

Abstract
The Rnd proteins (Rnd1, Rnd2 and Rnd3/RhoE) form a distinct branch of the Rho family of small GTPases. Altered Rnd3 expression causes changes in cytoskeletal organization and cell cycle progression. Rnd3 functions to decrease RhoA activity, but how Rnd3 itself is regulated to cause these changes is still under investigation. Unlike other Rho family proteins, Rnd3 is regulated not by GTP/GDP cycling, but at the level of expression and by posttranslational modifications such as prenylation and phosphorylation. We show here that, upon PKC agonist stimulation, Rnd3 undergoes an electrophoretic mobility shift and its subcellular localization becomes enriched at internal membranes. These changes are blocked by inhibition of conventional PKC isoforms and do not occur in PKCα-null cells or to a nonphosphorylatable mutant of Rnd3. We further show that PKCα directly phosphorylates Rnd3 in an in vitro kinase assay. Additionally, we provide evidence that the phosphorylation status of Rnd3 has a direct effect on its ability to block signaling from the Rho-ROCK pathway. These results identify an additional mechanism of regulation and provide clarification of how Rnd3 modulates Rho signaling to alter cytoskeletal organization.
Keywords: GTPase, phosphorylation, kinase, plasma membrane, stress fibers
Introduction
Members of the Rho family of small GTPases are involved in the regulation of cell growth and survival as well as organization of the actin cytoskeleton to control cell shape and cell motility [1]. These proteins act as molecular switches by cycling between an inactive GDP bound form and an active GTP bound form, the latter of which is then able to interact preferentially with effector molecules. The most thoroughly characterized proteins of this family of small GTPases are RhoA, Rac1 and Cdc42 [2]. Activation of Rho leads to formation of stress fibers and focal adhesions [3], while activation of Rac and Cdc42 lead to the formation of lamellipodia and filopodia, respectively [4, 5]. The Rnd family of proteins (Rnd1, Rnd2 and Rnd3/RhoE, also known as Rho6, Rho7 and Rho8, respectively) form a unique branch of the Rho family [6]. One striking difference between Rnd proteins and other members of the Rho family is their effect on the actin cytoskeleton. In contrast to RhoA, which upregulates stress fibers and focal adhesions in both epithelial cells and fibroblasts, expression of either Rnd1 or Rnd3 causes a decrease in stress fibers and the disappearance of focal adhesions, leading to cell rounding (hence “Rnd” for rounding) [7, 8]. Another striking difference in the Rnd family is that, unlike most other members of the Rho family, Rnd proteins lack intrinsic GTPase activity [7]. They are bound to GTP in vivo in a constitutively active state [9] due to amino acid residue substitutions at highly conserved positions critical for normal GTP hydrolysis [7, 9]. These results suggest that the activity of Rnd proteins is regulated not by GTP/GDP cycling, but at the level of expression and/or by post-translational modifications. Essentially all members of the Rho family, along with the Ras family, contain a CAAX motif (where C=cysteine, A=aliphatic residue and X=any amino acid) at their C-termini [10]. The CAAX motif is a crucial signal needed for these proteins to be post-translationally modified by isoprenylation, a permanent modification required for correct subcellular localization and for biological activity [11]. Rnd proteins contain both N- and C-terminal extensions, the latter of which contain conventional CAAX motifs. Rnd proteins terminate in a methionine in the “X” position and are thus farnesylated like Ras family proteins [9]. Farnesylation of Rnd proteins is required both for their membrane localization and for their ability to alter the cytoskeleton [12]. Several small GTPases of the Ras and Rho families have been shown to be substrates for phosphorylation on serine residues at their C-terminal regions immediately upstream of the CAAX motif, and these phosphorylation events have been demonstrated to have functional consequences. We have shown recently that the previously appreciated phosphorylation of the C-terminus of K-Ras4B [13] is directed by protein kinase C (PKC) at S181[14]. This phosphorylation causes K-Ras4B to translocate from the plasma membrane to the mitochondria, resulting in the biological consequence of enhanced apoptosis [14]. We reasoned that the location and function of Rnd3 might also be regulated in a similar manner by phosphorylation of a C-terminal serine residue. We have shown previously that Rnd3 binds to and is a substrate for ROCK1, and that this phosphorylation regulates its stability as well as its localization [15]. The consensus motifs for ROCK1 and PKC are similar; thus, Rnd3 might be a target of both of these kinases.
