycle consisted of 2 minutes incubation at 100°C that was immediately followed by 2 minutes in a dry-ice/ethanol bath. Between each freeze-thaw cycle, the cell suspensions were incubated for 1 minute in an Ultrasonic Cleaning bath. Proteins in the disrupted cell suspension were digested with proteinase K (Qiagen, Hilden, Germany) during a 1 hour incubation at 55°C. Further purification of the total DNA extract was performed using the Wizard DNA purification kit (Promega, Madison, WI, USA).
T-RFLP analysis of 16S rRNA genes
For analysis of terminal restriction fragment polymorphisms (T-RFLP) of 16S rRNA genes, internal regions of 16S rRNA genes in each sample were amplified in two separate reactions using fluorescently-labeled primer pairs, 8fm-926r and 49f-926r (based on Escherichia coli sequence). Primers 8fm, 49f, and 926r were labeled with VIC, NED, and 6-carboxy-fluorescein (6-FAM), respectively (Applied Biosystems, Foster City, CA). PCR was performed as previously described (Zhou et al., 2004).
The profiles of terminal restriction fragments from the vaginal microbial communities were determined as follows. A mixture of the two fluorescently labeled amplicons was equally divided and separately digested with MspI or HaeIII, and the digested products were recombined. The resulting mixture had 6 fluorescently labeled terminal restriction fragments from the use of 3 fluorophores and 2 restriction enzymes. This allowed for high resolution of the microbial communities. T-RFLP profiles were determined using an ABI PRISM 3100 DNA Analyzer, GeneScan software (Applied Biosystems) and CST ROX 25-1000 (BioVentures, Inc., Murfreesboro, Tenn.) as an internal standard.
Cluster analysis of T-RFLP data
Cluster analysis of the T-RFLP community profiles for Japanese women from this study and White and Black women from our previous study was performed to identify similar communities and the number of clusters. The data was first standardized by defining a threshold (baseline) and identifying the true peaks (Abdo et al., 2006). The fragments were then binned based upon length; those fragments within 2 bp in length were binned together and represented by their average length. The peak areas of binned fragments from the same samples were summed. Second, the Pearson correlation distances between T-RFLP profiles were calculated, these were hierarchically clustered based on Ward’s linkage method and a dendrogram was constructed. Third, the number of clusters was determined using methods previously described (Zhou et al., 2007).
Clone library construction and 16S rRNA gene sequence analysis
The samples used to construct clone libraries were chosen using a ‘coverage sampling approach’ (Abdo et al., 2006). This approach provided a way to identify the fewest samples necessary to describe 85% of the phylotype diversity within each cluster. The 16S rRNA genes in each sample identified using this approach were amplified using primers 8fm-926r without fluorescent labels and cloned as previously described (Zhou et al., 2004; Zhou et al., 2007). Approximately 100 clones of each sample were randomly chosen from each library and the cloned DNA fragments were partially sequenced using an ABI 3730 Prism DNA Analyzer. Phylogenetic analysis of cloned 16S rRNA gene sequences from the numerically dominant microbial populations was done to determine the composition of bacterial communities in each sample. High quality sequences with less than 3% uncalled bases and more than 500 bp long were analyzed using high-throughput methods (Brown et al., 2007) to identify similar sequences among the eubacterial type strains found in the Ribosomal Database Project (RDP) and GenBank databases.
Differences in the distribution of vaginal community among the different racial groups
There were two ways chosen to show the distribution of vaginal microbial communities among the various racial groups, and both of them were based on the T-RFLP community profiles. First, the distance matrix based on Pearson correlation distance analysis was constructed, and subjected to multidimensional-scaling to graphically present the similarity among the bacterial communities for each sample and each racial group. Second, we evaluated differences in the species rank abundance of microbial communities of women in each racial group. Pearson’s chi-squared test and Fisher’s exact test were used to assess whether differences in the distribution of microbial community types among Japanese, White and Black women were statistically significant.
Results
Classification of vaginal microbial communities based on T-RFLP and sequence data
The number of different kinds of bacterial communities in the vaginas of Japanese women was determined by comparing T-RFLP profiles and by sequence analysis of cloned 16S rRNA genes. We performed cluster analysis on the profiles of T-RFLPs and constructed a dendrogram in which the neighboring communities had similar species composition and structure (Figure 1). We categorized the communities into nine clusters (designated as C1-9). Community types were further defined by sequence analysis of the 16S rRNA genes in samples that were representative of each cluster. Clusters that had similar species composition were combined and assigned to a single group. For example, clusters C2 and C4 (Figure 1) were both dominated by Lactobacillus crispatus and were combined into a single group; namely group II (Tables 1, 2). Likewise, the communities with multiple non-Lactobacillus species found in C8 and C9 were combined into group III. After combining communities that were similar in terms of species composition there were seven different groups of communities (groups I to VII). These communities were also found in our previous study of vaginal microbiota in White and Black women (Zhou et al., 2007).
Figure 1.

Dendrogram constructed by cluster analysis (Ward’s method) based on similarity in the T-RFLP profiles among vaginal microbial communities in Japanese, White, and Black women. The samples from White, Black and Japanese women are designated with an open circle, closed circle and red triangle, respectively. The red line shows the grouping baseline. Big triangles labeled with various colors indicate clusters. The clusters are designated with a “C” followed by a number. Asterisks indicate the samples from each group used to construct clone libraries of 16S rRNA genes.
