

1. Introduction
Magnetism in medicine has had a long and interesting history. In the 10th century A.D., Egyptian physician and philosopher Avicenna prescribed a grain of magnetite dissolved in milk for the accidental swallowing of rust reasoning that magnetite would render the poisonous iron inert by attracting it and accelerating its excretion through the intestine.1 A thousand years later on July 3, 1977, “Indomitable”, the little machine that could, labored for five hours to produce one image, an event that used magnetism to change the landscape of modern medicine.2 Looking at the homemade superconducting magnet constructed from 30 miles of niobiumtitanium wire that now resides in its rightful place at the Smithsonian Institution, it is incredible to comprehend how in a mere 30 years magnetic resonance imaging (MRI) has gone from its crude, almost ugly, human scan to where physicians can now regularly order MRIs off their menu of diagnostic tools because of its exquisite anatomical resolution, routinely down to 0.5 to 1 mm.
When the field was first reviewed in this journal in 1987,3 only 39 papers were found in Medline with keywords “gado-“ and “MRI”.4 Today, this same search on PubMed pulls out over 250,000 records, of which a significant component has been development of MR contrast agents. The human body is essentially a super-sized water bottle, with about two-thirds of its weight consisting of water. Water's hydrogen atoms are able to act as microscopic compass needles that stand “at attention” when placed in a strong magnetic field. When submitted to pulses of radio waves, their magnetic alignment is disrupted and the differences in how they relax to the previous state are used to generate images. Contrast agents can act to catalyze the process of the return to the ground relaxed state. Now commonplace in the clinic, paramagnetic or superparamagnetic metal ions are administered in 40–50% of the 7–10 million MR examinations per year.5 These image-enhancing contrast agents add significant morphological and functional information to unenhanced MR images, allowing for enhanced tissue contrast, characterization of lesions, and evaluation of perfusion and flow-related abnormalities. In this review, we will introduce small molecule agents, but focus primarily on macromolecular MR contrast agents, particularly those containing gadolinium (Gd3+) that are assembled or based in part on these same small molecules. A brief discussion on iron oxide and manganese (Mn2+) agents is also provided.
2. Relaxation Theory and Mechanisms
While a detailed explanation of relaxation theory can be found in a number of excellent articles,6–8 we will reintroduce the essentials because of their importance in understanding how contrast agents work. The signal-to-noise ratios in MRI depend on the density of protons present in the region of interest and the degree of polarization of the nuclear spin states. When placed in a magnetic field, a slight majority of protons will orient in the direction of the magnetic field and precess at a Larmor resonance frequency related to the strength of the magnetic field. Relaxation is measured in two directions, longitudinal and transverse. Longitudinal or spin-lattice relaxation is defined by the time constant T1 and occurs in the direction of the main magnetic field. Signals related to T1 relaxation are obtained after excitation by an RF pulse at the Larmor frequency as the proton's dipole moment vector begins to realign or relax back to its ground state of alignment with the main magnetic field. Transverse or spin-spin relaxation corresponds to vector dephasing in the plane perpendicular to the main magnetic field and is characterized by T2. T1 represents the time required for the magnetization vector to be restored to 63% of its original magnitude and T2, a 37% decrease in net signal. T2 is always equal to or shorter than T1. Inhomogeneity in the static magnetic field and spin-spin relaxation has an effect on the transverse magnetization and is characterized by:
| (1) |
where T2' is a time constant arising from magnetic field inhomogeneity and T2* is the spin-spin time constant that takes into account these issues. T2* is always less than T2. Signals received from spin vectors are used to produce images by the superimposition of magnetic gradients which define the spatial location of the signal. Tissue types vary in their relaxation properties, and thus MRI is used to reconstruct images of structures such as organs and lesions and to evaluate perfusion and flow-related abnormalities.
Though it is possible to obtain images distinguishing tissues types by manipulation of pulse sequences alone, MRI is best optimized by use of contrast agents that dramatically highlight anatomic and pathologic features of interest. Paramagnetic ions decrease the proton relaxation time of bound water molecules. Thus, unlike other diagnostic media such as radionuclide, optical, and X-ray agents, MR contrast agents are themselves not a source of a signal and are not directly visualized, but rather affect the surrounding water molecules that in turn directly influence the signal. Paramagnetic species decrease T1 and T2, increasing longitudinal (spin-lattice) and transverse (spin-spin) relaxation of solvent nuclei. The observed solvent relaxation, (1/Ti)obs, is the sum of the intrinsic diamagnetic solvent relaxation rate in absence of the paramagnetic species, (1/Ti)d, and the additional paramagnetic contribution, (1/Ti)p.
| (2) |
In the absence of solute-solute interactions, the solvent relaxation rate is linearly dependent on the concentration of the paramagnetic ion, cagent:
| (3) |
where (Ri)agent is the relaxivity of the paramagnetic agent, typically defined in units of mM−1s−1. The effect of the agent is dependent on the distance from the ion and the diffusion of solvent molecules. Water interaction with the metal ion is classified into three types: 1) primary coordination sphere, 2) hydrogen-bonded molecules in the secondary coordination sphere, and 3) bulk water that translationally diffuses past the metal (Figure 1).3
Figure 1.

Relaxation coordination spheres of water: inner-sphere, secondary-sphere, and bulk water.
Inner-sphere relaxation is the enhancement found in the first coordination sphere. If the time of interaction is long compared to the time of diffusion, second coordination sphere water molecules demonstrate similar relaxation to the first sphere. However, typically enhancement in the second coordination sphere and bulk water is grouped together as outer-sphere relaxation. Thus, the total paramagnetic relaxation enhancement is:
| (4) |
2.1 Inner-Sphere Relaxation: Solomon-Bloembergen-Morgan (SBM) Equations
The inner-sphere contributions to longitudinal and transverse relaxation are a function of the mole fraction of metal ion per solvent molecule (Pm), the number of bound water (or solvent) nuclei per metal ion or the hydration number (q), and the average residence time of the solvent molecule in the complex (τm or 1/kex the reciprocal of the solvent exchange rate).3–4,9–10
| (5) |
| (6) |
The “m” subscript refers to the solvent molecule in the inner-sphere, and Δωm is the difference in Larmor frequencies between the inner coordination sphere and the bulk solvent reference. The relaxation times of the bound water molecules (T1,2 m) are further defined by the Solomon–Bloembergen–Morgan (SBM) equations4,8,11 which represent the sum of dipole-dipole (“through-space”) and scalar (contact or “through-bonds”) contributions:
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
Here, γI is the nuclear gyromagnetic ratio, g is the electronic g-factor, μB is the Bohr magneton, r is the proton-metal ion distance, ωI and ωs are the proton and electron Larmor precession frequencies, respectively, A/ħ is the electron – nuclear hyperfine coupling constant, and S is the total electron spin of the metal ion. The dipole–dipole and scalar correlation times τci and τei that modulate relaxation are defined by:
| (12) |
| (13) |
T1e and T2e are the electronic longitudinal and transverse relaxation times of the metal ion, τm is the water residence time, and τR is the rotational tumbling or correlation time of the entire metal-water complex.
Nuclear or electron Larmor frequency is directly related to the magnetic field, B, by the gyromagnetic ratio, γ:
| (14) |
Thus, all these equations describe relaxation as a function of magnetic field. The same is also true for the electronic relaxation rates. Equations 8–11 are only valid for ions with electronic spin S > ½, where inner-sphere collisions lead to zero field splitting (ZFS) of the electron spin levels. This ZFS modulates electronic relaxation rates by the following functions:
| (15) |
| (16) |
| (17) |
where the constant B is related to the magnitude of the transient ZFS, τs0 is the electronic relaxation time at zero field, τv is a correlation time for the modulation of this transient ZFS, and Δ is the trace of the ZFS tensor.
2.2 Limitations to the SBM Equations
While the Solomon-Bloembergen-Morgan (SBM) equations are the most commonly used approach to describe relaxation theory; however, there are a few points about the SBM methods to consider with caution. T1e and T2e are difficult parameters to determine independently because of their field dependence. Equations 15 and 16 are only valid as a mono-exponential electronic relaxation process under the limit of extreme narrowing, where ωs2τv2  1.6 Outside the extreme narrowing condition, electronic relaxation becomes multi-exponential for an ion such as Gd3+.12 A number of groups 6–7,13–20 have shown that the SBM equations are invalid in the “low-field” region when the energy of the ZFS interaction is larger than that of the Zeeman energy of the interaction between the magnetic moment of the molecule and the applied magnetic field. In the Zeeman or SBM limit, the electron spin precesses about the axis of the external magnetic field. In the ZFS limit the electron spin precesses about the principal axis of the ZFS tensor and the nuclear relaxation is strongly dependent upon the angle between the electron spin–nuclear spin vector and the ZFS tensor axis. The symmetry of the molecule also plays a role, i.e. rhombicity in the ZFS can greatly reduce nuclear relaxation. Qualitatively, the magnetic field dispersion profiles of nuclear relaxation generated using low-field theories look similar to those generated using SBM.
1.6 Outside the extreme narrowing condition, electronic relaxation becomes multi-exponential for an ion such as Gd3+.12 A number of groups 6–7,13–20 have shown that the SBM equations are invalid in the “low-field” region when the energy of the ZFS interaction is larger than that of the Zeeman energy of the interaction between the magnetic moment of the molecule and the applied magnetic field. In the Zeeman or SBM limit, the electron spin precesses about the axis of the external magnetic field. In the ZFS limit the electron spin precesses about the principal axis of the ZFS tensor and the nuclear relaxation is strongly dependent upon the angle between the electron spin–nuclear spin vector and the ZFS tensor axis. The symmetry of the molecule also plays a role, i.e. rhombicity in the ZFS can greatly reduce nuclear relaxation. Qualitatively, the magnetic field dispersion profiles of nuclear relaxation generated using low-field theories look similar to those generated using SBM.
Another point of discussion is that of anisotropic rotation. Strategies to increase the rotational correlation time τR include incorporation of a metal chelate on to a macromolecule such as polymer or dendrimer In these cases, relaxation is a function of both the overall motion of the macromolecule and its fast internal motion, i.e. side chain rotations. Lipari and Szabo21 have derived expressions that account for the fast motion by a second spectral density term.
2.3 Outer-Sphere Relaxation
The SMB theory can also be applied to describe second coordination sphere relaxation enhancement. Protons that are hydrogen-bonded to the contrast agent relax via a dipole–dipole interaction with the paramagnetic species, and consequently, their relaxation can be described by equations 5, 6, 8, and 10 with the relevant parameters denoted with a prime (e.g., q`, r`, τm'). However, because the number of second-sphere water molecules and the ion–H distances are unknown, second coordination relaxation is difficult to quantify. Furthermore, τm is very short and the likely limiting parameter in determining T1m.
Outer-sphere relaxation is most often described by translational diffusion of the water molecules past the metal complex. This contribution to relaxation is approached based upon a rigid-sphere model (Hwang and Freed model)4,22–24 where the water molecules and metal complex are treated as hard spheres.
| (18) |
| (19) |
| (20) |
| (21) |
| (22) |
where γI and γS are the nuclear and electron gyromagnetic ratios, NA is Avogadro's number, M is the concentration of the metal ion, a is the distance of closest approach between the protons and the paramagnetic complex, D is the sum of the diffusion constants of water and the complex, ωI and ωS are the proton and electron Larmor angular velocities, and τD is a diffusional correlation time. In the spectral density function, j(ω), Re stands for “the real part of”.
Second coordination sphere relaxation contribution is difficult to measure and the separation of the two contributions in a q = 0 chelate has not been observed, where q is the number of water molecules bound to the paramagnetic center. In fact, inner-sphere relaxivity is often determined by subtracting the relaxivity of a q = 0 complex such as [Gd3+(TTHA)]3− from the observed r1 with the assumption that it is a reasonable estimation of outer-sphere plus second-sphere relaxivity.25–26
Detailed discussions about outer-sphere relaxation are in the cited references. Second sphere relaxivity is not well characterized and outer sphere relaxivity can vary from paramagnetic complex to complex. As with inner-sphere models, SBM equations have limitations with regards to describing electronic relaxation in the low-field limit. Suffice to say, outer-sphere relaxivity is complex and typically the focus is placed on inner-sphere relaxation when developing Gd3+-based MR contrast agents.
2.4 Relaxation Theory – Lessons Learned
Overall relaxivity is a weighted average of relaxation rates from three local proton environments, with the principal contribution from within the inner hydration sphere of the ion. From the equations listed previously, it is evident that relaxation enhancement by paramagnetic ions on their surrounding protons is a compound effect of a number of factors. The most commonly used MR agents are Gd3+ based wherein due to the nature of its ionic bonding, the hyperfine coupling constant, A/ħ, is quite small. This coupling makes scalar relaxation (1/T1SC, equation 9) inefficient and inner-sphere relaxation more dependent on dipole-dipole relaxation (1/T1DD, equation 8). The key variables, thus, are τm, τR, q, r, T1e, and T2e. Increasing the hydration number, q, increases inner-sphere relaxivity (equation 5), but it is often accompanied by a decrease in thermodynamic stability and/or kinetic inertness of chelated Gd3+ associated with toxicity issues (vide infra) and may lead to the formation of ternary complexes with endogenous ligands such as phosphates and carbonates. Decreasing the distance between the water proton and the unpaired electron spin, r, has a large effect on relaxivity because of the 1/r6 dependence noted in equation 8. Gd3+- water oxygen distances range from 2.41 to 2.50 Å for monomeric complexes in the solid state (vide supra), and even a decrease of 0.2 Å would result in a 60% increase in relaxivity. The challenge with this distance r, however, is that it is a difficult parameter to both measure and control. The difficulties of modeling and determining electronic relaxation times T1e, and T2e were described in the previous sections, and so that leaves τm and τR.
Water residence time, τm, is the term used to describe the fast exchange between metal-coordinated water molecules and water in the bulk solvent. If exchange among protons in the shells is rapid, they all exhibit similar relaxation behavior. Studies have been conducted to improve the rate of water exchange4, but the vast majority of efforts have been directed at lengthening the rotational correlation time, τR. Increased steric hindrance and hydrodynamic size slows the rotation of larger molecules and increases τR. Thus, relaxivity is improved and there is more enhancement per unit dose of the paramagnetic ion. While rotational correlation times can be estimated in a number of ways,4 if there a good estimate of the viscosity, η, the Debye-Stokes theory can be used for a spherical molecule of radius a:
| (23) |
where k is the Boltzmann constant and T the absolute temperature. It is important to note that in microheterogeneous solutions, macroscopic translational viscosity may differ from rotational microviscosity which is a parameter that is not well understood. Additionally, for molecules with a long τR and in high magnetic fields, the Curie spin relaxation mechanism may contribute to the normal dipole-dipole mechanism, but it is negligible at the low fields used in MRI (typically at 1.5-3T, however higher field instruments, e.g. 8T, are becoming available).4,27
3. Some “Gado” Please
Because of its seven unpaired 4f electrons, the lanthanide ion Gd3+ (atomic number = 64, standard atomic weight = 157.25) is by far the most frequently chosen paramagnetic ion for MRI. Advances in MRI for faster scans and higher resolution have required more rapid pulsing and thus have favored T1-weighted imaging and use of contrast enhancers such as Gd3+. Two other lanthanide ions, dysprosium (Dy3+) and holmium (Ho3+) have larger magnetic moments than Gd3+ because they have orbital contributions to electron angular momentum. However, their asymmetric electronic ground state shifts solute resonance frequencies without line broadening.28 The 9 f-electrons of Dy3+ for instance, distribute themselves among the 7 f-orbitals leaving the ground state highly anisotropic, the net moment part spin/part orbital, and the spin-orbit interactions large. This reduces the electronic relaxation time (increases relaxation rate) 100-fold and has a large effect on proton resonance frequency. Meanwhile, the symmetry of the electronic S-state of Gd3+ makes it a broadening “relaxer” whose major effect is to increase longitudinal and transverse relaxation rates of the solute without shifting proton resonance frequencies. With its seven electrons forming a half-filled f-shell, Gd3+ has an isotropic S-ground state with no net orbital momentum and little spin-orbit interaction.28 This configuration leads to long electronic relaxation times, or slower relaxation rates.
What prevents Gd3+ from being directly administered is its high toxicity in free form. Gd3+ is chemically similar to Ca2+ in size (Gd3+ radius = 1.05–1.11 Å, Ca2+ radius = 1.00–1.06 Å), bonding, coordination and donor atom preference.29 Acutely, neuromuscular transmission arrest can occur by Gd3+ ions interfering with calcium-ion passage through muscle cells and calcium flow in bone epiphyses and nerve tissue cells.30 Chronically, accumulation can be found in bone and liver with a biological half-life of several weeks.31 Further complications can occur by transmetallation where Gd3+ also can replace endogenous metals, such as zinc.30
To sequester and render the ion nontoxic, a number of chelating agents have been developed. These highly stable complexation cages have a greater affinity for Gd3+ than other metals commonly present in vivo such as Zn2+, Ca2+, or Cu2+. Furthermore, after chelation renal excretion increases ~550-fold as compared with free Gd3+.32
4. Chelating Agents
There are currently eight clinically approved gadolinium-based contrast agents (Table 1, Figure 2): Magnevist® (gadopentetate dimeglumine, Gd-DTPA), Dotarem® (gadoterate, Gd-DOTA), ProHance® (gadoteridol, Gd-HP-DO3A), Gadovist® (gadobutrol, Gd-BT-DO3A), Omniscan® (gadodiamide, Gd-DTPA-BMA), OptiMARK® (gadoversetamide, Gd-DTPA-BMEA), MultiHance® (gadobenate dimeglumine, Gd-BOPTA), and Eovist®/Primovist® (Gd-EOB-DTPA). The chelates fall into two classes: cyclic and acyclic. The macrocyclic chelates, e.g. Dotarem® and ProHance®, are derivatives of 1,4,7,10-tetraazacyclododecane (cyclen).4 The cyclen-based tetraacetic acid derivative complex with gadolinium, Gd3+-DOTA is formulated as its N-methylglucamine salt. Two neutral macrocyclic derivatives of 1,4,7-tricarboxymethyl-1,4,7,10-tetraazacyclododecane (DO3A) are gadoteridol and gadobutrol. They are characterized by substitution of one carboxylate with a hydroxyl donor group. The second class of acyclic chelates is comprised of derivatives of polyaminocarboxylic acids such as diethylenetriaminepentaacetic acid (DTPA). Gd3+-DPTA was approved for clinical use in adult patients in 1988 and has since become the most commonly used MR contrast agent. Two diamide derivatives of DTPA were also approved for human use: Gd3+-DTPA-BMA and Gd3+-DTPA-BMEA. By reacting the dianhydride of DTPA with an amine (methyl amine or methoxyethyl amine, respectively), two carboxylates were replaced with two amide oxygen donors. This reaction strategy resulted in neutrally charged chelates that remain highly water soluble. They were developed in part to lower the osmolality of aqueous solutions.33 Chelating agents do reduce the number of coordinated water molecules in comparison to free metal ion. For example, Gd3+ and Gd3+-DTPA have approximately 8–9 and 1 coordinated water molecules, and the corresponding relaxivities are 7.0 and 2.0, respectively, at 37°C, 20 MHz, and 0.5 T.34 However, other factors also determine the in vivo efficacy of an agent in obtaining quality images, namely clearance rate and route of excretion.
Table 1.
| Gd-DTPA | Gd-DTPA-BMA | Gd-DOTA | Gd-HP-DO3A | Gd-DO3A-butrol | Gd-BOPTA | Gd-DTBA-BMEA | Gd-EOB-DTPA** | |
|---|---|---|---|---|---|---|---|---|
| Generic Names | Gadopentetate dimeglumine | Gadodiamide | Gadoterate meglumine | Gadoteridol | Gadobutrol | Gadobenate dimeglumine | Gadoversetamide | |
| Trademark | Magnevist® | Omniscan® | Dotarem® | ProHance® | Gadovist® | MultiHance® | OptiMARK® | Primovist® (Europe), Eovist® (USA) |
| Cyclic/Acyclic | acyclic | acyclic | cyclic | cyclic | cyclic | acyclic | acyclic | acyclic |
| Molecular Weight | 547 | 573 | 558 | 558 | 604 | 711 | 661 | 682 |
| r1/r1* (mM−1s−1) (20 MHz, 310 K) | 3.8/3.9 | 3.8/4.8 | 3.5/4.3 | 3.7/5.6 | 3.7/5.6 | 4.8/9.7 | 4.1 (308K) | 5.5 |
| τm (ns) (310K) | 143 | 967 | 122 | 176 | 176 | 140 | 1320 (308K) | 82 |
| τR (ps) (310K) | 54 | 65 | 217 | 57 | 89 | 71 (308K) | 86 | |
| Thermodynamic stability constant (log Keq) (0.5 mol/L) | 22.1 | 16.9 | 25.8 | 23.8 | 21.8 (1 mol/L) | 22.6 | 16.6 | 23.46 (0.25 mol/L) |
| Osmolality (Osm/kg) | 1.96 | 0.65 | 1.35 | 0.63 | 1.6 | 1.97 | 1.11 | 0.688 |
| Viscosity (mPa s at 37°C) | 2.9 | 1.4 | 2.0 | 1.3 | 4.96 | 5.3 | 2.0 | 1.19 |
| Approval | USA, EU, Japan | USA, EU, Japan | EU | USA, EU, Japan | EU, Canada | USA, EU | USA | USA, EU |
| Approved doses (mmol/kg) for body imaging | 0.1 | 0.1–0.3 | 0.1 | 0.1–0.3 | Not approved | Liver: 0.05 | 0.1 | 25 μmol/kg or 0.1 ml/kg |
| Approved doses (mmol/kg) for CNSa imaging | 0.1–0.2 | 0.1–0.3 | 0.1–0.3 | 0.1–0.3 | 0.1–0.3 | 0.1 | 0.1 | Not approved |
| Approved doses (mmol/kg) for MR angiography | 0.1–0.3b | 0.1–0.3 | 0.2 | Not approved | 0.1–0.15 (Imaging of 1 field of view) 0.2–0.3 (Imaging of >1 field of view) | Not approved | Not approved | Not approved |
| Approved doses (mmol/kg) for children | 0.1 | From 6 months: 0.1 | 0.1 | From 2 years and above: 0.1; 6 months-2 years: caution; <6 months: contra-indicated | Not approved <18 years | Not approved <18 years | Not approved <18 years | Not approved <18 years |
CNS: central nervous system
approved for whole body imaging and for doses of 0.1–0.3 mmol/kg but does not have a trial-based approval for MR angiography
Figure 2.

Commercially available Gd3+ chelate MR agents.
Clearance is dependent on a number of properties such as size, shape, surface charge and chemical makeup of the agent. Gd3+ chelates are generally excreted unchanged by passive glomerular filtration. They are typically hydrophilic, extracellular-fluid markers with low molecular masses of ~500 Da. These agents are rapidly cleared from the intravascular space through capillaries and into the interstitial space, but do not cross an intact blood-brain barrier. The biological elimination half-life is approximately 1.5 h41 with no detectable biotransformation, decomposition, or serum protein binding. When observed in mice and rats after 14 days, residual whole body Gd3+ for acyclic agents was found to be higher than macrocyclic agent with the order from least to most being: Gd3+-HP-DO3A≈ Gd3+-DOTA = Gd3+-DTPA< Gd3+-DTPA-BMA.42 For Gd3+-DPTA, 90% of the injected dose is cleared by renal filtration and vessel leakage in less than an hour.43 For patients with normal renal function, rapid clearance improves the safety profile. The converse of that same rapid clearance is that it can pose a challenge for conducting time-dependent imaging studies or obtaining highly resolved images.
Two Gd3+ chelates with almost double the relaxivity of the above mentioned chelates are also available: Gd3+-BOPTA and Gd3+-EOB-DTPA. These agents are eliminated through both the renal and hepatobiliary pathways with 2–4% hepatic uptake of the injected dose for Gd3+-BOPTA and 50% for Gd3+-EOB-DTPA.30 Thus, they can be used both as conventional extracellular contrast agents within minutes after injection and also to enhance normal liver parenchyma in a later, delayed phase (40–120 min post-administration). Tumor nodules typically lack functional hepatocytes and remain un-enhanced in these MR images, allowing for increased sensitivity and specificity in the detection and characterization of liver tumors.44 Additionally, Gd3+-BOPTA may have potential for MR angiography (MRA) due to weak and transient protein binding.30
While these agents are the approved and most commonly used chelates, the contents of Table 1 are by no means a complete list. Since 1995, a body of work has been published based off the structure of Gd3+(TREN-1-Me-3,2-HOPO)(H2O)2 (Figure 3).45 Hexadentate hydroxypyridinone (HOPO) based chelates bind high numbers of water molecules, at least doubling relaxivity, while also maintaining high stability. TREN-bis-HOPO-terephthalamide (TAM) chelates demonstrated the best relaxometric and solubility properties,46 and when their biodistribution in mice was evaluated at 1 hr after i.v. injection, accumulation was found in the liver.47 To make their synthesis more straightforward, the TREN (tris-(2-aminoethyl)-amine) scaffold was replaced with a triazacyclononane (TACN) derivative.46 This TACN ligand cap allowed for a hydration number of 3, compared to the q = 1 of commercial agents. Given that it is difficult to introduce new functionalities in the heterocyclic pyridinone ring of HOPO, a recent modification has been the use of 2-hydroxy-2H-isoquinolin-1-one (1,2-HOIQO) 3-carboxylic acid instead of the cyclic hydroxamic acid units. The TREN-1,2-HOIQO chelate forms mononuclear complexes with Fe3+ and one-dimensional coordination polymers with lanthanide(III) cations, including Gd3+.48
Figure 3.