Experimental
Antibodies and reagents
Antibodies detected HA (HA.11 clone 16B12) and Myc (clone 9E11) [Covance]; β-actin (clone AC-74), FLAG (M2) and phorbol myristic acid (PMA) [Sigma]; GFP (clone 3E6) [Molecular Probes]; PKCα (clone 3) [BD Biosciences]; RhoE (clone 4) [Upstate Technologies]; phospho-MARCKS (Ser152/156) and phosphoserine PKC substrate [Cell Signaling Technology]; and total MYPT1 and P-MYPT1 [Millipore]. Anti-Rnd3 anti-sera has been described previously [16]. Other reagents included ionomycin and Y-27632 [Calbiochem], Bryostatin-1 and Gö-6976 [BIOMOL Research Laboratories] and calf intestinal phosphatase (CIP) [New England Biolabs].
Molecular constructs
Rnd3 expression constructs were generated by inserting the full length human Rnd3 cDNA into the Bam-HI sites of pCGN-hyg [17] and pEGFP-C1 (Clontech) or into the BamHI and EcoRI sites of pGEX-2T. Rnd3-SAAX (STVM), Rnd3-S240A, Rnd3-S240E and Rnd3-S7,11,240A mutants were generated using the QuickChange Mutagenesis Kit (Stratagene). Full length wild type and kinase-deficient (K368R) rat PKCα cDNA (a generous gift from William Davis, University of North Carolina at Chapel Hill [UNC-CH]) were PCR amplified and inserted into the XhoI and HindIII sites of both pEGFP-C1 and pCMV-3b to generate GFP-PKCα and Myc-PKCα expression constructs, respectively. The FLAG-Rnd3 expression construct was generated by inserting full length human wild type Rnd3 cDNA into the EcoRI and XhoI sites of pHIT-FLAG3 (a generous gift from Yanping Zhang, UNC-CH). Generation of FLAG-Rnd3-S7A, S11A, S210A, T214A, S218A, S222A, S240A (henceforth termed Rnd3-All A) has been described previously [15]. To generate the GFP-Rnd3-All A expression construct, the Rnd3 open reading frame from FLAG-Rnd3-All A was inserted into the HindIII and SalI sites of pEGFP-C3. To generate the GST-Rnd3-All A expression construct, the Rnd3 open reading frame from FLAG-Rnd3-All A was inserted into the EcoRI and XhoI sites of pGEX-4T. All sequences were verified by the Genome Analysis Facility at UNC-CH.
Cell culture and transfections
NIH 3T3 cells were maintained in high glucose Dulbecco's modified Eagle medium (DMEM-H) (Gibco/Invitrogen) containing 10% calf serum (Invitrogen) and penicillin-streptomycin (P/S, Invitrogen) at 37° C in a humidified atmosphere of 10% CO2. Isolation of PKCα +/+ and -/- mouse embryonic fibroblasts has been described previously [18]. These cells were cultured in DMEM-H without sodium pyruvate (Sigma) containing 10% fetal calf serum (FCS), glutamine and P/S (Invitrogen), and maintained at 37° C in a humidified atmosphere of 5% CO2. Expression vectors were transfected into NIH 3T3 cells using TransIT-LT1 transfection reagent (Mirus) according to the manufacturer's instructions. Expression vectors were transfected into PKCα mouse embryonic fibroblasts using LipofectAMINE and Plus reagents (Invitrogen) according to the manufacturer's instructions.
Calf Intestinal Phosphatase Treatment Assay
Equal amounts of lysate (devoid of phosphatase inhibitors) from NIH 3T3 cells expressing HA-Rnd3 (treated with or without 100nM PMA for 10 minutes) were incubated in phosphatase buffer (100mM NaCl, Tris-HCl pH 7.9, 10mM MgCl2 and 1 mM DTT) with or without 20 units of calf intestinal phosphatase at 37° C for 1 hour. Lysates were resolved on 12% SDS-PAGE, transferred to Immobilon PVDF (Millipore) and blotted with anti-HA antibody.