Table 1.
Species composition of vaginal communities in healthy Japanese women.
| Group (% clones) |
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ib |
II |
III |
IV |
V |
VI |
VII |
||||||||||||||||
| J30 | J35 | J40 | J45 | J13 | J17 | J37 | J25 | J63 | J65 | J47 | J79 | J1 | J5 | J46 | J38 | J67 | J77 | J81 | J4 | J20 | J83 | |
| Phylotypea | nc=96 | n=96 | n=85 | n=96 | n=67 | n=−64 | n=96 | n=75 | n=92 | n=94 | n=94 | n=90 | n=90 | n=91 | n=80 | n=94 | n=89 | n=96 | n=92 | n=63 | n=91 | n=82 |
| Lactobacillus iners | 98d | 100.0 | 82.4 | 92.7 | 20.3 | 3.3 | 8.8 | 30.0 | 39.7 | 80.2 | ||||||||||||
| L. crispatus | 17.6 | 7.3 | 100.0 | 78.0 | 98.9 | 98.7 | 100.0 | 100.0 | 10.0 | 1.1 | 60.3 | 4.4 | ||||||||||
| L. jensenii | 1.7 | 1.3 | 6.6 | 100.0 | ||||||||||||||||||
| L. gasseri | 98.0 | 82.0 | 44.0 | 98.9 | ||||||||||||||||||
| L. vaginalis | 1.1 | |||||||||||||||||||||
| L. aviarius | 2.0 | |||||||||||||||||||||
| Aerococcus sp. | 3.2 | 10.0 | ||||||||||||||||||||
| Anaerobranca sp. | 2.2 | |||||||||||||||||||||
| Anaerococcus sp. | 2.2 | |||||||||||||||||||||
| Atopobium vaginae | 41.1 | 48.4 | 36.2 | 1.0 | ||||||||||||||||||
| Bergeyella sp. | 3.3 | |||||||||||||||||||||
| Bifidobacterium breve | 1.0 | |||||||||||||||||||||
| Bradyrhizobium sp. | 1.1 | |||||||||||||||||||||
| Chryseobacterium sp. | 1.1 | |||||||||||||||||||||
| Clostridium sp. | 13.3 | 2.2 | ||||||||||||||||||||
| Dialister sp. | 2.0 | 3.3 | 4.4 | 1.0 | ||||||||||||||||||
| Enterococcus faecalis | 1.1 | |||||||||||||||||||||
| Finegoldia magna | 1.1 | |||||||||||||||||||||
| Gardnerella vaginalis | 15.6 | |||||||||||||||||||||
| Gemella palaticanis | 4.4 | 1.1 | ||||||||||||||||||||
| Lachnospiraceae sp. | 1.1 | |||||||||||||||||||||
| Lachnospira sp. | 57.9 | 1.1 | ||||||||||||||||||||
| Leptotrichia sp. | 2.5 | |||||||||||||||||||||
| Megasphaera sp. | 1.1 | 4.4 | 6.2 | |||||||||||||||||||
| Mobiluncus mulieris | 18.9 | |||||||||||||||||||||
| Neisseria sp. | 1.1 | |||||||||||||||||||||
| Peptoniphilus sp. | 1.1 | |||||||||||||||||||||
| Peptostreptococcus sp. | 1.1 | 7.7 | ||||||||||||||||||||
| Prevotella sp. | 15.7 | 11.0 | 7.5 | 2.2 | ||||||||||||||||||
| Staphylococcus sp. | 2.0 | |||||||||||||||||||||
| Streptococcus sp. | 95.0 | 3.3 | 5.9 | 56.0 | 8.8 | |||||||||||||||||
| Veillonella sp. | 1.2 | |||||||||||||||||||||
| Novele | 13.2 | 6.4 | ||||||||||||||||||||
Clones were assigned to phylotypes by comparing their 16S rRNA gene sequences to those of known organisms. The species name was used if the sequence similarity to a type species was >97%; the genus only was used if the sequence similarity was <97%, but >90%. The clones were described as novel if the sequence similarity of clones to known organisms was < 90%.
‘G’ indicates group defined based on T-RFLP profiles and phylogenetic analysis of partial 16S rRNA gene sequences from clone libraries prepared from samples representative of each cluster. Group I, IV, V, VI and VII designate C6, C7, C3, C1 and C5 respectively (see Figure 1). Group II merges C2 and C4 and Group III merges C8 and C9.
The number of clones analyzed per woman.
Relative abundances of populations in each clone library.
‘Novel’ includes various phylotypes within the phylum Firmicutes.
Table 2.
