

1. Introduction
New peptide-based probes to facilitate the molecular imaging of disease are rapidly evolving due to implementation of combinatorial chemistry and bacteriophage (phage) display. Phage display is a powerful technique that allows vast sequence space screening, providing a means to improve peptide affinity and generate unique peptides that bind any given target. Since its inception in 1985, many thousands of peptides have been isolated and investigated using phage display. Such peptides are being explored in vaccine development, enzyme inhibition, inflammation, plant pathology, cardiovascular disease, cancer, etc. The purpose of this review is to analyze and describe those peptides obtained from phage display that have been used successfully in the past five years in both radio- and optical in vivo tumor imaging. New tumor targeting agents are required to advance cancer diagnosis and treatment and phage display selected peptides may be an attractive means to obtain such agents. Unfortunately, the vast majority of the hundreds of peptides selected against tumor antigens have not been shown to function as cancer molecular imaging agents in vivo. Recently, progress has been made in translation of the peptides from in vitro to in vivo applications. Not only have the peptides displayed on phage been employed in vivo as tumor imaging agents, but the phage themselves have been used in imaging with a number of labeling platforms. The integration of phage as not only vehicles for peptide discovery but also as a nanomaterial has wide-ranging applications. Phage display technology is emerging as a powerful, economical, rapid, and efficacious approach to develop new agents for the molecular imaging and diagnosis of cancer.
2. Peptides as Molecular Imaging Probes
Molecularly imaging probes have aided in our understanding of fundamental biological processes, disease pathologies and pharmaceutical development. 1–3 Molecular imaging agents that would allow real time visualization of biomolecules and interactions involved in disease, that could also facilitate diagnosis or response to therapy are of particular interest. The targeted molecular imaging of disease processes, particularly tumor growth and metastasis, has been a focus of many investigations for over the last ten years. 4–9 Enormous progress has been made in both imaging probe discovery and in vivo detection of cancer. In fact, antibodies, nanoparticles, and peptides are being developed for the specific detection of primary and disseminated disease. Undoubtedly antibodies and their fragments are the most common biological targeting vehicles for the specific delivery of an imaging modality to disease sites. Radionuclides, fluorophores, and biotin/streptavidin labeled antibodies have been used successfully to image cancers including those of the breast, prostate, ovary and others. Radiolabeled antibodies have been employed in cancer imaging and therapy for over thirty years. 10 A classic example is Oncoscint™, an 111In-labeled monoclonal antibody specific for the tumor-associated glycoprotein 72 (TAG-72), which has been used to detect colorectal metastasis lesions. 11 Another example is 111In-diethylenetriaminepentaacetic acid (DTPA)-Trastuzumab, an ErbB-2-targeting antibody that has been used for imaging metastatic breast cancer. 12 Unfortunately, many patients experienced hematological adverse effects and resistance problems after antibody administration with this antibody. 13 Antibodies also exhibit long biodistribution times and slow tumor penetration and clearance rates through the hepatobiliary system. 14 Antibody pre-targeting strategies have been developed to evade long circulation times, and antibody fragments have been produced to improve clearance characteristics.15
Peptides may offer fundamental advantages over antibodies for in vivo molecular imaging and diagnosis because of their rapid blood clearance, tissue penetration, increased diffusion, non-immunogenic nature, and straightforward synthesis. 16,17 Their small size may reduce or eliminate side effects that often occur with antibody or antibody fragment-based imaging or therapy applications. However, peptide constructs tagged with a radionuclide or fluorophore may only serve as viable molecular imaging agents if they retain sufficient target affinity. A limited set of regulatory peptides that bind receptors overexpressed on tumors exhibit high affinity (sub-nanomolar) and are being pursued in imaging and therapy studies. The classic example is Octreotide, an eight amino acid cyclized peptide that binds the somatostatin receptor. 111In-DTPA-Octreotide (OctreoScan™) has been used successfully to image somatostatin receptor positive tumors in humans. 18 Other natural peptides being explored include gastrin releasing peptide (GRP) for use in the detection and treatment of prostate, breast, and pancreatic cancer19 and α-melanocyte stimulating hormone (α-MSH) derivatives for the detection and therapy of malignant melanoma. 20 The CCMSH peptide was cyclized by 188Re or 99mTc for melanoma single photon emission tomography (SPECT) imaging and therapy (Figure 1) 21–24. The 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) was conjugated to the CCMSH peptide for radiolabeling while the CCMSH was cyclized by non-radioactive Re. The peptide was labeled with alpha-particle emitting 212Pb/212Bi for melanoma therapy studies25 and also with β+-emitting radionuclides for positron emission tomography (PET) imaging. 26,27
Figure 1.

SPECT imaging of melanocortin-1 receptor binding α-MSH peptide, CCMSH, in melanoma-bearing mice. Whole-body and transaxial images of 99mTc-(Arg11)CCMSH (A and B, respectively) and 111In-DOTA-Re(Arg11)CCMSH (D and C, respectively) in B16/F1 flank melanoma-bearing C57 mice at 2 h after injection. Whole-body and transaxial images of 99mTc-(Arg11)CCMSH (E and F, respectively) and 111In-DOTA-Re(Arg11)CCMSH (H and G, respectively) in B16/F10 pulmonary metastatic melanoma-bearing C57 mice 2 h after injection. Reprinted with permission from reference number 24 (Figure 2), Copyright 2007 Society of Nuclear Medicine.
3. Isolating Phage Display-derived Peptides
Development of cancer diagnostic and imaging (or therapeutic) compounds has customarily relied on isolation from: 1) natural product extracts, 2) screening synthetic compound databases, 3) structure-based rational design, or 4) antibody engineering. 28 An alternative resource for peptide-based imaging agents is through the screening of bacteriophage (phage) display libraries. Phage display was developed by George P. Smith at the University of Missouri in 1985, and was initiated by the finding that Escherichia coli (E. coli) filamentous phage could be engineered to display foreign amino acid sequences on the tips of certain phage coat proteins (cp) without compromising phage infectivity or propagation ability. 29 Filamentous phage self-assemble into ~900 nm × 6 nm rod-like protein-encapsulated structures, within which is the genetic information to dictate its own production. The genetic encoding of a library allows re- synthesis and screening of molecules with a desired binding activity. Amplification of interacting molecules in subsequent rounds of affinity selection can yield peptides that bind to almost any given target. The expression of foreign sequences on phage is not restricted to small peptides, as antibody fragments, receptors and enzymes have been displayed. 30,31 Typically however, in a phage display library, foreign small peptides (5–45 amino acids) are incorporated into the N-terminus of minor coat protein III (cpIII) of fd phage so that at most five copies of the peptide are displayed. 29 Foreign peptides have also been fused to the major coat protein VIII (cpVIII) of phage so that hundreds or thousands of copies of the peptide can be displayed. 32 Each phage clone displays a single peptide, but a library as a whole may represent 109 peptides, collectively. Peptide libraries are displayed as linear or constrained sequences. Constrained libraries possess two cysteine residues flanking a random segment of peptide, thus facilitating disulfide bond formation. Lytic phage libraries including T7 phage 33 and bacterial display libraries 34 have also been generated. Surface exposure of the foreign peptide on the phage is key to successful screening in that it allows vast numbers of peptides to be easily surveyed for clones whose displayed peptides bind specifically to any given molecular target. Screening of a phage display library typically involves passing the library over the desired target molecules either in vitro, with cultured cells, in situ, or in vivo, capturing bound clones, and washing away unbound phage (Figure 2). Bound phage are often recovered by acid or competitive elution. Captured phage retain infectivity and can be propagated and cloned by infecting fresh host bacteria. The primary structure of the foreign peptide can be elucidated easily by sequencing the peptide-coding sequence in the viral DNA. The affinity selection process is iterative, normally a minimum of four rounds of selection are required to obtain phage enriched for binding.
Figure 2.

Affinity selection using phage display libraries. A phage display library is typically selected against unwanted non-specific binders before four-five rounds of positive selection. Positive selection can be performed against an immobilized target antigen or tissue culture cells in vitro, in situ, or in vivo. After the last round of selection, validation of phage binding the desired target is performed in vitro and/or in vivo.
However, the success of a phage library affinity selection is critically dependent on the stringency employed in all rounds of screening. Affinity selection methods have been adapted in recent years to increase one’s chances of obtaining a peptide that binds with good affinity to the desired target. For example, it is quite common to complete an affinity selection screen of a phage display library, only to find that predominantly selected clones have a much better affinity for the target support (i.e. plastic) than the target. Thus, stringent washing and phage elution conditions are critical. Further, non-relevant phage can be propagated throughout the selection scheme if they possess superior growth advantages over rare phage that bind the desired target. One means to improve elution of specific phage from the target is to competitively elute with excess free target or ligand. In cultured cell/in situ and in vivo selections, the target is not known so nonspecific elution procedures are utilized (Figure 2). These nonspecific elutions consist of extreme pH, high ionic strength, reducing agents, and detergents. Recent reports suggest that these common nonspecific elution methods may not always be strong enough to disrupt phage-target interactions. Thus, the best binding phage may be lost in the affinity selection procedure. Ultrasound has shown improved elution of phage from the target in vitro. 35 Another means to improve the success of affinity selection is to better control the washing conditions throughout the experiment. For in vitro selections, more stringent and controlled washing conditions including selection and washing on macroporous gels, or chromato-panning, 36 has been proposed. Soh and co-workers have recently developed a microfluidic device involving fabricated ferromagnetic structures to allow for trapping and release of magnetic beads, which facilitates controlled washing using high fluidic forces. 37
4. Phage Display-derived Tumor-targeting Peptides
4.1 Overview of Selected Peptides
Since its inception almost thirty years ago, phage display has been utilized in vitro, in situ, and in vivo (Figure 2), and thousands of published papers have reported on the isolation of peptides that bind a myriad of targets. 38–57 Phage display libraries have been used most productively to obtain peptide mimics cross-reactive with antibody molecules that could be used for vaccine development. Examples include peptides that bind the anti-mucin antibody C595, 58 the prostate specific membrane antigen antibody 4G5, 59 and the ErbB-2 antibody Trastuzumab. 60,61 Limited phase I clinical trials are analyzing the T-cell response of many of these peptide-based antibody mimetics.
Far fewer studies have been published on the isolation of phage display peptides that target cancer-associated antigens. Of the roughly two thousand published phage display papers describing peptides to a given target, approximately 8% of these bind to tumors or tumor vasculature (Table 1). The paucity of tumor-targeting peptides in general, is likely a reflection of the complicated events involved in tumorigenesis and a lack of well-defined tumor markers. Nevertheless, peptides have been isolated using in vitro phage display that target several tumor-associated antigens ranging from receptors, vasculature components, lipids, and carbohydrates (Table 1).
Table 1.
Tumor Targeting Peptides from Phage Display
 |
Houimel, M.; Schneider, P.; Terskikh, A.; Mach, J. P. Int. J. Cancer 2001, 92, 748.
Urbanelli, L.; Ronchini, C.; Fontana, L.; Menard, S.; Orlandi, R.; Monaci, P. J. Mol. Biol. 2001, 313, 965.
Stortelers, C.; Souriau, C.; van Liempt, E.; van de Poll, M. L.; van Zoelen, E. J. Biochemistry2002, 41, 8732.
Arap, M. A.; Lahdenranta, J.; Mintz, P. J.; Hajitou, A.; Sarkis, A. S.; Arap, W.; Pasqualini, R. Cancer Cell2004, 6, 275.
Chen, L.; Hnatowich, D.; Rusckowski, M. J. Nucl. Med. Meeting Abstracts 2007, 48, 310P.
Rusckowski, M.; Gupta, S.; Liu, G.; Dou, S.; Hnatowich, D. J. Cancer Biother. Radiopharm. 2007, 22, 564.
Nothelfer, E. M.; Zitzmann-Kolbe, S.; Garcia-Boy, R.; Kramer, S.; Herold-Mende, C.; Altmann, A.; Eisenhut, M.; Mier, W.; Haberkorn, U. J. Nucl. Med. 2009, 50, 426.
Laakkonen, P.; Akerman, M. E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R. M.; Ruoslahti, E. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9381.
Kridel, S. J.; Chen, E.; Kotra, L. P.; Howard, E. W.; Mobashery, S.; Smith, J. W. J. Biol. Chem. 2001, 276, 20572.
Pan, W.; Arnone, M.; Kendall, M.; Grafstrom, R. H.; Seitz, S. P.; Wasserman, Z. R.; Albright, C. F. J. Biol. Chem. 2003, 278, 27820.
Ke, S. H.; Coombs, G. S.; Tachias, K.; Corey, D. R.; Madison, E. L. J. Biol. Chem. 1997, 272, 20456.
Yang, S. Q.; Craik, C. S. J. Mol. Biol. 1998, 279, 1001.