Live cell imaging
To visualize the effects of PKC activation on Rnd3 localization in real time, NIH 3T3 cells were transiently transfected with GFP-Rnd3. After 24 h, cells were treated with either bryostatin-1 (100 nM) or PMA (100 nM) and ionomycin (500 μg/mL). Live cell images were captured on a Zeiss 510 LSM confocal microscope at 20× magnification and analyzed using LSM 5 Image browser software (Zeiss). To evaluate a role for different PKC isoforms in modulating Rnd3 localization, NIH 3T3 cells were transiently transfected with GFP-Rnd3 as above. After 24 h, cells were incubated with either DMSO vehicle or the PKC inhibitor Gö-6976 (2.5 μM). After 3 h, cells were treated with PKC agonists PMA (100 nM) and ionomycin (500 μg/mL), and live cell images were captured by confocal microscopy at 5 minute intervals as described above.
In vitro kinase assay
GST alone, GST-Rnd3-WT and GST-Rnd3-All A were purified using glutathione sepharose beads. His-tagged vinculin tail (aa 881-1135) was a generous gift from Sharon Campbell, UNC-CH. Purified proteins were incubated with or without recombinant PKCα in kinase buffer supplemented with PKC lipid activator (Upstate Technologies) in the presence of 32P-labeled phosphate. The kinase reaction was incubated at 40° C for 30 minutes and the reactions were stopped by addition of 2× Laemmli sample buffer. Samples were resolved by SDS-PAGE, stained with Coomassie blue and developed by autoradiography.
Western blot analysis
Cells were washed with PBS, lysed in RIPA lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS and 0.5% sodium deoxycholate and supplemented with Complete Protease Inhibitor Cocktail tablets (Roche) along with phenyl-methyl sulfonyl fluoride (PMSF) and sodium pervanadate and spun down to remove insoluble material. 2× Laemmli sample buffer was added to equivalent amounts of cellular lysates which were then resolved on 12% SDS-PAGE and transferred to Immobilon PVDF membranes. Membranes were blocked in 5% nonfat dry milk in TBS-Tween-20 and probed with appropriate primary antibodies followed by anti-mouse or rabbit IgG-horseradish peroxidase (HRP)-conjugated secondary antibody (Amersham Biosciences). Membranes were then incubated in SuperSignal West Dura Extended Duration substrate (Pierce) and the signal developed on HyBlot CL autoradiography film (Denville Scientific Inc.).
Results
Rnd3 subcellular localization is altered upon PKC activation
Inspection of Rnd protein sequences revealed a consensus PKC site at serine 240 in the C-terminal hypervariable membrane-targeting domain immediately upstream of the CAAX prenylation motif, similar to the arrangement seen in the C-terminus of K-Ras4B. We previously found that this site can be phosphorylated by ROCK1 [15], which shares a similar consensus sequence to that of PKC. We reasoned that the subcellular localization of Rnd3 might also be regulated by a combination of prenylation and C-terminal phosphorylation. We therefore decided to use PKC agonists to test the effects of PKC activation on Rnd3 subcellular localization. To visualize these effects in living cells, we treated NIH 3T3 mouse fibroblast cells transiently expressing GFP-tagged Rnd3 with the non-phorbol, PKC-specific agonist bryostatin-1. Live cell images were taken before and ten minutes after treatment. As shown in Figure 1A, treatment with bryostatin-1 caused rapid loss of Rnd3 from the plasma membrane and enrichment in the cytosol and internal membranes. To determine whether this response was unique to bryostatin-1 or a reproducible consequence of activating PKC, we also treated cells with the phorbol ester PMA (phorbol myristate acetate) and the calcium ionophore ionomycin. NIH 3T3 cells expressing GFP-Rnd3 were treated with PMA + ionomycin, and live cell images were taken at 5 minute increments. As shown in Figure 1B, treatment with PMA + ionomycin, like bryostatin-1, also caused the loss of Rnd3 from the plasma membrane and enrichment in the cytosol and internal membranes, as we reported previously [15] for PMA treatment of HeLa cells. The change in Rnd3 subcellular localization after treatment with distinct types of PKC agonists indicates that PKC activity is inversely correlated with Rnd3 plasma membrane binding, and further suggests both that activated PKC may cause phosphorylation of Rnd3 itself and that the phosphorylation state of Rnd3 has direct consequences on its cellular localization. To begin to elucidate the identity of the specific PKC family member(s) responsible for the change in Rnd3 subcellular localization after stimulation with broad-based PKC activators, we employed Gö6976 that selectively inhibits conventional but not novel PKCs [19]. NIH 3T3 cells expressing GFP-Rnd3 as above were treated with either Gö6976 or DMSO vehicle alone for 3 h prior to stimulation with PMA + ionomycin. As shown in Figure 1C, Gö6976 but not the vehicle control blocked alterations in Rnd3 localization. This result indicates that at least one conventional PKC isoform is involved in the regulation of the subcellular location of Rnd3. Because NIH 3T3 cells express only the alpha isoform of conventional PKCs [20], it is likely that PKCα is the major isoform responsible for the effects seen with Rnd3.