HOPO- and HOIQO-based chelating agents.
Numerous analogs of these chelates have been synthesized, but reviewing their synthesis and characterization is beyond the scope of this review. For the following sections, our discussion focuses on those agents currently in use in humans.
4.1 Dosage
Because clearance is rapid, quick T1-weighted imaging is typically required with these agents to maximize enhancement. The recommended dosage of gadolinium chelates for visualization of lesions with abnormal vascularity in body tissue (excluding the heart) and in the central nervous system (brain, spine, and associated tissues) is 0.1–0.3 mmol/kg. Larger doses allow for better enhancement and discrimination of lesions from healthy tissue. The agents that have been approved for MRA can be administered at larger dosages (Table 1).30 For hepatic imaging, Gd3+-BOPTA and Gd3+-EOB-DTPA have been approved at lower dosages of 50 and 25 μmol/kg, respectively, though Gd3+-BOPTA can be used for CNS imaging at 0.1 mmol/kg (0.2 ml/kg of a 0.5 M solution). Four of these agents have been approved for administration in children as no significant adverse clinical events or vital sign trends have been observed. In Europe, from day one after birth, Gd3+-DTPA and Gd3+-DOTA can be given in doses up to 0.2 mmol/kg for CNS studies. Gd3+-DTPA-BMA is approved in Europe for infants from 6 months of age at a dose of 0.1 mmol/kg, and 0.1 mmol/kg Gd3+-HP-DO3A can be injected in children of 2 years and above.
In 2005, the Contrast Media Safety Committee of European Society of Urogenital Radiology (ESUR) evaluated the use of gadolinium-based agents in pregnant and lactating women.49 The recommendation was that when MR was deemed necessary, gadolinium media could be given to pregnant women with no need for follow up neonatal tests. Further studies demonstrated that minimal amounts (<0.04% of the injected dose) of gadolinium were found in human breast milk 24 h after administration in the mother.50 The amount in the gut of a nursing child after intravenous administration of a Gd3+ contrast agent to the mother is 100-fold less than the permitted dose for the infant.50 Furthermore, very small amounts of Gd3+ contrast agents are absorbed when they enter through the gut. Although instructions for use state to delay breast-feeding for 24–72 hrs after agent administrations, the Committee's recommendation was to continue normally.49
4.2 Adverse Reactions and Toxicity
Gd3+ chelates are tolerated well at both standard and high doses, with no clinically relevant difference amongst these agents. Adverse events, mostly mild and transient, are observed with an incidence of less than 2%.30 These may include nausea, headache, vomiting and pain, warmth and localized edema at the injection site. Anaphylactic reactions have been reported with a prevalence of 0.0002 – 0.001%,5 but mostly in patients with a history of respiratory difficulties or respiratory allergic disease. The major concern is for patients with compromised kidney function who may develop nephrogenic systemic fibrosis (NSF).
NSF, first described in 2000,51 is a systemic disorder characterized by widespread tissue fibrosis that can develop rapidly, confining patients to a wheelchair within a few weeks. Increased tissue deposition of collagen is observed with thickening and hardening of the skin of extremities. Involvement of other tissues such as lung, skeletal muscle, heart, diaphragm, and esophagus can occur,52 and while the disease sometimes stabilizes, it rarely spontaneously remits. No effective treatment exists, and so prevention is the currently the only approach.53
Of the more than 200 cases identified in the last decade, NSF is almost exclusively found among patients with advanced kidney disease.54 Since it was first proposed that gadolinium agents might be associated with NSF,55 much literature has been published supporting this relationship. It is theorized that lowered renal clearance of gadolinium increases tissue exposure to the metal and its dissociation from the chelate.56 Though the actual mechanism remains unclear, the result is an inflammatory reaction and fibrosis.57–59 A meta-analysis of the controlled studies examining gadolinium agents and the development of NSF, suggests a causal relationship.56
The FDA and American College of Radiology (ACR)'s recommendation is to withhold all gadolinium-based agents from patients with Stage 4–5 chronic kidney disease (CKD). If patients with severe CKD need gadolinium contrast media, the FDA recommends prompt haemodialysis following contrast administration,60 while the ACR only feels this is warranted in patients who are already on dialysis. For patients not already on haemodialysis, the recommendation is to consider the risks of initiating haemodialysis against that of developing NSF.61 For Stage 3 or moderate CKD patients, the data was not sufficient to make any recommendations. Both the FDA and ACR have given their recommendation across the board for all Gd3+ based agents assuming that NSF is not linked to one specific agent. While there are suggestions that Gd3+-DTPA-BMA administration may lead to a greater risk of NSF, currently there is no solid evidence to compare it relatively with the other gadolinium agents.
4.3 Motexafin Gadolinium
In the context of Gd3+ agents, motexafin gadolinium (MGd) deserves mention. MGd is an amphiphilic texaphyrin, a class of synthetic, aromatic macrocycles that resemble expanded porphryins, first prepared in 1988 by Sessler et al.62 The macrocyclic skeleton of this agent surrounds the Gd3+ that is coordinated by 5 pyrrole- and imine-derived nitrogens. In the presence of oxygen, MGd is reduced by various metabolites and forms reactive oxygen species by redox cycling.63 It selectively localizes in tumors and targets oxidative stress proteins such as metallothioneins and thioredoxin reductase. Oxidative stress impairs metabolism, alters metal ion homeostasis, and makes the cell more vulnerable to apoptosis. Why both texaphyrins and porphryins demonstrate tumor selectivity is not well understood, but in vitro uptake of the agent is temperature dependent, increases at lower pH, and is inhibited by serum proteins.64 Tumor response to radiation and chemotherapy is enhanced by MGd, and it may intrinsically be cytotoxic. International randomized studies in brain metastasis patients reported that in combination with MGd, radiation therapy improves time to neurological progression (15.4 months with and 10.0 months without MGd).65 MGd is being evaluated in a number of clinical trials for monotherapy and in combination with radiation and/or chemotherapy and monoclonal antibodies for various carcinomas including lymphomas, leukemia, lung cancer, renal cell cancer, and glioblastoma.63 Based off a Phase I trial, the maximum tolerated single dose is 22.3 mg/kg with dose-limiting reversible renal toxicity at 29.6 mg/kg.66 The noted adverse effects were diarrehea, nausea, vomiting, albuminuria, and reversible discoloration of skin, urine and sclera.
5. From Small Molecule to Macromolecular Agents
Low molecular weight agents have been the pioneers in improving MR contrast. They do have limits in vivo, though, particularly with rapid elimination restricting timing for studies and extravasation out of the vasculature reducing contrast from surrounding tissue. Macromolecular metal-chelate complexes, sometimes known as blood pool agents or macromolecular contrast media (MMCM), are larger agents with a molecular weight greater than 30kDa that were originally designed to address these issues. Their size limits extravasation through healthy vascular endothelium, but favors enhanced permeability and retention (EPR) in leaky vasculature that may be present where there is a pathology such as cancer67 or arthritic inflammatory response.68 Furthermore, because of increased steric hindrance, these agents have greater relaxivity than low molecular weight agents such as Magnevist and Dotarem. As was described, slower molecular tumbling increases rotational correlation time, τR, resulting in more enhancement per unit dose of the paramagnetic ion. Additionally, multiple chelates and metal ions can be appended to a macromolecular platform thereby also increasing enhancement and reducing dose of agent needed for satisfactory image acquisition.
In order to attach paramagnetic ions to larger structures, a class of chelates known as bifunctional chelates have been developed based on DTPA and DOTA. These chelating agents are typically modified to have an electrophilic group that is available for conjugation to nucleophile groups on biomolecules. For example, these functional groups include anhydride, bromo- or iodoacetamide, isothiocyanate, N-hydroxysuccinimide (NHS) ester, and maleimide. In cases where the biomolecules contain only electrophilic functionality, such as a carboxylic acid group, the common strategy is to use cross-linking agents that provide a link between the two moieties or introduce functionality that makes conjugation more amenable.
A plethora of MR macromolecular contrast agents have been reported over the last 30 years, ranging from protein- to polymer- to dendrimer-based molecules. As reviewed by Venditto, et al69 and references therein, these agents typically have diameters greater than 1–2 nm to reduce renal excretion as compared to low molecular weight agents such as Magnevist. At 8 nm, observations have been made that hepatic uptake begins to dominate clearance routes, and by 10–12 nm the reticuloendothelial excretion route becomes the dominant route for clearance. Increased retention times and limited extravasations affect the biodistribution profile of such agents.
6. Dendrimers in MRI
6.1 Synthesis and Structure
The use of dendrimers as scaffolds for MR contrast agents has generated a tremendous amount of interest and several reviews69–72 have been written describing their synthesis and applications since the first dendrimer-based contrast agents were reported in 1994.43 The principle behind the massive potential of this class of molecules in the development of diagnostic agents lies in that the synthetic chemistry used to construct them permits the “controlled occupation of space in three-dimensions as a function of size, shape and disposition of desired organic functionality”.73 The use of simple starting reagents, reaction conditions of high yields, and relatively easy purification procedures allow the precise size determination of monodisperse products based on generation number G, e.g., generation 3 is termed G3. Furthermore, the multivalent surface of the final product allows one to tailor the molecule for specific applications (Table 2).
Table 2.
Dendrimer Generation (G) and Terminal Amines (Z)
| G | Z |
|||
|---|---|---|---|---|
| Am | EDA | DAB | CYS | |
| 0 | 3 | 4 | 4 | 4 |
| 1 | 6 | 8 | 8 | 8 |
| 2 | 12 | 16 | 16 | 16 |
| 3 | 24 | 32 | 32 | 32 |
| 4 | 48 | 64 | 64 | 64 |
| 5 | 96 | 128 | 128 | 128 |
| 6 | 192 | 256 | 256 | 256 |
A dendrimer consists of a “core” from which sub-units emanate from in a branch-like fashion. Two general strategies are employed in the synthesis of dendrimers: a convergent approach, in which branches of desired generation are linked to a central core, and a divergent approach, in which subsequent branches originate and emanate from a central core, the chemistries of which are reviewed in thorough detail elsewhere.74–76 The convergent approach was first demonstrated by Hawker and co-workers in the synthesis of a series of dendritic polyether macromolecules based on the monomer 3,5-dihydroxybenzyl alcohol grafted onto a multi-functional core.77 Size-exclusion chromatography experiments demonstrated that the G5 member of this series exhibited a polydispersity index (PDI) less than 1.03. PDI, the ratio of the weight average molecular weight to the number average molecular weight, is a measure of the distribution of molecular mass in a sample. Jayaraman and co-workers used this same approach in the development of a new family of dendrimers with an aliphatic polyether backbone exhibiting PDIs less than 1.01.78 These examples demonstrate that the convergent approach permits a high degree of control in producing dendrimers of a very narrow molecular weight distribution.
The divergent approach was made possible by Buhleier and co-workers when they first demonstrated the synthesis of unidirectional branched polyamines in a “cascade-like” manner.79 Using a monoamine or diamine as a starting point, generations were produced by repetitive reaction with acrylonitrile to form “branches” with terminal nitrile groups, followed by reduction to the amine, permitting the “growth” of succeeding generations. In an analogous manner, Newkome demonstrated the unidirectional synthesis of branched polyalcohols known as “arborols”.80 The ability to grow branches in a “cascade-like” manner was used by Tomalia and co-workers to produce dendrimers possessing three-dimensional, radial symmetry, a class of molecules since called “Starburst” dendrimers.81 Generations were produced by the repeated reaction of either an ammonia (Am) or ethylenediamine (EDA) initiator core with an acrylate ester via Michael addition, followed by amidation of the resulting ester with alkylene diamine. Hence, these dendrimers also came to be known as poly(amidoamine) or PAMAM dendrimers. As a result of the three-dimensional growth of these structures, the number of terminal amines increases exponentially with generation number. However, the monodispersity of the final products was slightly affected detrimentally by unwanted side-reactions caused by dendrimer fragmentation, bridging, or incomplete removal of unreacted reagents at each generation sequence. Nevertheless, the PAMAM dendrimers have enjoyed an almost monopolistic usage in the development of dendrimer-based MR contrast agents, as described below. Systematic investigations of the atomistic structure of EDA-core PAMAM dendrimers up to G11 have also been performed to determine theoretical limits for uniform growth of successive generations.82 Poly(propylene imine) (or PPI) dendrimers based on a diaminobutane (DAB) core have also been synthesized,83–84 the first five generations of which were found to have a polydispersity index of 1.002.85 More recently, a family of PAMAM dendrimers with a cystamine (CYS) core was synthesized, which provide a versatile platform for producing novel shapes and terminal functionalities through redox chemistry at the disulfide core.86
Owing to the structural complexity of dendrimers, a system of nomenclature for this class of molecules should clearly express what the core, repeat and terminal units are. For cascade polymers having the same repeat unit throughout the structure (such as PAMAM dendrimers), Newkome et al. suggest75 that these may be represented by the formula,
| (24) |
where G is the generation number, Nb the branch multiplicity of the repeat unit, and Nc the branch multiplicity from the core. From this, the number of terminal groups Z can be calculated using Z = NcNbG. A name can then be assigned using the general formula,
| (25) |
where n denotes the number of repetitions of that unit. Applying these rules, a 2nd generation Am-core PAMAM dendrimer is then represented by the formula
and its name written as
Though this system of nomenclature is articulate, for brevity in this review we will use where appropriate a shorthand nomenclature which involves stating in sequence the kind of dendrimer (PAMAM vs. PPI), core, generation number, and terminal chelate. For example, a 2nd generation Am-core PAMAM dendrimer with terminal amines functionalized with the chelating ligand DOTA will be written simply as “PAMAM-Am-G2-DOTA”.
Furthermore, some confusion in the literature exists regarding the assignment of G to PPI dendrimers, and as a result the formula for Z may not apply. For the purpose of this review, we define G0 of the PAMAM and PPI dendrimers not as the initiator core, but as the functionalized core possessing terminal amines (Figure 4). It is important to stress this point as any meaningful comparison between increasing generations of PAMAM and PPI dendrimers in their use as contrast agents depends of the number of terminal amines (Z) available for functionalization with a paramagnetic chelate.
Figure 4.

Synthesis of G0 Am-, EDA-, DAB- and CYS-core dendrimers.
6.2 Solution Studies
The first report of dendrimer-based MR contrast agents described the conjugation of G2 and G6 PAMAM-Am dendrimers with Gd3+-1B4M.43 Due to their large molecular weight (and hence, a large molecular tumbling rate, τR), these agents exhibited very high longitudinal relaxivities. In terms of molar relaxivity, the G6 dendrimer was found be ~6 times that of Gd3+-DTPA alone. Owing to the potential usefulness of these compounds, an improved synthesis was reported more recently involving non-aqueous conjugation chemistry.87 Langereis and co-workers reported the synthesis of a series of G0, G2 and G4 PPI dendrimers conjugated with Gd3+-DTPA and found that both molecular and ionic relaxivities also increased as a function of generation number.88 Analogously, Margerum reported that the measured relaxivities of PAMAM-Am-DO3A dendrimers ranging from G2 to G5, and higher generations of PAMAM-EDA dendrimers conjugated with Gd3+-DOTA, ranging from G5 to G10 synthesized by Bryant and co-workers increased with increasing molecular weight.89,90 However, Bryant observed that molar relaxivities achieved a saturation limit beyond G7. Toth and co-workers performed a series of variable temperature and pressure 17O NMR experiments on Gd3+-DO3A labeled PAMAM-Am dendrimers of lower generation (specifically, G3 to G5), to study the effects of water exchange and rotational dynamics on the relaxivity of these agents.91 Their measurements showed that while τR increases approximately by a fourth with each increase in generation, the water exchange rate constants kex remain the same for all the systems studied, sacrificing any theoretical increase in molar relaxivity. They concluded by stating that these systems possess rotational correlation times long enough for the rate of water exchange to affect the over-all relaxivity of the dendrimer, and that further improvements would entail not just increasing molecular weight, but designing chelate systems which promote the dissociation step of water molecules bound to the paramagnetic Gd3+ ion. Furthermore, their results demonstrated that conjugation of the macrocyclic chelate to the large dendrimer did not affect the rate of water exchange at the metal center, suggesting that the kex value determined for any monomeric chelate should apply to any future dendrimeric conjugate.
To this effect, Laus and co-workers synthesized a series of higher generation (G5 to G9) PAMAM-EDA dendrimers conjugated with a novel ligand, ethylenepropylenetriamine pentaacetic acid (EPTPA).92 17O NMR experiments have shown that Gd3+-EPTPA possesses a water exchange rate ten-fold greater than that of Gd3+-DTPA.93 This is attributed to steric crowding around the Gd3+, thereby accelerating the dissociation of bound solvent molecules. The relaxivities of the systems measured increased from G5 to G7 (37 °C, 30 MHz), demonstrating the beneficial effect of using chelates with faster water exchange rates. However, the trend was found to decrease upon reaching G9. Relaxivity measurements at different pHs suggest that protonation of the tertiary amines of the dendrimer results in a more rigid and open structure, thereby improving relaxivity. Hence, it was rationalized that even with faster water exchange kinetics, the over-all relaxivity of higher generation dendrimers is affected also by internal motion. Similarly, Rudovsky and co-workers reported a series of PAMAM-EDA dendrimers ranging from G1 to G4 conjugated with a Gd3+-DO3A derivative, 1, 4, 7, 10-tetraazacyclododecane-4,7,10-triacetic-(methyl(4-aminophenylmethyl)phosphinic acid), (DO3A-PABn).94–95 1H nuclear magnetic resonance dispersion (NMRD) and VT-17O NMR measurements have shown that Gd3+-DO3A-PABn possesses an optimally short water residence time and a higher than expected relaxivity, due to steric crowding and the formation of a secondary hydration sphere by the bulky phosphinate group.96 As expected, measured relaxivities of these systems increased with generation number, and protonation of the tertiary amines of the dendrimer backbone resulted in a further increase in relaxivity. Furthermore, it was also demonstrated that formation of adducts with positively charged polycations, such as poly(Arg) and poly(Lys), increased relaxivity by reducing the internal motion in these dendrimers, which are negatively charged. The formation of adducts did not affect the water exchange rate, and relaxivities remained stable up to pH 9.5 for the poly(Lys) adduct, and pH 12 for the poly(Arg) adduct. A further report by Lebduskova and co-workers described the enhanced relaxivity of a PAMAM-EDA-G5 dendrimer conjugated with a DTPA-based chelate containing one phosphinate group, DTTAP, which also cited the benefits of faster water exchange and the role of the secondary hydration sphere.97 More recently, Ali and co-workers described a PAMAM-EDA-G5 dendrimer conjugated with a DOTA-like tetra-phosphonate ligand DOTA-4AmP sensitive to pH changes, whose relaxivity more than doubles when pH changed from pH 9 to pH 6.98 A comparison of relaxometric properties of these dendrimers is summarized below (Table 3).
Table 3.
Relaxivites of various paramagnetically-labeled PAMAM dendrimers.
| Dendrimer | Core | ionic r1 (mM−1s−1) | molec. r1 (mM−1s−1) | Field Strength | temp (°C) | pH | ref |
|---|---|---|---|---|---|---|---|
| PAMAM-G2-DTPA | Am | 21.3 ± 0.3 | 234 | 25 MHz | 20 | 7.4 | 43 |
| PAMAM-G3-DO3A | Am | 14.8 ± 0.4 | 25 MHz | 37 | 89 | ||
| PAMAM-G4-DO3A | Am | 16.9 ± 0.4 | 20 MHz | 37 | 89 | ||
| PAMAM-G5-DO3A | Am | 18.8 ± 0.2 | 25 MHz | 37 | 89 | ||
| PAMAM-G6-DTPA | Am | 34 ± 4 | 5800 | 25 MHz | 20 | 7.4 | 43 |
| PPI-G0-DTPA | DAB | 14.4 ± 0.2 | 45.6 | 1.5 T | 20 | 5.8 | 116 |
| PPI-G2-DTPA | DAB | 15.2 ± 0.2 | 243 | 1.5 T | 20 | 5.8 | 116 |
| PPI-G4-DTPA | DAB | 19.3 ± 0.2 | 1234 | 1.5 T | 20 | 5.8 | 116 |
| PAMAM-G5-DOTA | EDA | 30 | 2880 | 20 MHz | 23 | 7.4 | 90 |
| PAMAM-G7-DOTA | EDA | 35 | 13300 | 20 MHz | 23 | 7.4 | 90 |
| PAMAM-G9-DOTA | EDA | 36 | 47520 | 20 MHz | 23 | 7.4 | 90 |
| PAMAM-G10-DOTA | EDA | 36 | 66960 | 20 MHz | 23 | 7.4 | 90 |
| PAMAM-G1-DO3A-PABn | EDA | 14.8 | 20 MHz | 25 | 7.5 | 94 | |
| 10.1 | 20 MHz | 37 | 7.5 | 94 | |||
| PAMAM-G2-DO3A-PABn | EDA | 19.7 | 20 MHz | 25 | 7.5 | 94 | |
| 14.1 | 20 MHz | 37 | 7.5 | 94 | |||
| PAMAM-G4-DO3A-PABn | EDA | 25.8 | 20 MHz | 25 | 7.5 | 94 | |
| 18.6 | 20 MHz | 37 | 7.5 | 94 | |||
| PAMAM-G5-DTTAP | EDA | 26.8 | 20 MHz | 37 | 6.25 | 97 | |
| PAMAM-G5-EPTPA | EDA | 25.1 | 20 MHz | 20 | 6.0 | 92 | |
| 17.1 | 20 MHz | 37 | 6.0 | 92 | |||
| PAMAM-G7-EPTPA | EDA | 35.8 | 20 MHz | 25 | 6.0 | 92 | |
| 25.6 | 20 MHz | 37 | 6.0 | 92 | |||
| PAMAM-G9-EPTPA | EDA | 29.2 | 20 MHz | 25 | 6.0 | 92 | |
| 24.2 | 20 MHz | 37 | 6.0 | 92 |
Novel ideas include the synthesis of G0 and G2 PPI dendrimers functionalized with Yb3+-DOTAM as a pH sensitive paramagnetic chemical exchange saturation transfer (PARACEST) agent, in which the maximum effect was observed with decreasing pH from the mononuclear chelate to the G2 dendrimer.99 PARACEST agents (see Section LipoCEST) have been gaining more interest in molecular imaging since paramagnetic ions induce large shifts in the resonances of neighboring nuclei which can visualized at will by proper choice of irradiation frequency.100 The different chelates used to functionalize dendrimers are summarized below (Figure 5).
Figure 5.

BFCAs conjugated to dendrimers.
Lastly, a series of Gd3+-chelate-core branched-alcohol dendrimers was synthesized via a convergent approach.101 Uni-directional amino-substituted arborols of increasing length and branching were conjugated to a Gd3+ chelate possessing a DOTA-like ligand with peripheral carboxylate groups. Placing the Gd3+ ion at the center of a macromolecular structure was proposed to effectively couple the local motion of the Gd3+-OH2 vector with the rotation of the entire assembly, resulting in an increased relaxivity (Table 3, Figure 6).102 17O-NMR measurements indeed show that a greater length and degree of arborol branching of the complex in comparison with the parent compound correlates with a slower rotational correlation time, τR. However, the largest of these complexes, having the largest number of methyl and methylene groups, exhibited the slowest water exchange rate kex, thereby compromising any further theoretical gain in relaxivity.
Figure 6.

Gd3+ chelate at barycenter of dendrimer.
6.3 Biodistribution
6.3.1 Passive distribution
The most important property which determines the biodistribution of dendrimer-based MR agents is their size, which in turn is determined by a) the nature of the central core and interior architecture, and b) the generation number, G.
PAMAM-Am-G2-DTPA and PAMAM-Am-G6-DTPA were the first dendrimer-based MR contrast agents evaluated for use in magnetic resonance angiography (MRA).43 These agents possessed enhancement half-lives double and ten-times longer than Gd3+-DTPA, respectively, as measured in mice. An early dose-response study described the use of PAMAM-Am-G5-DO3A in visualizing vasculature in rabbits, reporting a minimum effective dose of 0.02 mmol/kg and a maximal contrast enhancement produced at a dose of 0.03 mmol/kg.103 In MRA experiments involving a series of PAMAM-Am-DO3A (G2 to G5), the blood clearance half-lives of these agents was observed to increase with increasing generation number.89 Gadomer-17 (Schering AG, Berlin, Germany), also known as Gd3+-DTPA-24-cascade polymer,104 in comparison with Gd3+-DTPA, polyLys-DTPA and Gd3+-DTPA-albumin,105–107 was shown to visualize intratumoral vasculature exhibiting high vascular permeability108 and acute myocardial ischemia,109 and that a dose of 0.033 mmol/kg of PAMAM-Am-G6-1B4M in mice was sufficient to visualize intratumoral vasculature as small as 100-μm in diameter.110 Furthermore, Gadomer-17, which is a dendrimer consisting of a trimesoyl triamide core with branched lysine amino acids104 was used to image vasculature in dogs; MRA images showed that a 0.1 mmol/kg of Gadomer-17, with 24 Gd3+-DOTA units, produced more enhanced contrast than a 0.3 mmol/kg dose of Gd3+-DTPA.111
Sato112, Kobayashi113–114, and Yordanov115 and co-workers embarked on systematic studies of the biodistribution of PAMAM-EDA-1B4M chelating Gd3+dendrimers, ranging from G3 to G10, for use in MRA studies for the visualization of both normal and tumoral vasculature. Their results show that smaller generations (G3 to G5) exhibit rapid clearance from the body and a high glomerular filtration rate, though G5 and G6 were retained long enough to visualize normal fine vasculature up to a 200 μm limit. Higher-generation dendrimers G7 to G9 were found to have less renal uptake than G6, with G8 and G9 exhibiting a much higher hepatic accumulation. Furthermore, G8 was found to visualize intratumoral vessels in a more stable manner over time than G6, due to the increased vascular permeability of fast-growing cancer cells. In summary, the authors indicate G7 as the best candidate for visualizing intratumoral vasculature since it was retained in blood circulation the longest; the low liver uptake and slow glomerular filtration may permit longer image acquisition times. The highest generation dendrimer studied, G10, was found to precipitate at physiological pH.
In a similar fashion, Langereis and co-workers studied the biodistribution of a range of lower to intermediate generation PPI-DTPA dendrimers (from core to G4).116 All the agents studied exhibited renal clearance, though higher generations prolonged blood retention. Furthermore, G2 and G4 exhibited a lesser tendency to leak from tumoral vasculature into the tumor, whereas core and G0 were found to do so rapidly. Lastly, the largest dendrimer studied, G4, was found to have a lowest detectable concentration around 80 nM, more than two orders of magnitude lower than that of Gd3+-DTPA.
Kobayashi and co-workers also embarked on systematic comparisons of biodistribution based on the nature of the PAMAM core. Results show that between PAMAM-Am-G6-1B4M and PAMAM-EDA-G6-1B4M, the latter exhibited longer blood retention and slower renal uptake, making it a better blood pool agent.117 In addition, PPI-G4-1B4M was found to exhibit a significant amount of hepatic uptake in comparison with PAMAM-EDA-G4-1B4M due to its relatively higher hydrophobicity,118 and has been demonstrated to visualize both normal liver parenchyma and micrometastatic tumors of 0.03-mm diameter in mice.119 Due to problems of prolonged retention in blood and poor clearance, a comparison study between dendrimers of different cores and sizes was performed to determine which possessed the best renal excretion properties, citing PAMAM-EDA-G2-1B4M, PPI-G3-1B4M and PPI-G2-1B4M as the best candidates for clinical studies,120 with PPI-G2-1B4M found to be the best agent for functional kidney imaging and early diagnosis of renal damage.121 Furthermore, control over circulation and excretion properties was demonstrated with conjugation of the dendrimers with polyethylene glycol (PEG),122 co-injection with lysine,123 or biotinylation of the dendrimer followed by an avidin chase.124
Higher generation dendrimers were found to be more suitable for MR lymphangiography applications. For example, PAMAM-EDA-G8-1B4M, with its large size and therefore low vascular permeation, was found to be retained inside lymphatic compartments, permitting discrimination between infection and proliferative or neoplastic swelling.114 A comparison between dendrimers of different cores showed this same dendrimer-based agent was more suitable for imaging lymphatic vessels while PPI-G5-DTPA better visualized lymph nodes.125 Lastly, advances in bioconjugation chemistry permitted the synthesis of a dendrimer-based fluorescent-MRI multi-modal probe capable of visualizing sentinel lymph nodes in mice.126–127 More recently, PAMAM-EDA-G8-1B4M was also evaluated as a CT-MR probe administered in conjunction with convection-enhanced delivery (CED) of therapy to the brain, though the effect of dendrimer size and core in this area of use has yet to be determined.128
6.3.2 Targeted agents
Several attempts have been made to improve the selectivity of dendrimer-based MR agents by synthesizing targeted bioconjugates. Wu and co-workers were amongst the first to describe the synthesis of a set of antibody-labeled (mAb 2E4) dendrimers, PAMAM-Am-G2-DOTA and PAMAM-Am-G2-CHXB, which were efficiently labeled with 90Y, 111In, 212Bi, and Gd3+, without loss of immunoreactivity, as potential tools for either directed radiotherapy or MR imaging.129 Kobayashi and co-workers also demonstrated that conjugation of PAMAM-EDA-G4-1B4M with OST7, a murine monoclonal IgG1, did not compromise immunoreactivity. Furthermore, in addition to specific accumulation in tumor sites, the antibody-dendrimer construct had better blood clearance behavior than the simple 1B4M-labeled antibody.130 PAMAM-Am-G4-DTPA conjugated with folic acid has been successfully shown to selectively label ovarian cancer tumors over-expressing the high-affinity folate receptor (hFR).131–134 PAMAM-EDA-G3 was consecutively conjugated with cyclic-RGD, a fluorescent dye, and Gd3+-1B4M to selectively visualize integrin αVβ3, a marker for angiogenesis.135 Though in vitro results were initially promising, the approach met with limited success in vivo.
6.3.3 Cell transfection
Lastly, several attempts have been made to develop strategies for the intracellular delivery of contrast agents. Solution studies of dendrimer- and non-dendrimer-based contrast agents in combination with commercially available cell transfection agents found that adduct formation reduced the relaxivity of the Gd3+-based agents (by blocking water coordination sites), but that adduct dissociation was a function of pH, suggesting a further capability of these systems as a pH switch.136 Successful cell delivery was reported by Kobayashi and co-workers using a bioconjugate construct composed of PAMAM-EDA-G6 labeled with biotin, Gd3+-1B4M, and lastly avidin, which was found to accumulate in SHIN3 tumor cells (human ovarian cancer) 50 times greater than mononuclear Gd3+-DTPA.137 Zhu and co-workers employed a three-step pre-targeting approach to visualize Her-2/neu xenografts in mice: biotinylated trastuzumab was first administered to label the tumors, followed by an avidin chase, and lastly a biotinylated PAMAM-G4-DTPA dendrimer. Though only limited selective MR enhancement was observed in the tumor xenografts, the bioconjugate construct was retained in tumors due to the EPR effect.138 Also, Xu and co-workers described the use of a cysteamine-core dendrimer to produce a multi-modal dendrimer-based agent, employing rather clever chemistry. PAMAM-CYS-G2 was first conjugated with 1B4M-DTPA[Gd3+], after which the disulfide core of the dendrimer was cleaved to allow for site-specific conjugation with biotin. Up to 4 of these bioconjugate constructs formed an adduct with fluorescently-labeled avidin, and multi-modal imaging techniques confirmed the accumulation of this supramolecular construct in mice bearing ovarian cancer tumors.139
7. Linear Polymers in MR imaging
Synthetic linear polymers have also been studied and tested as potential core platforms for creating macromolecular MR contrast media, citing characteristic advantages similar to those of dendrimers, namely, that polymer chemistry is certainly established enough to exercise control over polydispersity and molecular weight, a wide enough variety of monomers exist to produce polymers of minimal or no immunogenicity, and that polymers can be made to respond to environmental changes which are diagnostic of physiological phenomena.
7.1 Poly-L-lysine
By far the most studied linear polymer in MR imaging is poly-L-lysine. Poly-L-lysine is commercially available in a wide variety of molecular weights, and conjugation with DTPA takes place on the ε-amino group of lysine. Two labeling methods have been described, using either DTPA dianhydride or DTPA-OSu ester, the latter method displaying better conjugation efficiencies up to 100% on poly-L-lysine (38.5 kDa).140 DOTA has also been conjugated to polylysine via a mixed anhydride method.141 Complexation with Gd3+ resulted in polymers possessing a longitudinal relaxivity r1 three times greater than that of the monomeric chelate142 independent of polymer chain length.140 Pharmacokinetic studies have shown it to be well-tolerated in vivo, as reflected by a high LD50, and that clearance occurs primarily through the kidney, requiring at least a day to clear completely in rat and rabbit models.142–143 However, it was also shown that Gd3+-DTPA-polylysine formulations of higher molecular weight clear slower from the blood in comparison with smaller polymers, resulting in prolonged and constant tissue enhancement over a 1 hour period.144
These positive characteristics have since led to a series of in vivo studies employing Gd3+-DTPA-polylysine. As a possible blood pool agent, it was tested in MRA in rabbits to monitor blood flow in the extremities,145 and to distinguish normal myocardium from peripheral ischemic zones in cats.146 Gd3+-DTPA-polylysine has also been conjugated to human serum albumin to improve its blood pool behavior, based on other efforts to develop Gd3+-labeled albumin as an MR contrast agent (section 4.1).141 Gd3+-DTPA-polylysine has also been used to visualize pulmonary disease states exhibiting abnormal blood flow,147–149 and has also been shown to accumulate in tumours resulting in tumour tissue enhancement lasting for several days in a rat model.150
7.2 Polyethyleneglycol (PEG)
Polyethyleneglycol (PEG) was a most likely candidate for use as a platform for macromolecular MR contrast agents since it has for many years been used to covalently modify biomolecules and small-molecule drugs in order to prevent their recognition by the immune system and facilitate solubility and clearance, as reviewed elsewhere.151 For example, PEGylated bovine serum albumin was observed to possess virtually no immunogenicity when injected into rabbits, thereby prolonging its blood circulation time.152 17O NMR studies of Gd3+-DTPA-labeled PEG showed that the water exchange rate kex between bulk and bound water molecules on the paramagnetic center is identical between the polymer and the monomeric chelate, indicating a large degree of flexibility in the polymer chain.153 Functionalized PEG for conjugation chemistry is also available in a wide range of molecular weights, and Gd3+-DTPA-PEG of molecular weights greater than 20 kD have been shown to exhibit good blood pool enhancement dynamics while smaller conjugates demonstrate faster tumour enhancement in rabbits.154
Gd3+-labeled conjugates based on combinations of both polylysine and PEG have also been reported in the literature (Figure 7), as a strategy to improve solubility in blood and reduce the immunogenicity of polylysine. A prototype was reported by Bogdanov and co-workers, which exhibited a blood half-life of 14 hours and constant vascular enhancement for two hours.155 This concept was developed more thoroughly by Fu and co-workers who described the synthesis and characterization of a series of Gd3+-labeled polylysine dendrimers of different generations linked by PEG cores of varying length.156 These compounds exhibited good water solubility, good stability in both buffer and plasma, narrow size dispersity, and longitudinal relaxivities approximately three times that of the monomeric chelate. These polylysine-based agents have recently been used as contrast agents in MRI to visualize and distinguish cancerous from normal soft tissue in rat models.157
Figure 7.

DTPA- and DOTA-conjugated polylysine (PL), polyethylene glycol (PEG), and mixed PL-PEG species.
7.3 Other Linear Polymers
The wide variety of monomers and resulting polymers either commercially available or easily synthesized permits the evaluation of many other possible polymer-based macromolecular contrast agents. Cavagna and co-workers reported that the synthetic polypeptide polyaspartate containing ~220 monomers was capable of chelating as many as 40 mol Gd3+ per mole polyaspartate, though no comment was made about the stability of the resulting polychelate.158 Allen and co-workers described the use of ring-opening metathesis polymerization (ROMP) to produce a polymer incorporating the ligand hydroxypyridonate (HOPO) in its backbone, capable of chelating Gd3+ with high stability.159 Indeed, DTPA dianhydride itself has been used in copolymerization with different kinds of α,ω-diamines, to form polymers with Gd3+ chelating units along the polymer backbone (Figure 8). For example, DTPA has been co-polymerized with tartaric acid to produce a polymer with increased hydrophilicity and reduced toxicity.160 In contrast, DTPA has been co-polymerized with alkyldiamines of different alkyl chain lengths, resulting in macromolecular structures exhibiting relaxivities similar to those of dendrimers.161 It was hypothesized that intramolecular hydrophobic interactions between the alkyl chains resulted in the formation of rigid structures; indeed, variable-temperature, multiple-field 17O NMR and electron paramagnetic resonance studies have shown that the relaxivity behavior of these polymers is more characteristic of rigid globular micellar structures rather than of a linear system.162 Another report described the synthesis of polysuccinimide derivates containing PEG, as a hydrophilic component, and hexadecylamine, as a hydrophobic component, co-polymerized with DTPA, towards the development of biocompatible micellar MR agents with improved in vivo stability.163 Ladd and co-workers also reported a systematic study of DTPA copolymers which relate molecular weight, polymer rigidity, metal content, viscosity and chelate stability in the design of polymer-based blood pool agents.164
Figure 8.