Fong, S.; Doyle, M. V.; Goodson, R. J.; Drummond, R. J.; Stratton, J. R.; McGuire, L.; Doyle, L. V.; Chapman, H. A.; Rosenberg, S. Biol. Chem. 2002, 383, 149.
Shao, R.; Xiong, C.; Wen, X.; Gelovani, J. G.; Li, C. Mol. Imaging 2007, 6, 417.
Martens, C. L.; Cwirla, S. E.; Lee, R. Y.; Whitehorn, E.; Chen, E. Y.; Bakker, A.; Martin, E. L.; Wagstrom, C.; Gopalan, P.; Smith, C. W.; et al. J. Biol. Chem. 1995, 270, 21129.
Kelly, K.; Alencar, H.; Funovics, M.; Mahmood, U.; Weissleder, R. Cancer Res. 2004, 64, 6247.
Spear, M. A.; Breakefield, X. O.; Beltzer, J.; Schuback, D.; Weissleder, R.; Pardo, F. S.; Ladner, R. Cancer Gene Ther. 2001, 8, 506.
Mintz, P. J.; Cardo-Vila, M.; Ozawa, M. G.; Hajitou, A.; Rangel, R.; Guzman-Rojas, L.; Christianson, D. R.; Arap, M. A.; Giordano, R. J.; Souza, G. R.; Easley, J.; Salameh, A.; Oliviero, S.; Brentani, R. R.; Koivunen, E.; Arap, W.; Pasqualini, R. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2182.
Bockmann, M.; Drosten, M.; Putzer, B. M. J. Gene Med. 2005, 7, 179.
Kennel, S. J.; Mirzadeh, S.; Hurst, G. B.; Foote, L. J.; Lankford, T. K.; Glowienka, K. A.; Chappell, L. L.; Kelso, J. R.; Davern, S. M.; Safavy, A.; Brechbiel, M. W. Nucl. Med. Biol. 2000, 27, 815.
4.2 In Vitro Selected Peptides
To date, the majority of in vitro selected peptides have not been demonstrated to image tumors in vivo. Peptides that bind in vitro to the proteins CRIP-1 (cysteine-rich intestinal protein), 62 ephrin receptor, 63,64 heat shock protein 90, 65,66 MDM2/p53, 67 interleukin-11 receptor, 68 and prostate specific antigen (PSA) 69 have been reported. Numerous vasculature and lymphatic binding peptides and peptide motifs have also been identified including those that bind human vasculature endothelium, 70 integrins, 57,71 and growth factor receptors such as fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), insulin-like growth factor-1 (IGF-1), and transforming growth factor-β (TGF-β). 48,72–76 Peptides that bind the carbohydrate antigens sulfated Lewis A 77 and Thomsen-Friedenreich (TF), 49 both aberrantly expressed in a variety of adenocarcinomas, have been isolated and well-characterized. Nevertheless, the majority of the aforementioned peptides, while behaving well in in vitro assays, have not been reported to function in vivo.
P30, with the sequence HGRFILPWWYAFSPS, selected against TF antigen is a peptide that does not work well in vivo. 49,78 TF antigen is a Galβ1-3GalNAc disaccharide expressed on the surfaces of most adenocarcinomas including those of the breast and prostate. TF is involved in adhesion of cancer cells to the endothelium and the peptide P30 has been shown to inhibit adhesion of MDA-MB-435 breast and DU-145 prostate human cancer cells to human umbilical vein endothelial cells (Figure 3). Furthermore, TF functions to dock cancer cells to the endothelium by interacting with endothelium-expressed galectin-3, which binds TF. 5,6,79 However, the TF-binding peptide P30 has not been shown to bind to or image tumors in vivo.
Figure 3.

Dose-dependent inhibition of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI)-labeled (A) and acridine orange-labeled (B–G) DU-145 human prostate carcinoma cell adhesion to the endothelium by synthetic TF antigen-specific P30 peptide but not by control peptide (H). In B–H, numbers at the bottom indicate the concentration of the peptide tested. I and J, maximal inhibitory effect on adhesion of MDA-MB-435 human breast carcinoma (I) and DU-145 human prostate carcinoma (J) cells to the endothelium achievable with anti-TF antigen monoclonal antibody and P-30 peptide; bars, SD. Reprinted with permission from reference number 5 (Figure 2), Copyright 2001 American Association for Cancer Research, Inc.
Combinatorial affinity maturation experiments have been performed on a small number of these peptides, including P30, in an attempt to improve affinity and in vivo binding. Affinity maturation is an in vitro process that purportedly mimics in vivo recombination and selection used by the immune system to compensate for sparse representation in the initial library of sequences related to the optimum sequence. Such affinity maturation studies were performed with the TF-targeting peptide P30 consensus sequence WYAW/FSP in which a 15 amino acid phage library was constructed with random flanking amino acids and reselected against TF. Many of the second-generation peptides had ten-fold improved TF affinity (~30 nM) and increased binding to TF-positive carcinoma cells, but did not bind TF-expressing tumors in vivo. 80,81
4.3 Cultured Cell/In Situ Selected Peptides
Phage display has also been performed using cultured human carcinoma cells or in situ with laser captured microdissected cancer cells 82 resulting in many new peptides as potential imaging probes (Figure 2). Peptides have been selected against cultured human B-cell lymphoma, 83–85 breast, 86 cervical, 87 colon, 4,88,89 gastric, 90 glioblastoma, 91 hepatic, 92 lung, 93,94 neuroblastoma, 95 prostate, 54,96,97 and thyroid 98 carcinoma cells. While the peptides bind the cultured cell lines used in selection, their ability to bind the corresponding xenografted tumors in vivo has not markedly improved over those peptides selected against a purified antigen (Table 1). As shown in Table 1, approximately 20% of the published peptides that reportedly target tumor antigens, vasculature, or lymphatics, have been shown to bind to, image, or reduce tumor growth in vivo. In fact, prior to 2007, the success rate of peptides to function in vivo was much less. The meager performance of phage display selected peptides in vivo may be due to the method of selection, hydrophobic nature of most peptides displayed on phage, poor affinity, and/or inability of the peptides to function outside the phage framework.
4.4 In Vivo Selected Peptides
It has been argued that in vivo phage display selection procedures theoretically offer an advantage over in vitro or in situ screening procedures in that phage can be selected in the complicated milieu of the animal based on desired pharmacokinetic properties including delivery and tumor accumulation. Such a selection does not rely on knowledge of the target. Further, the selected peptides can allow identification of the target antigen using database search analyses and biochemical approaches such as affinity chromatography and mass spectrometry. More than ten years ago Pasqualini and Ruoslahti performed phage peptide library selections in vivo and isolated peptides with motifs that targeted the vasculature of various organs and tumors, most notably RGD and NGR motifs. 99–102 Tissue-specific peptides have been identified that bind brain, breast, lung, fat, pancreas, skin, etc. 100,103 A variety of RGD-containing peptides that bind αvβ3 integrin have been described by numerous groups. 57,104–112 Vascular endothelial lymphatic-targeting motifs have also been described using in vivo phage display. 57,113 Tumor vasculature receptors targeted by some of these peptides have been identified including interleukin 11 receptor, 100 aminopeptidase P 114 and the EphA4 receptor. 115 However, in vivo phage display has predominately identified peptides that bind to tumor vasculature components and not directly to tumor cells (Table 1). Nevertheless, many of these peptides are showing great promise not only in vitro but also in targeting in vivo in a variety of disease models. 116
Discovery of tumor-cell surface targeting agents in addition to tumor vasculature targeting agents is also being pursued. Procedures have recently been devised to obtain phage that extravasate the vasculature and home to tumor cell-associated antigens in vivo. 117–119 Key steps to the in vivo selection of tumor-targeting phage included pre-clearing of libraries of vasculature-targeting phage in vivo by isolating phage in the circulation, appropriate biodistribution times, 120 detergent extraction of phage from target tissues, and large scale mass propagation and amplification of phage. 119 In one study, prostate tumor-homing peptides were isolated from PC-3 and PC-3M human prostate tumor xenografts in vivo. In these studies, 1 × 109 infectious units of a precleared random peptide library fused to either cpIII (f3-15mer) or cpVIII (f88-15mer and cysteine constrained f88/cys6) were injected into human prostate tumor-bearing SCID mice for one hour. Animals were perfused, sacrificed, tumors were removed and washed, and phage were eluted in 0.5% CHAPs detergent buffer and the entire phage population were allowed to infect host E. coli for a round of amplification. The selections were repeated four times. Representative sequences of displayed peptides that were selected after the final round from the cys6 library are shown in Table 2. Micropanning experiments of individual phage clones with cultured prostate carcinoma cells and excised prostate tumors indicated that ~20% of the selected phage bound prostate carcinomas but not normal prostate. 119 The phage clones F11 and G1 bound well to PC-3 human prostate cultured carcinoma cells and xenografted tumors, but not to normal cells or tissue. Binding of AlexaFluor (AF) 680-labeled G1 phage to cultured PC-3 prostate carcinoma cells is shown in Figure 4.
Table 2.
In Vivo Phage Display Selected Peptides Against PC-3 Prostate Tumors
| f88-Cys6 Library Selection in PC-3M Tumor Bearing SCID Mice | f88-Cys6 Library Selection in PC-3 Tumor Bearing SCID Mice | ||
|---|---|---|---|
| Phage Clone | Sequence | Phage Clone | Sequence |
| A1 | TDVSCKNHKGACCSTN | E1 | INTECAGLGLVCKPHT |
| A2 | HIEPCVPGWVGCNSLI | E2 | NKSKCRCRQNACKQLI |
| A3 | EKFFCNTVNRGCTGPQ | E3 | CTSSCKPHSQSCKEKT |
| A4 | HMQQCKKRTTRCKVQS | E4 | NKKQCKTVLKMCHRRV |
| A7 | IKNNCGPVWEVCVQYP | E5 | DRPHCLKTWNICTSYY |
| A8 | KNLTCWNEEYQCGWKV | E6 | MKRECKNRCALCKSER |
| A9 | TMNWCNHNPMTCGSQF | E7 | IVPGCSKTERGCSYQS |
| A12 | PTIMCKKQEKLCRLRM | E10 | KPSPCSSFKSHCVRRD |
| B3 | PTKRCVRQDEICNNKR | E11 | AKYYCEELVNHCTSAQ |
| B4 | PQEMCTCMARGCRWKT | F1 | GDLRCRITKQKCEQQC |
| B9 | DETPCQHYGNCCTLIL | F8 | ETIMCIRYRCDCPLPH |
| B10 | YKCLCAAGASTCQPGP | F11 | GPAHCKRTISQCQTNE |
| B12 | VQRSCIQAPKECYTDK | G2 | DEWHCKFNGAVCTSMR |
| C1 | KIDSCEWTAKYCISEI | G1 | IAGLATPGWSHWLAL |
| C2 | PDQLCQPEESECGTLP | G5 | QRVTCDMAENCCPKTS |
| C5 | NVMACSCTHGWCITKT | G10 | PPRLCQGMRGTCSGNQ |
| C8 | ESMQCETSQNKCLTTR | G12 | CACICPCNPAFCTVAV |
| C9 | LKTRCISNSPHCNYSD | H1 | KMPECHEQQEYCDGDR |
| C10 | NKSQCSKWRASCDIPR | H2 | QKEHCILHTANCGRIT |
| C11 | TRNPCKKAKMVCEEWP | H4 | TNTNCGTDLEPCVSTM |
| D2 | DQRACKNSILTCMKAG | H6 | HDKQCLTAKDRCGTIK |
| D6 | LSNCCETPCAYCYLSP | H12 | ASCECNPHPRHCGETR |
Figure 4.

Binding of AF680 labeled phage and peptide to PC-3 human carcinoma cells and control HEK 293 cells. Slides containing fixed PC-3 or human embryonic kidney (HEK293) cells were incubated with AlexaFluor680 (AF680)-labeled phage (1×1011 virion/mL) or biotinylated peptide (20μM) at room temperature for 1 h in the dark. Binding of peptides was detected using NeutrAvidin-Texas Red.
In vivo selections often do not result in a predominant phage clone, but rather hundreds of thousands of different phage clones displaying unique sequences that need to be analyzed for binding. This highlights the difficulty in predicting which displayed peptide sequence from an isolated phage clone, once chemically synthesized, will function in vivo. High throughput, peptide database and search algorithms, and mass DNA sequencing approaches 121,122 may facilitate deconvolution of functional and specific phage for corresponding peptide synthesis.