Figure 1.
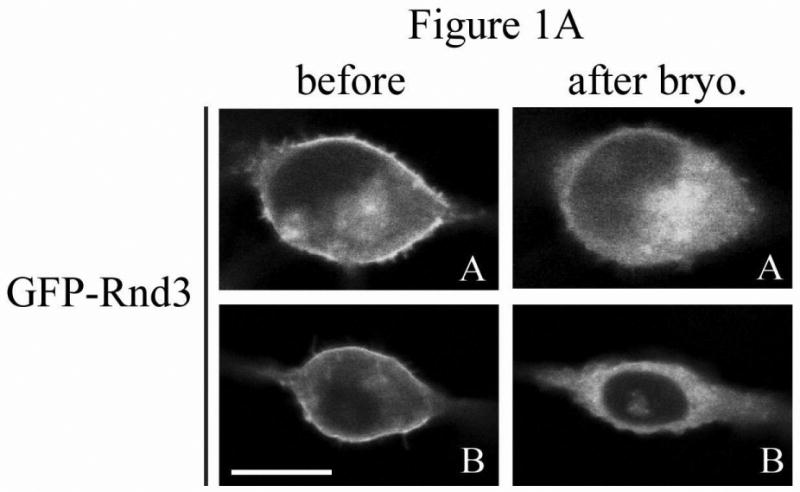
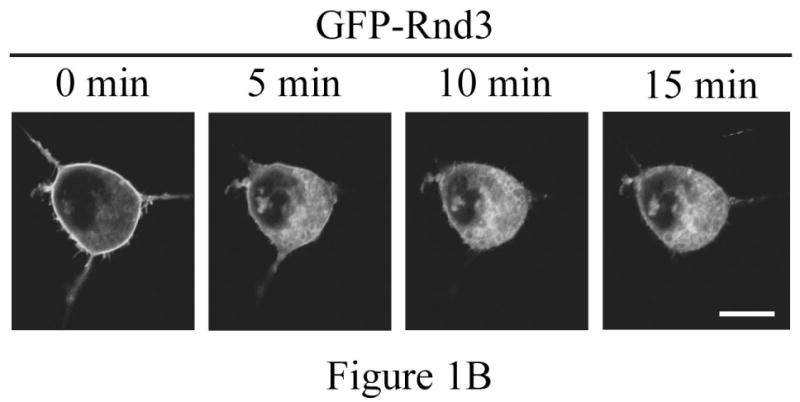
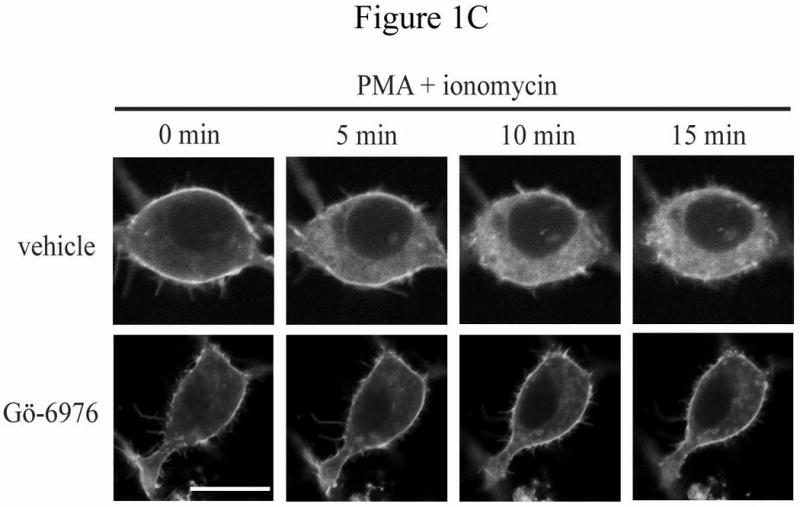
Rnd3 localization is altered after PKC activation. (A) PKC agonist bryostatin-1 causes loss of Rnd3 from the plasma membrane. NIH 3T3 cells transiently expressing GFP-Rnd3 were treated with bryostatin-1 (100 nM). Representative live images of two separate cells before (left panel) and 10 min after (right panel) addition of agonist are shown. (B) Activation of PKC by using PMA + ionomycin also causes loss of Rnd3 from the plasma membrane. NIH 3T3 cells transiently expressing GFP-Rnd3 were treated with PMA (100 nM) + ionomycin (500 μg/mL). Live images are shown of a single cell visualized at 5 min increments. (C) Inhibitor of conventional PKCs blocks Rnd3 translocation. NIH 3T3 cells transiently expressing GFP-Rnd3 were treated with DMSO vehicle or Gö-6976 (2.5 μM) to inhibit conventional PKCs for 3 h prior to stimulation with PMA (100 nM) + ionomycin (500 μg/mL). Live images are shown of single cells visualized at 5 min increments. Scale bar is 20 μm.