Examples of DTPA-copolymers of α,ω–diamines.
7.4 Targeted and Functional Polymers
Furthermore, by careful selection of co-polymer, the nature of the polymer bond, or even the metal chelate itself, polymer macromolecular contrast agents can either be designed with an intrinsic controlled biodistribution or to reflect particular physiological phenomena. For example, DTPA and sulfadiazine were incorporated into polyaspartamide, and then labeled with Gd3+ to produce a tumor specific polymer contrast agent, exhibiting preferential uptake in, and significant MR enhancement of hepatoma in a mouse model.165 Similarly, N-(2-hydroxypropyl)methacrylamide was co-polymerized with mannosamine and then labeled with Gd3+-DOTA to produce a contrast agent specific for mannose receptors over-expressed in activated macrophages.166 More recently, polydiamidopropanoyl dendrimer was labeled with multiple Gd3+ chelates and then conjugated with a peptide nucleic acid as a MR hybridization probe capable of hybridizing with specific mRNA.167
Bogdanov and co-workers described the novel strategy called MR signal amplification, or MRamp, which is based on enzyme-mediated polymerization of a paramagnetic monomer into oligomers exhibiting high relaxivity (Figure 9).168 This strategy was demonstrated by labeling E-selectin expressed on endothelial cells with an anti-E-selectin antibody conjugated with peroxidase. Subsequent administration of phenol-functionalized Gd3+-chelates resulted in the formation of polymetallic oligomeric species of high molecular weight and increased relaxivity. While the method was sensitive enough to detect nanomolar amounts of peroxidase, clearance of the resulting oligomers was not discussed. A reverse strategy was reported by Lu and coworkers in which Gd3+-labeled polyglutamic acid with a biodegradable disulfide spacer is broken down in the presence of endogenous blood plasma thiols, to facilitate clearance from the blood via renal filtration (Figure 10).169 Similarly, Wen and co-workers reported a polyglutamic acid based MR contrast agent which degrades in the presence of cathepsin B, a lysosomal enzyme.170 Gd3+-labeled polyglutamic acid has been tested for visualization of human breast cancer xenografts in mice, with the larger molecular weight polymer construct exhibiting better accumulation in tumour.171 Mohs and co-workers also reported Gd3+-labeled PEG-L-cystine copolymers which are also broken down in the presence of endogenous thiols,172 and that variations in PEG length had little effect on the relaxivity of the polymer.173
Figure 9.

MRamp polymerization of paramagentic chelates.
Figure 10.

Paramagnetic polymer with biodegradable spacer.
A pH sensitive polymer MR contrast agent was also reported by Mikawa and co-workers174, composed of Gd3+-DTPA conjugated to a polycation, poly[2-(diethylamino)ethyl methacrylate], which exhibited an increase in relaxivity when the pH is decreased from 7.2 to 5. Pathological states present different microenvironments in comparison with normal states, such as lowered pH in a lesion, and therefore a pH sensitive MR contrast agent would be useful in detecting these physiological states in a non-invasive manner. Lastly, a Eu3+-labeled small polymeric CEST agent has been described in the literature, which makes use of the paramagnetic chemical exchange saturation transfer (CEST) mechanism conferred by the presence of the lanthanide. Though small in size, the authors suggest that the CEST effect will permit detection of the agent even at low concentrations; furthermore, the small size of the polymer will facilitate its clearance via the kidney.175
8. Protein-based MR agents
8.1 Albumin Covalently-Bound to Gd3+-DTPA
Paramagnetically-labeled albumin has received significant attention over the last few years and much has been done towards its development as an intravascular probe. Initial biodistribution experiments involving the monomeric chelate Gd3+-DTPA showed that after 5 minutes post-injection, as much as 80% of administered contrast agent had been cleared from intravascular space, an effect directly related to low molecular weight.176 In a comparison study, the enhancement due to paramagnetically-labeled albumin persists for an hour, while that of Gd3+-DTPA completely disappears within that same time frame.177 As stated earlier, longer blood retention times are desirable as they permit both sufficient accumulation of contrast agent in sites of interest and longer acquisition times.
After Lauffer and co-workers reported a protocol for the direct reaction of DTPA-dianhydride with a variety of proteins and subsequent labeling with Gd3+,178 the method was soon applied to human serum albumin and procedures were determined to control the number of paramagnetic chelates, ranging from nine to 19 chelates per albumin.179–180 Solution studies showed that albumin-(Gd3+-DTPA)19 possesses a longitudinal relaxivity r1 of 14.8 mM−1 s−1, a value three times that of the monomeric chelate when measured under the same conditions, a result of the larger molecular weight of the contrast agent and hence its higher rotational correlation time τR.180 However, a study by Paajanen and co-workers in which Gd3+-labeled albumin was compared with larger molecular weight conjugates Gd3+-labeled IgG and fibrinogen found that not only was a wide range of chelate numbers possible for all proteins studied, but that the measured relaxivities for all three were relatively the same.181 Furthermore, since conjugation of the protein with DTPA via this method requires amide bond formation with one of the acetates of DTPA, Sherry and co-workers raised a caveat early on citing thermodynamic measurements which indicate a compromise of chelate stability.182
In addition to long blood retention times, initial biodistribution studies of Gd3+-labeled albumin reported enhancement intensity increases over 100% as observed in myocardium and liver with albumin-Gd3+-DTPA at concentrations one third that of Gd3+-DTPA, which produced enhancement increases much less than 100%.177 These results lead to a series of in vivo tests to evaluate the performance of paramagnetically-labeled albumin as a contrast agent in tissue exhibiting a high degree of vascularization. In addition to using albumin-Gd3+-DTPA to determine blood plasma volume by MRI techniques,183 it was also used to measure capillary permeability by monitoring the leakage rate of contrast agent from plasma to interstitial water or tissue plasma under normal conditions184 or when pharmacologically-induced.185 A similar concept was employed in measuring CO2-induced changes in cerebral blood volume186 and monitoring inflammation in arthritis.187 Disease states characterized by regions of reduced blood pool, such as ischemia of the kidney188 and myocardium,189–192 have also been visualized, as well as their reperfusion.
Albumin-Gd3+-DTPA has also been used to in contrast-enhanced imaging of cancerous tissue, having a different histological profile from normal tissue and abnormal capillary permeability.193 Indeed Daldrup and co-workers performed a series of imaging studies to correlate histologic tumor grade, ranging from benign to highly malignant, with MR enhancement. Their results show that correlation was possible only when albumin-Gd3+-DTPA was used, in comparison with the monomeric chelate which fails to distinguish between tumor grades.194 Similar techniques were used to determine histologic tumor grade in prostate195 and breast196–197 cancer models. Furthermore, albumin-Gd3+-DTPA was also demonstrated to be an effective probe for measuring increases in capillary density, thereby suggesting its use in estimating angiogenic activity.198 Monitoring changes in tumor capillary permeability under pharmacological stress199 or irradiation200 have also been reported, as well as the use of albumin-Gd3+-DTPA as a surrogate imaging tracer for convection-enhanced delivery of tumor-targeted toxins into rat brain.201
8.2 MS-325
In spite of its initial success, covalently-labeled albumin suffers from several undesirable traits. Elimination of the agent is slow, incomplete, and has been shown to remain in circulation for more than a week, eventually accumulating in liver and bone. In addition, albumin is also potentially immunogenic, and the combined risk of poor elimination and in vivo degradation have confined its use as a model prototype MR contrast agent in animal studies.202
MS-325 is a blood pool contrast agent which reversibly binds to albumin in a non-covalent fashion. The monomeric chelate is composed of C-functionalized Gd3+-DTPA derivative conjugated to a cyclohexyl diphenyl group via a phosphodiester linkage. Its solution properties and structure have been studied extensively,203 demonstrating superior stability in comparison with Gd3+-DTPA at physiological pH.204 Binding with human serum albumin is close to 100% with a constant of about 6100 ± 2130 M−1,205 and upon binding with HSA this agent exhibits a six- to ten-fold increase in relaxivity due to a large increase in rotational correlation time,206–207 although the relaxivity enhancement has recently been found to be dependent on the species of albumin used.208 This phenomenon has since been referred to as receptor-induced magnetization enhancement (RIME). While the hydrophobic group provides its affinity for albumin, the phosphodiester linkage is essential for preventing its accumulation in liver and increasing its plasma half-life to 155 min (versus 36 min for monomeric Gd3+-DTPA).209 Furthermore, biodistribution studies in cynomologous monkeys have demonstrated its efficient clearance via the renal pathway with up to 90% of administered dose eliminated after 24 hours post-injection and complete elimination by 72 hours.210 However, a study by Corot and co-workers compared the bolus and steady state phases of MS-325 with two other contrast agents, namely, an ultrasmall superparamagnetic iron oxide and P792 (a macromolecular derivative of Gd3+-DOTA). They report that in the bolus phase, tissue distribution of MS-325 is characteristic of a monomeric chelate, which extravagates into the surrounding tissue and is cleared by the kidney. The high concentrations of contrast agent in the bolus exceed that of available albumin. But after a minute post-injection, the steady-phase is achieved and 75% of MS-325 exists in the albumin bound form.211
MS-325 advanced into clinical studies. After preliminary concentration studies in comparison with monomeric chelates and iron particles,212 MS-325 was first evaluated as a contrast agent for the imaging of peripheral and carotid vasculature in humans, and to establish patient tolerance. The study reported that the long-circulation time of the agent permitted the imaging of different zones of interest. The dose required to produce an enhanced MR image was less than half that required to produce the same quality of image using simple monomeric Gd3+-DTPA. In addition, vessels as small as 1 mm in diameter were visualized.213 The success of this first study led to its clinical evaluation as a contrast agent in carotid imaging214 and the diagnosis aortoiliac occlusive disease,215–217 and was reported as safe and effective in these applications.
The use of MS-325 has also been evaluated in the detection of proteinuria in rat kidney,218 and in the determination of capillary permeability in rat breast tumor.219
8.3 Other Albumin-Affinity Agents
The success of MS-325 has prompted further investigation into developing other lipophilic RIME Gd3+ chelates (Figure 11). These include the use of the acyclic ligand 4-carboxy-5,8,11-tris(carboxymethyl)-1-phenyl-2-oxa-5,8,11-triazatridecan-13-oic acid (BOPTA),220–222 and the macrocyclic and acyclic analogues of benzyloxymethyl substituted DOTA/DTPA (DOTA/DTPA-BOM).223–224 The stability constants of Gd3+-chelates based on these ligands show that the presence of the aromatic group does little to affect the stability of the chelate. Furthermore, increasing the number of these substituents also increases affinity to human serum albumin, with the cyclic Gd3+-BOM chelate of three substituents exhibiting a relaxivity of 53.2 ± 0.7 mM−1s−1. However, the theoretical maximum of enhancement is not achieved due to a reduction in the exchange rate of the coordinated water molecule upon adduct formation with HSA. Another Gd3+ chelate employing the macrocyclic ligand 3,6,10,16-tetraazabicyclo[10.3.1]hexadecane-3,6,10-tris(methane-phosphonic) acid (PCTP-[13]) has an aromatic group as part of the carbon backbone of the ligand, providing the chelate with sufficient lipophilic character for HSA binding.225 Furthermore, enhancement is due not only to adduct formation, but also by the formation of a secondary hydration sphere around the chelate, caused by the methylene phosphonate arms of the ligand, resulting in outer-sphere relaxation effects. In other examples, PEG was introduced as a spacer between the chelate and the aromatic group, in order to increase the solubility of the complexes and exploit the beneficial effects of a large molecular weight. Though these demonstrated strong binding with HSA, the expected enhancement was not achieved,226 perhaps due to the flexibility of the PEG linker, and the displacement of water coordinated to the paramagnetic center upon protein adduct formation. The synthesis of a Gd3+-chelate based on a DTPA derivative with no aromatic substituents, 4-pentylbicyclo[2.2.2]octane-1-carboxyl-di-aspartyl-lysine-derived-DTPA (MP-2269), has also been described.227 Preliminary animal studies have demonstrated its use in the visualization of vasculature, and a 70% clearance after 24 hours via the hepatobiliary pathway. In contrast with the PEGylated complexes described earlier, 17O NMR studies of this chelate show that neither the presence of the hydrophobic chain nor adduct formation with albumin significantly affect the water exchange rate between bound and bulk solvent molecules.228 Acyclic ligands based on 3,6,10-tri(carboxymethyl)-3,6,10-triazadodecanedioic acid (TTDA) and its derivatives have also been described in which the aromatic groups are covalently linked via amide bonds.229 In addition to possessing affinity to HSA, Gd3+ chelates based on these ligands also exhibit decreased water exchange rates, a function of reduced steric crowding around the coordination site, and reduced charge effect since the chelates are neutral. The Gd3+ complex of (4S)-4-(4-ethoxybenzyl)-3,6,9-tris(carboxylatomethyl)-3,6,9-triazaundecandioic acid (EOB), constituted of a DTPA derivative covalently linked with a lipophilic ethoxybenzyl moiety, was initially developed as a contrast agent for hepatobiliary imaging.230–232 However, comprehensive MR studies have shown it also possess affinity for serum proteins,232 with the S isomer possessing a higher affinity for HSA than the R isomer.233 Also, multi-metallic complexes with affinity for albumin have been reported234–235 which deliver a higher payload of paramagnetic ions per protein molecule, and thereby reducing the minimal dose required for the observation of significant enhancement. Finally, enzyme-activatable pro-RIME agents have also been described in the literature. Nivorozhkin and co-workers report the use of a DTPA derivative functionalized with aromatic moieties for HSA binding, but which are masked by lysine residues which inhibit protein binding.236 Upon exposure to the enzyme thrombin-activatable fibrinolysis inhibitor (TAFI), the lysine residues are cleaved, resulting in a 100% increase in relaxivity at 37°C in the presence of HSA. The authors propose this as a strategy to detect disease states associated with certain protease activities. More recently, Hanaoka and co-workers reported a similar Gd3+-DTPA-based reporter agent employing galactopyranose as a masking group.237 Exposure to β-galactosidase, commonly used to monitor gene expression, cleaves the masking group, resulting in a 57% increase in relaxivity in the presence of HSA.
Figure 11.

Ligands used for albumin-affinity MR agents.
8.4 Other Protein-Binding Agents
Indeed there are many examples in the literature of Gd3+-based MR contrast agents which form adducts with proteins, and which have been shown to visualize specific tissues of interest or dynamic physiological phenomena. Anelli and co-workers described the synthesis of a Gd3+-DTPA derivative covalently-linked to sulfonamide and possessing a strong affinity for carbonic anhydrase (KA of 15,000 ± 5,000 M−1); the resulting adduct measured to have a relaxivity of 25.8 mM−1s−1.238 Since carbonic anhydrase is ubiquitously expressed in various tissues, the authors proposed use of this agent to visualize compartments outside the blood pool. Similarly, Tomaselli and co-workers described a Gd3+-DTPA derivative covalently-linked to cholanoic acid, and its adduct with liver bile acid binding protein characterized by multi-dimensional NMR techniques.239 Work by De Leon-Rodriguez and co-workers described a Gd3+-DOTA derivative conjugated with a 20 amino acid peptide sequence that binds to the yeast transcription repressor protein Gal80 with high affinity (KA = 5 × 105 M−1) and specificity, resulting in a 10-fold increase in image enhancement.240 Other peptide-functionalized Gd3+contrast agents, EP-1873 (a DTPA derivative)241 and EP-2104R (a DOTA derivative),242 were designed to bind strongly to fibrin, a protein abundant in arterial thrombi and associated with a variety of pulmonary disease states. EP-1873 has been shown to visualize thrombus formation in rabbits, and EP-2104R to possess affinity over wide range of fibrins and excellent specificity over fibrinogen and serum albumin. An area with much potential in its own right, the synthesis and applications of DOTA-peptide conjugates has recently been reviewed elsewhere.243
Larger constructs involve labeling of proteins with paramagnetic reporters. Aime and co-workers described the encapsulation of about 10 neutral Gd3+ complexes within an apoferritin cavity. As with similarly constructed ensomes, the assembly was measured to have a much higher relaxivity in comparison with the free monomeric chelate; in this particular example, the value increased as much as 20-fold.244 Molecular biological techniques have also been used to rationally design a multivalent protein containing evenly spaced lysine residues along the protein backbone, and capable of conjugating an average of 8 to 9 Gd3+-chelates.245 Similar techniques were employed in the creation of a series proteins designed with Gd3+ binding sites using amino acid residues and water molecules as coordinating ligands, and which exhibit a 20-fold increase in relaxivity in comparison with small-molecule contrast agents.246
More sophisticated strategies take advantage of known strong protein-protein interactions to visualize physiological phenomena. For example, Gustafsson and co-workers describe the conjugation of bovine serum albumin modified with maleic acid (mal-BSA) with as many as 22 Gd3+-DOTA chelates via a thioether linkage, which forms an adduct with scavenger receptor class A (SR-A) protein, present in high numbers on macrophages and therefore a convenient diagnostic marker for vascular lesion formation.247 Langereis,248 Dirksen249 and co-workers have developed a target-specific multivalent contrast agent based on the strong interaction between biotin and avidin. The authors conjugated a cyclic NGR peptide sequence, a specific ligand for aminopeptidase CD 13 over-expressed by angiogenic endothelial cells, with Gd3+-DTPA and biotin, which then forms an adduct with avidin in a 4:1 fashion. Again, adduct formation was accompanied by a dramatic increase in relaxivity. In a similar fashion, Jung,250 Neves251 and co-workers employed this strategy in the biotinylation of the C2A domain of the protein synaptotagmin I, known to bind to phosphatidyl serine expressed on the surface of apoptotic cells, followed by an administration of Gd3+-labeled avidin. This supramolecular aggregate has since been used to successfully visualize by MR the dynamic apoptotic response of tumors in mice when treated with etoposide.252
8.5 Antibody-Based Agents
Efforts in producing antibody-based MR agents have been met with only modest success. Employing facile antibody-labeling methods first described by Hnatowich towards the development of radiolabeled probes,253 Paik and co-workers reported a study which investigated the factors influencing DTPA conjugation of monoclonal antibodies with DTPA-dianhydride. Labeling experiments with 111In3+ demonstrated that increasing the number of metal ions on the antibody results in a loss of immunoreactivity.254 However, bovine IgG labeled with an average of four Gd3+ ions was measured to have a longitudinal relaxivity r1 of 26 mM−1 s−1, roughly six times that of monomeric Gd3+-DTPA.178 Unfortunately, imaging of animals injected with Gd3+-labeled monoclonal antibodies resulted in virtually none or only modest enhancement of tumor sites.255–256 Indeed, this result suggests that the potential of labeled monoclonal antibodies in tumor imaging lies in γ-camera and PET imaging, rather than in MRI, which has relatively low sensitivity. Local tissue concentrations greater than 5 × 10−7 M Gd3+ would have to be achieved for significant enhancement to occur,256 implying the need either for the use of higher field strengths in MR imaging, or higher Gd3+ loading of antibodies without compromising immunoreactivity. In contrast, melanoma257 and human rectal carcinoma258 were successfully visualized with antibodies directly labeled with Gd3+-DTPA, suggesting that the detrimental effects of direct conjugation of DTPA onto an antibody apply on a case-to-case basis.
Interestingly, Curtet and co-workers reported the labeling of monoclonal antibody 19–9, specific for human gastrointestinal cancer, with as many as 16 to 25 Gd3+ ions with only a slight loss of immunoreactivity,259–260 confirming that different antibodies react differently when conjugated with DTPA. Though this agent demonstrated good visualization of tumor, it unfortunately suffered from poor clearance characteristics and was shown to accumulate in liver at high levels even at five days post-injection.
Even higher numbers of Gd3+-loading were achieved with the synthesis of Gd3+-labeled poly-L-lysine-antibody conjugates. Work by Shreve described a conjugate of this type incorporating as many as 30 Gd3+ chelates. Unfortunately, not only did this construct present only a modest improvement in relaxivity (5.6 mM−1 s−1), indicating a high degree of rotational movement in the polymer, it was found to have an immunoreactivity reduced by as much as 70%.261 Manabe and co-workers achieved a poly-L-lysine-IgG1 conjugate containing up to 42.5 mol DTPA per mol antibody, with only a 10% loss of immunoreactivity.262 Göhr-Rosenthal and co-workers reported even higher numbers of Gd3+ in a poly-L-lysine-mAb (RA96) conjugate incorporating an average of 65 Gd3+ chelates, exhibiting a relaxivity of 15.86 mM−1 s−1, but with a 30% loss of immunoreactivity. Though MR enhancement of tumor was achieved, this bioconjugate also suffered from poor clearance characteristics and was found to accumulate not only in tumor, but also in the liver, spleen, kidney, and bone.263 To further circumvent the undesirable effects produced by direct labeling of antibodies, Artemov and co-workers reported the use of a pre-targeting strategy involving the primary administration of biotinylated anti-HER-2/neu antibody followed by the administration of Gd3+-labeled avidin.264 In vivo studies in mice demonstrated selective enhancement of breast cancer tumors for those samples pretreated with biotinylated antibody, and a complete clearance of contrast after 48 hours. Furthermore, the relatively small size of these modular components permitted extravasation into the interstitia of the tumors, as confirmed by an analogous fluorescence-based experiment.
9. Carbohydrate-Based MR contrast agents
The potential of dextran as a platform for MR contrast agents was suggested by its use as plasma volume expander for over 50 years, an indication of its low toxicity. Similar to PEG, conjugation of antibodies with low molecular weight dextran has been shown to reduce the protein's natural immunogenicity in addition to prolonging blood circulation.265–266 Indeed, many dextran-based therapeutics exist in the literature, and have been reviewed elsewhere.267 Furthermore, the metabolic function of sugars and their roles in cell-signaling have also suggested their use as targeted drug delivery agents.
9.1 Dextran, Starch and Inulin
9.1.1 Synthesis and Solution Properties
Initial attempts to conjugate dextran with DTPA involved reaction of DTPA dianhydride with hydroxyl groups along the sugar polymer backbone to form ester linkages. However, a study by Gibby and co-workers in which a series of dextrans of increasing molecular weights ranging from 17 kDa to 150 kDa were evaluated found this method resulted in cross-linking producing polydisperse products.268 Similar chemistry could be applied to carboxymethyl dextran, inulin and hydropropyl starch.269–270 The ester bond was found to be relatively robust against hydrolysis, possessing a half-life of 21 hours (37 °C, pH 7.4),269 although concerns were raised as to the stability of the resulting Gd3+ chelates since in the cross-linked polymer, carboxylates of the DTPA moiety were used for ester bond formation. To circumvent this, strategies were employed which involved conjugating the carbohydrate backbone with DTPA through a diamine linker which serves as a “tether” for amide, thioester or thiourea bond formation,270–273 and to reduce the degree of cross-linking (Figure 12). The relaxivities of the resulting Gd3+-DTPA-labeled dextrans were invariably found to be more than twice that of the monomeric chelate, though it was also found that differences in length of the “tether” or increasing the molecular weight of the dextran had no bearing on the relaxivity of the conjugate.274–276 It was suggested that any theoretical increase in relaxivity as a result of an increase in molecular weight is off-set by the rapid internal motion of the glucose units of the polymer.274 On the other hand, Gd3+-DO3A-labeled hydroxyethyl starch and inulin were found to have relaxivities greater than four and five times that of monomeric Gd3+-DTPA, respectively.277–279
Figure 12.

(A) Cross-linked and (B) amide-linked DTPA-dextran.
9.1.2 Biodistribution
In a study by Gibby and co-workers involving cross-linked Gd3+-labeled dextrans of different molecular weights, bioconjugates larger than 100 kDa were found to exhibit enhancement of the intravascular space and kidney, and only moderate enhancement of the liver in rats. Furthermore, the polymer was found to accumulate in the bladder within 70 minutes, where it was metabolized and excreted completely with urine within 24 hours.280 A 75 kDa Gd3+-DTPA-dextran conjugate bearing 15 Gd3+ atoms was then tested as an intravascular MR contrast agent in rats, and the agent similarly found to enhance imaging of vasculature in rats for a period of one hour post-injection.281 However, when tested for imaging acute myocardial infarction in pigs, the same agent failed to visualize the areas of infarction in vivo; discrimination between healthy and diseased tissue was possible only in ex vivo MR exams.282 More recently, a 165 kDa dextran conjugate labeled with as many as 187 Gd3+-DTPA chelates was synthesized and evaluated as a blood pool agent and used for tumor delineation in rabbits.283
Gd3+-labeled carboxymethyl dextran (CMD) has also been tested in vivo. A 40 kDa CMD was found to enhance vasculature in a stable and prolonged manner in rabbit models, including the distal part of the aorta and renal arteries.274,284 Similarly, a 52 kDa CMD-Gd3+-DOTA conjugate was demonstrated to be an effective agent for visualizing myocardial perfusion, the aortic arch and abdominal vasculature of pigs.285–286 Gd3+-labeled carboxymethyl hydroxyethyl starch, bearing 35 Gd3+-DO3A units (72 kDa) was also employed as an MRA contrast agent in rats, capable of visualizing leaky vasculature of tumors.277
Furthermore, Gd3+-labeled dextran has found a unique function in embryology, as a contrast agent for MR microscopy. Indeed, a developing frog embryo was injected with Gd3+-dextran at the 16-cell stage, and cell movements during the gastrulation and neurulation stages were visualized over a period of several days.287 This idea was extended by co-labeling dextran with both Gd3+-DTPA and rhodamine, and cell movements were correlated by both MR and fluorescence microscopy.288 This same multi-modal imaging agent was also used to successfully track the movements and distribution of neural stem cell transplants in rats.289
9.2 Other Carbohydrate-Based Agents
Other carbohydrate-based agents have been reported which make use of the inherent chemical and biochemical properties of glycoconjugates in general, employing novel and clever synthetic strategies, which may prove useful in an MRI setting. Convergent synthetic strategies have been employed to produce dendritic MR contrast agents by conjugating branched carbohydrate branches onto a paramagnetic core. For example, Takahashi and co-workers produced branched amino glycoside wedges of four and twelve glucose moieties which were reacted with DTPA dianhydride to form a glycodendrimer.290 Similarly, Fulton and co-workers reported C-4 symmetric glycoconjugates in which four branched carbohydrate wedges, with twelve terminal glucose or galactose resides, were grafted onto a DOTA core (Figure 13).291 As in the case with similarly constructed paramagnetic arborols,101 the resulting Gd3+-chelate exhibited a high relaxivity which was a result not only of secondary hydration sphere effect, but also of enhanced motional coupling due to the Gd3+ ion residing at the barycenter of the macromolecular structure.
Figure 13.

Glycodendrimers.
Gd3+-labeled oligosaccharides have also been reported which show some promise in MRI applications. NMS60 is an agent composed of chitotriitol conjugated to three Gd3+-DTPA moieties. The synthesis and characterization and NMS60 and analogues have been described in detail.292 Though small by macromolecular standards (2.1 kDa), NMS60 is still large enough to avoid fast diffusion from vascular to interstitial space and be useful as a blood pool agent; it has been used to visualize arterial vasculature in a canine model293 and to delineate tumors implanted in rabbits294 even an hour post-injection.
Finally, targeted carbohydrate-based MR contrast agents have been synthesized and characterized. These take advantage of the strong carbohydrate-protein interactions which occur in vivo and serve as markers for either particular tissues or specific physiological phenomena. For example, a bioconjugate composed of a polylysine backbone co-functionalized with Gd3+-DTPA and galactose residues has been demonstrated as a multivalent probe with strong affinity for the lectin asialoglycoprotein receptor (ASGPR) expressed specifically on liver hepatocytes.295 In tests with rats implanted with liver-implanted mammary adenocarcinoma, tumor regions were distinguished from healthy tissue. Similar multivalent glycoconjugates specific for ASPGR were synthesized and the structure of the resulting lanthanide chelates, based on either macrocyclic DOTA296 or acyclic DTPA297 ligands, were described in detail. Furthermore, the macrocyclic analogue has been shown to accumulate in rat liver implanted with human hepatocyte carcinoma cell line Hep G2, and that ASPGR is highly selective for the galactosyl analogue over the glycosyl analogue.298
10. Liposomes and Micelles in MR imaging
10.1 Supramolecular Assembly and Solution Properties
Liposomes have long been used in drug delivery applications, and the idea of using these structures to deliver MR contrast media was not long in coming. The ease of preparation, use of simple reagents, the ability to control size of these supramolecular assemblies and their membrane permeabilities, all contributed in producing a vast wealth of literature which explores not only the intrinsic properties of these systems, but which also introduce novel functions such as sensitivity to pH, temperature, and the design of targeted agents. Over the last twenty years, several reviews have been published which provide an account of the development of liposomes in MR imaging.299–304 In-depth calculations performed very early on provide a theoretical understanding of their behavior, verifying both established experiments and prompting the investigation of improved systems.305–306 To date, the examples which exist in the literature can roughly be classified into the following groups: ensomes, memsomes, micelles and, more recently, lipoCEST agents (Figure 14).
Figure 14.

Schematic representations of (A) ensome, (B) memsome, (C) micelle, (D) combined ensome-memsome, and (E) shrunken lipoCEST agent.
10.1.1 Ensomes
Initial attempts at producing liposome-based MR contrast agents involved the encapsulation of water-soluble paramagnetic species within the aqueous interior of the liposome; the resulting systems were hence referred to as “ensomes”. Encapsulated species included MnCl2307 or Mn2+ complexed with either DTPA308 or albumin,309 and small-molecule Gd3+ complexes.310–312 Although these agents shortened the T1 relaxation times of bulk water solvent, it was observed that the relaxivities of these liposomes were greatly reduced in comparison with that of free metal chelate; encapsulation of these agents within the interior of the liposome effectively “shields” bulk solvent water molecules from the inner-sphere coordination with the metal centers. Liposome size, membrane composition, and water-membrane permeability all influence the relaxivity of an ensome, as these affect the rate of water exchange across the lipid membrane, the mechanisms of which were described in detail by Pütz and co-workers.306 Indeed, relaxivity was found to be linearly dependent on the surface-area-to-volume ratio of a vesicle; the smaller the ensome, the higher the relaxivity.310 Other systems have been found to respond to pH, although the mechanisms of these have not been described in detail.311–313 Also, increasing temperature also increased permeability of the liposome membrane, resulting in an increase in relaxivity.314
10.1.2 Memsomes
The limitation of water exchange across the lipid membrane is circumvented by incorporating the metal chelate into the membrane itself by conjugating the metal-binding site to the hydrophilic heads of the membrane molecules (Figure 15), thereby providing solvent water molecules direct access to the inner-sphere coordination with the metal centers; these systems, in turn, are referred to as “memsomes”. First examples include Gd3+-DTPA derivatives which contain hydrophobic alkyl chains linked by either an amide or an ester bond).315–318 Incorporation of metal chelates into a large supramolecular assembly increases its effective molecular size and hence its rotational correlation time in solution (i.e., large τR). Indeed when Gd3+-DTPA-stearylamide and Gd3+-DTPA-stearylester were incorporated into the lipid membrane of a liposome, the resulting memsomes were found to exhibit relaxivities two to five times that of free chelate in solution (depending on the field strength), and as much as six to seven times that of memsomes encapsulating similar chelates. However, increasing the size of the vesicles did not result in an increase in relaxivity, since any further gain due to increased molecular size is off-set by the rate of diffusion of the paramagnetic chelate across the membrane. That is, if the attachment of the paramagnetic chelate to the membrane is not rigid, variation in size of the vesicle has no bearing on the relaxivity of the memsome.318 To this end, Storrs and co-workers reported the synthesis of paramagnetic polymerized liposomes (PPLs), in which the alkyl chains of the liposome are photochemically cross-linked. Though the relaxivity of this system was measured to be three times that of Gd3+-DTPA (under the same field strength), it was found to depend not so much on particle size, but on the length of the linker used.319 Gløgård and co-workers also described a memsome in which the Gd3+ chelate was conjugated to the liposome via a disulfide bond susceptible to radical-induced cleavage. In the presence of dithiothreitol, the chelates were cleaved from the liposome, reducing the measured relaxivity by half.320 The aggregation behavior of amphiphilic Gd3+-chelates has also been shown to be pH dependent. Vaccaro and co-workers reported the synthesis of a Gd3+-DTPA derivative incorporating two C18 alkyl chains which self-assembles into micelles at neutral pH, but which re-distributes into liposomes as the pH is decreased,321 suggesting a design for pH-responsive MR contrast agents which switch between aggregation states. However, since the acetate arms of DTPA are conjugated to the long-chain alkyl groups, this structural change will have a direct impact on the stability of the resulting Gd3+ chelates and may result in toxicity issues due to possible decomplexation in vivo.
Figure 15.