5. Labeling Peptides for Molecular Imaging
5.1 Radiolabeling Peptides
Peptides have been labeled with radionuclides, fluorophores, and other tags for molecular imaging. Radiolabeled peptides have shown the most success in imaging tumors in animals. The efficacy of radiolabeled peptides as imaging agents is closely associated with the radionuclide and chelate employed. One of the commonly employed radionuclides for use in SPECT imaging is 99mTc due to its nuclear properties (6 h half-life, 140 keV gamma emission) and ready availability. 123,124 Most hospitals are equipped with SPECT imaging equipment and personnel trained in image acquisition and interpretation. Development of radiopharmaceuticals based on 99mTc has been advanced by the use of “click” 99mTc-carbonyl chemistry. 125 Another widely used radioisotope for labeling peptides that can be used in diagnostic SPECT is 111In, which has a 2.8 day half-life, and emits gamma photons with energies of 173 and 247 KeV. 18,126
PET coupled with the β+-emitting tracer fluorine-18 2-deoxy-2-fluoro-D-glucose (18F-FDG) is commonly used in studying cell metabolism and disease. 127 18F-FDG is currently the primary PET tracer utilized for imaging processes with increased glucose metabolism including cancer. 128 PET has some advantageous over SPECT in that it is 1–2 orders of magnitude more sensitive and is quantitative.127 PET tracers are being developed that target specific receptors or antigens on tumors, since the uptake of 18F-FDG is not increased in all cancer cells. 64Cu-labeled complexes are being explored because the radiometal has favorable properties for PET imaging and therapy due to its half-life (t1/2= 12.7 h) and two different decay modes (β+ 17.4%, β− 39%). 126,129 Another PET isotope showing recent promise in imaging is Gallium-68 (68Ga). 68Ga is a short-lived positron emitter with high specific activity that can be routinely made from a 68Ge/68Ga generator 130,131 and appended to peptides. The 68 minute short half-life of 68Ga is ideal for the pharmacokinetics of radiopharmaceuticals with low molecular weight such as peptides.
5.2 Bioluminescent and Fluorescent Labeling
Non-invasive real-time in vivo imaging of biological processes and diseases including cancer is receiving much attention of late. Traditional methods for imaging including x-ray, magnetic resonance imaging, SPECT, and PET while suited for large masses or whole body imaging, have their limitations for monitoring more microscopic processes in real-time. Because of this, considerable effort has been placed on developing bioluminescent and fluorescent imaging systems. The use of bioluminescence to tag cells, pathogens, genes, and proteins can provide insights into biochemical mechanisms in the living organism. 8,132,133 Advances in instrumentation for detecting weak optical signals coupled with a better appreciation of the optical properties of tissue have allowed for detection and quantification of signal in vivo. 7 In bioluminescent imaging, cells produce bioluminescent proteins such as luciferase and generate light with appropriate substrates. 8 Analyses of the light produced in vivo reveals the spatial and temporal distribution of the biological process under investigation including cell growth, adhesion, apoptosis, metastasis etc. In fluorescent imaging, an external source of light is required for excitation of the fluorophore. Wide ranges of fluorescent molecules have been expressed in cells. 133 Additionally, Nanoparticles and quantum dots (Qdots) have been used for in vivo imaging. 134 The near-infrared fluorophores (NIRFs) with long emission wavelengths theoretically provide optimal optical images in vivo. NIRFs that emit between 700nm and 900nm have shown the most promise because tissue absorption from chromophores and non-specific autofluorescence are minimal at these wavelengths. Accepted NIRFs used in in vivo imaging in animals include AF680 and Cy5.5 (680nm absorption, 720nm emission) IRDye800CW (774nm absorption, 789nm emission), and IC-green (780nm absorption, 820nm emission). 135,136 Various studies have shown the efficacy of optical imaging probes using these fluorophores for breast cancer diagnosis and imaging of lymphatics. 136,137 However, the fluorescent imaging of deep tissue targets is challenging because it is difficult to accurately deconvolute the light signal produced from deep in tissues due to interference from bodily materials that alter light scattering and emitted light absorption.
Because no single imaging modality is ideal, efforts are also focused on developing multimodal imaging agents that take advantage of the strengths of each imaging technique employed. 138 For instance, Gelovani and co-workers labeled an RGD peptide with the NIRF IRDye800 and 111In for dual optical and SPECT imaging of human melanoma xenografts. 139 While both optical and gamma scintigraphy were able to detect the tumor, the optical images were superior in terms of resolution and detection of superficial tumors, and SPECT images were better at detecting deeper tumors.
6. Phage Display-selected Peptides Used in In Vivo Molecular Imaging of Cancer
6.1 Success Stories
While the vast majority of publications on phage display-selected peptides have only reported the use of peptides in vitro, there are a small number of phage display peptides that have been radiolabeled and used in SPECT and PET imaging and/or tagged with biotin or a fluorophore for use in optical imaging of cancer in vivo. As highlighted in Table 1, various peptides isolated from phage display screens are showing promise as potential targeted in vivo radio- and optical- imaging probes. Peptides used successfully to image tumor vasculature or lymphatics include αvβ3-integrin-binding RGD-containing peptides, 57,104–107,109,111,112 gastric cancer endothelium-binding CGNSNPKSC peptide, 140 vascular cell adhesion molecule-1-targeting (VCAM-1) VHSPNKK peptide, 50 matrix metalloproteinase (MMP)-2/9-targeting CTTHWGFTLC and SGKGPRQITAL peptides, 141,142 urokinase plasminogen activator (uPA)-binding SGRSA peptide 143 and urokinase-type plasminogen activator receptor (uPAR)-homing FSRYLWS peptide. 144
In one study, a T7 phage display library was selected against rhabdomyosarcoma cultured cell lines and an RGD-containing αvβ3-binding peptide and a lymphatic binding peptide with similarity to LyP-1 were isolated. The Lyp-1-like peptide CMGNKRSAKRPC was biotinylated and used in vivo to demonstrate that the peptide localized to neuroblastoma tumor vasculature. 57 In another study, a FSRYLWS variant peptide, which binds to loop three of uPAR domain III, was labeled via DOTA with 64Cu and used to PET image U87MG glioblastoma tumors in rodents. Tumor accumulation was 8.1 %ID/g at 6 h post-injection. 145 uPA and uPAR are key players in MMP degradation of extracellular matrix degradation in the tumor environment and are promising tumor targets for prostate cancer diagnosis and therapy.
Most of the peptides identified from phage display selections that have been used in molecular imaging in vivo bind vasculature components. However, proteins overexpressed on tumors have also been targeted by phage display-selected peptides for in vivo imaging. The ErbB-2-targeting peptide KCCYSL, 55,146 melanin-targeting peptide NPNWGPR, 147 hepsin/prostate cancer targeting peptide IPLVVPLGGSCK, 54 plectin-1/pancreatic ductal adenocarcimoa targeting peptide KTLLPTP, 53 and the galectin-3-targeting peptide ANTPCGPYTHDCPVKP, 148,149 have been used monovalently, on phage, or nanoparticles to image tumors in vivo. An example of a linear peptide functioning to in vivo image tumors is the galectin-3-binding peptide ANTPCGPYTHDCPVKP. Galectin-3 binds terminal galactopyranose residues on carbohydrates, most notably those of TF antigen (Figure 3). TF-galectin-3 association triggers re-localization of galectin-3 to cell surfaces of tumor cell-endothelial cell contact. The ANTPCGPYTHDCPVKP peptide, isolated from a cysteine-constrained library engineered in George Smith’s laboratory, 148 inhibited TF-galectin-3 interaction by ~50%. Its ability to localize to tumor-endothelium expressing galectin-3 was demonstrated by in vivo biodistribution and SPECT imaging studies with an 111In-DOTA-version of the peptide in human MDA-MB-435 breast tumor-bearing mice. In vivo biodistribution studies revealed that tumor uptake was 1.2 ± 0.24, 0.75 ± 0.05, and 0.6 ± 0.04 (mean ± SD) percent injected dose per gram (%ID/g) at 0.5, 1.0, and 2.0 h post injection of the radiotracer, respectively. However, high kidney uptake (~22%ID/g) was observed at 2 h. SPECT/CT studies with 111In-DOTA-glysergly(GSG)-ANTPCGPYTHDCPVKP showed good tumor uptake and contrast in the tumor-bearing mice (Figure 5). Specificity of peptide binding was demonstrated by successful blocking (52%) of in vivo tumor uptake of 111In-DOTA(GSG)-peptide in the presence of its non-radiolabeled counterpart at 2 h post injection.
Figure 5.
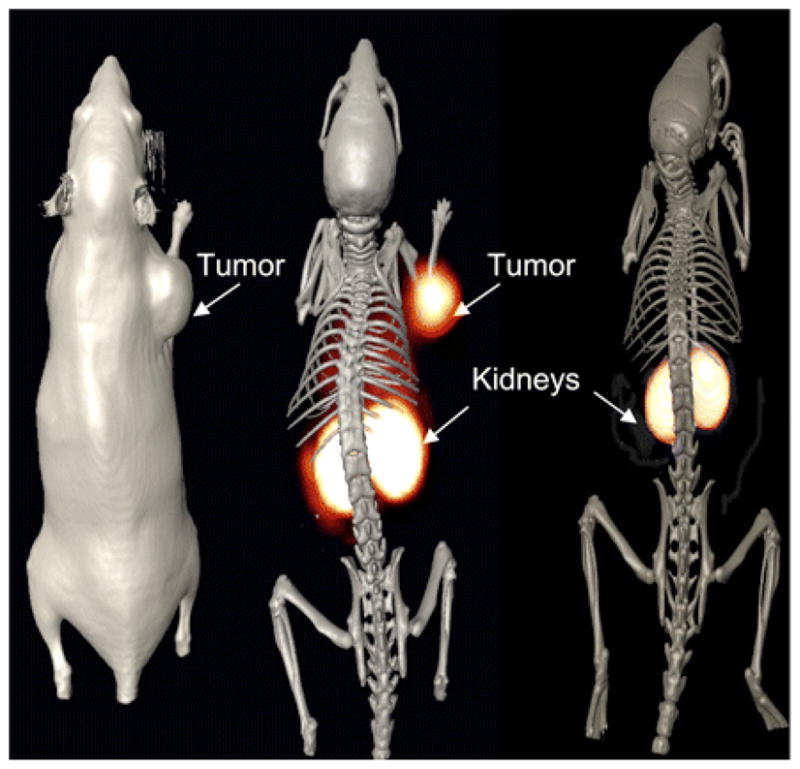
Tumor imaging with 111In-DOTA(GSG)-ANTPCGPYTHDCPVKR peptide. MDA-MB-435 breast tumor–xenografted SCID mice were injected in tail vein with 11.1 MBq of 111In-DOTA(GSG)-ANTPCGPYTHDCPVKR peptide and imaged in MicroCAT II (Siemens Medical Solutions) dedicated small-animal SPECT/CT scanner equipped with high-resolution 2-mm pinhole collimator. SPECT images were fused with conventional CT images to validate regions of increased radiolabeled ligand uptake. At left is volume-rendered CT image; at center, coregistered SPECT/CT radioligand uptake image of galectin-3–avid peptide; and at right, SPECT/CT image of scrambled peptide. Imaging was performed 2 h after injection. Reprinted with permission from reference number 149 (Figure 5), Copyright 2008 Society of Nuclear Medicine.
One of the best examples of a phage display-selected peptide used successfully in tumor imaging is the ErbB-2-targeting peptide KCCYSL. KCCYSL was obtained from a six amino acid fUSE5-cpIII phage library generated in George P. Smith’s laboratory. 29,146 ErbB-2 is a member of the epidermal growth factor receptor (EGFR) family of transmembrane receptor tyrosine kinases. While no known ligand or growth factor has been identified for ErbB-2, it heterodimerizes with other family members activating several signaling pathways resulting in increased cancer cell adhesion, growth, angiogenesis, and cell survival. 150,151 ErbB-2 has received much attention both as a biomarker for breast and prostate cancer and as a target for specific cancer imaging and therapeutic agents. Antibodies such as Trastuzumab (Herceptin®; Genetech, Inc.) have been developed to target and/or block the action of ErbB-2 and have been radiolabeled with a myriad of radionuclides for cancer imaging and therapy. 152,153 KCCYSL was shown to bind the extracellular domain (ECD) of ErbB-2 with 295nM affinity and bound cultured carcinoma cell lines that express ErbB-2 including breast, prostate, and ovarian cancer cell lines. The ErbB-2-targeting peptide KCCYSL may function as a mimic of a CCY/F motif present in EGF-like domains of ErbB family member ligands. 146 The KCCYSL peptide was conjugated with DOTA via a GSG spacer and radiolabeled with 111In for the SPECT imaging of ErbB-2-positive tumors.55 The 111In-DOTA-GSG-KCCYSL peptide bound ErbB-2-expressing human MDA-MB-435 breast carcinoma cells in vitro and competition studies with nonradiolabeled peptide revealed an IC50 value of 42.5 ± 2.76 nmol/L (Figure 6). Biodistribution studies showed rapid tumor uptake and whole body clearance of 111In-DOTA-GSG-KCCYSL in human breast carcinoma-bearing SCID mice, and SPECT/CT studies demonstrated that the breast tumor was readily visualized by the radiolabeled peptide conjugate at 2 h post injection (Figure 7). The only non-target organ uptake was in the kidneys.