Comparison of vaginal microbiota in different racial groups.
| Phylotypea | Group (% clones) |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ib |
II |
III |
IV |
V |
VI |
VII |
|||||||||||||||
| Bc | W | J | B | W | J | B | W | J | B | W | J | B | W | J | B | W | J | B | W | J | |
| Lactobacillus iners | 84.4d | 90.1 | 93.3 | 0.8 | 0.3 | 3.4 | 4.4 | 4.0 | 0.0 | 2.7 | 1.7 | 14.0 | 1.3 | 38.5 | 59.4 | 60.0 | 25.5 | ||||
| L. crispatus | 0.4 | 1.1 | 6.2 | 97.4 | 86.4 | 95.9 | 5.0 | 0.5 | 25.4 | 0.3 | 13.8 | 25.8 | 32.4 | 40.1 | |||||||
| L. jensenii | 0.2 | 0.6 | 1.8 | 6.2 | 0.5 | 3.6 | 0.4 | 2.9 | 3.3 | 80.9 | 34.4 | 100.0 | |||||||||
| L. gasseri | 0.7 | 0.1 | 1.4 | 21.2 | 66.7 | 70.3 | 80.7 | 8.9 | |||||||||||||
| L. vaginalis | 0.2 | 0.5 | |||||||||||||||||||
| L. coleohominis | 0.4 | 0.2 | 0.9 | ||||||||||||||||||
| L. salivarius | 0.1 | ||||||||||||||||||||
| L. aviarius | 0.5 | ||||||||||||||||||||
| Actinobaculum sp. | 0.2 | ||||||||||||||||||||
| Aerococcus sp. | 1.5 | 0.6 | 0.5 | 0.3 | 0.4 | 1.5 | 4.4 | 0.6 | |||||||||||||
| Anaerobranca sp. | 0.2 | 1.4 | 0.7 | 0.6 | |||||||||||||||||
| Anaerococcus sp. | 0.3 | 0.0 | 10.6 | 1.1 | 2.3 | 0.5 | 0.0 | ||||||||||||||
| Atopobium vaginae | 3.2 | 1.6 | 2.6 | 1.4 | 0.0 | 25.7 | 30.7 | 41.9 | 0.6 | 0.3 | |||||||||||
| Bergeyella sp. | 0.8 | ||||||||||||||||||||
| Bifidobacterium breve | 0.5 | ||||||||||||||||||||
| Bradyrhizobium sp. | 0.6 | ||||||||||||||||||||
| Catonella sp. | 0.0 | 2.3 | |||||||||||||||||||
| Chryseobacterium sp. | 0.0 | 0.3 | |||||||||||||||||||
| Clostridium sp. | 0.2 | 0.2 | 5.2 | ||||||||||||||||||
| Corynebacterium sp. | 1.8 | ||||||||||||||||||||
| Dialister sp. | 0.3 | 2.3 | 1.0 | 2.7 | 3.8 | 1.4 | 1.5 | 0.3 | |||||||||||||
| Eggerthella hongkongensis | 0.8 | ||||||||||||||||||||
| Enterococcus faecalis | 0.5 | 0.4 | 0.3 | ||||||||||||||||||
| Escherichia coli | 3.5 | 0.0 | |||||||||||||||||||
| Finegoldia magna | 0.1 | 0.7 | 0.7 | 0.3 | 1.2 | ||||||||||||||||
| Garanulicatella elegans | 0.4 | ||||||||||||||||||||
| Gardnerella vaginalis | 7.8 | 0.5 | 4.5 | 0.5 | |||||||||||||||||
| Gemella palaticanis | 0.4 | 0.3 | 2.8 | 7.1 | 2.2 | 2.6 | 0.4 | ||||||||||||||
| Lachnospiraceae sp. | 0.7 | 4.8 | 0.6 | 0.8 | 3.7 | ||||||||||||||||
| Lachnospira sp. | 29.0 | 0.3 | |||||||||||||||||||
| Leptotrichia sp. | 9.7 | 0.8 | |||||||||||||||||||
| Megasphaera sp. | 1.6 | 1.8 | 6.7 | 0.5 | 4.8 | 6.4 | 3.9 | ||||||||||||||
| Micromonas sp. | 0.8 | 0.4 | 5.0 | 1.4 | 11.4 | 1.1 | |||||||||||||||
| Mobiluncus mulieris | 0.2 | 0.3 | 6.3 | ||||||||||||||||||
| Mycoplasma sp. | 9.6 | ||||||||||||||||||||
| Neisseria sp. | 0.6 | ||||||||||||||||||||
| Peptococcus niger | 1.4 | 0.4 | |||||||||||||||||||
| Peptoniphilus sp. | 0.7 | 3.1 | 0.7 | 0.4 | |||||||||||||||||
| Peptostreptococcus sp. | 8.0 | 5.7 | 2.9 | ||||||||||||||||||
| Prevotella sp. | 0.3 | 1.1 | 11.4 | 0.6 | 2.5 | ||||||||||||||||
| Pseudomonas sp. | 1.5 | ||||||||||||||||||||
| Shigella sp. | 2.5 | ||||||||||||||||||||
| Staphylococcus sp. | 2.6 | 1.0 | |||||||||||||||||||
| Streptococcus sp. | 0.3 | 1.0 | 2.1 | 36.2 | 49.2 | 17.2 | 0.4 | 15.5 | 46.2 | 0.6 | 4.4 | 11.9 | |||||||||
| Veillonella sp. | 0.2 | 0.1 | 0.1 | 19.3 | 0.8 | 2.5 | 0.4 | ||||||||||||||
| Novele | 4.4 | 0.9 | 0.5 | 46.8 | 9.8 | 36.6 | 17.6 | 6.5 | 4.8 | 1.1 | 2.0 | 1.2 | |||||||||
| Number of women (per race) | 22 | 26 | 14 | 12 | 23 | 23 | 21 | 6 | 10 | 7 | 4 | 8 | 4 | 5 | 8 | 2 | 5 | 6 | 1 | 6 | 4 |
| Number of women (per group) | 62 | 58 | 37 | 19 | 17 | 13 | 11 | ||||||||||||||
Clones were assigned to phylotypes by comparing their 16S rRNA gene sequences to those of known organisms. The species name was used if the sequence similarity to a type species was >97%; the genus only was used if the sequence similarity was <97%, but >90%. The clones were described as novel if the sequence similarity of clones to known organisms was < 90%.