DTPA-derivatives (A) stearylamide, (B) stearylester and (C) phosphatidylethanolamine.
10.1.3 Micelles
Amphiphilic Gd3+-chelates have also been shown to self-assemble into micelles of low critical micelle concentration (CMC). In a study involving a series of amphiphilic Gd3+-chelates of increasing alkyl chain length, only those chelates with 10 carbons or more exhibited micelle formation, as evidenced by the measured relaxivities dependent on the CMC.322 17O-NMR measurements of micelles constituted of a Gd3+-DOTA derivative linked to a C12 alkyl chain demonstrate that since the Gd3+-chelates face outward toward the solvent, the exchange rate kex of water between the bulk solvent and the inner-coordination sphere of the chelate is conserved, i.e. identical to that of the free chelate. That is, similar to the case of memsomes, the relaxivity of micelles is not limited by access of solvent to the paramagnetic center, the chief limitation of ensomes. In addition, the supramolecular assembly as a whole exhibits a high rotational correlation time τR which results in a relaxivity comparable to that of lower generation dendrimers or Gd3+ chelates with non-covalent protein binding.323 Furthermore, incorporation of cholesterol into the hydrophobic interior of the micelle resulted in a more rigid structure and a 10% increase in relaxivity. To circumvent the limitation of rapid diffusion of metal chelate across the micellar surface, improvements would have to come in the form of using chelates with better properties. For example, Hovland and co-workers found that micelles formed from an amphiphilic Gd3+-chelate based on a derivative of 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,6,9-triacetate (PCTA-[12]) had improved relaxivities due to a faster water exchange rate kex relative to analogous chelates such as Gd3+-DO3A.324 Micellar structures whose relaxivities and aggregation states are responsive to pH have also been reported,325 as well as micelles based on Gd3+-chelates conjugated to biodegradable polymers proposed as potential platforms for drug-delivery systems.326
10.1.4 LipoCEST agents
More recently, an innovative approach to liposome-based MR imaging takes advantage of the chemical exchange saturation transfer (CEST) effect. Saturation transfer occurs when protons of the contrast agent undergoing exchange with the bulk solvent are selectively irradiated at their absorption frequency, resulting in a decrease in intensity of the bulk signal. The effect was first demonstrated by Balaban and co-workers, and applied to naturally-occurring metabolites.327 The idea was soon extended to ensomes incorporating non- Gd3+ lanthanide chelates. The first example of a lipoCEST agent was reported by Aime and co-workers, in which a Tm3+ complex was encapsulated within a liposome. Two different water resonances were observed, an intense signal corresponding to the bulk solvent, and a less intense peak shifted downfield by 3.1 ppm, corresponding to intraliposomal water in slow exchange with the bulk through the lipid membrane. Irradiation at this frequency resulted in saturation transfer detectable at liposomal concentrations as low as 90 pM.328 Zhao and co-workers demonstrated that the lipoCEST effect is also dependent on the size of the liposome, that increased lipoCEST contrast is observed in smaller ensomes due to their larger surface-to-volume ratio and faster exchange of water across the lipid membrane.329
Even more interestingly, an ensome containing Gd3+-HPDO3A was osmotically shrunk by increasing the ionic strength of the extra-liposomal solvent, resulting in the contraction of the liposomes from spheres to oblate vesicles. The shrinking of the vesicles resulted in an increase in relaxivity relative to the spherically-shaped liposome; however, the change in shape was also accompanied by a downfield shift of the intraliposomal water resonance from the bulk by 7 ppm.330 Indeed, even greater increases in paramagnetic-induced shift were observed in osmotically-shrunken ensomes containing polymetallic Tm3+ chelates, as large as 28.2 ppm downfield in an ensome containing a trinuclear Tm3+ complex, due to contributions from both dipolar and magnetic susceptibility effects.331 Similar large shifts were observed in osmotically-shrunken liposomes incorporating Tm3+ chelates in both the liposomal interior and the lipid membrane (as it were, a combined ensome-memsome system).332 Mixed-metal systems involving Dy3+- and Tm3+-chelates extended the window of irradiation frequencies from δ = ± 4 ppm (for spherical lipoCEST agents) to +30 < δ < −45 ppm, the shift direction determined by the magnetic anisotropy of the membrane-incorporated chelate.333–334 Taking advantage of the large frequency separation between intraliposomal water resonances for spherical and osmotically-shrunken liposomes (3 ppm and 15 ppm downfield from the bulk solvent, respectively in systems containing Tm3+ chelates), multiple CEST visualization of these two agents was demonstrated on bovine muscle tissue with no interference.335 More recently, Langereis and co-workers described a temperature sensitive lipoCEST agent incorporating both a Tm3+ chelate, as a CEST reporter, and NH4PF6, as a 19F-NMR probe: below the melting temperature TM of the liposome, the CEST effect is observed due to the presence of the paramagnetic chelate. However, above TM, the contents of the liposome leak out of the vesicle, switching off the CEST effect but switching on the 19F resonance.336
10.2 Biodistribution
10.2.1 Passive distribution
The high aqueous solubility and low molecular weight of early Gd3+-based MR contrast agents results in their rapid clearance. Though sufficient for visualizing renal vasculature, longer circular times are required for contrast agent to accumulate in other organs of interest. Simple ensomes and memsomes incorporating paramagnetic species have invariably been reported to accumulate in the liver and spleen by a passive targeting mechanism.308,315–317,337–339 Indeed, early on an ensome encapsulating Gd3+-DTPA was shown to enhance contrast between normal and tumor liver tissue, with the smallest liposomes under study producing the greatest contrast enhancement.340 Simple ensomes have been tested in hyperthermia protocols to monitor contrast agent and/or drug delivery to liver tissue upon heating beyond the TM of the liposomal preparation.341–342 More recently, Gd3+-DTPA-lipid derivatives were found to accumulate in atherosclerotic plaques.343–345
The relatively large molecular weight of liposomes also increases their circulation in blood. However, Gd3+ chelates have been shown to undergo decomplexation in vivo if retained in the body for extended periods,346 and structural modifications were performed to establish control over clearance rates. Kabalka and co-workers synthesized a series of liposomes composed of Gd3+-DTPA-stearyl derivatives conjugated through a variety of linkages, and found that the stearylamide derivative was completely retained in the liver for over 11 days, while half of the stearylester derivative had cleared in the same time period. In addition, the liver clearance half-life of the stearylthiol derivative was only two days. However, the reverse trend was observed with respect to serum stability.317 Another derivative, Gd3+-DTPA conjugated to phosphatidylethanolamine, was found to have a liver clearance half-life of 24 hours.337–338 Paramagnetic polymerized liposomes were found to re-circulate in the body without immediate clearance by either the kidneys or the liver,347 and liposomes conjugated with polyethylene glyocol (PEG) exhibited long blood circulation times, permitting longer acquisition times for the visualization of fine vasculature.348–349 Furthermore, biotinylation of PEG-coated liposomes also permits controlled avidin-mediated blood clearance.350
10.2.2 Targeted Delivery and Cell Labeling
The design of liposomes conjugated with targeting vectors extends the utility of these agents for visualizing a greater number of histological types by monitoring known physiological responses. Several “mixed micelles” have been reported in the literature incorporating amphiphilic Gd3+-chelates and a bioactive peptide conjugated to an alkyl chain.351–353 However, their use in vivo has yet to be demonstrated. Larger constructs involve liposomes conjugated with antibodies, which also take advantage of emergent multi-modal imaging protocols. Mulder and co-workers described the preparation of PEGylated paramagnetic and fluorescent immunoliposomes to monitor the over-expression of E-selectin in human umbilical endothelial cells (HUVEC) when treated with tumor necrosis factor α (TNFα).354 Erdogan and co-workers also described the use of a similar system exhibiting specificity for nucleosomes on cancer cell surfaces in in vivo murine experiments.355 These examples illustrate the combined advantage of using targeting moieties of high specificity in tandem with PEG-functionalization, which permits blood circulation times sufficiently long enough for the contrast agent to accumulate in the tissue of interest.
Finally, novel liposomal formulations are designed to deliver paramagnetic and fluorescent reporters directly into cells. Together with the increasing interest in cell-based therapies is the need to develop non-invasive methods for monitoring the fate of transplanted cells and their tissue biodistribution in vivo. Though incubation of a paramagnetic and fluorescent PPL with T47D breast cancer cells lines demonstrated cellular uptake of agent,356 a novel lipid formulation containing 40 mol% N'-cholesteryloxycarbonyl-3,7-diazanonane-1,9,-diamine (CDAN), 30 mol% dioleoylphosphatidylethanolamine (DOPE) and 30 mol% Gd3+-DOTA-cholesterol conjugate, coined as “MAGfect”, has shown better cell transfection properties for both contrast agent and the delivery of plasmid DNA.357–358 Also, a cationic liposome conjugated with hyaluronic acid has also been shown to efficiently label cells via a CD44 receptor-mediated uptake mechanism.359
11. Viral Particles
Viral capsids are natural biological vectors, but only when control was established over their chemical properties were they considered in depth as potential platforms for paramagnetic reporters (Figure 16). Douglas and co-workers first established a methodology for encapsulating polyoxometallate species within a virion – the cowpea chlorotic mottle virus (CCMV) – by pH modulation.360 In addition, work by Wang,361 Raja362 and co-workers demonstrated that the covalent labeling of cowpea mosaic virus (CPMV) did not compromise the integrity of the viral particle; both the interior of the virion through cysteine residues, and the exterior through lysine residues, were conjugated with fluorescent reporter dyes. The same groups also managed to covalently modify the surface of the virion with PEG in order to reduce its immunogenicity.
Figure 16.

Labeling strategies of viral particles: (A) direct encapsulation, (B) conjugation via external lysines, (C) “click” chemistry, (D) functionalization of external lysines, (E) functionalization of internal tyrosines.
Based on these encouraging results, Allen and co-workers investigated the uptake of Gd3+ ions in CCMV through Ca2+ binding sites on the protein components of the virion.363 They achieved a loading of 140 Gd3+ ions per viral particle, and the assembly was measured to have a longitudinal ion relaxivity r1 of 202 mM−1 s−1, a value five times higher than that of MS-325 bound to albumin206 and ten times that even of Gd3+-labeled higher generation PAMAM dendrimers.90 However, spectrophotometric measurements showed that the Gd3+-labeled particle had a KD = 31 μM, disqualifying it for biological applications. To avoid the danger of possible “leakage” of free Gd3+ in vivo, Anderson and co-workers used a stronger chelator and covalently labeled bacteriophage MS2 viral particles with Gd3+-DTPA, achieving a bioconjugate containing 514 Gd3+-DTPA moieties and exhibiting a total relaxivity of 7200 mM−1 s−1 per particle and an ion relaxivity of 14.0 mM−1 s−1 (1.5 T).364 Similarly, Prasuhn and co-workers covalently labeled CPMV particles using “click chemistry”; lysine residues on CPMV were first derivatized with azides, which were then reacted with alkyne-functionalized DOTA. Reaction with Gd3+ resulted in 80 ± 20 Gd3+ ions per virion.365 Also, Hooker,366 Datta367 and co-workers labeled the bacteriophage MS2 with bis(HOPO)-TAM, either at the interior of the capsid through tyrosine residues, or at the exterior through lysine residues, by functionalization of these residues with an aldehyde, followed by conjugation with the ligand via oxime condensation. The externally modified virion was measured to have an ion relaxivity of 30.7 mM−1 s−1, while that of the internally modified virion was 41.6 mM−1 s−1, which are among the highest values reported so far for viral-based macromolecular contrast agents. Finally, Vasalatiy and co-workers reported the labeling of adenovirus particles (AdCMVLuc) with Tm3+ and 177Lu3+ chelates of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra(methylcarbonylamide) (DTMA), the former a PARACEST agent and the latter a radioactive reporter, while retaining the capability of the virus to infect cells and express luciferase.368 Though between 630 and 1960 ligands could be loaded onto the adenovirus, a limit of ~800 attached ligands was determined to maintain a 75–80% bioactivity.
12. Gadofullerenes and Gadonanotubes
Efforts to develop fullerenes as a platform for MR contrast agents began after the successful encapsulation of Gd3+ into the endohedral fullerene C82 was first described.369 This endohedral metallofullerene was designated as Gd@C82, “@” indicating the incorporation of Gd3+ into the interior of the fullerene. Synthesis of Gd@C82 was achieved through a modification of the traditional arc synthesis by using Gd2O3-impregnated graphite rods. However, this species was highly insoluble in water, Mikawa and co-workers attempted to increase its hydrophilicity by hydroxylating the exterior of the fullerene to produce Gd@C82(OH)n, the first example of what are now called Gd-fullerenols, and which was also found to have an in vitro relaxivity 20 times higher than that of Gd3+-DTPA when measured at 1 Tesla.370 This high relaxivity is a surprising observation considering that there is no direct interaction between bulk solvent water molecules and the gadolinium ion. However, electron energy loss spectroscopy (EELS) experiments show that this is due to the paramagnetic electronic structure of the metallofullerene itself, a result of a 3-electron transfer from gadolinium to the fullerene cage;371–372 and hence, the protons of many water molecules H-bonded across the metallofullerene surface undergo relaxation simultaneously.
Soon after, the synthesis of a whole series of lanthanide-based fullerenols incorporating La3+, Ce3+, Dy3+ and Er3+ in addition to the previously-established Gd-fullerenol was reported.373 To further improve water-solubility and provide sites for bioconjugation, Shu and co-workers modified the surface of Gd@C82(OH)n with β-alanine residues, to yield the compound Gd@C82O6(OH)16(NHCH2CH2COOH)8.374 The synthesis of endohedral trimetallic fullerenes has also been reported,375 the Gd3+ analogue of which has been modified with PEG groups on the surface, Gd3N@C80[DiPEG5000(OH)n], to further improve water solubility.376
However, higher yields of material were made available for study when Bolskar and co-workers developed a strategy for separating Gd@C60, a major component of the arc synthesis sublimate, by first extracting out Gd@C82 (a minor product) with o-dichlorobenzene, followed by successive reductive and oxidative treatment of the remaining endohedral material to separate the majority Gd@C60 fraction.377 Further improvements to this synthetic procedure have recently been reported.378 The surface of Gd@C60 has also been modified with hydroxyl and carboxylic acid groups, yielding Gd@C60(OH)n and Gd@C60[C(COOH)2]10, and these derivatives were observed to form aggregates as the pH of the solution is decreased from 9 to 7, resulting in large increases in relaxivity,377,379–380 as the formation of aggregates results in an increase in rotational correlation time, τR. Indeed, similar pH-dependent aggregation was also observed with the larger metallofullerene Gd@C82O6(OH)16(NHCH2CH2COOH)8.381 However, when relaxivity measurements of these metallofullerenes were taken in phosphate-buffered saline (PBS) solution, the presence of salt, in particular phosphate, disrupted the aggregation leading to a reduction in relaxivity.382 Variable-temperature and multiple-field 17O and 1H NMR studies have shown that, in the aggregated form, the major contribution to relaxivity enhancement was due to outer-sphere effects caused by water molecules within the interstitial spaces of the aggregates in exchange with the bulk solvent, whereas in the non-aggregate form, inner-sphere relaxation predominates as a result of proton exchange between the bulk and protonated OH or COOH sites.383 Gd@C60[C(COOH)2]10 was also found to label mammalian cells with close to 100% efficiency without the use of a transfecting agent, suggesting its use as an in vivo probe for tracking mammalian cells.384 Future perspectives on the use of gadofullerene MRI contrast agents in a clinical setting have been discussed in more detail in a recent review.385
Investigations into using carbon nanotubes impregnated with Gd3+, now referred to as gadonanotubes, are still in their infancy, the basic synthesis of which involves the chemical “cutting” of singe-wall carbon nanotubes (SWNTs) into ultra-small (US) tubes, followed by sonication with aqueous GdCl3.386 These gadonanotubes were found to have relaxivities nearly 40 times larger than current contrast agents measured at field strengths employed in a clinical setting (20–60 MHz).387 And similar to gadofullerenes, their relaxivity was found to be pH-dependent, exhibiting a dramatic increase when pH is decreased from 7.4 to 7.0, suggesting its use as a highly sensitive probe for monitoring small changes in pH under physiological conditions.388 Indeed surface chemistry has been performed on these agents, and water-soluble derivatives functionalized with a variety of amino acids and even cyclic RGD peptide have been reported towards the development of these gadonanotubes as targeted bioconjugate MR agents.389
At this time, only a few gadofullerenes have been tested in vivo. Gd@C82(OH)40 was found to selectively accumulate in the reticular-endothelial system (RES) and was retained in liver and spleen 24 hours post-injection,370 but Gd@C60[C(COOH)2]10 showed enhanced renal uptake as opposed to liver, and excretion via the bladder within 1 hour post-injection in a mouse.377 Furthermore, Gd3N@C80[DiPEG5000(OH)n] was used to visualize infusions into rat brain.376 Hence, too few examples exist at present to definitely state how safe these agents are in a clinical setting.
13. Superparamagnetic Iron Oxide
Thus far we have described T1 positive contrast agents. A discussion of MR nanomaterials would be incomplete without mentioning superparamagnetic iron oxide nanoparticles (SPIOs), a category of T2 MR agents which provide negative contrast for in vitro and in vivo cellular and molecular imaging. SPIOs present an important advantage in their huge magnetic moments which can increase proton relaxivities ten-fold.390 The typical SPIO agent has a T2 relaxivity of 100 mM−1s−1 and T relaxivity of 30 mM−1s−1, substantially larger than Gd3+-DTPA's T2 relaxivity of 6 mM−1s−1 and T1 relaxivity of 4 mM−1s−1.391
SPIO agents were recently reviewed in exhaustive depth in this journal,392 and so their synthesis and applications such as hyperthermia and drug delivery will not be repeated here, but rather focus on comparing them to T1 macromolecular agents. SPIO particles are crystalline structures with the general formula of Fe3+2O3M2+O, wherein M2+ is a divalent metal ion such as iron, manganese, nickel, cobalt, or magnesium. Because of the ubiquitous presence of iron in living tissue, the ferrous ion Fe2+ is the most common divalent ion used in SPIOs to make magnetite (Fe3O4 or Fe3+[Fe2+Fe3+]O4). Indeed, magnetite is particularly suitable for manipulation in MR as it has been isolated from certain birds, fish, and bacteria in which its interaction with the magnetic field of the earth has been found to play a critical role in navigation.393 Maghemite forms from the oxidation of magnetite and is similar, but possesses cation vacancies. Both magnetite and maghemite have practically identical magnetic and relaxation properties. In fact, rarely will a pure magnetite sample be found, particularly in the relevant biomedical applications. Maghemite/magnetite particles have face-centered cubic packing (a.k.a. cubic close packed) of oxygen that allows fast electron hopping or continuous exchange of electrons between iron ions occupying interstitial tetrahedral (surrounded by 4 oxygen atoms) and octahedral (surrounded by 8 oxygen atoms) sites (Figure 17A). All tetrahedral holes are filled by Fe3+ ions and octahedral sites by the remaining Fe3+ and Fe2+ (or vacancies).394 The magnetic properties of this material arise from the ferrimagnetic alignment of the iron ions. The tetrahedral Fe3+ ions are aligned in one direction and all the octahedral ions in the opposite (Figure 17B). In magnetite, the octahedral and tetrahedral Fe3+ ions cancel each other and the magnetic moment is due to uncompensated octahedral Fe2+ ions. In maghemite, the moment arises from uncompensated octahedral Fe3+ ions. Compared to single paramagnetic ions, each vectorized SPIO particle has a huge magnetic moment, one of its advantages for MR applications.
Figure 17.
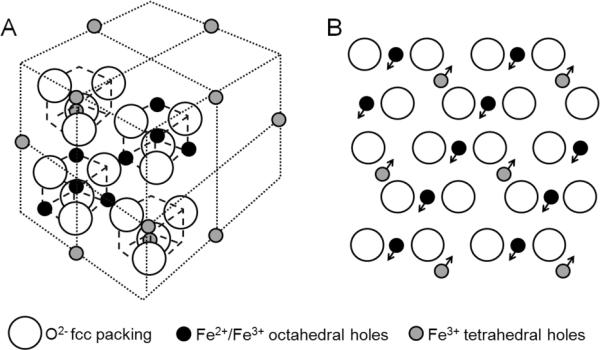
(A) Crystalline structure of magnetite. (B) Ferrimagnetic alignment observed from [1,1,1] plane.
13.1 Understanding Superparamagnetism
The magnetic moment of magnetite/maghemite preferentially aligns on what are known as easy directions or anisotropy axes. For large materials, Weiss domains of uniformly oriented magnetic momenta can be observed between which the alignments are different (Figure 18A).395 The diameters of SPIO-based particles used for MRI are well within the size of a Weiss domain, and are thus single-domain with a unique magnetic moment. Even while in ambient field conditions superparamagnets are almost fully saturated along the anisotropy axis. However, the magnetic moment of the entire crystal very rapidly jumps from one axis direction to another, time-averaging to a net zero magnetization. These fluctuations in magnetization that have a characteristic Néel relaxation time occur because the thermal energy is sufficient to overcome the magnetic anisotropy energy barrier.396
Figure 18.
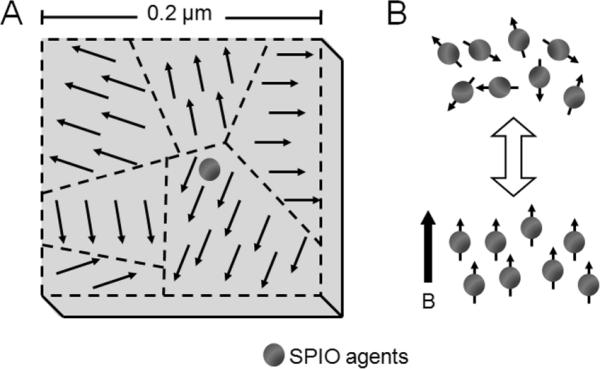
(A) Weiss domains in a large magnetite crystal in comparison to a typical SPIO agent. (B) The magnetic alignment of SPIO particles in the absence and presence of an external magnetic field.
In the absence of an external magnetic field, the effective magnetic anisotropy energy Eα serves as an energy barrier for blocking the flips of magnetic moments and can be approximated by:
| (26) |
where Keff is the effective magnetic anisotropy energy constant per unit volume, and V is the volume of magnetic nanocrystal. As this relation implies, a larger particle radius very rapidly increases the anisotropy energy. Thermal activation overcomes the anisotropy energy barrier when Eα is comparable with the thermal activation energy:397–398
| (27) |
Here, kB is the Boltzmann constant, TB is the critical blocking temperature, and the constant β, which typically varies between 25 and 34,398 represents the ratio between anisotropy and thermal energy when the relaxation time of a given particle is similar to the characteristic measuring time of the experiment. At temperatures below TB, thermal fluctuations do not dominate and the magnetic moments “freeze” in random orientations, while above TB a stable bulk magnetization cannot be established and the material demonstrates superparamagnetism.396
Thus, in the absence of an applied magnetic field, the direction of crystallite magnetization is free to rotate with thermal motion and randomly orient to average a net zero magnetization. The material behaves similarly to a paramagnet, except that instead of each individual atom being independently influenced by an external magnetic field, the magnetic moment of the entire crystallite tends to align with the applied field. Furthermore, the resultant magnetic moment after an external field is applied is much greater for a superparamagnet than for a paramagnet.399–400 When removed from the applied field, the magnetic orientation again randomizes with no hysteresis from the previous alignment (Figure 18B). This behavior applies up to the Néel and Curie temperatures, and above them, the material exhibits normal paramagnetic behavior.
13.2 Superparamagnetic Relaxation
In the previous sections, relaxation by paramagnetic ions, such as Gd3+-based agents, was described where the primary focus was on inner-sphere contribution and how macromolecular complexes enhance relaxivity by increasing the rotational correlation time, τR. A key difference between those agents and superparamagnetic particles is that for the latter the inner-sphere contribution is minor, if not negligible, when compared to the dominant outer-sphere relaxation. SPIO relaxation is also based on the theory described in Section 2, with relaxation due to the fluctuating dipole-dipole interaction between the water proton's nuclear magnetic spins and the superparamagnet's global magnetic moment. The key differences that we will discuss, however, relate to the electronic relaxation of the agent itself and its subsequent effect on proton relaxation.
The global electronic relaxation of superparamagnetic particles occurs by two mechanisms: 1) internal magnetic dipole Néel relaxation, and 2) bulk Brownian relaxation (Figure 20A). The external magnetic field supplies energy to overcome the anisotropic energy barrier and allows internal magnetic moments to rotate away from the anisotropy axis. The energy is then dissipated when the particle moment relaxes to its equilibrium orientation and is known as Néel relaxation. It is characterized by the Arrhenius law:
| (28) |
where T is the absolute temperature and the pre-exponential factor τo is also an expression of the anisotropy energy.401 The second type of relaxation is due to bulk rotational Brownian motion within a carrier liquid. With the energy barrier defined by viscous rotational friction in the surrounding liquid, the magnetic particle rotates as a whole because of the torque exerted on the magnetic moment by the external magnetic field. The Brown relaxation time τB is
| (29) |
where η is the viscosity of the surrounding liquid and V the hydrodynamic volume of the particle.402 Thus, the global magnetic relaxation rate of the colloid τeff is
| (30) |
Figure 20.