Figure 6.
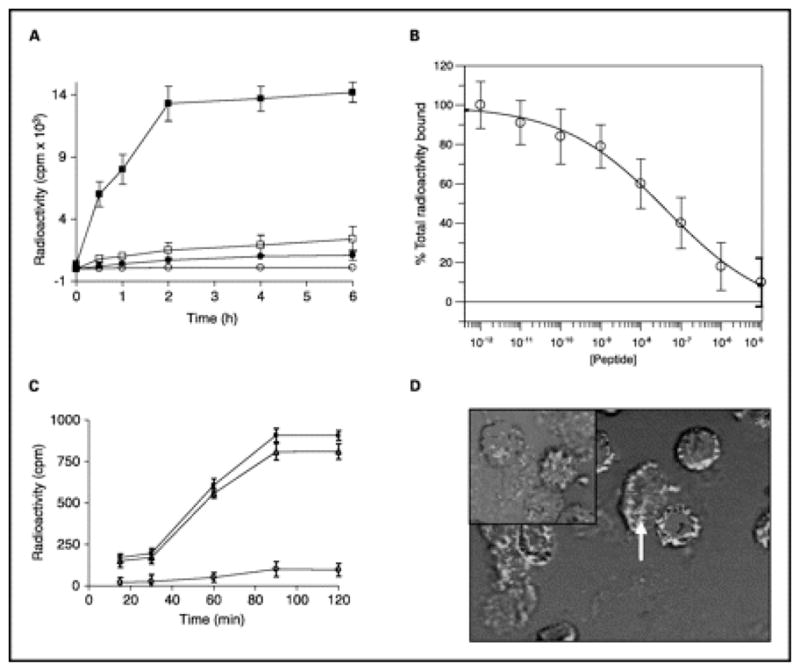
The ErbB-2 receptor binding properties of the radiolabeled 111In-DOTA(GSG)-KCCYSL. A, ~1.0 × 106 cells per well were incubated at 37°C for different time intervals with 5 × 104 cpm radioligand. Whereas significant radioligand binding to human MDA-MB-435 breast carcinoma cells was (■), minimal binding was observed with K-562 human chronic myeloid leukemia cells (●). Little binding of a radiolabeled scrambled peptide KYLCSC was observed with MDA-MB-435 (□) or K-562 (○) cell lines. Points, mean of three replicates; bars, SD. P < 0.001. B, displacement of 111In-DOTA(GSG)-KCCYSL peptide by its nonradiolabeled counterpart. MDA-MB-435 cells were incubated with 6 × 104 cpm radioligand and increasing concentrations of the nonradioactive peptide. The IC50 value obtained was 42.5 ± 2.76 nmol/L. Points, mean of three replicas; bars, SD. C, determination of percent internalized radioactivity in human MDA-MB-435 breast carcinoma cells. Cells (3 × 105 per tube) were incubated at 37°C with 111In-DOTA-(GSG) KCCYSL (4 × 104 cpm). The total (■), surface-bound (△), and internalized (○) radioactivity (cpm) as a function of time is depicted. Points, mean of two replicates; bars, SD. P < 0.001. D, surface binding and internalization of 5-carboxyfluorescein (FAM)-(GSG)-KCCYSL peptide. MDA-MB-435 cells were incubated with 0.5 μmol/L fluorescent peptide for 45 min at 37°C. After washing, the cells were fixed in paraformaldehyde and analyzed by confocal microscopy with an excitation/emission wavelength of 490/520 nm. The majority of the peptide was surface bound. Arrow, potential internalized peptide. Inset, analysis with FAM(GSG)-KYLCSC peptide indicated no binding. Reprinted with permission from reference number 55 (Figure 4), Copyright 2007 American Association for Cancer Research, Inc.
Figure 7.
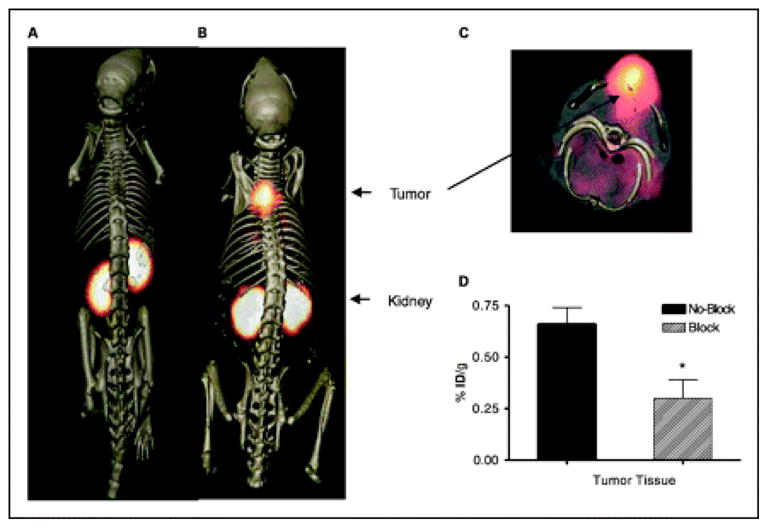
Tumor imaging with 111In-DOTA(GSG)-KCCYSL peptide. MDA-MB-435 breast tumor–xenografted SCID mice were injected in the tail vein with 11.1 MBq of 111In-DOTA(GSG)-KCCYSL or 111In-DOTA(GSG)-KYLCSC scrambled peptide and imaged in a microSPECT scanner. The SPECT images were fused with conventional microCT images to validate regions of increased radiolabeled ligand uptake. A, coregistered microSPECT/CT radioligand uptake image with 111In-DOTA(GSG)-KYLCSC; B, coregistered microSPECT/CT image with 111In-DOTA(GSG)-KCCYSL; C, microSPECT/CT image axial view focusing on tumor uptake of the radioligand. D, in vivo blocking studies with 111In-DOTA(GSG)-KCCYSL in MDA-MB-435 breast tumor–xenografted SCID mice. Fifteen minutes after injection of the nonradiolabeled In-DOTA(GSG)-KCCYSL (10–5-10–12 mol/L) peptide, 0.11 MBq of radiolabeled counterpart was injected and the blocking efficiency was evaluated after 2 h. A 50% block of the radiolabeled peptide binding to the tumor tissue was observed. Columns, mean of three animals for each experiment; bars, SD. *, P < 0.001. Reprinted with permission from reference number 55 (Figure 5), Copyright 2007 American Association for Cancer Research, Inc.
6.2 Kidney Uptake of Radiolabeled Peptides
Although radiolabeled peptides are excellent targeting moieties, their short plasma half-life and high renal retention will need to be improved for translation into humans, especially if employed as therapeutic agents. Radiolabeled peptides as well as small antibody fragments are cleared from the kidney, which is the preferred route of elimination of a radiopharmaceutical. High kidney uptake has been observed for most of the commonly studied radiolabeled peptides, 154–156 and is also evident with phage display-selected peptides (Figures 5 and 6). For imaging purposes, renal uptake greatly reduces the sensitivity of detection if the tumor is in the vicinity of the kidney. For therapy, renal accumulation of radiolabeled molecules limits the maximum tolerated dose that can be administered without radiation-induced nephrotoxicity. Thus, lowering the renal uptake would allow for administration of higher tumor radiation doses and improved therapeutic response. Numerous approaches have been investigated to alter renal retention of radiopharmaceuticals. Standard approaches of changing peptide sequence and charge, radionuclide, and/or chelate have shown limited success. Coadministration of basic compounds, particularly lysine or arginine can reduce radioactive uptake in the kidney by on average 50%, 155–157 however high doses of these amino acids can cause serious side effects including arrhythmia, hyperkalemia, and nephrotoxicity. 158 More recently, plasma substitutes that cause low molecular weight proteinuria by decreasing tubular reabsorption have been investigated with similar results as amino acid infusions. 159 Additional means to reduce kidney uptake are thus, warranted.
6.3 Peptides in Tumor Reduction
Not only have phage display-selected peptides been used in cancer imaging, but they have also been used in vivo to reduce tumor growth. The ability of unlabeled peptides to functionally modulate tumor growth and spread should positively impact future studies aimed at developing peptide-based cancer therapeutics. Potentially harmful radiation-induced damage to non-target organs will be avoided if unlabeled peptides can be employed. Previous studies have relied on chemotherapeutics and/or antibodies (which often cause immune response problems). There have been a few reports of the use of phage display-selected peptides in vivo to lessen tumor growth (Table 1). In a 2008 study, a protein kinase (CK2) inhibitor cyclic peptide CIGB-300 blocked CK2 phosphorylation and reduced cervical tumor growth in heterotransplanted nude mice. Further, the safety of CIGB-3000 administration was evaluated in 31 women with cervical cancer. The peptide was well tolerated and reported to reduce cervical lesions in the majority of patients. 160 In 2009, Chang and co-workers reported that a non-small cell lung cancer (NSCLC)-targeting peptide TDSILRSYDWTY, when coupled to liposomes with doxorubicin or vinorelbine, increased the efficacy of the chemotherapeutics and survival rates of NSCLC-xenografted mice 94. Thus, a promising application of phage display peptides may not only be for imaging of molecular processes and diseases but inhibition of steps involved in disease progression.
7. Phage as Molecular Imaging Agents
7.1 Radiolabeled Phage
Phage displaying tumor-homing peptides have shown recent promise as tumor imaging agents. In fact, there are more reports of peptide-displaying phage used in tumor imaging then the corresponding synthesized peptides (Table 1). There are many attributes of phage that uniquely suit them as in vivo imaging agents and biological nanoparticles. Filamentous phage self-assemble into long (~900 nm × 6 nm) rod-like protein-encapsulated structures, within which is the genetic information to dictate their own production. Phage can be covalently attached to numerous tags such as biotin or fluorophores such as AF680 while simultaneously expressing multiple copies of foreign vasculature or tumor-targeting peptides. This results in signal amplification. Phage have also been physically well-characterized, are resistant to harsh conditions, and are non-pathogenic. Studies have shown that fd phage are well tolerated at high concentrations in vivo and generate only a weak immune response after weeks of administration. 120 Given this, they may be superior to that of commonly employed inorganic nanoparticles for in vivo imaging.
A few laboratories have been developing phage into agents for use in SPECT and PET radioimaging. 119,161–163 Initial studies demonstrated that 99mTc-labeled phage could be used in vivo to image bacterial infections in mice. 161,162 This approach has been extended to tumor imaging in vivo. For tumor imaging, it would seem that the most straightforward approach would be to covalently couple a radiometal bifunctional chelator onto the phage for direct labeling. However, phage radiolabeled with 99mTc via a mercaptoacetyltriglycine chelate demonstrated high liver uptake of radioactivity (10–40%ID/g). This finding is consistent with the long clearance time of approximately 72 h of phage through the reticuloendothelial system (RES). 120 Hence, implementation of a pretargeting system with phage may allow for phage clearance before administration of the radiolabel. Both two-step and three-step pretargeting strategies with biotinylated phage have been reported for the SPECT imaging of cancer. 163,164 The in vivo selected prostate tumor-targeting phage clone G1 (Tables 1, 2; Figure 4) was used in a two-step system with biotinylated phage in combination with 111In-radiolabeled diethylene triamine pentaacetic acid (DTPA)-streptavidin. Results were compared to a three-step pretargeting method utilizing biotinylated phage, avidin, and 111In-bisbiotin. Biodistribution studies revealed there was high radioactive uptake in the two-step pretargeting approach in the liver, kidneys, and intestines at 24 h, which may be attributed to the pharmacokinetics of 111In-streptavidin. However, there was less than 1%ID/g of radioactivity in the tumor. In contrast, there was good (~3%ID/g) uptake of radiolabel in the tumor using the three-step approach and less kidney uptake (5%ID/g at 1 h). Very little radioactivity accumulated in other organs including the liver. Taken together, these results suggest that a three-step pretargeting approach may yield the best phage-based radioimaging probe. The ability of G1 phage to act as SPECT imaging agents of human PC-3 prostate tumor heterotransplants in mice using both the two-step and three-step methods is shown in Figure 8.164 As shown, SPECT images of G1 phage using the two-step approach did not show tumoral accumulation, whereas the prostate tumor was clearly imaged using the three-step scheme.
Figure 8.