‘G’ indicates group defined based on T-RFLP profiles and phylogenetic analysis of partial 16S rRNA gene sequences from clone libraries prepared from samples representative of each cluster. Group I, IV, V, VI and VII designate C6, C7, C3, C1 and C5 respectively (see Figure 1). Group II merges C2 and C4 and Group III merges C8 and C9.
‘B’ ‘W’ and ‘J’ represent Black, Caucasian and Japanese women.
Mean relative abundances of populations in each clone library.
‘Novel’ includes various phylotypes within the phylum Firmicutes, most of them belong to novel phylotype of Clostridiales.
Species composition of vaginal communities
The 16S rRNA genes sequences from 19 clone libraries representing each of the major groups of Japanese women were identified and their relative abundances were calculated. The numerically important bacterial phylotypes that constituted >1% of a community in Japanese women are summarized in Table 1.
These data were compared to those obtained the analysis of vaginal communities in White and Black women (57 clone libraries; Zhou et al., 2007). The mean relative abundances of populations in vaginal communities of women in all three racial groups are presented in Table 2. Among the Japanese women, most of the vaginal communities were dominated by species of Lactobacillus (group I, II and V to VII), and these communities were found in about 75.3% (54/72) of the women. Only four species of Lactobacillus (L. iners, L. crispatus, L. gasseri and L. jensenii) were found to be common in Japanese women, which was similar to the findings for White and Black women. Of the Lactobacillus-dominated communities of Japanese women, L. crispatus was the most common dominant species. The data showed that 40.2% (29/72) of the Japanese women had microbial communities with high numbers of L. crispatus (groups II and VI). In most cases, L. crispatus constituted from 78–100% of the clones (group II), and in a few of cases, L. crispatus represented less than 50% of the sequenced clones (group VI, mean values). The second most common Lactobacillus species in Japanese women was L. iners, which occurred at high frequency in 27.8% (20/72) of the women sampled (groups I and VI). In Group I, L. iners was the predominant species, but group VI also contained high proportions of several other Lactobacillus species including L. crispatus and L. jensenii. Vaginal communities of groups III and IV found in Japanese, White and Black women exhibited greater species evenness and lower numbers of lactobacilli (Table 1 and 3). Group IV was characterized by communities with high frequencies of Atopobium (20–50%), and included various other species of strict or facultative anaerobes, for example, lactobacilli, Clostridium sp., Dialister sp., Gemella sp., Lachnospiraceae sp., Leptotrichia sp., Megasphaera sp., Micromonas sp., Mobiluncus mulieris, Peptinophilus sp., Prevotella sp. Veillonella sp. and some novel microorganisms that belong to the order Clostridiales. In contrast, group III contained diverse species of anaerobes other than lactobacilli and Atopobium. The dominant members included Streptococcus sp., novel microorganisms, Veillonella sp., Lachnospira sp. Gardnerella vaginalis. Anaerococuus sp., Peptostreptococcus sp. Gemella sp., Lachnospiraceae sp., Megasphaera sp., Micromonas sp., Mycoplasma sp. Peptostreptococcus sp. Prevotella sp., Escherichia coli and Shigella sp.
Table 3.
Composition of vaginal microbiota in Groups III and IV.