(A) Néel and Brownian components of electronic relaxation, and (B) effects of Neel and Curie spin relaxation on proton nuclear relaxation.
For large particles (r > 7.5nm), τB is shorter than τN because of their direct versus exponential dependence on volume. Thus, the viscous rotation of the particle becomes the dominant mechanism determining the global relaxation. For smaller particles, the primary process becomes Néel relaxation (Figure 19). These two mechanisms refer to the relaxation of the electronic moments of the crystal and not the nuclear relaxation of water protons.
Figure 19.
The effects of Néel and Brownian relaxation in relation to magnetite crystal radius (according to Rosensweig403).
The superparamagnetic agent's effect on proton nuclear relaxation occurs by two mechanisms (Figure 20B). The first is by the electronic moment fluctuations as a result of Néel relaxation. The second results from diffusion into the inhomogeneous nonfluctuating magnetic field created by the mean crystal moment. Described by Gueron404 and Vega and Fiat405, the thermal average of an electron spin (the molecular susceptibility or Curie spin) that is aligned along the applied magnetic field B0 relaxes protons with an effect that increases quadratically with the external field and is proportional to the square of the magnetic moment of the molecule.406 Curie spin relaxation is primarily a T2 effect found at high fields and is modulated by reorientation and exchange but not by the fluctuations of the electron magnetic moment.
For large particles in low fields, both water diffusion and Néel relaxation effects are significant and so superparamagnetic relaxation is generally governed by Freed's equations with τS1 = τN. At high fields, the magnetic moment of large particles is locked onto the external magnetic field direction and Néel relaxation is no longer possible. Thus, τN drops out and nuclear relaxation is reduced to Ayant equations where τD is the primary correlation time. At intermediate fields, both the mean and the fluctuating magnetic moments contribute to the induced nuclear relaxation and a combination of Freed and Ayant equations are used to model the relaxation. For further discussion on these equations please refer to the previously mentioned review article.392
For very small particles, crystal anisotropy energy is low and so particle magnetic moment locking on anisotropy axes is reduced. This results in the observation of low field dispersion in the NMRD profile of ultrasmall SPIOs, whereas there is no dispersion for larger SPIOs. Various models that account for anisotropy energy differently have been used to describe this occurrence.392
Aggregated particles have very different relaxation properties both in terms of the agglomeration's magnetic field distribution on its surrounding environment and on the internal moments of the crystal itself. Transverse relaxivity appears to increase initially to a maximum and then decrease, while longitudinal relaxivity continuously decreases with increasing aggregate size.407 Furthermore, compartmentalization of the particles within cells,408–410 and the type of cell play key roles in relaxivity.411 With transverse relaxivity of superparamagnetic particles dropping 2–3 fold when in cells, the effect must be accounted for when analyzing the agents by MRI.
13.3 Current SPIOs
In order to stabilize colloidal ferrofluids, it is necessary to functionalize the iron oxide surface.392 Particularly in the physiological pH range, uncoated USPIOs do not have a strong enough surface charge to maintain electrostatic repulsion and thus will flocculate in suspension, forming large aggregates.412 Surface functionalization can provide a strong negative zeta potential, allowing the particles to both remain in stable colloidal suspension as well as mimic the surface charge of typical biomolecules. Monomeric stabilizers such as carboxylates, phosphates, and sulfates can be used to form electrostatic layers.392 Thicker layers of material also provide steric hindrance to prevent particle-particle interaction. Silica392,412–414 and gold392,415–416 are the two inorganic materials that have been used to coat iron oxide particles. The most commonly used, however, are organic materials which include polymeric structures such as dextran, carboxymethylated dextran, carboxydextran, polyvinyl alcohol, polyethylene glycol, alginate, starch, arabinogalactan, glycosaminoglycan, sulfonated styrene–divinylbenzene, chitosan, poloxamers, and polyoxamines.392
Superparamagnetic iron oxide contrast agents with their full functionalization are loosely categorized into two groups classed by size (inclusive of coatings):
-
(1)
SPIOs: >60nm
-
(a)
large oral SPIOs: 300nm – 3.5 μm
-
(b)
standard SPIOs (SSPIO): 60 – 150 nm
-
(a)
-
(2)
ultrasmall SPIOs (USPIO): <40 nm
-
(a)
monocrystalline iron oxide nanoparticles (MION)
-
(i)
cross-linked iron oxide (CLIO)
-
(i)
-
(a)
Table 4 provides a list of commercially available SPIOs and their properties.
Table 4.
| Agent | Particle Size | Classification | Surface Coating | Relaxivity (mM−1s−1) (37°C, 0.47 T) | Target Organs | Dosea | Mode of Admin | Generic Name | Trade Name | Developing Company | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OMP | 3.5μm | Oral SPIO | polystyrene | GI lumen | 0.5 g/L, 400–600mL | p.o. | Ferristene | Abdoscan® | Amersham/GE Healthcare | discontinued | |
| AMI-121 | 300nm | Oral SPIO | siloxane | GI lumen | 52.5 mg Fe (=0.94mmol Fe) | p.o. | Ferumoxsil | Lumirem® | Guerbet | approved in US and Europe | |
| Gastromark® | Advanced Magnetics/ AMAG Pharmaceuticals |
||||||||||
| AMI-25 | 80–150nm | SSPIO | dextran | r1 = 20.2 r2=101 | liver/spleen, perfusion, MRA |
15 μmol Fe/kg | slow infusion | Ferumoxide | Endorem® | Guerbet | approved in US and Europe, discontinued |
| Feridex IV® | Advanced Magnetics/ AMAG Pharmaceuticals |
||||||||||
| SHU 555A | 62nm | SSPIO | carboxy-dextran | r1 = 23.6 r2=179 | liver | <60kg = 0.45 mmol Fe, >60kg = 0.7mmol Fe | bolus injection | Ferrixan, Ferucarbotran | Resovist® | Bayer Schering Pharma | approved in Europe, Australia, Japan |
| SHU 555C | 21nm | USPIO | carboxy-dextran | r1 = 24 r2 = 60 | MRA | Supravist® | Bayer Schering Pharma | Phase III complete | |||
| AMI-227 | 20–40nm | USPIO | dextran | r1 = 23.3 r2 = 65 | lymph nodes, MRA | 14–45 μmol Fe/kg | slow infusion | Ferumoxtran | Sinerem® | Guerbet | not approved in US or Europe, discontinued |
| Combidex® | Advanced Magnetics/ AMAG Pharmaceuticals |
||||||||||
| AMI-228 | 30nm | USPIO | semisynthetic carbohydrate |
r1 = 38 r2 = 83 | iron replacement | 2 doses of 510 mg Fe | bolus injection | Ferumoxytol | Feraheme® | Advanced Magnetics/ AMAG Pharmaceuticals |
approved in US (for iron therapy) |
| NC100150 | 20nm | USPIO | PEG | r1 = 21.8 r2 = 35.3 | perfusion, MRA | 7–100 μmol Fe/kg | bolus injection | Feruglose | Clariscan® | Amersham/GE Healthcare | completed Phase II, discontinued |
Doses may vary with imaging sequences and target organs
Oral SPIO preparations used for contrast enhancement of the abdomen typically contain larger particles and are coated with a non-biodegradable and insoluble matrix to reduce aggregation. They are suspended in viscosity-increasing agents based on ordinary food additives such as starch and cellulose that prevent ingested iron absorption and particle aggregation to allow homogeneous contrast distribution throughout the bowel. These suspensions are well-tolerated by the patients and the intestinal mucosal membrane is not irritated.417–418
Standard superparamagnetic iron oxide agents are easily sequestered by reticuloendothelial system (RES) cells in the liver and spleen upon intravenous administration with minimal lymph node uptake. AMI-25 (Endorem® or Feridex®) consists of iron oxide crystals coated by dextran with a final diameter of 80–150 nm. The T2 and T1 relaxivities respectively are 98.3 and 23.9 mL−1s−1.419 With a blood half-life of 6 min, AMI-25 quickly accumulates in the liver (~80% of the injected dose) and spleen (5–10% of the injected dose) within minutes of injection. Peak concentrations of iron are found in the liver and spleen after 2 h and 4 h with half-lives of 3 and 4 days, respectively.419 The preparation is usually not administered in a bolus injection because of cardiovascular side effects and lumbar pain. The recommended mode of administration is a dose of 15 μmol Fe/kg in 100 mL of 5% glucose with a biphasic infusion (2 mL/min over 10 min and 4 mL/min over 20 min). SHU 555A (Resovist®) is a carbodextran SSPIO with 4.2 nm iron crystals and a hydrodynamic diameter of 62 nm. Its T2 and T1 relaxivities are 151.0 and 25.4 mM−1s−1 respectively. This particle shows no side effects after rapid intravenous injection and a dose of 8 μmol Fe/kg can be scanned within 30 min after administration.420 Due to smaller size and higher T1 relaxation, SHU 555A has also been used for MRA and dynamic T1-weighted MR imaging similar to gadolinium chelates.421
USPIOs typically have iron crystals of 5–12 nm and prolonged blood half-life that affords them the opportunity to eventually cross capillary walls and have more widespread tissue distribution. They can be delivered to the interstitium by non-specific vesicular transport and through transendothelial channels. Once in the interstitium, draining lymphatic vessels transport them to lymph nodes, thus making them suitable agents for MR lymphography (MRL). At low concentrations, these agents can be used for T1-weighted MRA, though high concentrations will lead to signal loss due to T2-shortening effects. AMI-227 (Sinerem® or Combidex®) is a 20–40 nm dextran-coated USPIO with a human blood pool half-life of more than 24 h.422 It can be used as a MRA agent during the early phase of intravenous administration 423–425 and MRL agent during late phase.426–427 To avoid hypotensive reactions or acute lumbar pain, AMI-227 is infused slowly over a period of 30 minutes. NC100150 (Clariscan®), a PEGylated USPIO, and HU 555C (Resovist®) are bolus-injectable agents developed for MRA and perfusion studies.428 NC100150 has been tested as a brain T2*-weight perfusion agent at a dose of 7 μmol Fe/kg,429 but because of some adverse events and improvements in MRI units allowing better MRA with low molecular weight contrast agents, the sponsor (GE Healthcare) discontinued development of this product. Monocrystalline iron oxide nanoparticles (MIONs) are a subclass of USPIOs that differ from other SPIOs in their monocrystallinity. Their small iron oxide diameter, typically 2–9 nm, allows them to pass through capillary endothelium while retaining their superparamagnetism. In vivo detection of the agent is feasible with MRI at concentrations as low as 1 μg Fe/g tissue.430–431 CLIOs are a specific form of MIONs stabilized by a cross-linked aminated dextran coating. USPIO size affords them great potential for receptor-directed and magnetically labeled cell probe MRI.
13.4 Applications
SPIO agents are endocytosed and metabolized by RES cells and thereafter incorporated into the normal metabolic iron pool.436 Eventually they are secreted as the body iron stores turn over. Smaller particles degrade faster into paramagnetic forms of iron. When injected at an appropriate rate, the toxicity of SPIO agents is low, and animal studies have disclosed no acute toxicity or chronic injury at doses more than 100 times the clinical effective dose.429 The amount of iron oxide required for clinical MRI is small when compared with actual physiological iron stores.417,420 The diameter of superparamagnetic agents greatly affects their localization in vivo, even without targeting ligands on the surface. Particles ranging in size from 60 to 150 nm rapidly appear in the liver and spleen,437 while USPIOs are optimal for prolonged blood circulation, can cross capillary walls, and are often extensively taken up by lymph nodes and bone marrow.437
Pharmacokinetic behavior of SPIOs is generally comparable in animals and humans.419,438–441 Hepatic RES Kupffer cells account for 80% of the uptake of the injected dose of AMI-25 (Feridex®).419,438–441 After intravenous injection, the agent has a blood half-life of 10 min and accumulates in the liver and spleen. The first clinical trials with SPIOs were for hepatic imaging439,441–443 where particle uptake by Kupffer cells in healthy liver tissue darkened the normal liver. The cancerous lesions appeared bright against the dark background as they did not take up the SPIOs. Similar studies followed where USPIOs were used to detect cancer metastasis in lymph nodes and bone marrow.427,444–446 USPIOs have also been successfully used to visualize inflammation in the brain after stroke,447 surrounding atherosclerotic plaques,448–450 and from transplant rejection451–454 when macrophages that have phagocytosed particles enter the regions. Another application of SPIOs that has not yet reached the clinic is specific cell tracking where nonphagocytotic cells are loaded with particles in vitro using cell-permeable peptides455–457 and transfection agents in combination with the negatively charged surface of magnetic nanoparticles136,458–460. Such a method has allowed the non-invasive tracking of stem cells455, neural transplants461, white blood cells462, T-cells463–465, and monocytes466 in animal studies.
While the above mentioned applications have used untargeted superparamagnetic agents, recent developments have been directed towards molecular imaging. Targeting agents that have been conjugated include antibodies, antibody fragments, oligosaccharides, proteins, peptides, peptidomimetics, and other small ligands. Laurent et al.392 have provided an excellent table that describes these targeting studies. The field has advanced incredibly where it is now possible to use iron oxide to detect down to single cells in vivo.467
14. Manganese-Based Agents
The first contrast agent for MRI was in fact not Gd3+ based, but instead centered on the use of Mn2+. With an administration of manganese salt, Lauterbur et al.468 found T1 enhancement, particularly in liver, kidney, and heart.469 Mn2+ is dominated by a dipole-dipole contribution to T1 and a strong scalar contribution to T2.8 Manganese chloride (MnCl2) can be administrated orally at a dosage of 0.8–1.6 g per patient470 where it reaches from the gastrointestinal tract to the liver through the portal system. However, interest in Mn-based agents has been sporadic and the development was significantly less than Gd3+-based agents because of concerns over free Mn2+ toxicity. Parkinsonism-like symptoms occur when the ion blocked normal calcium fluxes in the heart.469,471 To avoid cardiotoxicity, two strategies were considered: 1) administration in combination with Ca2+ ions472–473 to competitively reduce binding of Mn2+ to Ca2+ channels, and 2) chelation474–476 to control or modulate the concentration of free Mn2+; both approaches reduced toxicity.
The most well-known Mn2+ chelate is Mn-dipyridoxyl-diphosphate (MnDPDP, Mangafodipir, or Teslascan®, manufactured by GE Healthcare) (Figure 21). Whereas following oral intake, manganese accumulates only in the liver and bile,470 after intravenous injection, it is also found in the pancreas, kidneys, and cardiac muscle.477 T1 liver enhancement after intravenous infusion begins early 1–2 min post-injection, maximizes within 5–10 min, and persists for several hours allowing flexibility for patient scheduling when compared to Gd3+ chelates. No longer on the market, for human clinical imaging to detect hepatic lesions, the approved dosage of Mn-DPDP was 5 μmol/kg478 and approximately 15% was eliminated in the urine by 24h post-injection and 59% in the feces by 5 days.30 In a European phase III clinical trial, adverse events, such as nausea, headache, and pruritus, were observed in 7% of the 624 patients.478–479 Transient decrease in alkaline phosphatase levels and sensations of heat and flushing with high injection rates most likely related to peripheral vasodilatation have also been reported.
Figure 21.