Rnd3 is phosphorylated upon PKC activation
To evaluate the possible direct involvement of Rnd3 phosphorylation in modulating its location, we wished to determine whether Rnd3 itself becomes phosphorylated upon activation of PKC. NIH 3T3 cells transiently expressing HA-tagged Rnd3 were treated with PKC agonists as in Figure 1C. Cell lysates were collected, resolved on SDS-PAGE and immunoblotted with anti-HA antibody. As shown in Figure 2A, a slightly slower-migrating band consistent with post-translationally modified HA-Rnd3 appeared in lysates of cells stimulated with PMA, but not with DMSO vehicle. This result is consistent with phosphorylation of Rnd3 upon PKC activation. Furthermore, Gö6976 (lane 3) blocked this mobility shift, suggesting that it is mediated by a conventional PKC. Rnd3 can be a subject of ROCK-mediated phosphorylation [15, 21]. ROCK and conventional PKCs share a common phosphorylation recognition sequence [22] and these kinases can directly phosphorylate identical residues [23]. We therefore investigated whether ROCK-mediated Rnd3 phosphorylation was stimulated by treatment with PMA. As seen in Figure 2A (lane 2), pretreatment of NIH 3T3 cells with the ROCK-selective inhibitor Y-27632 failed to prevent the mobility shift of HA-tagged Rnd3 when cells were treated with PMA, although it did block ROCK activation as shown by abrogation of phospho-MYPT1 (data not shown). Next, to confirm that the slower migrating band represents a phosphorylation event, we treated cells with a PKC agonist followed by exposure of the lysates to calf intestinal phosphatase (CIP) and immunoblotted with anti-HA antibody as before. CIP treatment (Figure 2B, lane 3) abrogated the appearance of the slower migrating band present in the cells treated with PKC agonist alone (lane 2). Lastly, a SAAX mutant of Rnd3 was employed to demonstrate that proper membrane localization is required for the band shift seen upon PKC activation. Rnd3-SAAX migrated at a slower mobility on SDS-PAGE as compared to WT Rnd3 (Figure 2C), consistent with improper processing and membrane localization of the SAAX mutant. Furthermore, after PKC activation, the Rnd3-SAAX mutant did not show the band shift (lane 4) seen with WT Rnd3 (lane 2).
Figure 2.


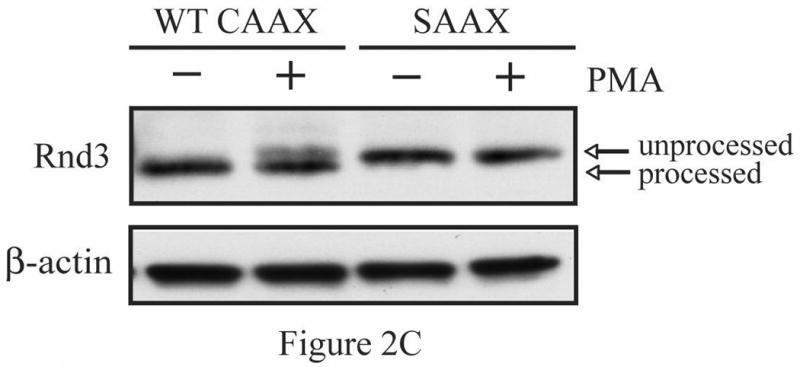
Activation of conventional PKCs causes a phosphorylation-dependent mobility shift of Rnd3. (A) NIH 3T3 cells expressing HA-Rnd3 were pretreated for 3 h with either DMSO vehicle, Y-27632 (10 μM) or Gö-6976 (2.5 μM). Cells were then treated with PMA (100 nM) for 10 min and cell lysates were resolved on SDS-PAGE. The slower migrating Rnd3 band (arrow) was seen in both the vehicle and the Y-27632 pretreated cells, but not in cells pretreated with the conventional PKC inhibitor Gö-6976. (B) Calf intestinal phosphatase (CIP) treatment causes disappearance of the slower migrating band of Rnd3 (arrow). NIH 3T3 cells transiently expressing HA-Rnd3 expression vector were treated with PMA (100 nM) + ionomycin (500 μg/mL). CIP was added to the cell lysate to reverse phosphorylation. Cell lysates were resolved on SDS-PAGE and blotted with anti-HA antibody. (C) A CAAX mutant of Rnd3 does not shift after activation of PKCs. NIH 3T3 cells expressing HA-tagged WT Rnd3 and a SAAX mutant were treated with PMA (100 nM) for 10 min and cell lysates were resolved on SDS-PAGE.