| Phylotypea | Group (% clones) |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| III |
IV |
|||||||||||||||||||||
| B8b | B14 | B20 | B37 | B45 | B49 | B50 | B56 | W13 | W27 | W68 | J47 | J79 | B19 | B38 | B48 | W39 | W63 | W69 | J1 | J5 | J46 | |
| n=83c | n=88 | n=84 | n=94 | n=87 | n=89 | n=41 | n=86 | n=69 | n=90 | n=83 | n=94 | n=90 | n=87 | n=90 | n=88 | n=61 | n=66 | n=94 | n=90 | n=91 | n=80 | |
| Lactobacillus iners | 31.8e | 1.2 | 2.4 | 2.9 | 5.5 | 3.6 | 4.6 | 3.4 | 3.0 | 2.0 | 3.3 | 8.8 | 30.0 | |||||||||
| L. crispatus | 10.0 | 1.5 | ||||||||||||||||||||
| L. gasseri | 63.6 | |||||||||||||||||||||
| Aerococcus sp. | 1.2 | 2.4 | 1.0 | 1.1 | 4.5 | 3.2 | 10.0 | |||||||||||||||
| Anaerobranca sp. | 1.2 | 9.8 | 2.2 | |||||||||||||||||||
| Anaerococcus sp. | 31.8 | 2.2 | 1.1 | 5.7 | 1.5 | |||||||||||||||||
| Atopobium vaginae | 1.2 | 1.2 | .4 | 3.4 | 6.7 | 4.9 | 1.2 | 4.3 | 29.9 | 27.8 | 19.3 | 31.0 | 15.4 | 45.7 | 41.1 | 48.4 | 36.2 | |||||
| Bifidobacterium breve | 1.0 | |||||||||||||||||||||
| Bradyrhizobium sp. | 1.1 | |||||||||||||||||||||
| Clostridium sp. | 1.2 | 13.3 | 2.2 | |||||||||||||||||||
| Corynebacterium sp. | ||||||||||||||||||||||
| Dialister sp. | 4.8 | 3.6 | 4.9 | 4.7 | 2.9 | 2.0 | 3.3 | 6.9 | 3.3 | 1.1 | 1.6 | 1.5 | 1.0 | 4.4 | ||||||||
| Eggerthella hongkongensis | 2.3 | |||||||||||||||||||||
| Enterococcus faecalis | 1.1 | |||||||||||||||||||||
| Escherichia coli | 27.6 | 0.0 | ||||||||||||||||||||
| Finegoldia magna | 2.2 | 2.2 | ||||||||||||||||||||
| Garanulicatella elegans | 1.1 | |||||||||||||||||||||
| Gardnerella vaginalis | 15.6 | 1.5 | ||||||||||||||||||||
| Gemella palaticanis | 15.7 | 6.7 | 4.3 | 12.2 | 4.8 | 4.4 | 7.8 | 1.1 | ||||||||||||||
| Lachnospiraceae sp. | 14.4 | 1.1 | 2.3 | 2.6 | 8.5 | |||||||||||||||||
| Lachnospira sp. | 57.9 | |||||||||||||||||||||
| Leptotrichia sp. | 29.0 | 2.5 | ||||||||||||||||||||
| Megasphaera sp. | 5.7 | 4.8 | 10.3 | 9.8 | 23.3 | 1.4 | 12.2 | 2.3 | 9.8 | 9.5 | 1.1 | 4.4 | 6.2 | |||||||||
| Micromonas sp. | 1.2 | 2.4 | 4.5 | 30.3 | 1.2 | 4.3 | 26.4 | 6.7 | 1.1 | 3.2 | ||||||||||||
| Mobiluncus mulieris | 2.3 | 18.9 | ||||||||||||||||||||
| Mycoplasma sp. | 74.7 | 2.4 | ||||||||||||||||||||
| Neisseria sp. | 1.1 | |||||||||||||||||||||
| Peptococcus niger | 4.3 | 1.1 | ||||||||||||||||||||
| Peptoniphilus sp. | 1.1 | 10.7 | 3.4 | 9.8 | 1.1 | 1.1 | 1.1 | |||||||||||||||
| Peptostreptococcus sp. | 2.4 | 30.3 | 29.3 | 2.3 | 1.2 | 15.9 | 1.1 | 7.7 | ||||||||||||||
| Prevotella sp. | 2.2 | 3.2 | 15.7 | 11.0 | 7.5 | |||||||||||||||||
| Shigella sp. | 20.2 | |||||||||||||||||||||
| Staphylococcus sp. | 2.0 | |||||||||||||||||||||
| Streptococcus sp. | 16.9 | 91.6 | 95.0 | 3.3 | ||||||||||||||||||
| Veillonella sp. | 1.1 | 57.8 | 2.3 | 7.5 | 1.2 | |||||||||||||||||
| Noveld | 57.9 | 73.7 | 52.2 | 76.1 | 22.6 | 24.3 | 67.3 | 29.4 | 31.0 | 36.7 | 42.1 | 19.6 | 33.3 | 13.2 | 6.4 | |||||||
Clones were assigned to phylotypes by comparing their 16S rRNA gene sequences to those of known organisms. The species name was used if the sequence similarity to a type species was >97%; the genus only was used if the sequence similarity was <97%, but >90%. The clones were described as novel if the sequence similarity of clones to known organisms was < 90%.
‘B’ ‘W’ and ‘J’ represent Black, Caucasian and Japanese women.
Number of clones analyzed per woman.
‘Novel’ includes various phylotypes within the phylum Firmicutes, most of them belong to novel phylotype of Clostridiales.
Relative abundance of population in clone library.
Distribution of vaginal community among the different racial groups
A multidimensional-scaling (MDS) analysis of T-RFLP data was employed to assess the variation of vaginal microbial communities among the different women and racial groups (Figure 2). Some communities were very similar and clustered together in the figure, while others had little similarity to any other communities and were scattered across the graph. A striking finding was the distribution of communities and the observation that, for all clustered data, women of all three racial groups were represented. There were no isolated communities, nor any clusters of communities that included only a specific racial group. Thus, we can conclude that all three racial groups share common vaginal community types.
Figure 2.

Two dimensional scatter plot constructed by multidimensional-scaling (MDS) analysis based on similarity in T-RFLP profiles among vaginal microbial communities in Japanese, White and Black women. The samples from White, Black and Japanese women are designated with green, black and red dots, respectively.
The rank abundances of vaginal community types in Japanese, White and Black women are shown in Figure 3. Although the types of vaginal microbial communities found in healthy Japanese women were similar to those of the other two racial groups, there were significant differences in the frequencies of communities in these groups (p < 0.01). Within groups III and IV, the incidence of Japanese women (24.7%) was lower than in Black women (40.5%), but higher than in White women (13.3%). These communities were not dominated by lactobacilli, but had comparatively equal proportions of other dominant species. On the other hand, communities dominated by multiple species of lactobacilli (group VI) were common in both Japanese and White women, but were rare in Black women.