Manganese chelates.
Given the theme of this specific review, what is most relevant is the use of Mn2+ in macromolecular structures. As with relaxation by Gd3+, increasing the rotational correlation time τR also increases the relaxivity of Mn2+. T1 relaxation of MnDPDP has been shown to increase by intracellular protein binding. In the rat myocardium, intracellular relaxivity by Mn2+ ions was 8 times and 36 times higher than Mn2+ aqua ions and MnDPDP in vitro (r1 = 56 mM−1 s−1 at 0.47 T).480 Later, Mn2+ complexes derivatized with benzyloxymethyl (BOM) substituents were synthesized to promote non-covalent interaction with human serum albumin (HSA).481 The two EDTA-based agents contained one coordinated water molecule and the third no directly coordinated molecules. They displayed relaxivity values smaller than Gd3+-DTPA (Table 5) however the exchange rates of the coordinated water was one order of magnitude higher than those reported for Gd3+ complexes with octadentate ligands. Relaxivity of the agents increased up to 15 times when bound to HSA.481 Building on this work, Troughton et al.482 synthesized a EDTA complex with a diphenylcyclohexyl moiety, like gadolinium-based MS-325, to again promote binding to serum albumin. MnL1 was also found to bind well to HSA and had double the water exchange rate (Table 5).
Table 5.
| Relaxivity (mM−1s−1)(20 MHz, 298 K, pH 7.4) | kex298 (s−1) at 298 K | ||
|---|---|---|---|
| Unbound | Bound to HSA | ||
| EDTA(BOM) | 3.6 ± 0.2 | 55.3 ± 2.5 | 0.93 × 108 |
| EDTA(BOM)2 | 4.3 ± 0.2 | 48.0 ± 2.3 | 1.3 × 108 |
| DO3A(BOM)3 | 1.6 ± 0.1 | 8.1 ± 0.4 | No coordinated water |
| L1 | 5.8 (37°C) | 48 (37°C) | 2.3 ± 0.9 × 108 |
These chelating agents were useful in providing MR contrast, however, work with them was mostly abandoned in favor of Gd3+-based agents partially due to the significantly larger doses needed for non-hepatic imaging applications. Searches of the literature will find a handful of macromolecular structures synthesized to sequester manganese. In the mid 1980s, Mn2+-DTPA and Mn2+-citrate were entrapped in multilamellar liposomes 308,483 and accumulation compared to free Mn2+-DTPA was dramatically increased in spleen and liver while relatively reduced in the heart and kidney. The work was then followed by encapsulation of Mn2+-labeled HSA in liposomes as MRI contrast agents.484 Enclosure in the liposome did not affect the relaxivity of the HSA-Mn2+ complex. More recently, Mn2+ has been incorporated in SPIO agents to increase magnetic moment and relaxivity and has now been used to target tumors by conjugating monoclonal antibody trastuzumab to the surface.485 Larger manganese structures currently being developed for MR that will release Mn2+ ions over time are metal-organic frameworks with trimesic acid (BTC) bridging ligands.486 Mn3(BTC)2(H2O)6 nanoparticles adopted a spiral rod morphology with diameters of 50–100 nm and lengths of 1–2 μm. They were further labeled with Rhodamine B dye for fluorescence imaging and c(RGDfK) peptide to target angiogenic cancers. The relaxivity of this particulate on a per Mn basis was r1= 4.0 and r2= 112.8 mM−1 s−1 at 9.4T, but the hope is that T1 enhancement will increase as Mn2+ leaches out of the structure. Initial in vitro and in vivo studies have begun, but much work must be done to further characterize this agent.
15. Conclusions
A decade after an excellent review by Caravan and co-workers was published in this same journal on Gd3+-based MR contrast agents,4 we have seen a tremendous amount of growth in the field. New macromolecular constructs have been added to the arsenal of already well-studied macromolecular and supramolecular platforms. These, in addition to maximizing the benefits of large rotational correlation times, optimal water residency times, and retention in blood plasma, also employ “smart” drug delivery strategies which improve pharmacokinetics and report on selective physiological phenomena.
While throughout this article the advantages of macromolecular agents have been highlighted, one observation that is clear at this point is that bigger is not necessarily better. The chemist would do well to keep in mind the real biological limits in designing macromolecular constructs for MR applications. Attempts to push rotational correlation times to their theoretical maximum by increasing molecular weight are all too often compromised either by non-optimal water residency times, poor solubility in water, or lack realistic usefulness in the actual intended or hypothesized application. Attempts to maximize Gd3+-loading onto targeted agents such as antibodies can result in reduced target affinity and selectivity. Furthermore, the pharmacokinetic behavior of these agents remains a challenge, and is determined on a case-to-case basis. Yet, the creation of assemblies that exist to just contain remarkably large numbers of Gd3+ continue to be pursued despite an apparent lack of acceptance that the accompanying sizes of such assemblies are truly limiting to their usefulness. Improvements in MR technology have certainly enhanced the sensitivity of the technique. While the lure of novel materials such as fullerenes and carbon nanotubes and advances of the chemistry to append additional functionality is readily available, the value added of using such platforms solely as platforms for the novelty is unclear. The toxicity of SWNTs and fullerenes, whole body clearance, and retention of the Gd3+ in vivo are issues that have barely begun to be addressed. The appending of functionality to the exterior of such structures fundamentally has to disrupt their exquisite structural stability so that again the issue of long-term in vivo stability remains to be addressed.
Much of these limitations apply across the agents discussed herein. The use of dendrimers as a core platform for the assembly of MRI contrast agents has been studied for less than 20 years. The number of types of dendrimers that have been reported for this application is less than 10. The potential diversity of core, repeating units, and internal and exterior functionality that might be applied to the creation of dendrimers is for practical purposes near infinite. These variations may be the subject of ongoing investigations, but standardized methods for comparison across the board have not been established and so systematically investigating what advantages might be gained through manipulating the possibilities remains an unknown. By and large, the existing studies have been based upon commercially available or previously reported dendrimers which does provide a base to begin some comparisons. In parallel, a similar condition and assessment of how little is really known and how vast the remaining opportunities are for dendrimers can be extended directly to most of the other platforms for assembling novel and potentially useful clinical agents. Equally challenging, however, are a host of issues that regulatory agencies face such as consistent characterization, reproducibility in production of clinical use materials, long-term toxicity, and the metrics for assessing these agents; those available for low molecular weight agents all too frequently simply do not apply. Thus, to state that this field remains in its infancy is probably conservative.
While the future is still bright for Gd3+-based contrast agents, new roads have been paved towards the development of alternatives which depend on different detection protocols, as is the case with lipoCEST agents, SPIOs, and other agents. The ranges of these possible MR agents is continuously expanding, and while there remains more than adequate room for more interesting chemistry, we are now also faced with the challenge of determining which agents are most suitable for particular medical protocols.
Acknowledgement
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Biographies
AUTHOR BIOGRAPHIES
Aaron Joseph L. Villaraza was born 1979 in Manila, Philippines. He received his B.S. in Chemistry from the University of the Philippines-Diliman in 2001 and taught at the same university until 2004. In 2008 he obtained his doctorate from Manchester University, UK, working under the supervision of Prof. Stephen Faulkner (currently at Oxford University) on the synthesis of heteropolymetallic luminescent lanthanide complexes. At present he is on a visiting scholar fellowship at the National Cancer Institute, working in the group of Dr. Brechbiel on the synthesis of polymer-protein bioconjugates for cancer imaging and therapy.
Ambika Bumb was born in 1984 in Rajasthan, India. She studied biomedical engineering in Atlanta, USA at the Georgia Institute of Technology and obtained her doctorate in 2008 from Oxford University, UK while on the Marshall Scholarship. For her graduate research, she focused on synthesizing and characterizing multimodal imaging nanoparticles and exploring their diagnostic applications in animal models. The work was conducted through the National Institutes of Health-Oxford Graduate Partnership Program under the collaborative guidance of Prof. Peter Dobson, Dr. Martin Brechbiel, Dr. Peter Choyke, and Prof. Lars Fugger. She is currently at the National Cancer Institute on a post-doctoral fellowship further pursuing her interests in developing imaging contrast agents and expanding their applications for drug delivery and hyperthermia treatment.
Martin W. Brechbiel received his B.A. in 1979 from Gettysburg College and a M.S. in 1982 from the University of Delaware under the guidance of Professor Harold Kwart. After working for FMC Corp, he joined the National Cancer Institute in 1984. Thereafter, he worked to develop novel bifunctional chelating agents for sequestering radionuclides and their conjugation to immunoproteins under the direction of Dr. Otto A. Gansow while simultaneously obtaining a Ph.D. from American University in 1988 with Professor Thomas Cantrell. He remained with the NCI and in 1997 was appointed Acting Section Chief of the Radioimmune & Inorganic Chemistry Section and was tenured at the Section Chief in 2001. His research group's activities span the range of continuing development of novel chelating agents for radionuclides, the development of contrast media for MRI, Electron Paramagnetic Resonance, and CT imaging, and the medicinal chemistry of novel metal complexes.
REFERENCES
- (1).Andrä W, Nowak H. Magnetism in Medicine: A Handbook. Wiley-VCH; Berlin: 2006. [Google Scholar]
- (2).Bernard V. Australas. Radiol. 1989;33:390. doi: 10.1111/j.1440-1673.1989.tb03320.x. [DOI] [PubMed] [Google Scholar]
- (3).Lauffer RB. Chem. Rev. 1987;87:901. [Google Scholar]
- (4).Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem. Rev. 1999;99:2293. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- (5).Shellock FG, Kanal E. J. Magn. Reson. Imaging. 1999;10:477. doi: 10.1002/(sici)1522-2586(199909)10:3<477::aid-jmri33>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- (6).Banci L, Bertini I, Luchinat C. Nuclear and Electron Relaxation. VCH; Weinheim: 1991. [Google Scholar]
- (7).Kowalewski J, Nordenskiold L, Benetis N, Westlund PO. Prog. Nucl. Magn. Reson. Spectrosc. 1985;17:145. [Google Scholar]
- (8).Bertini I, Luchinat C. Coord. Chem. Rev. 1996;150:1. [Google Scholar]
- (9).Luz Z, Meiboom S. J. Chem. Phys. 1964;40:2686. [Google Scholar]
- (10).Swift TJ, Connick RE. J. Chem. Phys. 1962;37:307. [Google Scholar]
- (11).Bloembergen N, Morgen LO. J. Chem. Phys. 1961;34:842. [Google Scholar]
- (12).Strandberg E, Westlund P-O. J. Magn. Reson. A. 1996;122:179. [Google Scholar]
- (13).Abernathy SM, Sharp RR. J. Chem. Phys. 1997;106:9032. [Google Scholar]
- (14).Abernathy SM, Miller JC, Lohr LL, Sharp RR. J. Chem. Phys. 1998;109:4035. [Google Scholar]
- (15).Sharp R, Abernathy SM, Lohr LL. J. Chem. Phys. 1997;107:7620. [Google Scholar]
- (16).Sharp RR. J. Chem. Phys. 1993;98:912. [Google Scholar]
- (17).Sharp RR. J. Chem. Phys. 1993;98:2507. [Google Scholar]
- (18).Sharp RR. J. Chem. Phys. 1993;98:6092. [Google Scholar]
- (19).Westlund P-O. J. Chem. Phys. 1998;108:4945. [Google Scholar]
- (20).Banci L, Bertini I, Briganti F, Luchinat C. J. Magn. Reson. 1986;66:58. [Google Scholar]
- (21).Lipari G, Szabo A. J. Am. Chem. Soc. 1982;104:4546. [Google Scholar]
- (22).Bennett HF, Brown RD, Koenig SH, Swartz HM. Magn. Reson. Med. 1987;4:93. doi: 10.1002/mrm.1910040202. [DOI] [PubMed] [Google Scholar]
- (23).Freed JH. J. Chem. Phys. 1978;68:4034. [Google Scholar]
- (24).Hwang L-P, Freed JH. J. Chem. Phys. 1975;63:4017. [Google Scholar]
- (25).Geraldes CFGC, Sherry AD, Cacheris WP, Kuan KT, Brown RD, Koenic SH, Spillers M. Magn. Reson. Med. 1988;8:191. doi: 10.1002/mrm.1910080209. [DOI] [PubMed] [Google Scholar]
- (26).Geraldes CFGC, Urbano AM, Alpoim MC, Sherry AD, Kuan KT, Rajagopalan R, Maton F, Muller RN. Magn. Reson. Imaging. 1995;13:401. doi: 10.1016/0730-725x(94)00117-l. [DOI] [PubMed] [Google Scholar]
- (27).Vega AJ, Fiat D. Mol. Phys. 1976;31:347. [Google Scholar]
- (28).Kellar KE, Fossheim SL, Koenig SH. Invest. Radiol. 1998;33:835. doi: 10.1097/00004424-199811000-00007. [DOI] [PubMed] [Google Scholar]
- (29).Evans C. Biochemistry of the Lanthanides. Plenum Press; New York: 1990. [Google Scholar]
- (30).Bellin M-F. Eur. J. Radiol. 2006;60:314. doi: 10.1016/j.ejrad.2006.06.021. [DOI] [PubMed] [Google Scholar]
- (31).Ersoy H, Rybicki FJ. J. Magn. Reson. Imaging. 2007;26:1190. doi: 10.1002/jmri.21135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chang C. Invest. Radiol. 1993;28:521. doi: 10.1097/00004424-199303001-00003. [DOI] [PubMed] [Google Scholar]
- (33).Zhang Z, S.A. N, T.J. M. Curr. Med. Chem. 2005;12:751. doi: 10.2174/0929867053507379. [DOI] [PubMed] [Google Scholar]
- (34).Wiener E, Narayanan VV. In: Advances in Dendritic Macromolecules. Newkome GR, editor. Vol. 5. Elsevier; Oxford: 2002. [Google Scholar]
- (35).Adzamli K, Periasamy MP, Spiller M, Koenig SH. Invest. Radiol. 1999;34:410. doi: 10.1097/00004424-199906000-00004. [DOI] [PubMed] [Google Scholar]
- (36).de Graaf RA, Brown PB, McIntyre S, Nixon TW, Behar KL, Rothman DL. Magn. Reson. Med. 2006;56:386. doi: 10.1002/mrm.20946. [DOI] [PubMed] [Google Scholar]
- (37).Kirchin MA, Runge VM. Top. Magn. Reson. Imaging. 2003;14:426. doi: 10.1097/00002142-200310000-00007. [DOI] [PubMed] [Google Scholar]
- (38).Laurent S, Botteman F, Elst LV, Muller RN. Eur. J. Inorg. Chem. 2004;3:463. [Google Scholar]
- (39).Laurent S, Elst LV, Muller RN. Contrast Media Mol. Imaging. 2006;1:128. doi: 10.1002/cmmi.100. [DOI] [PubMed] [Google Scholar]
- (40).Vander EL, Maton F, Laurent S, Seghi F, Chapelle F, Muller RN. Magn. Reson. Imaging. 1997;38:604. doi: 10.1002/mrm.1910380415. [DOI] [PubMed] [Google Scholar]
- (41).Oksendal AN, Hals PA. J. Magn. Reson. Imaging. 1993;3:157. doi: 10.1002/jmri.1880030128. [DOI] [PubMed] [Google Scholar]
- (42).Tweedle MF, Wedeking P, Kumar K. Invest. Radiol. 1995;30:372. doi: 10.1097/00004424-199506000-00008. [DOI] [PubMed] [Google Scholar]
- (43).Wiener E, Brechbiel MW, Brothers H, Magin RL, Gansow OA, Tomalia DA, Lauterbur PC. Magn. Reson. Med. 1994;31:1. doi: 10.1002/mrm.1910310102. [DOI] [PubMed] [Google Scholar]
- (44).Kirchin MA, Pirovano G, Venetianer C, Spinazzi A. J. Magn. Reson. Imaging. 2001;14:281. doi: 10.1002/jmri.1184. [DOI] [PubMed] [Google Scholar]
- (45).Xu J, Franklin SJ, Whisenhunt DW, Raymond KN. J. Am. Chem. Soc. 1995;117:7245. [Google Scholar]
- (46).Werner EJ, Avedano S, Botta M, Hay BP, Moore EG, Aime S, Raymond KN. J. Am. Chem. Soc. 2007;129:1870. doi: 10.1021/ja068026z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Thompson MK, Misselwitz B, Tso LS, Doble DMJ, Schmitt-Willich H, Raymond KN. J. Med. Chem. 2005;48:3874. doi: 10.1021/jm049041m. [DOI] [PubMed] [Google Scholar]
- (48).Seitz M, Pluth MD, Raymond KN. Inorg. Chem. 2007;46:351. doi: 10.1021/ic0614869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Webb JAW, Thomsen HS, Morcos SK. Eur. Radiol. 2005;15:1234. doi: 10.1007/s00330-004-2583-y. [DOI] [PubMed] [Google Scholar]
- (50).Kubik-Huch RA, Gottstein-Aalame NM, Frenzel T, Seifert B, Puchert E, Wittek S, Debatin JF. Radiology. 2000;216:555. doi: 10.1148/radiology.216.2.r00au09555. [DOI] [PubMed] [Google Scholar]
- (51).Thomsen HS, Morcos SK, Dawson P. Clin. Radiol. 2006;61:905. doi: 10.1016/j.crad.2006.09.003. [DOI] [PubMed] [Google Scholar]
- (52).Cowper S, Boyer P. Curr. Rheumatol. Rep. 2006;8:151. doi: 10.1007/s11926-006-0056-9. [DOI] [PubMed] [Google Scholar]
- (53).Knopp EA, Cowper SE. Semin. Dial. 2008;21:123. doi: 10.1111/j.1525-139X.2007.00399.x. [DOI] [PubMed] [Google Scholar]
- (54).Chewning RH, Murphy KJ. J. Vasc. Interv. Radiol. 2007;18:331. doi: 10.1016/j.jvir.2007.01.025. [DOI] [PubMed] [Google Scholar]
- (55).Grobner T. Nephrol. Dial. Transplant. 2006;21:1104. doi: 10.1093/ndt/gfk062. [DOI] [PubMed] [Google Scholar]
- (56).Agarwal R, Brunelli SM, Williams K, Mitchell MD, Feldman HI, Umscheid CA. Nephrol. Dial. Transplant. 2009;24:856. doi: 10.1093/ndt/gfn593. [DOI] [PubMed] [Google Scholar]
- (57).Boyd AS, Zic JA, Abraham JL. J. Am. Acad. Dermatol. 2007;56:27. doi: 10.1016/j.jaad.2006.10.048. [DOI] [PubMed] [Google Scholar]
- (58).Peak AS, Sheller A. Ann. Pharmacother. 2007;41:1481. doi: 10.1345/aph.1K295. [DOI] [PubMed] [Google Scholar]
- (59).Issa N, Poggio ED, Fatica RA, Patel R, Ruggieri PM, Heyka RJ. Cleve. Clin. J. Med. 2008;75:95. doi: 10.3949/ccjm.75.2.95. [DOI] [PubMed] [Google Scholar]
- (60).Information for Healthcare Professionals Gadolinium-Based Contrast Agents for Magnetic Resonance Imaging (marketed as Magnevist, MultiHance, Omniscan, Opt iMARK, ProHance) http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandP roviders/ucm142884.htm (accessed October 1, 2009)
- (61).Kanal E, Barkovich AJ, Bell C, Borgstede JP, Bradley WG, Jr., Froelich JW, Gilk T, Gimbel JR, Gosbee J, Kuhni-Kaminski E, Lester JW, Jr., Nyenhuis J, Parag Y, Schaefer DJ, Sebek-Scoumis EA, Weinreb J, Zaremba LA, Wilcox P, Lucey L, Sass N. Am. J. Roentgenol. 2007;188:1447. doi: 10.2214/AJR.06.1616. [DOI] [PubMed] [Google Scholar]
- (62).Sessler JL, Murai T, Lynch V, Cyr M. J. Am. Chem. Soc. 1988;110:5586. [Google Scholar]
- (63).Magda D, Miller RA. Semin. Cancer Biol. 2006;16:466. doi: 10.1016/j.semcancer.2006.09.002. [DOI] [PubMed] [Google Scholar]
- (64).Bohmer RM, Morstyn G. Cancer Res. 1985;45:5328. [PubMed] [Google Scholar]
- (65).Mehta M, Gervais R, Chabot P, Shapiro WR, Patchell RA, Glantz MJ, Recht L, Phan S, Smith JA, Renschler MF. J. Clin. Oncol. 2006;24:7014. (Meeting Abstracts) [Google Scholar]
- (66).Rosenthal DI, Nurenberg P, Becerra CR, Frenkel EP, Carbone DP, Lum BL, Miller R, Engel J, Young S, Miles D, Renschler MF. Clin. Cancer Res. 1999;5:739. [PubMed] [Google Scholar]
- (67).Maeda H. Adv. Enzyme Regul. 2001;41:189. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- (68).Wang D, Miller SC, Sima M, Parker D, Buswell H, Goodrich KC, Kopeckova P, Kopecek J. Pharm. Res. 2004;21:1741. doi: 10.1023/b:pham.0000045232.18134.e9. [DOI] [PubMed] [Google Scholar]
- (69).Venditto VJ, Regino CAS, Brechbiel MW. Mol. Pharm. 2005;2:302. doi: 10.1021/mp050019e. [DOI] [PubMed] [Google Scholar]
- (70).Fischer M, Vögtle F. Angew. Chem., Int. Ed. 1999;38:884. doi: 10.1002/(SICI)1521-3773(19990401)38:7<884::AID-ANIE884>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- (71).Langereis S, Dirksen A, Hackeng TM, van Genderen MHP, Meijer EW. New J. Chem. 2007;31:1152. [Google Scholar]
- (72).Stiriba S-E, Frey H, Haag R. Angew. Chem., Int. Ed. Engl. 2002;41:1329. doi: 10.1002/1521-3773(20020415)41:8<1329::aid-anie1329>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- (73).Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P. Polym. J. 1985;17:117. [Google Scholar]
- (74).Frechet JM. Science. 1994;263:1710. doi: 10.1126/science.8134834. [DOI] [PubMed] [Google Scholar]
- (75).Newkome GR, Moorefield CN, Vogtle F. Dendrimers and Dendrons: Concepts, Syntheses, Applications. Wiley-VCH; Weinheim: 2001. [Google Scholar]
- (76).Tomalia DA, M. NA, Goddard WA., III Angew. Chem., Int. Ed. Engl. 1990;29:138. [Google Scholar]
- (77).Hawker C, Frechet JMJ. J. Chem. Soc., Chem. Commun. 1990;15:1010. [Google Scholar]
- (78).Jayaraman M, Frechet JMJ. J. Am. Chem. Soc. 1998;120:12996. [Google Scholar]
- (79).Buhleier E, Wehner W, Vögtle F. Synthesis. 1978;1978:155. [Google Scholar]
- (80).Newkome GR, Yao Z, Baker GR, Gupta VK. J. Org. Chem. 1985;50:2003. [Google Scholar]
- (81).Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P. Polym. J. 1985;17:117. [Google Scholar]
- (82).Maiti PK, Cagin T, Wang G, Goddard WA. Macromolecules. 2004;37:6236. [Google Scholar]
- (83).de Brabander-van den Berg EMM, Meijer EW. Angew. Chem., Int. Ed. Engl. 1993;32:1308. [Google Scholar]
- (84).Wörner C, Mülhaupt R. Angew. Chem., Int. Ed. Engl. 1993;32:1306. [Google Scholar]
- (85).Hummelen JC, Van Dongen JLJ, Meijer EW. Chem. Eur. J. 1997;3:1489. [Google Scholar]
- (86).Tomalia DA, Huang B, Swanson DR, Brothers HM, Klimash JW. Tetrahedron. 2003;59:3799. [Google Scholar]
- (87).Xu H, Regino CAS, Bernardo M, Koyama Y, Kobayashi H, Choyke PL, Brechbiel MW. J. Med. Chem. 2007;50:3185. doi: 10.1021/jm061324m. [DOI] [PubMed] [Google Scholar]
- (88).Langereis S, de Lussanet QG, van Genderen MHP, Backes WH, Meijer EW. Macromolecules. 2004;37:3084. [Google Scholar]
- (89).Margerum LD, Campion BK, Koo M, Shargill N, Lai J-J, Marumoto A, Sontum PC. J. Alloys Compd. 1997;249:185. [Google Scholar]
- (90).Bryant LHJ, Brechbiel MW, Wu C, Bulte JWM, Herynek V, Frank JA. J. Magn. Reson. Imaging. 1999;9:348. doi: 10.1002/(sici)1522-2586(199902)9:2<348::aid-jmri30>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- (91).Tóth E, Pubanz D, Vauthey S, Helm L, Merbach AE. Chem. Eur. J. 1996;2:1607. doi: 10.1021/ic951492x. [DOI] [PubMed] [Google Scholar]
- (92).Laus S, Sour A, Ruloff R, Tóth E, Merbach AE. Chem. Eur. J. 2005;11:3064. doi: 10.1002/chem.200401326. [DOI] [PubMed] [Google Scholar]
- (93).Laus S, Ruloff R, Tóth E, Merbach AE. Chem. Eur. J. 2003;9:3555. doi: 10.1002/chem.200204612. [DOI] [PubMed] [Google Scholar]
- (94).Rudovsky J, Botta M, Hermann P, Hardcastle KI, Lukes I, Aime S. Bioconjugate Chem. 2006;17:975. doi: 10.1021/bc060149l. [DOI] [PubMed] [Google Scholar]
- (95).Rudovsky J, Hermann P, Botta M, Aime S, Lukes I. Chem. Commun. 2005;18:2390. doi: 10.1039/b418712a. [DOI] [PubMed] [Google Scholar]
- (96).Rudovsky J, Kotek J, Hermann P, Lukes I, Mainero V, Aime S. Org. Biomol. Chem. 2005;3:112. doi: 10.1039/b410103k. [DOI] [PubMed] [Google Scholar]
- (97).Lebduskova P, Sour A, Helm L, Toth E, Kotek J, Lukes I, Merbach AE. Dalton Trans. 2006;28:3399. doi: 10.1039/b517847a. [DOI] [PubMed] [Google Scholar]
- (98).Ali MM, Woods M, Caravan P, Opina ACL, Spiller M, Fettinger JC, Sherry AD. Chem. Eur. J. 2008;14:7250. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Pikkemaat JA, Wegh RT, Lamerichs R, van de Molengraaf RA, Langereis S, Burdinski D, Raymond AYF, Janssen HM, de Waal BFM, Willard NP, Meijer EW, Grüll H. Contrast Media Mol. Imaging. 2007;2:229. doi: 10.1002/cmmi.149. [DOI] [PubMed] [Google Scholar]
- (100).Aime S, Carrera C, Delli Castelli D, Crich SG, Terreno E. Angew. Chem., Int. Ed. 2005;44:1813. doi: 10.1002/anie.200462566. [DOI] [PubMed] [Google Scholar]
- (101).Fulton DA, O'Halloran M, Parker D, Senanayake K, Botta M, Aime S. Chem. Commun. 2005;4:474. doi: 10.1039/b413536a. [DOI] [PubMed] [Google Scholar]
- (102).Merbach AE, Toth E, editors. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. Wiley; Chichester: 2001. [Google Scholar]
- (103).Bourne MW, Margerun L, Hylton N, Campion B, Lai J-J, Derugin N, Higgins CB. J. Magn. Reson. Imaging. 1996;6:305. doi: 10.1002/jmri.1880060209. [DOI] [PubMed] [Google Scholar]
- (104).Weinmann HJ, Ebert W, Wagner S, Taupitz M, Misselwitz M, Schmitt-Wilich H. Proceedings of the IX International Workshop on Magnetic Resonance Angiography Valencia.1997. p. 355. [Google Scholar]
- (105).Adam G, Neuerburg J, Spüntrup E, Mühler A, Scherer K, Günther RW. J. Magn. Reson. Imaging. 1994;4:462. doi: 10.1002/jmri.1880040336. [DOI] [PubMed] [Google Scholar]
- (106).Roberts HC, Saeed M, Roberts TPL, Mühler A, Shames DM, Mann JS, Stiskal M, F. D, Brasch RC. J. Magn. Reson. Imaging. 1997;7:331. doi: 10.1002/jmri.1880070213. [DOI] [PubMed] [Google Scholar]
- (107).Tacke J, Adam G, Claßen H, Mühler A, Prescher A, Günther RW. J. Magn. Reson. Imaging. 1997;7:678. doi: 10.1002/jmri.1880070412. [DOI] [PubMed] [Google Scholar]
- (108).Su M-Y, Mühler A, Lao X, Nalcioglu O. Magn. Reson. Imaging. 1998;39:259. doi: 10.1002/mrm.1910390213. [DOI] [PubMed] [Google Scholar]
- (109).Roberts HC, Saeed M, Roberts TPL, Mühler A, Brasch RC. J. Magn. Reson. Imaging. 1999;9:204. doi: 10.1002/(sici)1522-2586(199902)9:2<204::aid-jmri8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- (110).Kobayashi H, Sato N, Kawamoto S, Saga T, Hiraga A, Ishimori T, Konishi J, Togashi K, Brechbiel MW. Magn. Reson. Med. 2001;46:579. doi: 10.1002/mrm.1230. [DOI] [PubMed] [Google Scholar]
- (111).Dong Q, Hurst DR, Weinmann HJ, Chevenert TL, Londy FJ, Prince MR. Invest. Radiol. 1998;33:699. doi: 10.1097/00004424-199809000-00026. [DOI] [PubMed] [Google Scholar]
- (112).Sato N, Kobayashi H, Hiraga A, Saga T, Togashi K, Konishi J, Brechbiel MW. Magn. Reson. Med. 2001;46:1169. doi: 10.1002/mrm.1314. [DOI] [PubMed] [Google Scholar]
- (113).Kobayashi H, Kawamoto S, Star RA, Waldmann TA, Brechbiel MW, Choyke PL. Bioconjugate Chem. 2003;14:1044. doi: 10.1021/bc034064l. [DOI] [PubMed] [Google Scholar]
- (114).Kobayashi H, Kawamoto S, Star RA, Waldmann TA, Tagaya Y, Brechbiel MW. Cancer Res. 2003;63:271. [PubMed] [Google Scholar]
- (115).Yordanov A, Kobayashi H, English SJ, Reijnders K, Milenic D, Krishna MC, Mitchell JB, Brechbiel MW. J. Mater. Chem. 2003;13:1523. [Google Scholar]
- (116).Langereis S, de Lussanet QG, van Genderen MHP, Meijer EW, Beets-Tan RGH, Griffioen AW, van Engelshoven JMA, Backes WH. NMR Biomed. 2006;19:133. doi: 10.1002/nbm.1015. [DOI] [PubMed] [Google Scholar]
- (117).Kobayashi H, Sato N, Kawamoto S, Saga T, Hiraga A, Laz Haque T, Ishimori T, Konishi J, Togashi K, Brechbiel MW. Bioconjugate Chem. 2001;12:100. doi: 10.1021/bc000075s. [DOI] [PubMed] [Google Scholar]
- (118).Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Ishimori T, Akita Y, Mamede MH, Konishi J, Togashi K, Brechbiel MW. Magn. Reson. Med. 2001;46:795. doi: 10.1002/mrm.1259. [DOI] [PubMed] [Google Scholar]
- (119).Kobayashi H, Saga T, Kawamoto S, Sato N, Hiraga A, Ishimori T, Konishi J, Togashi K, Brechbiel MW. Cancer Res. 2001;61:4966. [PubMed] [Google Scholar]
- (120).Kobayashi H, Kawamoto S, Jo S, Bryant LH, Brechbiel MW, Star RA. Bioconjugate Chem. 2003;14:388. doi: 10.1021/bc025633c. [DOI] [PubMed] [Google Scholar]
- (121).Kobayashi H, Jo S-K, Kawamoto S, Yasuda H, Hu X, Knopp MV, Brechbiel MW, Choyke PL, Star RA. J. Magn. Reson. Imaging. 2004;20:512. doi: 10.1002/jmri.20147. [DOI] [PubMed] [Google Scholar]
- (122).Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Ishimori T, Konishi J, Togashi K, W. BM. Magn. Reson. Med. 2001;46:781. doi: 10.1002/mrm.1257. [DOI] [PubMed] [Google Scholar]
- (123).Kobayashi H, Sato N, Kawamoto S, Saga T, Hiraga A, Ishimori T, Konishi J, Togashi K, Brechbiel MW. Magn. Reson. Med. 2001;46:457. doi: 10.1002/mrm.1214. [DOI] [PubMed] [Google Scholar]
- (124).Kobayashi H, Kawamoto S, Star RA, Waldmann TA, Brechbiel MW, Choyke PL. Bioconjugate Chem. 2003;14:1044. doi: 10.1021/bc034064l. [DOI] [PubMed] [Google Scholar]
- (125).Kobayashi H, Kawamoto S, Choyke PL, Sato N, Knopp MV, Star RA, Waldmann TA, Tagaya Y, Brechbiel MW. Magn. Reson. Med. 2003;50:758. doi: 10.1002/mrm.10583. [DOI] [PubMed] [Google Scholar]
- (126).Koyama Y, Talanov VS, Bernardo M, Hama Y, Regino CAS, Brechbiel MW, Choyke PL, Kobayashi H. J. Magn. Reson. Imaging. 2007;25:866. doi: 10.1002/jmri.20852. [DOI] [PubMed] [Google Scholar]
- (127).Talanov VS, Regino CAS, Kobayashi H, Bernardo M, Choyke PL, Brechbiel MW. Nano Lett. 2006;6:1459. doi: 10.1021/nl060765q. [DOI] [PubMed] [Google Scholar]
- (128).Regino CAS, Walbridge S, Bernardo M, Wong KJ, Johnson D, Lonser R, Oldfield EH, Choyke PL, Brechbiel MW. Contrast Media Mol. Imaging. 2008;3:2. doi: 10.1002/cmmi.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Wu C, Brechbiel MW, Kozak RW, Gansow OA. Bioorg. Med. Chem. 1994;4:449. [Google Scholar]
- (130).Kobayashi H, Sato N, Saga T, Nakamoto Y, Ishimori T, Toyama S, Tagashi K, Konishi J, Brechbiel MW. Eur. J. Nucl. Med. 2000;27:1334. doi: 10.1007/s002590000293. [DOI] [PubMed] [Google Scholar]
- (131).Konda S, Aref M, Brechbiel MW, Wiener EC. Invest. Radiol. 2000;35:50. doi: 10.1097/00004424-200001000-00006. [DOI] [PubMed] [Google Scholar]