SPECT/CT imaging studies of pretargeted 111In labeled streptavidin and biotin in SCID mice bearing human prostate PC-3 carcinoma tumors. SCID mice bearing human PC-3 prostate carcinoma tumors received tail vein injections of 1011 virions of biotinylated G1 phage. (A) Four hours post injection of the biotinylated G1 phage, mouse A received an injection 7.40 MBq of 111In-DTPA-SA for the purpose of two-step pretargeting by biotinylated G1 phage. The image was taken twenty four hours post injection of the radiolabel. (B & C) Mouse B and C received three-step pretargeting treatments. Four hours post injection of the biotinylated G1 phage both mice received an injection of avidin which was allowed to circulate and clear the body for twenty four hours. Mouse B then received a third injection 7.40 MBq of 111In-DOTA-biotin, while the third injection given to mouse C contained both cold In-DOTA-biotin and 111In-DOTA-biotin. All mice were euthanized before fifteen hours of scan data were obtained. Reprinted with permission from reference number 164 (Figure 5), Copyright 2009 Elsevier.
7.2 Fluorescent Labeled Phage
A review of the literature suggests that phage display has been most successfully used in molecular imaging by employing fluorescently-tagged phage or nanoparticle platforms. A benefit of the use of fluorescently labeled phage is that they can be used for long-term animal imaging studies, whereas radiolabeled phage may cause non-target organ damage over long periods of time.
Numerous approaches have been undertaken to directly modify phage for use in optical imaging. The cpVIII of phage has been modified using standard chemistry approaches to obtain FITC, AF647, AF680, and AF750, labeled phage that retain their target affinity. 165 K. Kelly et al. using intravital confocal microscopy demonstrated that phage displaying the VHSPNKK peptide (Table 1) imaged VCAM-1-expressing endothelial cells in a murine tumor necrosis factor-α (TNFα) induced inflammatory ear model. 166 In another study from their group, phage that bound secreted protein acidic and rich in cysteine (SPARC) upregulated in invasive cancer were obtained. Phage were selected against purified SPARC protein in vitro using a 7 amino acid random linear peptide library (New England Biolaboratories, Cambridge MA). Clone 23, with the displayed peptide sequence SPPTGIN was coupled using hydroxysuccinimide esters of VT680 and AF750 and used in surface reflectance imaging of Lewis lung carcinoma (LLC)-xenografted nude mice. The surface reflectance imaging (Figure 9) and fluorescent molecular tomography (FMT, not shown) clearly demonstrated increased tumor uptake compared to WT phage. 167
Figure 9.
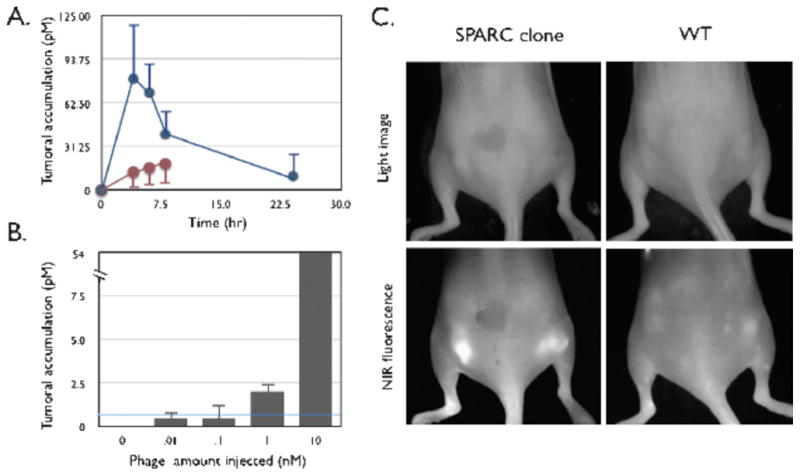
In vivo behavior of labeled phage. (A) Time course of tumor homing. Mice bearing subcutaneous bilateral LLC-derived tumors were coinjected through the tail vein with VT680-labeled SPARC-targeted phage and AF750-labeled wild-type phage (no insert) and imaged at 0, 2, 4, 6, and 24 hours after injection. Blue line: SPARC-targeted phage clone 23. Brown line: wild-type phage (no insert). (B) Detection threshold. Tumor-bearing mice were injected with increasing log doses of labeled phage and imaged 4 hours after injection. The line indicates detection threshold. (C) Reflectance imaging. Mice bearing subcutaneous bilateral tumors (LLC cells) were injected with either VT680-labeled wild-type phage (right) or VT680-labeled SPARC-targeted phage. Note the brightly fluorescent tumors in the near-infrared fluorescence channel of the SPARC-targeted phage clone [identical white light (WL) settings]. Reprinted with permission from reference number 167 (Figure 4), Copyright 2006 Neoplasia Press, Inc.
In vivo selected phage that extravasate the vasculature and target tumors in vivo have been also used successfully in optical imaging. The ability of one such phage clone, G1, (Table 2) to image PC-3 tumors was investigated using AF680-labeled phage. The in vivo distribution of the G1 phage showed tumoral accumulation as early as 5 minutes and peaked from 4 to 6 h. To determine the usefulness of G1 phage as an in vivo imaging agent surface reflectance imaging with AF680 labeled G1 phage was performed. AF680 labeled phage could be imaged in the PC-3 derived tumors in SCID mice as early as 1 h post injection peaking at 4 h. At 4 h post-injection of AF680 G1 phage, a 4.5 fold increase in the fluorescent signal within the tumor compared to that of the normal tissue was observed (Figure 10). 119
Figure 10.

Optical imaging of prostate tumor-targeting phage in vivo. Phage displaying the prostate carcinoma-targeting peptide G1, IAGLATPGWSHWLAL, (left top panel) labeled with AF680 were injected into the tail vein of PC-3 human prostate tumor xenografted mice. The animals were imaged 1, 4, 24 hours post phage injection. The only signal detected was from the tumor on the right shoulder of the mouse injected with prostate tumor-selected phage (blue image). Reprinted with permission from reference number 119 (Figure 5), Copyright 2006 Neoplasia Press, Inc.
7.3 Chimeric Phage and Nanoparticles
An intriguing application of phage is in the integration of tumor targeting and genetic (viral) imaging in order to deliver and image specific transgenes. Pasqualini, Arap, and co-workers developed chimeric phage (P) displaying the RGD-4C integrin-targeting sequence CDCRGDCFC and adeno-associated virus (AAV) constructs (AAVP vectors) which were evaluated for tumor targeting and imaging, and also Herpes Simplex Virus thymidine kinase (HSVtk) gene expression in a mouse model of human soft-tissue sarcoma. PET imaging was performed with 18F-FDG and the nucleoside analogue 2′-18F-fluoro-2″deoxy-1-β-D-arabino-furanosyl-5-ethyl-uracil (18F-FEAU), a substrate for HSVtk (Figure 11). 168,169 These studies demonstrated the simultaneous specific targeting of RGD-4C expressing cells, noninvasive imaging of HSVtk reporter genes, and drug response monitoring using a PET-based imaging probe. Further, these studies suggest that dual targeting AAV/phage vectors can be used to image gene expression and drug response prediction, which may be highly beneficial in humans.
Figure 11. Predicting and Monitoring Drug Response in a Preclinical Model of Human Sarcoma.
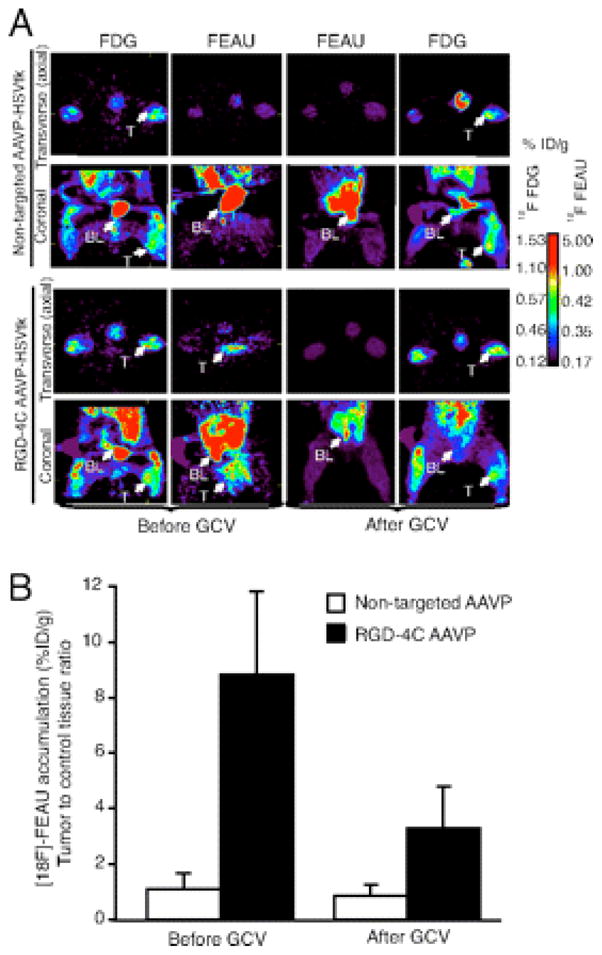
PET imaging of HSVtk transgene expression was performed in sarcoma-bearing rats after i.v. delivery of RGD-4C targeted AAVP or nontargeted control. The first GCV treatment cycle was initiated at 24 h after [18F]-FEAU administration and imaging to enable the molecular–genetic imaging of the corresponding drug response. (A) Cohorts of nude rats bearing human SKLMS1-derived xenografts (n = 8) received a single i.v. dose (3 × 1012 TU) of RGD-4C AAVP-HSVtk or control nontargeted AAVP-HSVtk. PET imaging of [18F]-FEAU was performed after AAVP administration (day 9) and then again after drug treatment with GCV (day 15). PET imaging of [18F]-FDG was performed (day 8) and then again after the second [18F]-FEAU (day 16). PET imaging with [18F]-FDG and with [18F]-FEAU are presented (before and after treatment with GCV) as indicated. Transverse (axial) and coronal sections are shown. A standard calibration scale is provided, and correspondence of [18F]-FDG and [18F]-FEAU PET imaging is indicated. (B) Relative sarcoma expression of HSVtk as assessed by repetitive PET imaging with [18F]-FEAU before and after initiation of cytotoxic drug treatment with GCV. A mesenchymal-derived normal tissue (muscle) served to normalize the tumor-to-control reporter transgene expression ratio. Reprinted with permission from reference number 168 (Figure 2), Copyright, 2008 National Academy of Sciences, USA.
Phage display selected peptides have also been conjugated to nanoparticles and used to image cancer. For example, Kelly et al used phage display with cultured cells to identify peptides specific for pancreatic ductal adenocarcinoma (PDAC). Two clones, 15 (TMAPSIK) and 27 (LLPSGKP) demonstrated good affinity and specificity for the target PDAC cells. To test these plectin-1 binding peptides as potential diagnostic agents for PDAC, the phage were labeled with the fluorochromes VT680 and Texas Red then injected into mice from the Kras p53 PDAC model. This technique enabled the imaging of PDAC in mice. Peptide 27 was synthesized and attached to a magnetofluorescent nanoparticle (NP) (crosslinked iron oxide CLIO-Cy5.5) (PTP-NP) and the resulting MRI/optical imaging agent was tested in PDAC tumor-bearing mice. Intravital confocal microscopy detected discrete areas of fluorescence in the abdomen of the mice 24 h after administration (Figure 12). Biodistribution studies demonstrated specific uptake in tumor, with also some liver, kidney, spleen, and lung uptake. The agent was present in the tumor tissue rather than the vasculature since a vasculature agent administered before injection did not colocalize. 53
Figure 12.

Fluorescence imaging of PDAC using PTP-NP or Control-NP. (A) Schematic of conjugation of PTP to NP. Control-NP is synthesized the same way with substitution of control peptide for PTP. (B) Intravital confocal microscopy of early pancreatic lesions imaged using PTP-NP (red, top) or control-NP (red, bottom) and AF750-labeled bloodpool agent (blue). (C) Low-magnification view of pancreatic fluorescence shows distribution of PTP-NP in distinct areas of the pancreas. White light overlay provides anatomic correlation (left). Dotted line outlines the pancreas. (D) Biodistribution of PTP-NP and control-NP. Reprinted with permission from reference number 53 (Figure 5), Copyright 2008 PLoS Medicine.
8. Prospects and Challenges
Given the progress made in the past few years in utilizing phage display-selected peptides for the in vivo tumor imaging and diagnosis of cancer in animals, it is predicted that similar peptide constructs will be translated in the not too distant future into the clinic for use in humans. A major obstacle in efficacious use in humans is the high kidney uptake observed with radiolabeled peptides including those isolated using phage display 154–156. Even though radiopharmaceutical renal clearance is preferred over elimination through the RES, high kidney uptake can severely limit the effectiveness of a radiolabeled peptide in diagnostic cancer imaging especially if the tumor (or metastases) is in the vicinity of the kidneys. A better understanding of the mechanisms of radiolabeled peptide renal retention in combination with novel molecular genetic and combinatorial approaches to identify peptide signatures retained in the kidney may be warranted. In addition to progress made with phage display-selected peptides, phage displaying the cancer-avid peptides have been utilized in vivo and are particularly well suited to a wide-range of modifications and imagining modalities. While their use in animals is gaining widespread acceptability, the use of phage in humans still faces obstacles including immunogenicity and clearance problems, especially since phage are extremely large and clear through the reticuloendothelial system. A combination of peptide and organic nanoparticle sciences may be an attractive approach to address some of the challenges.