Figure 3.
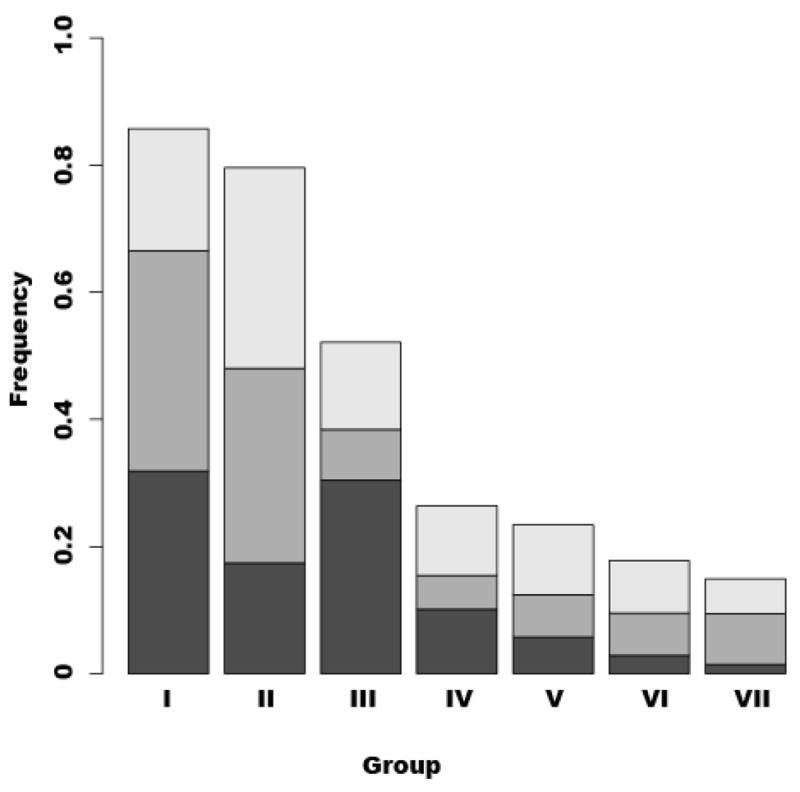
The rank abundances of vaginal communities found in White, Black and Japanese women. Each column was designated with each group (community types; see Tables 1–3). The proportion of Black and White women of North America, and Japanese women are shown as dark gray, gray and light gray in each column, respectively.
It should be noted that the age range in the Japanese women differed slightly from those of White and Black women. The White and Black women sampled were on average younger than the Japanese women. However, our previous studies have shown that there are not significant differences in the distribution of community types between young (13–18y) and older age groups (19–40y) (Yamamoto et al., 2009).
Discussion
In this study, we found that the types of vaginal microbial communities found in apparently healthy Japanese women resembled those of White and Black women from North America. In total, there were seven major kinds of microbial communities identified in Japanese women, and the numbers of community types were the same as those in Black and White women. As with White and Black women, most vaginal communities were dominated by lactobacilli, and only four species of Lactobacillus (L. iners, L. crispatus, L. jensenii and L. gasseri) were commonly found. Comparable results were reported by Shi et al. (Shi et al., 2009) who analyzed the vaginal community composition of a limited number of Chinese women. Taken together these findings suggest there may be limited differences among women in different racial/ethnic groups in terms of the bacterial species found in the vagina. Moreover, it suggests that host-determined factors, but not race per se, exert selective pressures that permit persistent colonization of the vagina by only a limited number of different kinds of bacterial species. Studies could be done in the future to directly test whether here are differences in the vaginal communities in women of the same ethnic group that reside in two widely separated geographical regions.
How host factors might govern the composition of vaginal bacterial communities is not well understood. However, recent work has shown that the host immune system may influence human-microbial symbioses, and differences in cell surface receptors may dictate adhesion of beneficial microorganisms in the human gastrointestinal tract (Mazmanian et al., 2005; Dethlefsen et al., 2007). It is thought that specialized tissues and cells actively sample the intestinal content and initiate local immune responses that help to confine and shape the microbial diversity of the human intestine (Macpherson et al., 2005). McFall-Ngai recently proposed that adaptive immunity plays a role in recognizing and managing complex community composition of beneficial microbes in vertebrates (McFall-Ngai, 2007). Similar selectivity may occur in the human urogenital tract, in which case the local vaginal immune system may play an important role in determining the composition of vaginal microbial communities.
Complex food webs exist in most microbial communities in which populations occupy different trophic levels (Azam & Malfatti, 2007), and this is likely to be true for the vagina as well. This means that one or more populations are primary consumers, while others consume their metabolites, and so on. This results in a ‘network’, wherein the number and strength of these ecological interactions determine the stability and resilience of the community (Azam & Malfatti, 2007). Different lactic acid bacteria such as L. crispatus, L. iners, Streptococcus sp. and Atopobium sp. are known to have different nutritional needs, and thus ecological interactions, competition for resources, and cross-feeding among species may determine whether the physiological needs of a given species can be met (Falsen et al., 1999; Rodriguez Jovita et al., 1999). Conceptually, this may be important since vaginal communities that differ in species composition may be more or less resilient to various kinds of disturbances (Peterson et al., 1998) such as sexual intercourse, douching, spermicides, and (in women of reproductive age) changes in the environment due to menses. If these communities differ in their responses to disturbance, it could as well affect an individual’s ability to resist invasive infectious agents.