- (132).Konda SD, Aref A, Wang S, Brechbiel MW, Wiener EC. MAGMA. 2001;12:104. doi: 10.1007/BF02668091. [DOI] [PubMed] [Google Scholar]
- (133).Konda SD, Wang S, Brechbiel MW, Weiner EC. Invest. Radiol. 2002;37:199. doi: 10.1097/00004424-200204000-00005. [DOI] [PubMed] [Google Scholar]
- (134).Wiener EC, Konda S, Shadron A, Brechbiel M, Gansow O. Invest. Radiol. 1997;32:748. doi: 10.1097/00004424-199712000-00005. [DOI] [PubMed] [Google Scholar]
- (135).Boswell CA, Eck PK, Regino CAS, Bernardo M, Wong KJ, Milenic DE, Choyke PL, Brechbiel MW. Mol. Pharm. 2008;5:527. doi: 10.1021/mp800022a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Kalish H, Arbab AS, Miller BR, Lewis BK, Zywicke HA, Bulte JWM, Bryant LH, Jr, Frank JA. Magn. Reson. Med. 2003;50:275. doi: 10.1002/mrm.10556. [DOI] [PubMed] [Google Scholar]
- (137).Kobayashi H, Kawamoto S, Saga T, Sato N, Ishimori T, Konishi J, Ono K, Togashi K, Brechbiel MW. Bioconjugate Chem. 2001;12:587. doi: 10.1021/bc010002o. [DOI] [PubMed] [Google Scholar]
- (138).Zhu W, Okollie B, Bhujwalla ZM, Artemov D. Magn. Reson. Med. 2008;59:679. doi: 10.1002/mrm.21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Xu H, Regino CAS, Koyama Y, Hama Y, Gunn AJ, Bernardo M, Kobayashi H, Choyke PL, Brechbiel MW. Bioconjugate Chem. 2007;18:1474. doi: 10.1021/bc0701085. [DOI] [PubMed] [Google Scholar]
- (140).Spanoghe M, Lanens D, Dommisse R, Van der Linden A, Alderweireldt F. Magn. Reson. Imaging. 1992;10:913. doi: 10.1016/0730-725x(92)90445-6. [DOI] [PubMed] [Google Scholar]
- (141).Sieving PF, Watson AD, Rocklage SM. Bioconjugate Chem. 1990;1:65. doi: 10.1021/bc00001a008. [DOI] [PubMed] [Google Scholar]
- (142).Schuhmann-Giampieri G, Schmitt-Wilich H, Frenzel T, Press WR, Weinmann HJ. Invest. Radiol. 1991;26:969. doi: 10.1097/00004424-199111000-00008. [DOI] [PubMed] [Google Scholar]
- (143).Van Hecke P, Marchal G, Bosmans H, Johannik K, Jiang Y, Vogler H, Van Ongeval C, Baert AL, Speck U. Magn. Reson. Imaging. 1991;9:313. doi: 10.1016/0730-725x(91)90417-k. [DOI] [PubMed] [Google Scholar]
- (144).Vexler VS, Clément O, Schmitt-Willich H, Brasch RC. J. Magn. Reson. Imaging. 1994;4:381. doi: 10.1002/jmri.1880040325. [DOI] [PubMed] [Google Scholar]
- (145).Marchal G, Bosmans H, Van Hecke P, Speck U, Aerts P, Vanhoenacker P, Baert AL. Am. J. Roentgenol. 1990;155:407. doi: 10.2214/ajr.155.2.2115276. [DOI] [PubMed] [Google Scholar]
- (146).Lim TH, Lee DH, Kim YH, Park SW, Park PH, Seo DM, Kim ST, Lee TK, Mun CW. Radiology. 1993;189:765. doi: 10.1148/radiology.189.3.7694313. [DOI] [PubMed] [Google Scholar]
- (147).Berthezene Y, Vexler V, Jerome H, Sievers R, Moseley ME, Brasch RC. Radiology. 1991;181:773. doi: 10.1148/radiology.181.3.1947095. [DOI] [PubMed] [Google Scholar]
- (148).Berthezene Y, Vexler V, Price D, Wisner-Dupon J, Moseley ME, Aicher KP, Brasch RC. Invest. Radiol. 1992;27:346. doi: 10.1097/00004424-199205000-00004. [DOI] [PubMed] [Google Scholar]
- (149).Berthezene Y, Vexler V, Kuwatsuru R, Rosenau W, Muhler A, Clement O, Price DC, Brasch RC. Radiology. 1992;185:97. doi: 10.1148/radiology.185.1.1523341. [DOI] [PubMed] [Google Scholar]
- (150).Opsahl LR, Uzgiris EE, Vera DR. Acad. Radiol. 1995;2:762. doi: 10.1016/s1076-6332(05)80486-6. [DOI] [PubMed] [Google Scholar]
- (151).Zalipsky S. Adv. Drug Delivery Rev. 1995;16:157. [Google Scholar]
- (152).Abuchowski A, van Es T, Palczuk NC, Davis FF. J. Biol. Chem. 1977;252:3578. [PubMed] [Google Scholar]
- (153).Tóth E, van Uffelen I, Helm L, Merbach AE, Ladd D, Briley-Sæbø K, Kellar KE. Magn. Reson. Chem. 1998;36:S125. [Google Scholar]
- (154).Desser TS, Rubin DL, Muller HH, Qing F, Khodor S, Zanazzi G, Young SW, Ladd DL, Wellons JA, Kellar KE, Toner JL, Snow RA. J. Magn. Reson. Imaging. 1994;4:467. doi: 10.1002/jmri.1880040337. [DOI] [PubMed] [Google Scholar]
- (155).Bogdanov AA, Jr., Weissleder R, Frank HW, Bogdanova AV, Nossif N, Schaffer BK, Tsai E, Papisov MI, Brady TJ. Radiology. 1993;187:701. doi: 10.1148/radiology.187.3.8497616. [DOI] [PubMed] [Google Scholar]
- (156).Fu Y, Raatschen H-J, Nitecki DE, Wendland MF, Novikov V, Fournier LS, Cyran C, Rogut V, Shames DM, Brasch RC. Biomacromolecules. 2007;8:1519. doi: 10.1021/bm061141h. [DOI] [PubMed] [Google Scholar]
- (157).Cyran CC, Fu Y, Raatschen H-J, Rogut V, Chaopathomkul B, Shames DM, Wendland MF, Yeh BM, Brasch RC. J. Magn. Reson. Imaging. 2008;27:581. doi: 10.1002/jmri.21245. [DOI] [PubMed] [Google Scholar]
- (158).Cavagna F, Luchinat C, Scozzafava A, Xia Z. Magn. Reson. Med. 1994;31:58. doi: 10.1002/mrm.1910310109. [DOI] [PubMed] [Google Scholar]
- (159).Allen MJ, Raines RT, Kiessling LL. J. Am. Chem. Soc. 2006;128:6534. doi: 10.1021/ja061383p. [DOI] [PubMed] [Google Scholar]
- (160).Lucas RL, Benjamin M, Reineke TM. Bioconjugate Chem. 2008;19:24. doi: 10.1021/bc700375m. [DOI] [PubMed] [Google Scholar]
- (161).Kellar KE, Henrichs PM, Hollister R, Koenig SH, Eck J, Wei D. Magn. Reson. Med. 1997;38:712. doi: 10.1002/mrm.1910380506. [DOI] [PubMed] [Google Scholar]
- (162).Tóth E, Helm L, Kellar KE, Merbach AE. Chem. Eur. J. 1999;5:1202. [Google Scholar]
- (163).Lee HY, Jee HW, Seo SM, Kwak BK, Khang G, Cho SH. Bioconjugate Chem. 2006;17:700. doi: 10.1021/bc060014f. [DOI] [PubMed] [Google Scholar]
- (164).Ladd DL, Hollister R, Peng X, Wei D, Wu G, Delecki D, Snow RA, Toner JL, Kellar K, Eck J, Desai VC, Raymond G, Kinter LB, Desser TS, Rubin MDDL. Bioconjugate Chem. 1999;10:361. doi: 10.1021/bc980086+. [DOI] [PubMed] [Google Scholar]
- (165).Yan G-P, Liu M-L, Li L-Y. Bioconjugate Chem. 2005;16:967. doi: 10.1021/bc050026l. [DOI] [PubMed] [Google Scholar]
- (166).Zarabi B, Nan A, Zhuo J, Gullapalli R, Ghandehari H. Mol. Pharm. 2006;3:550. doi: 10.1021/mp060072i. [DOI] [PubMed] [Google Scholar]
- (167).Amirkhanov NV, Dimitrov I, Opitz AW, Zhang K, Lackey JP, Cardi CA, Lai S, Wagner NJ, Thakur ML, Wickstrom E. Biopolymers. 2008;89:1061. doi: 10.1002/bip.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Bogdanov AA, Jr., Matuszewski L, Bremer C, Petrovsky A, Weissleder R. Mol. Imaging. 2002;1:16. doi: 10.1162/15353500200200001. [DOI] [PubMed] [Google Scholar]
- (169).Lu Z-R, Wang X, Parker DL, Goodrich KC, Buswell HR. Bioconjugate Chem. 2003;14:715. doi: 10.1021/bc0340464. [DOI] [PubMed] [Google Scholar]
- (170).Wen X, Jackson EF, Price RE, Kim EE, Wu Q, Wallace S, Charnsangavej C, Gelovani JG, Li C. Bioconjugate Chem. 2004;15:1408. doi: 10.1021/bc049910m. [DOI] [PubMed] [Google Scholar]
- (171).Ye F, Ke T, Jeong E-K, Wang X, Sun Y, Johnson M, Lu Z-R. Mol. Pharm. 2006;3:507. doi: 10.1021/mp060052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Mohs AM, Wang X, Goodrich KC, Zong Y, Parker DL, Lu ZR. Bioconjugate Chem. 2004;15:1424. doi: 10.1021/bc049828r. [DOI] [PubMed] [Google Scholar]
- (173).Mohs AM, Zong Y, Guo J, Parker DL, Lu Z-R. Biomacromolecules. 2005;6:2305. doi: 10.1021/bm050194g. [DOI] [PubMed] [Google Scholar]
- (174).Mikawa M, Miwa N, Bräutigam M, Akaike T, Maruyama A. J. Biomed. Mater. Res. 2000;49:390. doi: 10.1002/(sici)1097-4636(20000305)49:3<390::aid-jbm12>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- (175).Wu Y, Zhou Y, Ouari O, Woods M, Zhao P, Soesbe TC, Kiefer GE, Sherry AD. J. Am. Chem. Soc. 2008;130:13854. doi: 10.1021/ja805775u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Strich G, Hagan PL, Gerber KH, Slutsky RA. Radiology. 1985;154:723. doi: 10.1148/radiology.154.3.3969477. [DOI] [PubMed] [Google Scholar]
- (177).Schmiedl U, Ogan MD, Moseley ME, Brasch RC. Am. J. Roentgenol. 1986;147:1263. doi: 10.2214/ajr.147.6.1263. [DOI] [PubMed] [Google Scholar]
- (178).Lauffer RB, Brady TJ. Magn. Reson. Imaging. 1985;3:11. doi: 10.1016/0730-725x(85)90004-9. [DOI] [PubMed] [Google Scholar]
- (179).Schmiedl U, Ogan M, Paajanen H, Marotti M, Crooks LE, Brito AC, Brasch RC. Radiology. 1987;162:205. doi: 10.1148/radiology.162.1.3786763. [DOI] [PubMed] [Google Scholar]
- (180).Ogan MD, Schmiedl U, Moseley ME, Grodd W, Paajanen H, Brasch RC. Invest. Radiol. 1987;22:665. [PubMed] [Google Scholar]
- (181).Paajanen H, Reisto T, Hemmila I, Komu M, Niemi P, Kormano M. Magn. Reson. Med. 1990;13:38. doi: 10.1002/mrm.1910130106. [DOI] [PubMed] [Google Scholar]
- (182).Sherry AD, Cacheris WP, Kuan K-T. Magn. Reson. Imaging. 1988;8:180. doi: 10.1002/mrm.1910080208. [DOI] [PubMed] [Google Scholar]
- (183).Kuwatsuru R, Shames DM, Mühler A, Mintorovitch J, Vexler V, Mann JS, Cohn F, Price D, Huberty J, Brasch RC. Magn. Reson. Med. 1993;30:76. doi: 10.1002/mrm.1910300112. [DOI] [PubMed] [Google Scholar]
- (184).Shames DM, Kuwatsuru R, Vexler V, Mühler A, Brasch RC. Magn. Reson. Imaging. 1993;29:616. doi: 10.1002/mrm.1910290506. [DOI] [PubMed] [Google Scholar]
- (185).Vexler VS, Berthezene Y, Wolfe CL, Sievers R, Dupon JW, Aicher K, Moseley ME, Brasch RC. Invest. Radiol. 1992;27:935. doi: 10.1097/00004424-199211000-00012. [DOI] [PubMed] [Google Scholar]
- (186).Moseley ME, Chew WM, White DL, Kucharczyk J, Litt L, Derugin N, Dupon J, Brasch RB, Norman D. Magn. Reson. Med. 1992;23:21. doi: 10.1002/mrm.1910230104. [DOI] [PubMed] [Google Scholar]
- (187).van Dijke CF, Kirk BA, Peterfy CG, Genant HK, Brasch RC, Kapila S. Radiology. 1997;204:825. doi: 10.1148/radiology.204.3.9280267. [DOI] [PubMed] [Google Scholar]
- (188).Vexler VS, Berthèzene Y, Clément O, Mühler A, Rosenau W, Moseley ME, Brasch RC. J. Magn. Reson. Imaging. 1992;2:311. doi: 10.1002/jmri.1880020311. [DOI] [PubMed] [Google Scholar]
- (189).Schmiedl U, Sievers RE, Brasch RC, Wolfe CL, Chew WM, Ogan MD, Engeseth H, Lipton MJ, Moseley ME. Radiology. 1989;170:351. doi: 10.1148/radiology.170.2.2911657. [DOI] [PubMed] [Google Scholar]
- (190).Sievers RE, Schmiedl U, Wolfe CL, Moseley ME, Parmley WW, Brasch RC, Lipton MJ. Magn. Reson. Imaging. 1989;10:172. doi: 10.1002/mrm.1910100203. [DOI] [PubMed] [Google Scholar]
- (191).Wolfe CL, Moseley ME, Wikstrom MG, Sievers RE, Wendland MF, Dupon JW, Finkbeiner WE, Lipton MJ, Parmley WW, Brasch RC. Circulation. 1989;80:969. doi: 10.1161/01.cir.80.4.969. [DOI] [PubMed] [Google Scholar]
- (192).Bremerich J, Wendland MF, Arheden H, Wyttenbach R, Gao DW, Huberty JP, Dae MW, Higgins CB, Saeed M. J. Am. Coll. Cardiol. 1998;32:787. doi: 10.1016/s0735-1097(98)00315-5. [DOI] [PubMed] [Google Scholar]
- (193).Wikstrom MG, Moseley ME, White DL, Dupon J, Winkelhake JL, Kopplin J, Brasch RC. Invest. Radiol. 1989;24:609. doi: 10.1097/00004424-198908000-00007. [DOI] [PubMed] [Google Scholar]
- (194).Daldrup H, Shames DM, Wendland M, Okuhata Y, Link TM, Rosenau W, Lu Y, Brasch RC. Am. J. Roentgenol. 1998;171:941. doi: 10.2214/ajr.171.4.9762973. [DOI] [PubMed] [Google Scholar]
- (195).Gossmann A, Okuhata Y, Shames DM, Helbich TH, Roberts TPL, Wendland MF, Huber S, Brasch RC. Radiology. 1999;213:265. doi: 10.1148/radiology.213.1.r99oc43265. [DOI] [PubMed] [Google Scholar]
- (196).van Dijke CF, Brasch RC, Roberts TP, Weidner N, Mathur A, Shames DM, Mann JS, Demsar F, Lang P, Schwickert HC. Radiology. 1996;198:813. doi: 10.1148/radiology.198.3.8628876. [DOI] [PubMed] [Google Scholar]
- (197).Turetschek K, Huber S, Floyd E, Helbich T, Roberts TPL, Shames DM, Tarlo KS, Wendland MF, Brasch RC. Radiology. 2001;218:562. doi: 10.1148/radiology.218.2.r01fe37562. [DOI] [PubMed] [Google Scholar]
- (198).Brasch R, Pham P, Shames D, Roberts T, van Dijke K, van Bruggen N, Mann J, Ostrowitzki S, Melnyk O. J. Magn. Reson. Imaging. 1997;7:68. doi: 10.1002/jmri.1880070110. [DOI] [PubMed] [Google Scholar]
- (199).Aicher KP, Dupon JW, White DL, Aukerman SL, Moseley ME, Juster R, Rosenau W, Winkelhake JL, Brasch RC. Cancer Res. 1990;50:7376. [PubMed] [Google Scholar]
- (200).Schwickert HC, Stiskal M, Roberts TP, van Dijke CF, Mann J, Muhler A, Shames DM, Demsar F, Disston A, Brasch RC. Radiology. 1996;198:893. doi: 10.1148/radiology.198.3.8628889. [DOI] [PubMed] [Google Scholar]
- (201).Murad GJA, Walbridge S, Morrison PF, Garmestani K, Degen JW, Brechbiel MW, Oldfield EH, Lonser RR. Clin. Cancer Res. 2006;12:3145. doi: 10.1158/1078-0432.CCR-05-2583. [DOI] [PubMed] [Google Scholar]
- (202).Barrett T, Kobayashi H, Brechbiel M, Choyke PL. Eur. J. Radiol. 2006;60:353. doi: 10.1016/j.ejrad.2006.06.025. [DOI] [PubMed] [Google Scholar]
- (203).Tyeklar Z, Dunham SU, Midelfort K, Scott DM, Sajiki H, Ong K, Lauffer RB, Caravan P, McMurry TJ. Inorg. Chem. 2007;46:6621. doi: 10.1021/ic7006843. [DOI] [PubMed] [Google Scholar]
- (204).Caravan P, Comuzzi C, Crooks W, McMurry TJ, Choppin GR, Woulfe SR. Inorg. Chem. 2001;40:2170. doi: 10.1021/ic001117r. [DOI] [PubMed] [Google Scholar]
- (205).Muller RN, Radüchel B, Laurent S, Platzek J, Piérart C, Mareski P, Elst LV. Eur. J. Inorg. Chem. 1999;11:1949. [Google Scholar]
- (206).Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JW, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. J. Am. Chem. Soc. 2002;124:3152. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- (207).Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg. Chem. 2007;46:6632. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- (208).Zech SG, Eldredge HB, Lowe MP, Caravan P. Inorg. Chem. 2007;46:3576. doi: 10.1021/ic070011u. [DOI] [PubMed] [Google Scholar]
- (209).McMurry TJ, Parmelee DJ, Sajiki H, Scott DM, Ouellet HS, Walovitch RC, Tyeklar Z, Dumas S, Bernard P, Nadler S, Midelfort K, Greenfield M, Troughton J, Lauffer RB. J. Med. Chem. 2002;45:3465. doi: 10.1021/jm0102351. [DOI] [PubMed] [Google Scholar]
- (210).Lauffer RB, Parmelee DJ, Ouellet HS, Dolan RP, Sajiki H, Scott DM, Bernard PJ, Buchanan EM, Ong KY, Tyeklár Z, Midelfort KS, McMurry TJ, Walovitch RC. Acad. Radiol. 1996;3:S356. doi: 10.1016/s1076-6332(96)80583-6. [DOI] [PubMed] [Google Scholar]
- (211).Corot C, Violas J, Robert P, Gagneur G, Port M. Invest. Radiol. 2003;38:311. [PubMed] [Google Scholar]
- (212).Lauffer RB, Parmelee DJ, Dunham SU, Ouellet HS, Dolan RP, Witte S, McMurry TJ, Walovitch RC. Radiology. 1998;207:529. doi: 10.1148/radiology.207.2.9577506. [DOI] [PubMed] [Google Scholar]
- (213).Grist TM, Korosec FR, Peters DC, Witte S, Walovitch RC, Dolan RP, Bridson WE, Yucel EK, Mistretta CA. Radiology. 1998;207:539. doi: 10.1148/radiology.207.2.9577507. [DOI] [PubMed] [Google Scholar]
- (214).Bluemke DA, Stillman AE, Bis KG, Grist TM, Baum RA, D'Agostino R, Malden ES, Pierro JA, Yucel EK. Radiology. 2001;219:114. doi: 10.1148/radiology.219.1.r01ap42114. [DOI] [PubMed] [Google Scholar]
- (215).Perreault P, Edelman MA, Baum RA, Yucel EK, Weisskoff RM, Shamsi K, Mohler ER. Radiology. 2003;229:811. doi: 10.1148/radiol.2293021180. [DOI] [PubMed] [Google Scholar]
- (216).Rapp JH, Wolff SD, Quinn SF, Soto JA, Meranze SG, Muluk S, Blebea J, Johnson SP, Rofsky NM, Duerinckx A, Foster GS, Kent KC, Moneta G, Middlebrook MR, Narra VR, Toombs BD, Pollak J, Yucel EK, Shamsi K, Weisskoff RM. Radiology. 2005;236:71. doi: 10.1148/radiol.2361040148. [DOI] [PubMed] [Google Scholar]
- (217).Goyen M, Edelman MA, Perreault P, O'Riordan E, Bertoni H, Taylor J, Siragusa D, Sharafuddin M, Mohler ER, III, Breger R, Yucel EK, Shamsi K, Weisskoff RM. Radiology. 2005;236:825. doi: 10.1148/radiol.2363040577. [DOI] [PubMed] [Google Scholar]
- (218).Zhang Y, Choyke PL, Lu H, Takahashi H, Mannon RB, Zhang X, Marcos H, Li KCP, Kopp JB. J. Am. Soc. Nephrol. 2005;16:1752. doi: 10.1681/ASN.2004110981. [DOI] [PubMed] [Google Scholar]
- (219).Turetschek K, Floyd E, Helbich T, Roberts TPL, Shames DM, Wendland MF, Carter WO, Brasch RC. J. Magn. Reson. Imaging. 2001;14:237. doi: 10.1002/jmri.1179. [DOI] [PubMed] [Google Scholar]
- (220).Uggeri F, Aime S, Anelli PL, Botta M, Brocchetta M, de Haeen C, Ermondi G, Grandi M, Paoli P. Inorg. Chem. 1995;34:633. [Google Scholar]
- (221).Cavagna FM, Maggioni F, Castelli PM, Dapra M, Imperatori LG, Lorusso V, Jenkins BG. Invest. Radiol. 1997;32:780. doi: 10.1097/00004424-199712000-00009. [DOI] [PubMed] [Google Scholar]
- (222).Prokop M, Schneider G, Vanzulli A, Goyen M, Ruehm SG, Douek P, Dapra M, Pirovano G, Kirchin MA, Spinazzi A. Radiology. 2005;234:399. doi: 10.1148/radiol.2342040023. [DOI] [PubMed] [Google Scholar]
- (223).Aime S, Botta M, Fasano M, Crich SG, Terreno E. J. Biol. Inorg. Chem. 1996;1:312. [Google Scholar]
- (224).Aime S, Chiaussa M, Digilio G, Gianolo E, Terreno E. J. Biol. Inorg. Chem. 1999;4:766. doi: 10.1007/s007750050349. [DOI] [PubMed] [Google Scholar]
- (225).Aime S, Botta M, Crich SG, Giovenzana GB, Pagliarin R, Piccinini M, Sisti M, Terreno E. J. Biol. Inorg. Chem. 1997;2:470. [Google Scholar]
- (226).Botta M, Quici S, Pozzi G, Marzanni G, Pagliarin R, Barra S, Crich SG. Org. Biomol. Chem. 2004;2:570. doi: 10.1039/b313677a. [DOI] [PubMed] [Google Scholar]
- (227).Wallace RA, Haar JPJ, Miller DB, Woulfe SR, Polta JA, Galen KP, Hynes MR, Adzamli K. Magn. Reson. Med. 1998;40:733. doi: 10.1002/mrm.1910400514. [DOI] [PubMed] [Google Scholar]
- (228).Tóth E, Connac F, Helm L, Adzamli K, Merbach AE. J. Biol. Inorg. Chem. 1998;3:606. [Google Scholar]
- (229).Ou M-H, Chen Y-M, Chang Y-H, Lu W-K, Liu G-C, Wang Y-M. Dalton Trans. 2007;26:2749. doi: 10.1039/b703211k. [DOI] [PubMed] [Google Scholar]
- (230).Schuhmann-Giampieri G, Schmitt-Willich H, Press WR, Negishi C, Weinmann HJ, Speck U. Radiology. 1992;183:59. doi: 10.1148/radiology.183.1.1549695. [DOI] [PubMed] [Google Scholar]
- (231).Reimer P, Rummeny EJ, Shamsi K, Balzer T, Daldrup HE, Tombach B, Hesse T, Berns T, Peters PE. Radiology. 1996;199:177. doi: 10.1148/radiology.199.1.8633143. [DOI] [PubMed] [Google Scholar]
- (232).Elst LV, Maton F, Laurent S, Seghi F, Chapelle F, Muller RN. Magn. Reson. Med. 1997;38:604. doi: 10.1002/mrm.1910380415. [DOI] [PubMed] [Google Scholar]
- (233).Elst LV, Chapelle F, Laurent S, Muller RN. J. Biol. Inorg. Chem. 2001;6:196. doi: 10.1007/s007750000195. [DOI] [PubMed] [Google Scholar]
- (234).Martin VV, Ralston WH, Hynes MR, Keana JFW. Bioconjugate Chem. 1995;6:616. doi: 10.1021/bc00035a017. [DOI] [PubMed] [Google Scholar]
- (235).Parac-Vogt TN, Kimpe K, Laurent S, Elst LV, Burtea C, Chen F, Muller RN, Ni Y, Verbruggen A, Binnemans K. Chem. Eur. J. 2005;11:3077. doi: 10.1002/chem.200401207. [DOI] [PubMed] [Google Scholar]
- (236).Nivorozhkin AL, Kolodziej AF, Caravan P, Greenfield MT, Lauffer RB, McMurry TJ. Angew. Chem., Int. Ed. 2001;40:2903. [PubMed] [Google Scholar]
- (237).Hanaoka K, Kikuchi K, Terai T, Komatsu T, Nagano T. Chem. Eur. J. 2008;14:987. doi: 10.1002/chem.200700785. [DOI] [PubMed] [Google Scholar]
- (238).Anelli PL, Bertini I, Fragai M, Lattuada L, Luchinat C, Parigi G. Eur. J. Inorg. Chem. 2000;4:625. [Google Scholar]
- (239).Tomaselli S, Zanzoni S, Ragona L, Gianolio E, Aime S, Assfalg M, Molinari H. J. Med. Chem. 2008;51:6782. doi: 10.1021/jm800820b. [DOI] [PubMed] [Google Scholar]
- (240).De Leon-Rodriguez LM, Ortiz A, Weiner AL, Zhang S, Kovacs Z, Kodadek T, Sherry AD. J. Am. Chem. Soc. 2002;124:3514. doi: 10.1021/ja025511v. [DOI] [PubMed] [Google Scholar]
- (241).Botnar RM, Perez AS, Witte S, Wiethoff AJ, Laredo J, Hamilton J, Quist W, Parsons EC, Jr., Vaidya A, Kolodziej AF, Barrett JA, Graham PB, Weisskoff RM, Manning WJ, Johnstone MT. Circulation. 2004;109:2023. doi: 10.1161/01.CIR.0000127034.50006.C0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. J. Am. Chem. Soc. 2008;130:6025. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- (243).De Leon-Rodriguez LM, Kovacs Z. Bioconjugate Chem. 2008;19:391. doi: 10.1021/bc700328s. [DOI] [PubMed] [Google Scholar]
- (244).Aime S, Frullano L, Crich SG. Angew. Chem., Int. Ed. 2002;41:1017. doi: 10.1002/1521-3773(20020315)41:6<1017::aid-anie1017>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- (245).Karfeld LS, Bull SR, Davis NE, Meade TJ, Barron AE. Bioconjugate Chem. 2007;18:1697. doi: 10.1021/bc700149u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Yang JJ, Yang J, Wei L, Zurkiya O, Yang W, Li S, Zou J, Zhou Y, Maniccia ALW, Mao H, Zhao F, Malchow R, Zhao S, Johnson J, Hu X, Krogstad E, Liu Z-R. J. Am. Chem. Soc. 2008;130:9260. doi: 10.1021/ja800736h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Gustafsson B, Youens S, Louie AY. Bioconjugate Chem. 2006;17:538. doi: 10.1021/bc060018k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Langereis S, Kooistra H-AT, Genderen M. H. P. v., Meijer EW. Org. Biomol. Chem. 2004;2:1271. doi: 10.1039/b402917h. [DOI] [PubMed] [Google Scholar]
- (249).Dirksen A, Langereis S, Waal B. F. M. d., Genderen M. H. P. v., Hackeng TM, Meijer EW. Chem. Commun. 2005;22:2811. doi: 10.1039/b502347e. [DOI] [PubMed] [Google Scholar]
- (250).Jung H.-i., Kettunen MI, Davletov B, Brindle KM. Bioconjugate Chem. 2004;15:983. doi: 10.1021/bc049899q. [DOI] [PubMed] [Google Scholar]
- (251).Neves AA, Krishnan AS, Kettunen MI, Hu D, de Backer MM, Davletov B, Brindle KM. Nano Lett. 2007;7:1419. doi: 10.1021/nl070126v. [DOI] [PubMed] [Google Scholar]
- (252).Krishnan AS, Neves AA, de Backer MM, Hu D-E, Davletov B, Kettunen MI, Brindle KM. Radiology. 2008;246:854. doi: 10.1148/radiol.2463070471. [DOI] [PubMed] [Google Scholar]
- (253).Hnatowich DJ, Layne WW, Childs RL, Lanteigne D, Davis MA, Griffin TW, Doherty PW. Science. 1983;220:613. doi: 10.1126/science.6836304. [DOI] [PubMed] [Google Scholar]
- (254).Paik CH, Ebbert MA, Murphy PR, Lassman CR, Reba RC, Eckelman WC, Pak KY, Powe J, Steplewski Z, Koprowski H. J. Nucl. Med. 1983;24:1158. [PubMed] [Google Scholar]
- (255).Unger EC, Totty WG, Neufeld DM, Otsuka FL, Murphy WA, Welch MS, Connett JM, Philpott GW. Invest. Radiol. 1985;20:693. doi: 10.1097/00004424-198510000-00008. [DOI] [PubMed] [Google Scholar]
- (256).Anderson-Berg WT, Strand M, Lempert TE, Rosenbaum AE, Joseph PM. J. Nucl. Med. 1986;27:829. [PubMed] [Google Scholar]
- (257).Shahbazi-Gahrouei D, Williams M, Rizvi S, Allen BJ. J. Magn. Reson. Imaging. 2001;14:169. doi: 10.1002/jmri.1168. [DOI] [PubMed] [Google Scholar]
- (258).Kuriu Y, Otsuji E, Kin S, Nakase Y, Fukuda K-I, Okamoto K, Hagiwara A, Yamagishi H. J. Surg. Oncol. 2006;94:144. doi: 10.1002/jso.20411. [DOI] [PubMed] [Google Scholar]
- (259).Curtet C, Tellier C, Bohy J, Conti ML, Saccavini JC, Thedrez P, Douillard JY, Chatal JF, Koprowski H. Proc. Natl. Acad. Sci. 1986;83:4277. doi: 10.1073/pnas.83.12.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Curtet C, Bourgoin C, Bohy J, Saccavini J-C, Thédrez P, Akoka S, Tellier C, Chatal J-F. Int. J. Cancer. 1988;41:126. doi: 10.1002/ijc.2910410728. [DOI] [PubMed] [Google Scholar]
- (261).Shreve P, Aisen AM. Magn. Reson. Imaging. 1986;3:336. doi: 10.1002/mrm.1910030220. [DOI] [PubMed] [Google Scholar]
- (262).Manabe Y, Longley C, Furmanski P. Biochim. Biophys. Acta. 1986;883:460. doi: 10.1016/0304-4165(86)90285-0. [DOI] [PubMed] [Google Scholar]
- (263).Göhr-Rosenthal S, Schmitt-Wilich H, Ebert W, Conrad J. Invest. Radiol. 1993;28:789. [PubMed] [Google Scholar]
- (264).Artemov D, Mori N, Ravi R, Bhujwalla ZM. Cancer Res. 2003;63:2723. [PubMed] [Google Scholar]
- (265).Fagnani R, Hagan MS, Bartholomew R. Cancer Res. 1990;50:3638. [PubMed] [Google Scholar]
- (266).Fagnani R, Halpern S, Hagan MS. Nucl. Med. Commun. 1995;16:362. doi: 10.1097/00006231-199505000-00008. [DOI] [PubMed] [Google Scholar]
- (267).Mehvar R. J. Control. Release. 2000;69:1. doi: 10.1016/s0168-3659(00)00302-3. [DOI] [PubMed] [Google Scholar]
- (268).Gibby WA, Bogdan A, Ovitt TW. Invest. Radiol. 1989;24:302. doi: 10.1097/00004424-198904000-00009. [DOI] [PubMed] [Google Scholar]
- (269).Armitage FE, Richardson DE, Li KCP. Bioconjugate Chem. 1990;1:365. doi: 10.1021/bc00006a001. [DOI] [PubMed] [Google Scholar]
- (270).Rongved P, Klaveness J. Carbohydr. Res. 1991;214:315. doi: 10.1016/0008-6215(91)80038-o. [DOI] [PubMed] [Google Scholar]
- (271).Bligh SWA, Harding CT, Sadler PJ, Bulman RA, Bydder GM, Pennock JM, Kelly JD, Clatham IA, Marriott JA. Magn. Reson. Med. 1991;17:516. doi: 10.1002/mrm.1910170222. [DOI] [PubMed] [Google Scholar]
- (272).Mann JS, Huang JC, Keana JFW. Bioconjugate Chem. 1992;3:154. doi: 10.1021/bc00014a010. [DOI] [PubMed] [Google Scholar]
- (273).Chu W-J, Elgavish GA. NMR Biomed. 1995;8:159. doi: 10.1002/nbm.1940080404. [DOI] [PubMed] [Google Scholar]
- (274).Meyer D, Schaefer M, Chambon C, Beaute S. Invest. Radiol. 1994;29:S90. doi: 10.1097/00004424-199406001-00030. [DOI] [PubMed] [Google Scholar]
- (275).Rebizak R, Schaefer M, Dellacherie E. Bioconjugate Chem. 1997;8:605. doi: 10.1021/bc970062n. [DOI] [PubMed] [Google Scholar]
- (276).Rebizak R, Schaefer M, Dellacherie E. Bioconjugate Chem. 1998;9:94. doi: 10.1021/bc9701499. [DOI] [PubMed] [Google Scholar]
- (277).Helbich TH, Gossman A, Mareski PA, Radüchel B, Roberts TPL, Shames DM, Mühler M, Turetschek K, Brasch RC. J. Magn. Reson. Imaging. 2000;11:694. doi: 10.1002/1522-2586(200006)11:6<694::aid-jmri17>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- (278).Corsi DM, Elst LV, Muller RN, van Bekkum H, Peters JA. Chem. Eur. J. 2001;7:64. doi: 10.1002/1521-3765(20010105)7:1<64::aid-chem64>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- (279).Lebduskova P, Kotek J, Hermann P, VanderElst L, Muller RN, Lukes I, Peters JA. Bioconjugate Chem. 2004;15:881. doi: 10.1021/bc049966g. [DOI] [PubMed] [Google Scholar]
- (280).Gibby WA, Billings J, Hall J, Ovitt TW. Invest. Radiol. 1990;25:164. doi: 10.1097/00004424-199002000-00012. [DOI] [PubMed] [Google Scholar]
- (281).Wang SC, Wikstrom MG, White DL. Radiology. 1990;175:483. doi: 10.1148/radiology.175.2.1691513. [DOI] [PubMed] [Google Scholar]
- (282).Wikstrom M, Martinussen HJ, Wikstrom G, Ericsson A, Nyman R, Waldenstrom A, Hemmingsson A. Acta Radiol. 1992;33:301. [PubMed] [Google Scholar]
- (283).Sirlin CB, Vera DR, Corbeil JA, Caballero MB, Buxton RB, Mattrey RF. Acad. Radiol. 2004;11:1361. doi: 10.1016/j.acra.2004.11.016. [DOI] [PubMed] [Google Scholar]
- (284).Loubeyre P, Canet E, Zhao S, Benderbous S, Amiel M, Revel D. Invest. Radiol. 1996;31:288. doi: 10.1097/00004424-199605000-00008. [DOI] [PubMed] [Google Scholar]
- (285).Casali C, Canet E, Obadia FJ, Benderbous S, Desenfant A, Revel D, Janier M. Acad. Radiol. 1998;5:S214. doi: 10.1016/s1076-6332(98)80109-8. [DOI] [PubMed] [Google Scholar]
- (286).Kroft LJM, Doornbos J, Benderbous S, De Roos A. J. Magn. Reson. Imaging. 1999;9:777. doi: 10.1002/(sici)1522-2586(199906)9:6<777::aid-jmri4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- (287).Jacobs RE, Fraser SE. Science. 1994;263:681. doi: 10.1126/science.7508143. [DOI] [PubMed] [Google Scholar]
- (288).Huber MM, Staubli AB, Kustedjo K, Gray MHB, Shih J, Fraser SE, Jacobs RE, Meade TJ. Bioconjugate Chem. 1998;9:242. doi: 10.1021/bc970153k. [DOI] [PubMed] [Google Scholar]
- (289).Modo M, Cash D, Mellodew K, Williams SCR, Fraser SE, Meade TJ, Price J, Hodges H. Neuroimage. 2002;17:803. [PubMed] [Google Scholar]
- (290).Takahashi M, Hara Y, Aoshima K, Kurihara H, Oshikawa T, Yamashita M. Tetrahedron Lett. 2000;41:8485. [Google Scholar]
- (291).Fulton DA, Elemento EM, Aime S, Chaabane L, Botta M, Parker D. Chem. Commun. 2006;10:1064. doi: 10.1039/b517997a. [DOI] [PubMed] [Google Scholar]
- (292).Tanaka H, Ando Y, Wada M, Takahashi T. Org. Biomol. Chem. 2005;3:3311. doi: 10.1039/b507824e. [DOI] [PubMed] [Google Scholar]
- (293).Bammer R, de Crespigny AJ, Howard D, Seri S, Hashiguchi Y, Nakatani A, Moseley ME. Magn. Reson. Imaging. 2004;22:619. doi: 10.1016/j.mri.2004.01.044. [DOI] [PubMed] [Google Scholar]
- (294).de Crespigny AJ, Howard D, D'Arceuil H, Muller H, Agoston AT, Seri S, Hashiguchi Y, Fujimoto C, Nakatani A, Moseley ME. Magn. Reson. Imaging. 1999;17:1297. doi: 10.1016/s0730-725x(99)00079-x. [DOI] [PubMed] [Google Scholar]
- (295).Vera DR, Buonocore MH, Wisner ER, Katzberg RW, Stadalnik RC. Acad. Radiol. 1995;2:497. doi: 10.1016/s1076-6332(05)80407-6. [DOI] [PubMed] [Google Scholar]
- (296).André JP, Geraldes CFGC, Martins JA, Merbach AE, Prata MIM, Santos AC, de Lima JJP, Tóth E. Chem. Eur. J. 2004;10:5804. doi: 10.1002/chem.200400187. [DOI] [PubMed] [Google Scholar]
- (297).Baía P, André JP, Geraldes CFGC, Martins JA, Merbach AE, Tóth E. Eur. J. Inorg. Chem. 2005;11:2110. [Google Scholar]
- (298).Prata MIM, Santos AC, Torres S, André JP, Martins JA, Neves M, García-Martín ML, Rodrigues TB, López-Larrubia P, Cerdán S, Geraldes CFGC. Contrast Media Mol. Imaging. 2006;1:246. doi: 10.1002/cmmi.111. [DOI] [PubMed] [Google Scholar]
- (299).Unger EC, Shen D-K, Fritz TA. J. Magn. Reson. Imaging. 1993;3:195. doi: 10.1002/jmri.1880030132. [DOI] [PubMed] [Google Scholar]
- (300).Unger E, Fritz T, Wu G, Shen D, Kulik B, New T, Crowell M, Wilke N. J. Liposom. Res. 1994;4:811. [Google Scholar]
- (301).Krause W, Klopp R, Leike J, Sachse A, Schuhmann-Giampieri G. J. Liposom. Res. 1995;5:1. [Google Scholar]