9. Conclusion
Combinatorial chemistry and phage display technologies provide robust means for the rapid discovery of tumor antigen-avid peptides. Using in vitro and in vivo phage display, new targeting peptides with capabilities to image tumors in living animals have been selected. In the last five years, many of these peptides have been radio- and/or optically- labeled for the imaging of a range of tumors in vivo. Phage bearing the tumor-avid peptide sequences have been modified so as to function as multimodal or multi-step imaging agents, as well. Thus, phage display technology has developed into a rapid, economical, and efficacious approach to the development of agents for the molecular imaging and diagnosis of cancer.
Acknowledgments
I would like to thank Marie Dickerson and Jessica Newton-Northup for contributions to this work. This work was supported in part by a Merit Review award from the Veterans Administration and by the US National Institutes of Health (Grants: P50 CA103130-01, 1R21CA134960 and CA137239).
10. Abbreviations
- AAV
adeno-associated virus
- AF
AlexaFluor
- α-MSH
α-melanocyte stimulating hormone
- CLIO
crosslinked iron oxide
- cp
coat protein
- CRIP-1
cysteine-rich intestinal protein
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DTPA
diethylenetriaminepentaacetic acid
- EGFR
epidermal growth factor receptor
- ECD
extracellular domain
- FGF
fibroblast growth factor
- 18F-FDG
fluorine-18 2-deoxy-2-fluoro-D-glucose
- 18F-FEAU
2′-18F-fluoro-2″deoxy-1-β-D-arabino-furanosyl-5-ethyl-uracil
- FMT
fluorescent molecular tomography
- GRP
gastrin releasing peptide
- HSVtk
Herpes Simplex Virus thymidine kinase
- IGF-1
insulin-like growth factor-1
- LLC
Lewis lung carcinoma
- MMP
matrix metalloproteinase
- NP
nanoparticle
- NIRFs
near-infrared fluorophores
- NSCLC
non-small cell lung cancer
- P
chimeric phage
- PDAC
pancreatic ductal adenocarcinoma
- PET
positron emission tomography
- phage
bacteriophage
- PBP
plectin-1 binding peptides
- PSA
prostate specific antigen
- Qdots
quantum dots
- RES
reticuloendothelial system
- SPARC
secreted protein acidic and rich in cysteine
- SPECT
single photon emission tomography
- TAG-72
tumor-associated glycoprotein 72
- TGF-β
transforming growth factor-β
- TF
Thomsen-Friedenreich
- TNFα
tumor necrosis factor-α
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- VCAM-1
vascular cell adhesion molecule-1-targeting
- VEGF
vascular endothelial growth factor
Biography
 Susan Deutscher graduated with a Bachelor of Science in Biology from Purdue University in 1980. She received her Ph.D. in Biochemistry at St. Louis University in the laboratory of Dr. Carlos Hirschberg in 1985 where she worked on carbohydrate-protein interactions. After this, she pursued Post-doctoral studies at Duke University in the Department of Microbiology and Immunology. She became a faculty member in the Department of Biochemistry at the University of Missouri in 1991 and is currently a Professor here and a Research Chemist at the Harry S. Truman Veterans Hospital in Columbia Missouri. Her laboratory is interested in the role of carbohydrate and protein-protein interactions in cancer. Work focuses on applying novel combinatorial bacteriophage display approaches and structural biochemistry to characterize these interactions.
Susan Deutscher graduated with a Bachelor of Science in Biology from Purdue University in 1980. She received her Ph.D. in Biochemistry at St. Louis University in the laboratory of Dr. Carlos Hirschberg in 1985 where she worked on carbohydrate-protein interactions. After this, she pursued Post-doctoral studies at Duke University in the Department of Microbiology and Immunology. She became a faculty member in the Department of Biochemistry at the University of Missouri in 1991 and is currently a Professor here and a Research Chemist at the Harry S. Truman Veterans Hospital in Columbia Missouri. Her laboratory is interested in the role of carbohydrate and protein-protein interactions in cancer. Work focuses on applying novel combinatorial bacteriophage display approaches and structural biochemistry to characterize these interactions.
References
- 1.Hood L, Heath JR, Phelps ME, Lin B. Science. 2004;306:640. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 2.Rudin M, Weissleder R. Nat Rev Drug Discov. 2003;2:123. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 3.Akerman ME, Chan WC, Laakkonen P, Bhatia SN, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99:12617. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly KA, Jones DA. Neoplasia. 2003;5:437. doi: 10.1016/s1476-5586(03)80046-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glinsky VV, Glinsky GV, Rittenhouse-Olson K, Huflejt ME, Glinskii OV, Deutscher SL, Quinn TP. Cancer Res. 2001;61:4851. [PubMed] [Google Scholar]
- 6.Glinsky VV, Glinsky GV, Glinskii OV, Huxley VH, Turk JR, Mossine VV, Deutscher SL, Pienta KJ, Quinn TP. Cancer Res. 2003;63:3805. [PubMed] [Google Scholar]
- 7.Pierce MC, Javier DJ, Richards-Kortum R. Int J Cancer. 2008;123:1979–90. doi: 10.1002/ijc.23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ray P, Wu AM, Gambhir SS. Cancer Res. 2003;63:1160. [PubMed] [Google Scholar]
- 9.Edwards WB, Xu B, Akers W, Cheney PP, Liang K, Rogers BE, Anderson CJ, Achilefu S. Bioconjugate Chem. 2008;19:192. doi: 10.1021/bc700291m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharkey RM, Goldenberg DM. J Nucl Med. 2005;46(Suppl 1):115S. [PubMed] [Google Scholar]
- 11.Kruglyak E, Freeman LM, Wadler S. Ann Oncol. 1997;8:301. doi: 10.1023/a:1008213100329. [DOI] [PubMed] [Google Scholar]
- 12.Goldenberg MM. Clin Ther. 1999;21:309. doi: 10.1016/S0149-2918(00)88288-0. [DOI] [PubMed] [Google Scholar]
- 13.Klastersky J. Curr Opin Oncol. 2006;18:316. doi: 10.1097/01.cco.0000228734.32261.62. [DOI] [PubMed] [Google Scholar]
- 14.Goldenberg DM. J Nucl Med. 2002;43:693. [PubMed] [Google Scholar]
- 15.Lewis MR, Wang M, Axworthy DB, Theodore LJ, Mallet RW, Fritzberg AR, Welch MJ, Anderson CJ. J Nucl Med. 2003;44:1284. [PubMed] [Google Scholar]
- 16.Reubi JC. Endocr Rev. 2003;24:389. doi: 10.1210/er.2002-0007. [DOI] [PubMed] [Google Scholar]
- 17.Behr TM, Gotthardt M, Barth A, Behe M. Q J Nucl Med. 2001;45:189. [PubMed] [Google Scholar]
- 18.Bakker WH, Krenning EP, Reubi JC, Breeman WA, Setyono-Han B, de Jong M, Kooij PP, Bruns C, van Hagen PM, Marbach P. Life Sci. 1991;49:1593. doi: 10.1016/0024-3205(91)90053-e. [DOI] [PubMed] [Google Scholar]
- 19.Smith CJ, Volkert WA, Hoffman TJ. Nucl Med Biol. 2003;30:861. doi: 10.1016/s0969-8051(03)00116-1. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Cheng Z, Miao Y, Jurisson SS, Quinn TP. Cancer. 2002;94:1196. doi: 10.1002/cncr.10284. [DOI] [PubMed] [Google Scholar]
- 21.Miao Y, Owen NK, Fisher DR, Hoffman TJ, Quinn TP. J Nucl Med. 2005;46:121. [PubMed] [Google Scholar]
- 22.Chen J, Cheng Z, Hoffman TJ, Jurisson SS, Quinn TP. Cancer Res. 2000;60:5649. [PubMed] [Google Scholar]
- 23.Chen J, Cheng Z, Owen NK, Hoffman TJ, Miao Y, Jurisson SS, Quinn TP. J Nucl Med. 2001;42:1847. [PubMed] [Google Scholar]
- 24.Miao Y, Benwell K, Quinn TP. J Nucl Med. 2007;48:73. [PubMed] [Google Scholar]
- 25.Miao Y, Hylarides M, Fisher DR, Shelton T, Moore H, Wester DW, Fritzberg AR, Winkelmann CT, Hoffman T, Quinn TP. Clin Cancer Res. 2005;11:5616. doi: 10.1158/1078-0432.CCR-05-0619. [DOI] [PubMed] [Google Scholar]
- 26.Wei L, Miao Y, Gallazzi F, Quinn TP, Welch MJ, Vavere AL, Lewis JS. Nucl Med Biol. 2007;34:945. doi: 10.1016/j.nucmedbio.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McQuade P, Miao Y, Yoo J, Quinn TP, Welch MJ, Lewis JS. J Med Chem. 2005;48:2985. doi: 10.1021/jm0490282. [DOI] [PubMed] [Google Scholar]
- 28.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 29.Smith GP. Science. 1985;228:1315. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 30.Atwell S, Wells JA. Proc Natl Acad Sci U S A. 1999;96:9497. doi: 10.1073/pnas.96.17.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbas CF, 3rd, Kang AS, Lerner RA, Benkovic SJ. Proc Natl Acad Sci U S A. 1991;88:7978. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith GP, Petrenko VA. Chem Rev. 1997;97:391. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 33.Brissette R, Goldstein NI. Methods Mol Biol. 2007;383:203. doi: 10.1007/978-1-59745-335-6_13. [DOI] [PubMed] [Google Scholar]
- 34.Bessette PH, Rice JJ, Daugherty PS. Protein Eng Des Sel. 2004;17:731. doi: 10.1093/protein/gzh084. [DOI] [PubMed] [Google Scholar]
- 35.Lunder M, Bratkovic T, Urleb U, Kreft S, Strukelj B. Biotechniques. 2008;44:893. doi: 10.2144/000112759. [DOI] [PubMed] [Google Scholar]
- 36.Noppe W, Plieva F, Galaev IY, Pottel H, Deckmyn H, Mattiasson B. BMC Biotechnol. 2009;9:21. doi: 10.1186/1472-6750-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Adams JD, Turner K, Cochran FV, Gambhir SS, Soh HT. Lab Chip. 2009;9:1033. doi: 10.1039/b820985e. [DOI] [PubMed] [Google Scholar]
- 38.Yip YL, Ward RL. Curr Pharm Biotechnol. 2002;3:29. doi: 10.2174/1389201023378472. [DOI] [PubMed] [Google Scholar]
- 39.Brown KC. Curr Opin Chem Biol. 2000;4:16. doi: 10.1016/s1367-5931(99)00045-9. [DOI] [PubMed] [Google Scholar]
- 40.Aina OH, Sroka TC, Chen ML, Lam KS. Biopolymers. 2002;66:184. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 41.Sehgal A. Curr Opin Drug Discov Devel. 2002;5:245. [PubMed] [Google Scholar]
- 42.Work LM, Nicklin SA, White SJ, Baker AH. Methods Enzymol. 2002;346:157. doi: 10.1016/s0076-6879(02)46055-7. [DOI] [PubMed] [Google Scholar]
- 43.Ladner RC. Q J Nucl Med. 1999;43:119. [PubMed] [Google Scholar]
- 44.Hoess RH. Chem Rev. 2001;101:3205. doi: 10.1021/cr000056b. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson F, Tarli L, Viti F, Neri D. Adv Drug Delivery Rev. 2000;43:165. doi: 10.1016/s0169-409x(00)00068-5. [DOI] [PubMed] [Google Scholar]
- 46.Manoutcharian K, Gevorkian G, Cano A, Almagro JC. Curr Pharm Biotechnol. 2001;2:217. doi: 10.2174/1389201013378671. [DOI] [PubMed] [Google Scholar]