Adhesins expressed by different strains of lactobacilli exhibit specificity in their ability to mediate adhesion to host epithelial cells, mucus, and extracellular matrices (ECM) (Velez et al., 2007). These proteins constitute a diverse group of molecules with important functions related to adherence, signaling, and interaction with the host immune system or environment (Dramsi et al., 2005). To date, several kinds of specific mucus adhesion proteins (extracellular mucus-binding proteins, MUB) have been identified and functionally characterized in lactobacilli (Velez et al., 2007). In addition, several Lactobacillus proteins belonging to the sortase-dependent protein family have been functionally characterized (Dramsi et al., 2005). These are all involved in the binding of a particular strain to human epithelial cells and mucus and may play a role in mediating colonization of the human vagina.
The vaginal bacterial communities of healthy women are not always dominated by Lactobacillus species. The results from this study on Japanese women confirms our previous findings that a fair proportion of women have vaginal communities dominated by lactic acid bacteria other than Lactobacillus such as Atopobium and Streptococcus, in addition to a number of populations of the order Clostridiales. This suggests that these various microbial species are naturally selected by their host and constitute bacteria normally found in the human vagina. Most of the communities that were dominated by these kinds of bacteria appear to have greater species diversity than those dominated by Lactobacillus species, thus we postulate that the ecology of these communities may differ from that of other kinds of vaginal communities.
Acknowledgments
We would like to thank Zaid Abdo, Christopher Williams and James Foster for their suggestions for data analysis, and Sanqing Yuan, Maria G. Schneider, Hyun-Joon La and Hyo-Jin Ahn for their technical assistance, Linda Rogers and Ivan Kluetz for editing the manuscript.
Funding
The project described was supported by grants P20RR16448 and P20RR016454 from the National Center for Research Resources (NCRR) of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. Financial support from the Procter & Gamble Company, Cincinnati, OH was used to design and conduct the study.
Footnotes
Disclosure of Interests
Dr. Forney has served as a consultant to the Procter & Gamble Co. on issues unrelated to this article.
Ethics Approval
Study protocol and informed consent document were reviewed and approved by ethics committee-Sogo Clinical Pharmacology Co., Ltd. Documented informed consent was obtained from all subjects prior to participation in this study.
References
- Abdo Z, Schuette U, Bent S, Williams C, Forney LJ, Joyce P. Statistical methods for characterizing diversity in microbial communities by analysis of terminal restriction fragment length polymorphism of 16S rRNA. Environ Microbiol. 2006;8:929–938. doi: 10.1111/j.1462-2920.2005.00959.x. [DOI] [PubMed] [Google Scholar]
- Azam F, Malfatti F. Microbial structuring of marine ecosystems. Nat Rev Microbiol. 2007;5:782–791. doi: 10.1038/nrmicro1747. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Wong M, Davis CC, Kanti A, Zhou X, Forney LJ. Preliminary characterization of the normal microbiota of the human vulva using cultivation-independent methods. J Med Microbiol. 2007;56:271–276. doi: 10.1099/jmm.0.46607-0. [DOI] [PubMed] [Google Scholar]
- Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S, Trieu-Cuot P, Bierne H. Sorting sortases: a nomenclature proposal for the various sortases of Gram-positive bacteria. Res Microbiol. 2005;156:289–297. doi: 10.1016/j.resmic.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Falsen E, Pauscual C, Sjoden B, Ohlen M, Collins MD. Phenotypic and phylogenetic characterization of a novel Lactobacillus species from human source: description of Lactobacillus iners sp. nov. Int J Syst Bacteriol. 1999;49:217–221. doi: 10.1099/00207713-49-1-217. [DOI] [PubMed] [Google Scholar]
- Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacterial vaginosis. N Engl J Med. 2005;353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- Hyman RW, Fukushima M, Diamond L, Kumm J, Giudice LC, Davis RW. Microbes on the human vaginal epithelium. PNAS. 2005;102:7952–7957. doi: 10.1073/pnas.0503236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5:104–112. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Geuking MB, McCoy KD. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology. 2005;115:153–162. doi: 10.1111/j.1365-2567.2005.02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- McClelland RS, Richardson BA, Graham SM, Masese LN, Gitau R, Lavreys L, Mandaliya K, Jaoko W, Baeten JM, Ndinya-Achola JO. A prospective study of risk factors for bacterial vaginosis in HIV-1-seronegative African women. Sex Transm Dis. 2008;35:617–623. doi: 10.1097/OLQ.0b013e31816907fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall-Ngai M. Adaptive immunity: care for the community. Nature. 2007;445:153. doi: 10.1038/445153a. [DOI] [PubMed] [Google Scholar]