- (302).Torchilin VP. Mol. Med. Today. 1996;2:242. doi: 10.1016/1357-4310(96)88805-8. [DOI] [PubMed] [Google Scholar]
- (303).Mulder WJM, Strijkers GJ, van Tilborg GAF, Griffioen AW, Nicolay K. NMR Biomed. 2006;19:142. doi: 10.1002/nbm.1011. [DOI] [PubMed] [Google Scholar]
- (304).Terreno E, Delli Castelli D, Cabella C, Dastrù W, Sanino A, Stancanello J, Tei L, Aime S. Chem. Biodiv. 2008;5:1901. doi: 10.1002/cbdv.200890178. [DOI] [PubMed] [Google Scholar]
- (305).Barsky D, B. P, Schulten K, Magin RL. Magn. Reson. Med. 1992;24:1. doi: 10.1002/mrm.1910240102. [DOI] [PubMed] [Google Scholar]
- (306).Pütz B, Barsky D, Schulten K. J. Liposom. Res. 1994;4:771. [Google Scholar]
- (307).Bacic G, Niesman MR, Bennett HF, Magin RL, Swartz HM. Magn. Reson. Med. 1988;6:445. doi: 10.1002/mrm.1910060410. [DOI] [PubMed] [Google Scholar]
- (308).Caride VJ, Sostman HD, Winchell RJ, Gore JC. Magn. Reson. Imaging. 1984;2:107. doi: 10.1016/0730-725x(84)90064-x. [DOI] [PubMed] [Google Scholar]
- (309).Navon G, Panigel R, Valensin G. Magn. Reson. Med. 1986;3:876. doi: 10.1002/mrm.1910030608. [DOI] [PubMed] [Google Scholar]
- (310).Tilcock C, Unger E, Cullis P, MacDougall P. Radiology. 1989;171:77. doi: 10.1148/radiology.171.1.2928549. [DOI] [PubMed] [Google Scholar]
- (311).Fossheim SL, Fahlvik AK, Klaveness J, Muller RN. Magn. Reson. Imaging. 1999;17:83. doi: 10.1016/s0730-725x(98)00141-6. [DOI] [PubMed] [Google Scholar]
- (312).Løkling K-E, Fossheim SL, Skurtveit R, Bjørnerud A, Klaveness J. Magn. Reson. Imaging. 2001;19:731. doi: 10.1016/s0730-725x(01)00380-0. [DOI] [PubMed] [Google Scholar]
- (313).Løkling J-E, Skurtveit R, Bjørnerud A, Fossheim SL. Magn. Reson. Med. 2004;51:688. doi: 10.1002/mrm.20009. [DOI] [PubMed] [Google Scholar]
- (314).Wang T, Hossann M, Reinl HM, Peller M, Eibl H, Reiser M, Issels RD, Lindner LH. Contrast Media Mol. Imaging. 2008;3:19. doi: 10.1002/cmmi.226. [DOI] [PubMed] [Google Scholar]
- (315).Kabalka G, Buonocore E, Hubner K, Moss T, Norley N, Huang L. Radiology. 1987;163:255. doi: 10.1148/radiology.163.1.3454163. [DOI] [PubMed] [Google Scholar]
- (316).Kabalka GW, Buonocore E, Hubner K, Davis M, Huang L. Magn. Reson. Med. 1988;8:89. doi: 10.1002/mrm.1910080111. [DOI] [PubMed] [Google Scholar]
- (317).Kabalka GW, Davis MA, Moss TH, Buonocore E, Hubner K, Holmberg E, Maruyama K, Huang L. Magn. Reson. Med. 1991;19:406. doi: 10.1002/mrm.1910190231. [DOI] [PubMed] [Google Scholar]
- (318).Tilcock C, Ahkong QF, Koenig SH, Brown RD, Davis M, Kabalka G. Magn. Reson. Imaging. 1992;27:44. doi: 10.1002/mrm.1910270106. [DOI] [PubMed] [Google Scholar]
- (319).Storrs RW, Tropper FD, Li HY, Song CK, Kuniyoshi JK, Sipkins DA, Li KCP, Bednarski MD. J. Am. Chem. Soc. 1995;117:7301. doi: 10.1002/jmri.1880050617. [DOI] [PubMed] [Google Scholar]
- (320).Gløgård C, Stensrud G, Aime S. Magn. Reson. Chem. 2003;41:585. [Google Scholar]
- (321).Vaccaro M, Accardo A, Tesauro D, Mangiapia G, Lof D, Schillen K, Soderman O, Morelli G, Paduano L. Langmuir. 2006;22:6635. doi: 10.1021/la053500k. [DOI] [PubMed] [Google Scholar]
- (322).Glogard C, Hovland R, Fossheim SL, Aasen AJ, Klaveness J. J. Chem. Soc., Perkin Trans. 2. 2000;5:1047. [Google Scholar]
- (323).André JP, Tóth E, Fischer H, Seelig A, Mäcke HR, Merbach AE. Chem. Eur. J. 1999;5:2977. [Google Scholar]
- (324).Hovland R, Glogard C, Aasen AJ, Klaveness J. Org. Biomol. Chem. 2003;1:644. doi: 10.1039/b211039c. [DOI] [PubMed] [Google Scholar]
- (325).Hovland R, Glogard C, Aasen AJ, Klaveness J. J. Chem. Soc., Perkin Trans. 2. 2001;6:929. [Google Scholar]
- (326).Zhang G, Zhang R, Wen X, Li L, Li C. Biomacromolecules. 2008;9:36. doi: 10.1021/bm700713p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Ward KM, Aletras AH, Balaban RS. J. Magn. Reson. Imaging. 2000;143:79. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- (328).Aime S, Delli Castelli D, Terreno E. Angew. Chem., Int. Ed. 2005;44:5513. doi: 10.1002/anie.200501473. [DOI] [PubMed] [Google Scholar]
- (329).Zhao JM, Har-el Y, McMahon MT, Zhou J, Sherry AD, Sgouros G, Bulte JWM, van Zijl PCM. J. Am. Chem. Soc. 2008;130:5178. doi: 10.1021/ja710159q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Aime S, Delli Castelli D, Lawson D, Terreno E. J. Am. Chem. Soc. 2007;129:2430. doi: 10.1021/ja0677867. [DOI] [PubMed] [Google Scholar]
- (331).Terreno E, Barge A, Beltrami L, Cravotto G, Delli Castelli D, Fedeli F, Jebasingh B, Aime S. Chem. Commun. 2008;5:600. doi: 10.1039/b715383j. [DOI] [PubMed] [Google Scholar]
- (332).Terreno E, Delli Castelli D, Violante E, Sanders HMHF, Sommerdijk AJM, Aime S. Chem. Eur. J. 2009;15:1440. doi: 10.1002/chem.200801766. [DOI] [PubMed] [Google Scholar]
- (333).Terreno E, Cabella C, Carrera C, Delli Castelli D, Mazzon R, Rollet S, Stancanello J, Visigalli M, Aime S. Angew. Chem., Int. Ed. 2007;46:966. doi: 10.1002/anie.200604027. [DOI] [PubMed] [Google Scholar]
- (334).Delli Castelli D, Terreno E, Carrera C, Giovenzana GB, Mazzon R, Rollet S, Visigalli M, Aime S. Inorg. Chem. 2008;47:2928. doi: 10.1021/ic702501y. [DOI] [PubMed] [Google Scholar]
- (335).Terreno E, Delli Castelli D, Milone L, Rollet S, Stancanello J, Violante E, Aime S. Contrast Media Mol. Imaging. 2008;3:38. doi: 10.1002/cmmi.225. [DOI] [PubMed] [Google Scholar]
- (336).Langereis S, Keupp J, van Velthoven JLJ, de Roos IHC, Burdinski D, Pikkemaat JA, Grull H. J. Am. Chem. Soc. 2009;131:1380. doi: 10.1021/ja8087532. [DOI] [PubMed] [Google Scholar]
- (337).Grant CWM, Karlik S, Florio E. Magn. Reson. Med. 1989;11:236. doi: 10.1002/mrm.1910110211. [DOI] [PubMed] [Google Scholar]
- (338).Karlik S, Florio E, Grant CWM. Magn. Reson. Med. 1991;19:56. doi: 10.1002/mrm.1910190106. [DOI] [PubMed] [Google Scholar]
- (339).Unger E, Shen D-K, Wu G, Fritz T. Magn. Reson. Imaging. 1991;22:304. doi: 10.1002/mrm.1910220229. [DOI] [PubMed] [Google Scholar]
- (340).Unger EC, Winokur T, MacDougall P, Rosenblum J, Clair M, Gatenby R, Tilcock C. Radiology. 1989;171:81. doi: 10.1148/radiology.171.1.2928550. [DOI] [PubMed] [Google Scholar]
- (341).Frich L, Bjørnerud A, Fossheim S, Tillung T, Gladhaug I. Magn. Reson. Med. 2004;52:1302. doi: 10.1002/mrm.20289. [DOI] [PubMed] [Google Scholar]
- (342).Viglianti BL, Abraham SA, Michelich CR, Yarmolenko PS, MacFall JR, Bally MB, Dewhirst MW. Magn. Reson. Med. 2004;51:1153. doi: 10.1002/mrm.20074. [DOI] [PubMed] [Google Scholar]
- (343).Frias JC, Williams KJ, Fisher EA, Fayad ZA. J. Am. Chem. Soc. 2004;126:16316. doi: 10.1021/ja044911a. [DOI] [PubMed] [Google Scholar]
- (344).Mulder WJM, Douma K, Koning GA, van Zandvoort MA, Lutgens E, Daemen MJ, Nicolay K, Strijkers GJ. Magn. Reson. Med. 2006;55:1170. doi: 10.1002/mrm.20883. [DOI] [PubMed] [Google Scholar]
- (345).Chen W, Vucic E, Leupold E, Mulder WJM, Cormode DP, Briley-Saebo KC, Barazza A, Fisher EA, Dathe M, Fayad ZA. Contrast Media Mol. Imaging. 2008;3:233. doi: 10.1002/cmmi.257. [DOI] [PubMed] [Google Scholar]
- (346).Unger EC, Fritz TA, Tilcock C, New TE. J. Magn. Reson. Imaging. 1991;1:689. doi: 10.1002/jmri.1880010613. [DOI] [PubMed] [Google Scholar]
- (347).Storrs RW, Tropper FD, Li HY, Song CK, Sipkins DA, Kuniyoshi JK, Bednarski MD, Strauss HW, Li KCP. J. Magn. Reson. Imaging. 1995;5:719. doi: 10.1002/jmri.1880050617. [DOI] [PubMed] [Google Scholar]
- (348).Bertini I, Bianchini F, Calorini L, Colagrande S, Fragai M, Franchi A, Gallo O, Gavazzi C, Luchinat C. Magn. Reson. Med. 2004;52:669. doi: 10.1002/mrm.20189. [DOI] [PubMed] [Google Scholar]
- (349).Ayyagari AL, Zhang X, Ghaghada KB, Annapragada A, Hu X, Bellamkonda RV. Magn. Reson. Imaging. 2006;55:1023. doi: 10.1002/mrm.20846. [DOI] [PubMed] [Google Scholar]
- (350).van Tilborg GAF, Strijkers GJ, Pouget EM, Reutelingsperger CPM, Sommerdijk NAJM, Nicolay K, Mulder WJM. Magn. Reson. Imaging. 2008;60:1444. doi: 10.1002/mrm.21780. [DOI] [PubMed] [Google Scholar]
- (351).Accardo A, Tesauro D, Roscigno P, Gianolio E, Paduano L, D'Errico G, Pedone C, Morelli G. J. Am. Chem. Soc. 2004;126:3097. doi: 10.1021/ja039195b. [DOI] [PubMed] [Google Scholar]
- (352).Mangiapia G, Accardo A, Lo Celso F, Tesauro D, Morelli G, Radulescu A, Paduano L. J. Phys. Chem. B. 2004;108:17611. [Google Scholar]
- (353).Morisco A, Accardo A, Gianolio E, Tesauro D, Benedetti E, Morelli G. J. Pept. Sci. 2009;15:242. doi: 10.1002/psc.1087. [DOI] [PubMed] [Google Scholar]
- (354).Mulder WJM, Strijkers GJ, Griffioen AW, van Bloois L, Molema G, Storm G, Koning GA, Nicolay K. Bioconjugate Chem. 2004;15:799. doi: 10.1021/bc049949r. [DOI] [PubMed] [Google Scholar]
- (355).Erdogan S, Medarova ZO, Roby A, Moore A, Torchilin VP. J. Magn. Reson. Imaging. 2008;27:574. doi: 10.1002/jmri.21202. [DOI] [PubMed] [Google Scholar]
- (356).Vuu K, Xie J, McDonald MA, Bernardo M, Hunter F, Zhang Y, Li K, Bednarski M, Guccione S. Bioconjugate Chem. 2005;16:995. doi: 10.1021/bc050085z. [DOI] [PubMed] [Google Scholar]
- (357).Oliver M, Ahmad A, Kamaly N, Perouzel E, Caussin A, Keller M, Herlihy A, Bell J, Miller AD, Jorgensen MR. Org. Biomol. Chem. 2006;4:3489. doi: 10.1039/b605394g. [DOI] [PubMed] [Google Scholar]
- (358).Kamaly N, Kalber T, Ahmad A, Oliver MH, So P-W, Herlihy AH, Bell JD, Jorgensen MR, Miller AD. Bioconjugate Chem. 2008;19:118. doi: 10.1021/bc7001715. [DOI] [PubMed] [Google Scholar]
- (359).Esposito G, Crich SG, Aime S. ChemMedChem. 2008;3:1858. doi: 10.1002/cmdc.200800234. [DOI] [PubMed] [Google Scholar]
- (360).Douglas T, Young M. Nature. 1998;393:152. [Google Scholar]
- (361).Wang Q, Lin T, Tang L, Johnson JE, Finn MG. Angew. Chem., Int. Ed. 2002;41:459. doi: 10.1002/1521-3773(20020201)41:3<459::aid-anie459>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- (362).Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE, Finn MG. Biomacromolecules. 2003;4:472. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- (363).Allen M, Bulte JWM, Liepold L, Basu G, Zywicke HA, Frank JA, Young M, Douglas T. Magn. Reson. Imaging. 2005;54:807. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- (364).Anderson EA, Isaacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Nano Lett. 2006;6:1160. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- (365).Prasuhn DEJ, Yeh RM, Obenaus A, Manchester M, Finn MG. Chem. Commun. 2007;12:1269. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- (366).Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Nano Lett. 2007;7:2207. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- (367).Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. J. Am. Chem. Soc. 2008;130:2546. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- (368).Vasalatiy O, Gerard RD, Zhao P, Sun X, Sherry AD. Bioconjugate Chem. 2008;19:598. doi: 10.1021/bc7002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Wilson LJ. Electrochem. Soc. Interface. 1999;8:24. [Google Scholar]
- (370).Mikawa M, Kato H, Okumura M, Narazaki M, Kanazawa Y, Miwa N, Shinohara H. Bioconjugate Chem. 2001;12:510. doi: 10.1021/bc000136m. [DOI] [PubMed] [Google Scholar]
- (371).Funasaka H, Sakurai K, Oda Y, Yamamoto K, Takahashi T. Chem. Phys. Lett. 1995;232:273. [Google Scholar]
- (372).Kato H, Suenaga K, Mikawa M, Okumura M, Miwa N, Yashiro A, Fujimura H, Mizuno A, Nishida Y, Kobayaashi K, Shinohara H. Chem. Phys. Lett. 2000;324:255. [Google Scholar]
- (373).Kato H, Kanazawa Y, Okumura M, Taninaka A, Yokawa T, Shinohara H. J. Am. Chem. Soc. 2003;125:4391. doi: 10.1021/ja027555+. [DOI] [PubMed] [Google Scholar]
- (374).Shu C-Y, Gan L-H, Wang C-R, Pei X-L, Han H-B. Carbon. 2006;44:496. [Google Scholar]
- (375).Ge Z, Duchamp JC, Cai T, Gibson HW, Dorn HC. J. Am. Chem. Soc. 2005;127:16292. doi: 10.1021/ja055089t. [DOI] [PubMed] [Google Scholar]
- (376).Fatouros PP, Corwin FD, Chen Z-J, Broaddus WC, Tatum JL, Kettenmann B, Ge Z, Gibson HW, Russ JL, Leonard AP, Duchamp JC, Dorn HC. Radiology. 2006;240:756. doi: 10.1148/radiol.2403051341. [DOI] [PubMed] [Google Scholar]
- (377).Bolskar RD, Benedetto AF, Husebo LO, Price RE, Jackson EF, Wallace S, Wilson LJ, Alford JM. J. Am. Chem. Soc. 2003;125:5471. doi: 10.1021/ja0340984. [DOI] [PubMed] [Google Scholar]
- (378).Raebiger JW, Bolskar RD. J. Phys. Chem. C. 2008;112:6605. [Google Scholar]
- (379).Toth E, Bolskar RD, Borel A, Gonzalez G, Helm L, Merbach AE, Sitharaman B, Wilson LJ. J. Am. Chem. Soc. 2005;127:799. doi: 10.1021/ja044688h. [DOI] [PubMed] [Google Scholar]
- (380).Sitharaman B, Bolskar RD, Rusakova I, Wilson LJ. Nano Lett. 2004;4:2373. [Google Scholar]
- (381).Shu C-Y, Zhang E-Y, Xiang J-F, Zhu C-F, Wang C-R, Pei X-L, Han H-B. J. Phys. Chem. B. 2006;110:15597. doi: 10.1021/jp0615609. [DOI] [PubMed] [Google Scholar]
- (382).Laus S, Sitharaman B, Toth E, Bolskar RD, Helm L, Asokan S, Wong MS, Wilson LJ, Merbach AE. J. Am. Chem. Soc. 2005;127:9368. doi: 10.1021/ja052388+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Laus S, Sitharaman B, Toth E, Bolskar RD, Helm L, Wilson LJ, Merbach AE. J. Phys. Chem. C. 2007;111:5633. [Google Scholar]
- (384).Sitharaman B, Tran LA, Pham QP, Bolskar RD, Muthupillai R, Flamm SD, Mikos AG, Wilson LJ. Contrast Media Mol. Imaging. 2007;2:139. doi: 10.1002/cmmi.140. [DOI] [PubMed] [Google Scholar]
- (385).Bolskar RD. Nanomedicine. 2008;3:201. doi: 10.2217/17435889.3.2.201. [DOI] [PubMed] [Google Scholar]
- (386).Sitharaman B, Kissell KR, Hartman KB, Tran LA, Baikalov A, Rusakova I, Sun Y, Khant HA, Ludtke SJ, Chiu W, Laus S, Toth E, Helm L, Merbach AE, Wilson LJ. Chem. Commun. 2005;31:3915. doi: 10.1039/b504435a. [DOI] [PubMed] [Google Scholar]
- (387).Sitharaman B, Wilson LJ. Int. J. Nanomedicine. 2006;1:291. [PMC free article] [PubMed] [Google Scholar]
- (388).Hartman KB, Laus S, Bolskar RD, Muthupillai R, Helm L, Toth E, Merbach AE, Wilson LJ. Nano Lett. 2008;8:415. doi: 10.1021/nl0720408. [DOI] [PubMed] [Google Scholar]
- (389).Mackeyev Y, Hartman KB, Ananta JS, Lee AV, Wilson LJ. J. Am. Chem. Soc. 2009;131:8342. doi: 10.1021/ja900918x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Coroiu I. J. Magn. Magn. Mater. 1999;201:449. [Google Scholar]
- (391).Jung CW, P. J. Magn. Reson. Imaging. 1995;13:661. doi: 10.1016/0730-725x(95)00024-b. [DOI] [PubMed] [Google Scholar]
- (392).Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L, Muller RN. Chem. Rev. 2008;108:2064. doi: 10.1021/cr068445e. [DOI] [PubMed] [Google Scholar]
- (393).Blakemore RP, Frankel R. Sci. Am. 1981;246:58. [Google Scholar]
- (394).Fleet M. Acta Crystallogr. B. 1981;37:917. [Google Scholar]
- (395).Weiss P. J. Phys. 1907;6:661. [Google Scholar]
- (396).Neel L. Ann. Geophys. 1949;5:99. [Google Scholar]
- (397).Kumar D, Narayan J, Kvit AV, Sharma AK, Sankar J. J. Magn. Magn. Mater. 2001;232:161. [Google Scholar]
- (398).Martinez B, Roig A, Obradors X, Molins E, Rouanet A, Monty C. J. Appl. Phys. 1996;79:2580. [Google Scholar]
- (399).Bean CP. J. Appl. Physiol. 1995;26:1381. [Google Scholar]
- (400).Bean CP, Livingston JD. J. Appl. Physiol. 1959;30:120S. [Google Scholar]
- (401).Dormann JL, Spinu L, Tronc E, Jolivet JP, Lucari F, D'Orazio F, Fiorani D. J. Magn. Magn. Mater. 1998;183:L255. [Google Scholar]
- (402).Zeng P, Kline TL, Wang J.-p., Wiedmann TS. J. Magn. Magn. Mater. 2009;321:373. [Google Scholar]
- (403).Rosensweig RE. J. Magn. Magn. Mater. 2002;252:370. [Google Scholar]
- (404).Gueron M. J. Magn. Reson. 1975;19:58. [Google Scholar]
- (405).Vega AJ, Fiat D. Mol. Phys. 1976;31:347. [Google Scholar]
- (406).Caravan P, Greenfield MT, Bulte JWM. Magn. Reson. Med. 2001;46:917. doi: 10.1002/mrm.1277. [DOI] [PubMed] [Google Scholar]
- (407).Roch A, Gossuin Y, Muller RN, Gillis P. J. Magn. Magn. Mater. 2005;293:532. [Google Scholar]
- (408).Bowen CV, Zhang X, Saab G, Gareau PJ, Rutt BK. Magn. Reson. Med. 2002;48:52. doi: 10.1002/mrm.10192. [DOI] [PubMed] [Google Scholar]
- (409).Hartung A, Lisy MR, Herrmann K-H, Hilger I, Schüler D, Lang C, Bellemann ME, Kaiser WA, Reichenbach JR. J. Magn. Magn. Mater. 2007;311:454. [Google Scholar]
- (410).Simon G, Bauer J, Saborovski O, Fu Y, Corot C, Wendland M, Daldrup-Link H. Eur. Radiol. 2006;16:738. doi: 10.1007/s00330-005-0031-2. [DOI] [PubMed] [Google Scholar]
- (411).Rad AM, Arbab AS, Iskander ASM, Jiang Q, Soltanian-Zadeh H. J. Magn. Reson. Imaging. 2007;26:366. doi: 10.1002/jmri.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Bumb A, Brechbiel MW, Choyke PL, Fugger L, Eggeman A, Prabhakaran D, Hutchinson J, Dobson PJ. Nanotechnology. 2008;19:335601. doi: 10.1088/0957-4484/19/33/335601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Alcalá MD, Real C. Solid State Ionics. 2006;177:955. [Google Scholar]
- (414).Woo K, Hong J, Ahn J-P. J. Magn. Magn. Mater. 2005;293:177. [Google Scholar]
- (415).Lyon JL, Fleming DA, Stone MB, Schiffer P, Williams ME. Nano Lett. 2004;4:719. [Google Scholar]
- (416).Lin J, Zhou W, Kumbhar A, Wiemann J, Fang J, Carpenter EE, O'Connor CJ. J. Solid State Chem. 2001;159:26. [Google Scholar]
- (417).Bach-Gansmo T. Acta Radiol. 1993;387:1. [PubMed] [Google Scholar]
- (418).Hahn PF, Stark DD, Lewis JM, Saini S, Elizondo G, Weissleder R, Fretz CJ, Ferrucci JT. Radiology. 1990;175:695. doi: 10.1148/radiology.175.3.2343116. [DOI] [PubMed] [Google Scholar]
- (419).Weissleder R, Stark DD, Engelstad BL, Bacon BR, Compton CC, White DL, Jacobs P, Lewis J. Am. J. Roentgenol. 1989;152:167. doi: 10.2214/ajr.152.1.167. [DOI] [PubMed] [Google Scholar]
- (420).Reimer P, Rummeny EJ, Daldrup HE, Balzer T, Tombach B, Berns T, Peters PE. Radiology. 1995;195:489. doi: 10.1148/radiology.195.2.7724772. [DOI] [PubMed] [Google Scholar]
- (421).Reimer P, Tombach B. Eur. Radiol. 1998;8:1198. doi: 10.1007/s003300050535. [DOI] [PubMed] [Google Scholar]
- (422).McLachlan SJ, Morris MR, Lucas MA, Fisco RA, Eakins MN, Fowler DR, Scheetz RB, Olukotun AY. J. Magn. Reson. Imaging. 1994;4:301. doi: 10.1002/jmri.1880040313. [DOI] [PubMed] [Google Scholar]
- (423).Stillman AE, Wilke N, Li D, Haacke M, McLachlan S. J. Comput. Assist. Tomogr. 1996;20:51. doi: 10.1097/00004728-199601000-00011. [DOI] [PubMed] [Google Scholar]
- (424).Stillman AE, Wilke N, Jerosch-Herold M. J. Magn. Reson. Imaging. 1997;7:765. doi: 10.1002/jmri.1880070425. [DOI] [PubMed] [Google Scholar]
- (425).Mayo-Smith WW, Saini S, Slater G, Kaufman JA, Sharma P, Hahn PF. Am. J. Roentgenol. 1996;166:73. doi: 10.2214/ajr.166.1.8571910. [DOI] [PubMed] [Google Scholar]
- (426).Anzai Y, Brunberg JA, Lufkin RB. J. Magn. Reson. Imaging. 1997;7:774. doi: 10.1002/jmri.1880070503. [DOI] [PubMed] [Google Scholar]
- (427).Anzai Y, Blackwell KE, Hirschowitz SL, Rogers JW, Sato Y, Yuh WT, Runge VM, Morris MR, McLachlan SJ, Lufkin RB. Radiology. 1994;192:709. doi: 10.1148/radiology.192.3.7520182. [DOI] [PubMed] [Google Scholar]
- (428).Saeed M, Wendland MF, Engelbrecht M, Sakuma H, Higgins CB. Eur. Radiol. 1998;8:1047. doi: 10.1007/s003300050512. [DOI] [PubMed] [Google Scholar]
- (429).Simonsen CZ, Ostergaard L, Vestergaard-Poulsen P, Rohl L, Bjornerud A, Gyldensted C. J. Magn. Reson. Imaging. 1999;9:342. doi: 10.1002/(sici)1522-2586(199902)9:2<342::aid-jmri29>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- (430).Weissleder R, Lee AS, Khaw BA, Shen T, Brady TJ. Radiology. 1992;182:381. doi: 10.1148/radiology.182.2.1732953. [DOI] [PubMed] [Google Scholar]
- (431).Weissleder R, Lee AS, Fischman AJ, Reimer P, Shen T, Wilkinson R, Callahan RJ, Brady TJ. Radiology. 1991;181:245. doi: 10.1148/radiology.181.1.1887040. [DOI] [PubMed] [Google Scholar]
- (432).Wang YX, Hussain SM, Krestin GP. Eur. Radiol. 2001;11:2319. doi: 10.1007/s003300100908. [DOI] [PubMed] [Google Scholar]
- (433).Magnetic Resonance - Technology Information Portal. http://www.mr-tip.com (accessed November 3, 2009)
- (434).Ferumoxytol (Feraheme) Injection. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm170316.htm (accessed November 3, 2009)
- (435).Corot C, Port M, Guilbert I, Robert P, Raynal I, Robic C, Raynand JS, Prigent P, Dencausse A, Idee JM. In: Molecular and Cellular MR Imaging. Modo MMJ, Bulte JWM, editors. Taylor & Francis Group, LLC; Boca Raton, FL: 2007. [Google Scholar]
- (436).Andrews NC. N. Engl. J. Med. 1999;341:1986. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- (437).LaConte L, Nitin N, Bao G. Mater. Today. 2005;8:32. [Google Scholar]
- (438).Bacon BR, Stark DD, Park CH, Saini S, Groman EV, Hahn PF, Compton CC, Ferrucci JT. J. Lab. Clin. Med. 1987;110:164. [PubMed] [Google Scholar]
- (439).Stark DD, Weissleder R, Elizondo G, Hahn PF, Saini S, Todd LE, Wittenberg J, Ferrucci JT. Radiology. 1988;168:297. doi: 10.1148/radiology.168.2.3393649. [DOI] [PubMed] [Google Scholar]
- (440).Saini S, Stark DD, Hahn PF, Wittenberg J, Brady TJ, Ferrucci JT. Radiology. 1987;162:211. doi: 10.1148/radiology.162.1.3786765. [DOI] [PubMed] [Google Scholar]
- (441).Weissleder R. Radiology. 1994;193:593. doi: 10.1148/radiology.193.3.7972790. [DOI] [PubMed] [Google Scholar]
- (442).Pouliquen D, L. I., Chouly C, Perdrisot R, Le Jeune JJ, Jallet P. Magn. Reson. Imaging. 1993;11:219. doi: 10.1016/0730-725x(93)90026-a. [DOI] [PubMed] [Google Scholar]
- (443).Pouliquen D, P. R., Ermias A, Akoka S, Jallet P, Le Jeune JJ. Magn. Reson. Imaging. 1989;7:619. doi: 10.1016/0730-725x(89)90530-4. [DOI] [PubMed] [Google Scholar]
- (444).Anzai Y, McLachlan S, Morris M, Saxton R, Lufkin RB. Am. J. Neuroradiol. 1994;15:87. [PMC free article] [PubMed] [Google Scholar]
- (445).Deserno WMLLG, Harisinghani MG, Taupitz M, Jager GJ, Witjes JA, Mulders PF, Hulsbergen van de Kaa CA, Kaufmann D, Barentsz JO. Radiology. 2004;233:449. doi: 10.1148/radiol.2332031111. [DOI] [PubMed] [Google Scholar]
- (446).Vassallo P, Matei C, Heston WD, McLachlan SJ, Koutcher JA, Castellino RA. Invest. Radiol. 1995;30:706. doi: 10.1097/00004424-199512000-00003. [DOI] [PubMed] [Google Scholar]
- (447).Saleh A, Schroeter M, Jonkmanns C, Hartung H-P, Modder U, Jander S. Brain. 2004;127:1670. doi: 10.1093/brain/awh191. [DOI] [PubMed] [Google Scholar]
- (448).Kool ME, Cappendijk VC, Cleutjens KBJM, Kessels AGH, Kitslaar PJEHM, Borgers M, Frederik PM, Daemen MJAP, Van Engelshoven JMA. Circulation. 2003;107:2453. doi: 10.1161/01.CIR.0000068315.98705.CC. [DOI] [PubMed] [Google Scholar]
- (449).Ruehm SG, Corot C, Vogt P, Cristina H, Debatin JF. Acad. Radiol. 2002;9:S143. doi: 10.1016/s1076-6332(03)80422-1. [DOI] [PubMed] [Google Scholar]
- (450).Schmitz SA, Taupitz M, Wagner S, Wolf KJ, Beyersdorff D, Hamm B. J. Magn. Reson. Imaging. 2001;14:355. doi: 10.1002/jmri.1194. [DOI] [PubMed] [Google Scholar]
- (451).Beckmann N, Cannet C, Fringeli-Tanner M, Baumann D, Pally C, Bruns C, Zerwes HG, Andriambeloson E, Bigaud M. Magn. Reson. Med. 2003;49:459. doi: 10.1002/mrm.10387. [DOI] [PubMed] [Google Scholar]
- (452).Kanno S, Lee PC, Dodd SJ, Williams M, Griffith BP, Ho C, Mentzer SJ, Egan TM, DeCamp MM. J. Thorac. Cardiovasc. Surg. 2000;120:923. doi: 10.1067/mtc.2000.110184. [DOI] [PubMed] [Google Scholar]
- (453).Kanno S, Yi-Jen Lin W, Lee PC, Dodd SJ, Williams M, Griffith BP, Ho C. Circulation. 2001;104:934. doi: 10.1161/hc3401.093148. [DOI] [PubMed] [Google Scholar]
- (454).Zhang Y, Dodd SJ, Hendrich KS, Williams M, Ho C. Kidney Int. 2000;58:1300. doi: 10.1046/j.1523-1755.2000.00286.x. [DOI] [PubMed] [Google Scholar]
- (455).Bulte JWM, Douglas T, Witwer B, Zhang SC, Strable E, Lewis BK, Zywicke H, Miller B, Van Gelderen P, Moskowitz BM, Duncan ID, Frank JA. Nat. Biotechnol. 2001;19:1141. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- (456).Josephson L, Tung CH, Moore A, Weissleder R. Bioconjugate Chem. 1999;10:186. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- (457).Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat. Biotechnol. 2000;18:410. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- (458).Arbab AS, Bashaw LA, Miller BR, Jordan EK, Lewis BK, Kalish H, Frank JA. Radiology. 2003;229:838. doi: 10.1148/radiol.2293021215. [DOI] [PubMed] [Google Scholar]
- (459).Arbab AS, Yocum GT, Kalish H, Jordan EK, Anderson SA, Khakoo AY, Read EJ, Frank JA. Blood. 2004;104:1217. doi: 10.1182/blood-2004-02-0655. [DOI] [PubMed] [Google Scholar]
- (460).Frank JA, Miller BR, Arbab AS, Zywicke HA, Jordan EK, Lewis BK, Bryant LH, Jr, Bulte JWM. Radiology. 2003;228:480. doi: 10.1148/radiol.2281020638. [DOI] [PubMed] [Google Scholar]
- (461).Hawrylak N, Ghosh P, Broadus J, Schlueter C, Greenough WT, Lauterbur PC. Exp. Neurol. 1993;121:181. doi: 10.1006/exnr.1993.1085. [DOI] [PubMed] [Google Scholar]
- (462).Bulte JWM, Ma LD, Magin RL, Kamman RL, Hulstaert CE, Go KG, The TH, De Leij L. Magn. Reson. Med. 1993;29:32. doi: 10.1002/mrm.1910290108. [DOI] [PubMed] [Google Scholar]
- (463).Anderson SA, Shukaliak-Quandt J, Jordan EK, Arbab AS, Martin R, McFarland H, Frank JA. Ann. Neurol. 2004;55:654. doi: 10.1002/ana.20066. [DOI] [PubMed] [Google Scholar]
- (464).Sundstrom JB, Mao H, Santoianni R, Villinger F, Little DM, Huynh TT, Mayne AE, Hao E, Ansari AA. J. Acquir. Immune Defic. Syndr. 2004;35:9. doi: 10.1097/00126334-200401010-00002. [DOI] [PubMed] [Google Scholar]
- (465).Yeh TC, Zhang W, Ildstad ST, Ho C. Magn. Reson. Med. 1993;30:617. doi: 10.1002/mrm.1910300513. [DOI] [PubMed] [Google Scholar]
- (466).Zelivyanskaya ML, Nelson JA, Poluektova L, Uberti M, Mellon M, Gendelman HE, Boskal MD. J. Neurosci. Res. 2003;73:284. doi: 10.1002/jnr.10693. [DOI] [PubMed] [Google Scholar]
- (467).Shapiro EM, Sharer K, Skrtic S, Koretsky AP. Magn. Reson. Med. 2006;55:242. doi: 10.1002/mrm.20718. [DOI] [PubMed] [Google Scholar]
- (468).Lauterbur PC, Dias MHM, Rudin AM. In: Frontiers of Biological Energetics. Dutton PL, Leigh JS, Scarpa A, editors. Academic Press; New York: 1978. [Google Scholar]
- (469).Mendonca-Dias MH, Gaggelli E, Lauterbur PC. Semin. Nucl. Med. 1983;13:364. doi: 10.1016/s0001-2998(83)80048-8. [DOI] [PubMed] [Google Scholar]
- (470).Thomsen HS, Loegager V, Noerrgaard H, Chabanova E, Moller J, Sonne J. Acad. Radiol. 2005;12:S21. doi: 10.1016/j.acra.2005.02.019. [DOI] [PubMed] [Google Scholar]
- (471).Wolf GL, Baum L. Am. J. Roentgenol. 1983;141:193. doi: 10.2214/ajr.141.1.193. [DOI] [PubMed] [Google Scholar]
- (472).Schaefer S, Lange RA, Kulkarni PV, Katz J, Parkey RW, Willerson JT, Peshock RM. J. Am. Coll. Cardiol. 1989;14:472. doi: 10.1016/0735-1097(89)90204-0. [DOI] [PubMed] [Google Scholar]
- (473).Schaefer S, Lange RA, Gutekunst DP, Parkey RW, Willerson JT, Peshock RM. Invest. Radiol. 1991;26:551. doi: 10.1097/00004424-199106000-00009. [DOI] [PubMed] [Google Scholar]
- (474).Pflugfelder PW, Wendland MF, Holt WW, Quay SC, Worah D, Derugin N, Higgins CB. Radiology. 1988;167:129. doi: 10.1148/radiology.167.1.3347711. [DOI] [PubMed] [Google Scholar]
- (475).Pomeroy OH, Wendland M, Wagner S, Derugin N, Holt WW, Rocklage SM, Quay S, Higgins CB. Invest. Radiol. 1989;24:531. doi: 10.1097/00004424-198907000-00004. [DOI] [PubMed] [Google Scholar]
- (476).Saeed M, Wagner S, Wendland MF, Derugin N, Finkbeiner WE, Higgins CB. Radiology. 1989;172:59. doi: 10.1148/radiology.172.1.2500678. [DOI] [PubMed] [Google Scholar]
- (477).Hustvedt SO, Grant D, Southon TE, Zech K. Acta Radiol. 1997;38:690. doi: 10.1080/02841859709172401. [DOI] [PubMed] [Google Scholar]
- (478).Torres CG, Lundby B, Tufte Sterud A, McGill S, Gordon PB, Strand Bjerknes H. Acta Radiol. 1997;38:631. doi: 10.1080/02841859709172393. [DOI] [PubMed] [Google Scholar]
- (479).Federle M, Chezmar J, Rubin DL, Weinreb J, Freeny P, Schmiedl UP, Brown JJ, Borrello JA, Lee JKT, Semelka RC, Mattrey R, Dachman AH, Saini S, Harms SE, Mitchell DG, Anderson MW, Halford Iii HH, Bennett WF, Young SW, Rifkin M, Gay SB, Ballerini R, Sherwin PF, Robison RO. J. Magn. Reson. Imaging. 2000;12:689. doi: 10.1002/1522-2586(200011)12:5<689::aid-jmri5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- (480).Nordhøy W, Anthonsen HW, Bruvold M, Brurok H, Skarra S, Krane J, Jynge P. Magn. Reson. Med. 2004;52:506. doi: 10.1002/mrm.20199. [DOI] [PubMed] [Google Scholar]
- (481).Aime S, Anelli P, Botta M, Brocchetta M, Canton S, Fedeli F, Gianolio E, Terreno E. J. Biol. Inorg. Chem. 2002;7:58. doi: 10.1007/s007750100265. [DOI] [PubMed] [Google Scholar]
- (482).Troughton JS, Greenfield MT, Greenwood JM, Dumas S, Wiethoff AJ, Wang J, Spiller M, McMurry TJ, Caravan P. Inorg. Chem. 2004;43:6313. doi: 10.1021/ic049559g. [DOI] [PubMed] [Google Scholar]
- (483).Parasassi T, Bombieri G, Conti F, Croatto U. Inorg. Chim. Acta. 1985;106:135. [Google Scholar]
- (484).Navon G, R. P, Valensin G. Magn. Reson. Med. 1986;3:876. doi: 10.1002/mrm.1910030608. [DOI] [PubMed] [Google Scholar]
- (485).Lee J-H, Huh Y-M, Jun Y-W, Seo J-W, Jang J-T, Song H-T, Kim S, Cho E-J, Yoon H-G, Suh J-S, Cheon J. Nat. Med. 2007;13:95. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]
- (486).Taylor KML, Rieter WJ, Lin W. J. Am. Chem. Soc. 2008;130:14358. doi: 10.1021/ja803777x. [DOI] [PubMed] [Google Scholar]