- 47.Nielsen UB, Marks JD. Pharm Sci Technol Today. 2000;3:282. doi: 10.1016/s1461-5347(00)00280-7. [DOI] [PubMed] [Google Scholar]
- 48.Skelton NJ, Chen YM, Dubree N, Quan C, Jackson DY, Cochran A, Zobel K, Deshayes K, Baca M, Pisabarro MT, Lowman HB. Biochemistry. 2001;40:8487. doi: 10.1021/bi0103866. [DOI] [PubMed] [Google Scholar]
- 49.Peletskaya EN, Glinsky VV, Glinsky GV, Deutscher SL, Quinn TP. J Mol Biol. 1997;270:374. doi: 10.1006/jmbi.1997.1107. [DOI] [PubMed] [Google Scholar]
- 50.Kelly KA, Nahrendorf M, Yu AM, Reynolds F, Weissleder R. Mol Imaging Biol. 2006;8:201. doi: 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
- 51.Fukuda MN, Ohyama C, Lowitz K, Matsuo O, Pasqualini R, Ruoslahti E, Fukuda M. Cancer Res. 2000;60:450. [PubMed] [Google Scholar]
- 52.Funovics M, Montet X, Reynolds F, Weissleder R, Josephson L. Neoplasia. 2005;7:904. doi: 10.1593/neo.05352. [DOI] [PubMed] [Google Scholar]
- 53.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, Depinho RA, Mahmood U, Weissleder R. PLoS Med. 2008;5:e85. doi: 10.1371/journal.pmed.0050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelly KA, Setlur SR, Ross R, Anbazhagan R, Waterman P, Rubin MA, Weissleder R. Cancer Res. 2008;68:2286. doi: 10.1158/0008-5472.CAN-07-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar SR, Quinn TP, Deutscher SL. Clin Cancer Res. 2007;13:6070. doi: 10.1158/1078-0432.CCR-07-0160. [DOI] [PubMed] [Google Scholar]
- 56.Thapa N, Kim S, So IS, Lee BH, Kwon IC, Choi K, Kim IS. J Cell Mol Med. 2008;12:1649. doi: 10.1111/j.1582-4934.2008.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Witt H, Hajdin K, Iljin K, Greiner O, Niggli FK, Schafer BW, Bernasconi M. Int J Cancer. 2009;124:2026. doi: 10.1002/ijc.24170. [DOI] [PubMed] [Google Scholar]
- 58.Fusenig NE, Boukamp P. Mol Carcinog. 1998;23:144. doi: 10.1002/(sici)1098-2744(199811)23:3<144::aid-mc3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 59.Zhu ZY, Zhong CP, Xu WF, Lin GM, Ye GQ, Ji YY, Sun B, Yeh M. Cell Res. 1999;9:271. doi: 10.1038/sj.cr.7290026. [DOI] [PubMed] [Google Scholar]
- 60.Riemer AB, Jensen-Jarolim E. Immunol Lett. 2007;113:1. doi: 10.1016/j.imlet.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riemer AB, Klinger M, Wagner S, Bernhaus A, Mazzucchelli L, Pehamberger H, Scheiner O, Zielinski CC, Jensen-Jarolim E. J Immunol. 2004;173:394. doi: 10.4049/jimmunol.173.1.394. [DOI] [PubMed] [Google Scholar]
- 62.Hao J, Serohijos AW, Newton G, Tassone G, Wang Z, Sgroi DC, Dokholyan NV, Basilion JP. PLoS Comput Biol. 2008;4:e1000138. doi: 10.1371/journal.pcbi.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koolpe M, Burgess R, Dail M, Pasquale EB. J Biol Chem. 2005;280:17301. doi: 10.1074/jbc.M500363200. [DOI] [PubMed] [Google Scholar]
- 64.Koolpe M, Dail M, Pasquale EB. J Biol Chem. 2002;277:46974. doi: 10.1074/jbc.M208495200. [DOI] [PubMed] [Google Scholar]
- 65.Kim Y, Lillo AM, Steiniger SC, Liu Y, Ballatore C, Anichini A, Mortarini R, Kaufmann GF, Zhou B, Felding-Habermann B, Janda KD. Biochemistry. 2006;45:9434. doi: 10.1021/bi060264j. [DOI] [PubMed] [Google Scholar]
- 66.Vidal CI, Mintz PJ, Lu K, Ellis LM, Manenti L, Giavazzi R, Gershenson DM, Broaddus R, Liu J, Arap W, Pasqualini R. Oncogene. 2004;23:8859. doi: 10.1038/sj.onc.1208082. [DOI] [PubMed] [Google Scholar]
- 67.Pazgier M, Liu M, Zou G, Yuan W, Li C, Li J, Monbo J, Zella D, Tarasov SG, Lu W. Proc Natl Acad Sci U S A. 2009;106:4665. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zurita AJ, Troncoso P, Cardo-Vila M, Logothetis CJ, Pasqualini R, Arap W. Cancer Res. 2004;64:435. doi: 10.1158/0008-5472.can-03-2675. [DOI] [PubMed] [Google Scholar]
- 69.Pakkala M, Jylhasalmi A, Wu P, Leinonen J, Stenman UH, Santa H, Vepsalainen J, Perakyla M, Narvanen A. J Pept Sci. 2004;10:439. doi: 10.1002/psc.557. [DOI] [PubMed] [Google Scholar]
- 70.Zhao X, Hu J, Huang R, Yang L. J Exp Clin Cancer Res. 2007;26:509. [PubMed] [Google Scholar]
- 71.Jager S, Jahnke A, Wilmes T, Adebahr S, Vogtle FN, Delima-Hahn E, Pfeifer D, Berg T, Lubbert M, Trepel M. Leukemia. 2007;21:411. doi: 10.1038/sj.leu.2404548. [DOI] [PubMed] [Google Scholar]
- 72.Hetian L, Ping A, Shumei S, Xiaoying L, Luowen H, Jian W, Lin M, Meisheng L, Junshan Y, Chengchao S. J Biol Chem. 2002;277:43137. doi: 10.1074/jbc.M203103200. [DOI] [PubMed] [Google Scholar]
- 73.Fan H, Duan Y, Zhou H, Li W, Li F, Guo L, Roeske RW. IUBMB Life. 2002;54:67. doi: 10.1080/15216540214308. [DOI] [PubMed] [Google Scholar]
- 74.Maruta F, Parker AL, Fisher KD, Hallissey MT, Ismail T, Rowlands DC, Chandler LA, Kerr DJ, Seymour LW. Cancer Gene Ther. 2002;9:543. doi: 10.1038/sj.cgt.7700470. [DOI] [PubMed] [Google Scholar]
- 75.Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, Mazie JC. EMBO J. 2000;19:1525. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michon IN, Penning LC, Molenaar TJ, van Berkel TJ, Biessen EA, Kuiper J. Biochem Biophys Res Commun. 2002;293:1279. doi: 10.1016/S0006-291X(02)00378-9. [DOI] [PubMed] [Google Scholar]
- 77.Hyun S, Lee EH, Park J, Yu J. Bioorg Med Chem Lett. 2008;18:4011. doi: 10.1016/j.bmcl.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Peletskaya EN, Glinsky G, Deutscher SL, Quinn TP. Mol Diversity. 1996;2:13. doi: 10.1007/BF01718695. [DOI] [PubMed] [Google Scholar]
- 79.Glinsky VV, Huflejt ME, Glinsky GV, Deutscher SL, Quinn TP. Cancer Res. 2000;60:2584. [PubMed] [Google Scholar]
- 80.Landon LA, Peletskaya EN, Glinsky VV, Karasseva N, Quinn TP, Deutscher SL. J Protein Chem. 2003;22:193. doi: 10.1023/a:1023483232397. [DOI] [PubMed] [Google Scholar]
- 81.Landon LA, Zou J, Deutscher SL. Mol Diversity. 2003;8:35. doi: 10.1023/b:modi.0000006897.40575.41. [DOI] [PubMed] [Google Scholar]
- 82.Kubo N, Akita N, Shimizu A, Kitahara H, Parker AL, Miyagawa S. J Drug Targeting. 2008;16:396. doi: 10.1080/10611860802088796. [DOI] [PubMed] [Google Scholar]
- 83.Ding H, Prodinger WM, Kopecek J. Bioconjug Chem. 2006;17:514. doi: 10.1021/bc0503162. [DOI] [PubMed] [Google Scholar]
- 84.McGuire MJ, Samli KN, Chang YC, Brown KC. Exp Hematol. 2006;34:443. doi: 10.1016/j.exphem.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 85.Choi JH, Lee WK, Han SH, Ha S, Ahn SM, Kang JS, Choi YJ, Yun CH. Int Immunopharmacol. 2008;8:852. doi: 10.1016/j.intimp.2008.01.037. [DOI] [PubMed] [Google Scholar]
- 86.Askoxylakis V, Zitzmann S, Mier W, Graham K, Kramer S, von Wegner F, Fink RH, Schwab M, Eisenhut M, Haberkorn U. Clin Cancer Res. 2005;11:6705. doi: 10.1158/1078-0432.CCR-05-0432. [DOI] [PubMed] [Google Scholar]
- 87.Robinson P, Stuber D, Deryckere F, Tedbury P, Lagrange M, Orfanoudakis G. J Mol Recognit. 2005;18:175. doi: 10.1002/jmr.723. [DOI] [PubMed] [Google Scholar]
- 88.Rasmussen UB, Schreiber V, Schultz H, Mischler F, Schughart K. Cancer Gene Ther. 2002;9:606. doi: 10.1038/sj.cgt.7700476. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Chen J, Hu Z, Hu D, Pan Y, Ou S, Liu G, Yin X, Zhao J, Ren L, Wang J. J Biomol Screening. 2007;12:429. doi: 10.1177/1087057106299164. [DOI] [PubMed] [Google Scholar]
- 90.Liang S, Lin T, Ding J, Pan Y, Dang D, Guo C, Zhi M, Zhao P, Sun L, Hong L, Shi Y, Yao L, Liu J, Wu K, Fan D. J Mol Med. 2006;84:764. doi: 10.1007/s00109-006-0064-2. [DOI] [PubMed] [Google Scholar]
- 91.Wu C, Lo SL, Boulaire J, Hong ML, Beh HM, Leung DS, Wang S. J Controlled Release. 2008;130:140. doi: 10.1016/j.jconrel.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 92.Du B, Qian M, Zhou Z, Wang P, Wang L, Zhang X, Wu M, Zhang P, Mei B. Biochem Biophys Res Commun. 2006;342:956. doi: 10.1016/j.bbrc.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 93.Zang L, Shi L, Guo J, Pan Q, Wu W, Pan X, Wang J. Cancer Lett. 2009;281:64. doi: 10.1016/j.canlet.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 94.Chang DK, Lin CT, Wu CH, Wu HC. PLoS One. 2009;4:e4171. doi: 10.1371/journal.pone.0004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Askoxylakis V, Mier W, Zitzmann S, Ehemann V, Zhang J, Kramer S, Beck C, Schwab M, Eisenhut M, Haberkorn U. J Nucl Med. 2006;47:981. [PubMed] [Google Scholar]
- 96.Romanov VI, Durand DB, Petrenko VA. Prostate. 2001;47:239. doi: 10.1002/pros.1068. [DOI] [PubMed] [Google Scholar]
- 97.Zitzmann S, Mier W, Schad A, Kinscherf R, Askoxylakis V, Kramer S, Altmann A, Eisenhut M, Haberkorn U. Clin Cancer Res. 2005;11:139. [PubMed] [Google Scholar]
- 98.Zitzmann S, Kramer S, Mier W, Hebling U, Altmann A, Rother A, Berndorff D, Eisenhut M, Haberkorn U. J Nucl Med. 2007;48:965. doi: 10.2967/jnumed.106.036699. [DOI] [PubMed] [Google Scholar]
- 99.Pasqualini R, Ruoslahti E. Nature. 1996;380:364. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 100.Arap W, Kolonin M, Trepel M, Lahdenranta J, Cardo-Vila M, Giordano RJ, Mintz PJ, Ardelt PU, Yao VJ, Vidal CI, Chen L, Flamm A, Valtanen H, Weavind LM, Hicks ME, Pollock RE, Botz GH, Bucana CD, Koivunen E, Cahill D, Troncoso P, Baggerly KA, Pentz RD, Do KA, Logothetis CJ, Pasqualini R. Nat Med. 2002;8:121. [Google Scholar]
- 101.Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99:1527. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolonin MG, Bover L, Sun J, Zurita AJ, Do KA, Lahdenranta J, Cardo-Vila M, Giordano RJ, Jaalouk DE, Ozawa MG, Moya CA, Souza GR, Staquicini FI, Kunyiasu A, Scudiero DA, Holbeck SL, Sausville EA, Arap W, Pasqualini R. Cancer Res. 2006;66:34. doi: 10.1158/0008-5472.CAN-05-2748. [DOI] [PubMed] [Google Scholar]
- 103.Nishimura S, Takahashi S, Kamikatahira H, Kuroki Y, Jaalouk DE, O’Brien S, Koivunen E, Arap W, Pasqualini R, Nakayama H, Kuniyasu A. J Biol Chem. 2008;283:11752. doi: 10.1074/jbc.M708849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 105.Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 106.Chen X, Sievers E, Hou Y, Park R, Tohme M, Bart R, Bremner R, Bading JR, Conti PS. Neoplasia. 2005;7:271. doi: 10.1593/neo.04538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cheng Z, Wu Y, Xiong Z, Gambhir SS, Chen X. Bioconjugate Chem. 2005;16:1433. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Haubner R, Wester HJ. Curr Pharm Des. 2004;10:1439. doi: 10.2174/1381612043384745. [DOI] [PubMed] [Google Scholar]