- Naruszewicz M, Johansson ML, Zapolska-Downar D, Bukowska H. Effect of Lactobacillus plantarum 299v on cardiovascular disease risk factors in smokers. Am J Clin Nutr. 2002;76:1249–1255. doi: 10.1093/ajcn/76.6.1249. [DOI] [PubMed] [Google Scholar]
- Nugent RP, Krohn MA, Hillier SL. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J Clin Microbiol. 1991;29:297–301. doi: 10.1128/jcm.29.2.297-301.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y, Nakai S, Tsukamoto T, Masumori N, Akaza H, Miyanaga N, Kitamura T, Kawabe K, Kotake T, Kuroda M, Naito S, Koga H, Saito Y, Nomata K, Kitagawa M, Aso Y. Habitual intake of lactic acid bacteria and risk reduction of bladder cancer. Urol Int. 2002;68:273–280. doi: 10.1159/000058450. [DOI] [PubMed] [Google Scholar]
- Parsonnet J, Goering RV, Hansmann MA, Jones MB, Ohtagaki K, Davis CC, Totsuka K. Prevalence of toxic shock syndrome toxin 1 (TSST-1)-producing strains of Staphylococcus aureus and antibody to TSST-1 among healthy Japanese women. J Clin Microbiol. 2008;46:2731–2738. doi: 10.1128/JCM.00228-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsonnet J, Hansmann M, Delaney M, Dubois A, Wieland-Alter W, Wissemann K, Wild J, Jones M, Seymour J, Onderdonk A. Prevalence of toxic shock syndrome toxin 1 producing Staphylococcus aureus and the presence of antibodies to this superantigen in menstruating women. J Clin Microbiol. 2005;43:4628–4634. doi: 10.1128/JCM.43.9.4628-4634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson G, Allen CR, Holling CS. Ecological resilience, biodiversity, and scale. Ecosystems. 1998;1:6–18. [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rayes N, Hansen S, Seehofer D, Muller AR, Serke S, Bengmark S, Neuhaus P. Early enteral supply of fiber and lactobacilli versus conventional nutrition: a controlled trial in patients with major abdominal surgery. Nutrition. 2002;18:609–615. doi: 10.1016/s0899-9007(02)00811-0. [DOI] [PubMed] [Google Scholar]
- Rodriguez Jovita M, Collins MD, Sjoden B, Falsen E. Characterization of a novel Atopobium isolate from the human vagina: description of Atopobium vaginae sp. nov. Int J Syst Bacteriol. 1999;49:1573–1576. doi: 10.1099/00207713-49-4-1573. [DOI] [PubMed] [Google Scholar]
- Rozanova GN, Voevodin DA, Stenina MA, Kushnareva MV. Pathogenetic role of dysbacteriosis in the development of complications of type 1 diabetes mellitus in children. Bull Exp Biol Med. 2002;133:164–166. doi: 10.1023/a:1015503006854. [DOI] [PubMed] [Google Scholar]
- Schwebke JR. Abnormal vaginal flora as a biological risk factor for acquisition of HIV infection and sexually transmitted diseases. J Infect Dis. 2005;192:1315–1317. doi: 10.1086/462430. [DOI] [PubMed] [Google Scholar]
- Shi Y, Chen L, Tong J, Xu C. Preliminary characterization of vaginal microbiota in healthy Chinese women using cultivation-independent methods. J Obstet Gynaecol Res. 2009;35:525–532. doi: 10.1111/j.1447-0756.2008.00971.x. [DOI] [PubMed] [Google Scholar]
- Tannock GW. Exploring the relationships between intestinal microflora and inflammatory conditions of the human bowel and spine. Antonie Van Leeuwenhoek. 2002;81:529–535. doi: 10.1023/a:1020517603993. [DOI] [PubMed] [Google Scholar]
- Velez MP, De Keersmaecker SC, Vanderleyden J. Adherence factors of Lactobacillus in the human gastrointestinal tract. FEMS Microbiol Lett. 2007;276:140–148. doi: 10.1111/j.1574-6968.2007.00908.x. [DOI] [PubMed] [Google Scholar]
- Verhelst R, Verstraelen H, Claeys G, Verscharaegen G, Delanghe J, Simaey LV, Ganck CD, Temmerman M, Vaneechoutte M. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol. 2004;4:1–11. doi: 10.1186/1471-2180-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali B, Pugliese C, Biagi E, Candela M, Turroni S, Bellen G, Donders GG, Brigidi P. Dynamics of vaginal bacterial communities in women developing bacterial vaginosis, candidiasis, or no infection, analyzed by PCR-denaturing gradient gel electrophoresis and real-time PCR. Appl Environ Microbiol. 2007;73:5731–5741. doi: 10.1128/AEM.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Zhou X, Williams CJ, Hochwalt A, Forney LJ. Bacterial populations in the vaginas of healthy adolescent women. J Pediatr Adolesc Gynecol. 2009;22:11–18. doi: 10.1016/j.jpag.2008.01.073. [DOI] [PubMed] [Google Scholar]
- Zhou X, Bent SJ, Schneider MG, Davis CC, Islam MR, Forney LJ. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology. 2004;150:2565–2573. doi: 10.1099/mic.0.26905-0. [DOI] [PubMed] [Google Scholar]
- Zhou X, Brown CG, Abdo Z, Davis CC, Hansmann MA, Joyce P, Foster JA, Forney LJ. Difference in the composition of vaginal microbial communities found in healthy Caucasian and Black women. The ISME Journal. 2007;1:121–133. doi: 10.1038/ismej.2007.12. [DOI] [PubMed] [Google Scholar]