- 109.Kwon S, Ke S, Houston JP, Wang W, Wu Q, Li C, Sevick-Muraca EM. Mol Imaging. 2005;4:75. doi: 10.1162/15353500200505103. [DOI] [PubMed] [Google Scholar]
- 110.Su ZF, Liu G, Gupta S, Zhu Z, Rusckowski M, Hnatowich DJ. Bioconjugate Chem. 2002;13:561. doi: 10.1021/bc0155566. [DOI] [PubMed] [Google Scholar]
- 111.Wang W, Ke S, Wu Q, Charnsangavej C, Gurfinkel M, Gelovani JG, Abbruzzese JL, Sevick-Muraca EM, Li C. Mol Imaging. 2004;3:343. doi: 10.1162/15353500200404148. [DOI] [PubMed] [Google Scholar]
- 112.Ye Y, Bloch S, Xu B, Achilefu S. J Med Chem. 2006;49:2268. doi: 10.1021/jm050947h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Laakkonen P, Zhang L, Ruoslahti E. Ann N Y Acad Sci. 2008;1131:37. doi: 10.1196/annals.1413.003. [DOI] [PubMed] [Google Scholar]
- 114.Essler M, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99:2252. doi: 10.1073/pnas.251687998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yao VJ, Ozawa MG, Trepel M, Arap W, McDonald DM, Pasqualini R. Am J Pathol. 2005;166:625. doi: 10.1016/S0002-9440(10)62283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sato M, Arap W, Pasqualini R. Oncology. Vol. 21. Williston Park, N.Y: 2007. p. 1346. [PubMed] [Google Scholar]
- 117.Newton J, Deutscher SL. Handb Exp Pharmacol. 2008:145. doi: 10.1007/978-3-540-77496-9_7. [DOI] [PubMed] [Google Scholar]
- 118.Newton JR, Deutscher SL. Methods Mol Biol. 2009;504:275. doi: 10.1007/978-1-60327-569-9_17. [DOI] [PubMed] [Google Scholar]
- 119.Newton JR, Kelly KA, Mahmood U, Weissleder R, Deutscher SL. Neoplasia. 2006;8:772. doi: 10.1593/neo.06331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zou J, Dickerson MT, Owen NK, Landon LA, Deutscher SL. Mol Biol Rep. 2004;31:121. doi: 10.1023/b:mole.0000031459.14448.af. [DOI] [PubMed] [Google Scholar]
- 121.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Nature. 2005;437:376. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J. Nat Methods. 2007;4:931. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 123.Jurisson SS, Lydon JD. Chem Rev. 1999;99:2205. doi: 10.1021/cr980435t. [DOI] [PubMed] [Google Scholar]
- 124.Bourguignon MH, Loc’h G, Maziere B. J Nucl Biol Med. 1994;38:573. [PubMed] [Google Scholar]
- 125.Mindt TL, Struthers H, Brans L, Anguelov T, Schweinsberg C, Maes V, Tourwe D, Schibli R. J Am Chem Soc. 2006;128:15096. doi: 10.1021/ja066779f. [DOI] [PubMed] [Google Scholar]
- 126.Anderson CJ, Welch MJ. Chem Rev. 1999;99:2219. doi: 10.1021/cr980451q. [DOI] [PubMed] [Google Scholar]
- 127.Adler LP, Crowe JP, al-Kaisi NK, Sunshine JL. Radiology. 1993;187:743. doi: 10.1148/radiology.187.3.8497624. [DOI] [PubMed] [Google Scholar]
- 128.Pelosi E, Messa C, Sironi S, Picchio M, Landoni C, Bettinardi V, Gianolli L, Del Maschio A, Gilardi MC, Fazio F. Eur J Nucl Med Mol Imaging. 2004;31:932. doi: 10.1007/s00259-004-1483-3. [DOI] [PubMed] [Google Scholar]
- 129.McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, Anderson CJ, Welch MJ. Nucl Med Biol. 1997;24:35. doi: 10.1016/s0969-8051(96)00157-6. [DOI] [PubMed] [Google Scholar]
- 130.Zhernosekov KP, Filosofov DV, Baum RP, Aschoff P, Bihl H, Razbash AA, Jahn M, Jennewein M, Rosch F. J Nucl Med. 2007;48:1741. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- 131.Breeman WA, Verbruggen AM. Eur J Nucl Med Mol Imaging. 2007;34:978. doi: 10.1007/s00259-007-0387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Doyle TC, Burns SM, Contag CH. Cell Microbiol. 2004;6:303. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- 133.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 134.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Bioconjugate Chem. 2004;15:79. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 135.Li C, Greenwood TR, Glunde K. Neoplasia. 2008;10:389. doi: 10.1593/neo.07856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kovar JL, Volcheck W, Sevick-Muraca E, Simpson MA, Olive DM. Anal Biochem. 2009;384:254. doi: 10.1016/j.ab.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sampath L, Kwon S, Ke S, Wang W, Schiff R, Mawad ME, Sevick-Muraca EM. J Nucl Med. 2007;48:1501. doi: 10.2967/jnumed.107.042234. [DOI] [PubMed] [Google Scholar]
- 138.Culver J, Akers W, Achilefu S. J Nucl Med. 2008;49:169. doi: 10.2967/jnumed.107.043331. [DOI] [PubMed] [Google Scholar]
- 139.Li C, Wang W, Wu Q, Ke S, Houston J, Sevick-Muraca E, Dong L, Chow D, Charnsangavej C, Gelovani JG. Nucl Med Biol. 2006;33:349. doi: 10.1016/j.nucmedbio.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 140.Hui X, Han Y, Liang S, Liu Z, Liu J, Hong L, Zhao L, He L, Cao S, Chen B, Yan K, Jin B, Chai N, Wang J, Wu K, Fan D. J Controlled Release. 2008;131:86. doi: 10.1016/j.jconrel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 141.Kuhnast B, Bodenstein C, Haubner R, Wester HJ, Senekowitsch-Schmidtke R, Schwaiger M, Weber WA. Nucl Med Biol. 2004;31:337. doi: 10.1016/j.nucmedbio.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 142.Chen J, Tung CH, Allport JR, Chen S, Weissleder R, Huang PL. Circulation. 2005;111:1800. doi: 10.1161/01.CIR.0000160936.91849.9F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K. Biochemistry. 2001;40:12157. doi: 10.1021/bi010662g. [DOI] [PubMed] [Google Scholar]
- 144.Goodson RJ, Doyle MV, Kaufman SE, Rosenberg S. Proc Natl Acad Sci U S A. 1994;91:7129. doi: 10.1073/pnas.91.15.7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li ZB, Niu G, Wang H, He L, Yang L, Ploug M, Chen X. Clin Cancer Res. 2008;14:4758. doi: 10.1158/1078-0432.CCR-07-4434. [DOI] [PubMed] [Google Scholar]
- 146.Karasseva N, Glinsky VV, Chen NX, Komatireddy R, Quinn TP. J Protein Chem. 2002;21:287. doi: 10.1023/a:1019749504418. [DOI] [PubMed] [Google Scholar]
- 147.Howell RC, Revskaya E, Pazo V, Nosanchuk JD, Casadevall A, Dadachova E. Bioconjugate Chem. 2007;18:1739. doi: 10.1021/bc060330u. [DOI] [PubMed] [Google Scholar]
- 148.Zou J, Glinsky VV, Landon LA, Matthews L, Deutscher SL. Carcinogenesis. 2005;26:309. doi: 10.1093/carcin/bgh329. [DOI] [PubMed] [Google Scholar]
- 149.Kumar SR, Deutscher SL. J Nucl Med. 2008;49:796. doi: 10.2967/jnumed.107.048751. [DOI] [PubMed] [Google Scholar]
- 150.Landgraf R. Breast Cancer Res. 2007;9:202. doi: 10.1186/bcr1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Holbro T, Hynes NE. Annu Rev Pharmacol Toxicol. 2004;44:195. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 152.Lub-de Hooge MN, Kosterink JG, Perik PJ, Nijnuis H, Tran L, Bart J, Suurmeijer AJ, de Jong S, Jager PL, de Vries EG. Br J Pharmacol. 2004;143:99. doi: 10.1038/sj.bjp.0705915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K, Sandstrom M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY. Cancer Res. 2007;67:2773. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]
- 154.Garcia Garayoa E, Ruegg D, Blauenstein P, Zwimpfer M, Khan IU, Maes V, Blanc A, Beck-Sickinger AG, Tourwe DA, Schubiger PA. Nucl Med Biol. 2007;34:17. doi: 10.1016/j.nucmedbio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 155.Behr TM, Sharkey RM, Juweid ME, Blumenthal RD, Dunn RM, Griffiths GL, Bair HJ, Wolf FG, Becker WS, Goldenberg DM. Cancer Res. 1995;55:3825. [PubMed] [Google Scholar]
- 156.Rolleman EJ, Valkema R, de Jong M, Kooij PP, Krenning EP. Eur J Nucl Med Mol Imaging. 2003;30:9. doi: 10.1007/s00259-002-0982-3. [DOI] [PubMed] [Google Scholar]
- 157.Racusen LC, Whelton A, Solez K. Am J Pathol. 1985;120:436. [PMC free article] [PubMed] [Google Scholar]
- 158.Mogensen CE. Solling Scand J Clin Lab Invest. 1977;37:477. doi: 10.3109/00365517709101835. [DOI] [PubMed] [Google Scholar]
- 159.van Eerd JE, Vegt E, Wetzels JF, Russel FG, Masereeuw R, Corstens FH, Oyen WJ, Boerman OC. J Nucl Med. 2006;47:528. [PubMed] [Google Scholar]
- 160.Perea SE, Reyes O, Baladron I, Perera Y, Farina H, Gil J, Rodriguez A, Bacardi D, Marcelo JL, Cosme K, Cruz M, Valenzuela C, Lopez-Saura PA, Puchades Y, Serrano JM, Mendoza O, Castellanos L, Sanchez A, Betancourt L, Besada V, Silva R, Lopez E, Falcon V, Hernandez I, Solares M, Santana A, Diaz A, Ramos T, Lopez C, Ariosa J, Gonzalez LJ, Garay H, Gomez D, Gomez R, Alonso DF, Sigman H, Herrera L, Acevedo B. Mol Cell Biochem. 2008;316:163. doi: 10.1007/s11010-008-9814-5. [DOI] [PubMed] [Google Scholar]
- 161.Rusckowski M, Gupta S, Liu G, Dou S, Hnatowich DJ. J Nucl Med. 2004;45:1201. [PubMed] [Google Scholar]
- 162.Rusckowski M, Gupta S, Liu G, Dou S, Hnatowich DJ. Nucl Med Biol. 2008;35:433. doi: 10.1016/j.nucmedbio.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Newton JR, Miao Y, Deutscher SL, Quinn TP. J Nucl Med. 2007;48:429. [PubMed] [Google Scholar]
- 164.Newton-Northup JR, Figueroa SD, Quinn TP, Deutscher SL. Nucl Med Biol. 2009;36:789. doi: 10.1016/j.nucmedbio.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jaye DL, Geigerman CM, Fuller RE, Akyildiz A, Parkos CA. J Immunol Methods. 2004;295:119. doi: 10.1016/j.jim.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 166.Kelly KA, Clemons PA, Yu AM, Weissleder R. Mol Imaging. 2006;5:24. [PubMed] [Google Scholar]
- 167.Kelly KA, Waterman P, Weissleder R. Neoplasia. 2006;8:1011. doi: 10.1593/neo.06610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hajitou A, Lev DC, Hannay JA, Korchin B, Staquicini FI, Soghomonyan S, Alauddin MM, Benjamin RS, Pollock RE, Gelovani JG, Pasqualini R, Arap W. Proc Natl Acad Sci U S A. 2008;105:4471. doi: 10.1073/pnas.0712184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hajitou A, Trepel M, Lilley CE, Soghomonyan S, Alauddin MM, Marini FC, 3rd, Restel BH, Ozawa MG, Moya CA, Rangel R, Sun Y, Zaoui K, Schmidt M, von Kalle C, Weitzman MD, Gelovani JG, Pasqualini R, Arap W. Cell. 2006;125:385. doi: 10.1016/j.cell.2006.02.042. [DOI] [PubMed] [Google Scholar]