Mutation of Rnd3 C-terminal residue S240 alone does not alter cell morphology, or Rnd3 PKC sensitivity or localization
We had identified serine 240, just upstream of the CAAX motif, as a potential PKC phosphorylation site similar to those found in some other small GTPases that regulate their localization and function. To determine if phosphorylation of serine 240 is required for Rnd3 function, we used site-directed mutagenesis to mutate the serine to a nonphosphorylatable alanine residue, thereby generating a putatively phospho-deficient Rnd3 protein, termed S240A. After sequence confirmation, we transiently expressed GFP-Rnd3-WT and GFP-Rnd3-S240A in NIH 3T3 cells. As anticipated, cells expressing empty GFP vector were flat and well spread, whereas cells expressing GFP-Rnd3-WT were rounded up. Most of the GFP-Rnd3 was located at the plasma membrane with additional cytosolic and perinuclear staining observed. Surprisingly, the morphology of cells expressing the putatively phospho-deficient S240A mutant was indistinguishable from that of cells expressing GFP-Rnd3-WT, and GFP-Rnd3-S240A was localized similarly to that of GFP-Rnd3-WT (data not shown). Staining with Texas Red phalloidin also revealed no change in stress fibers between cells expressing different Rnd3 proteins. These surprising results indicate that phosphorylation of S240 is not required for the ability of Rnd3 to cause cytoskeletal changes and cell rounding, and suggest that at least one other site in Rnd3 is also targeted by PKC. To determine whether the S240A mutation rendered Rnd3 PKC-insensitive, we evaluated whether it retained or lost the PKC-induced mobility shift on SDS-PAGE. Contrary to our initial hypothesis, but consistent with the cell morphology data, both the WT and the S240A mutant forms of Rnd3 displayed the same mobility on SDS-PAGE in the absence or presence of PKC activation (Figure 3A). Taken together, we concluded that phosphorylation of Rnd3 at serine 240 alone is not sufficient to regulate Rnd3 subcellular localization or to produce the mobility-shifted form of Rnd3. These results are consistent with our observations that the mobility shift is induced by phosphorylation of other residues, as indicated below.
Figure 3.
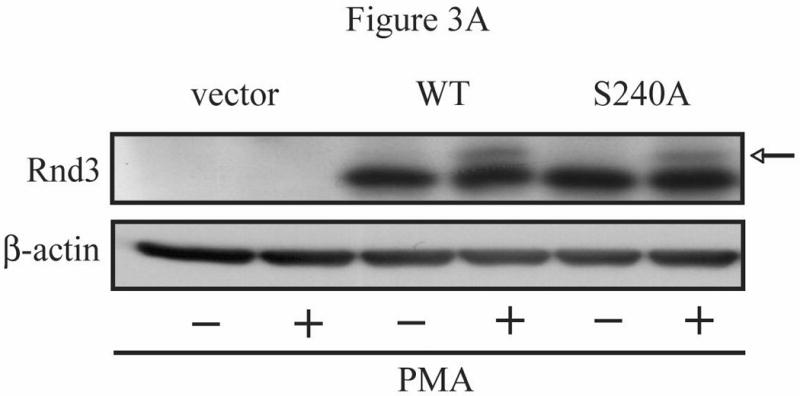

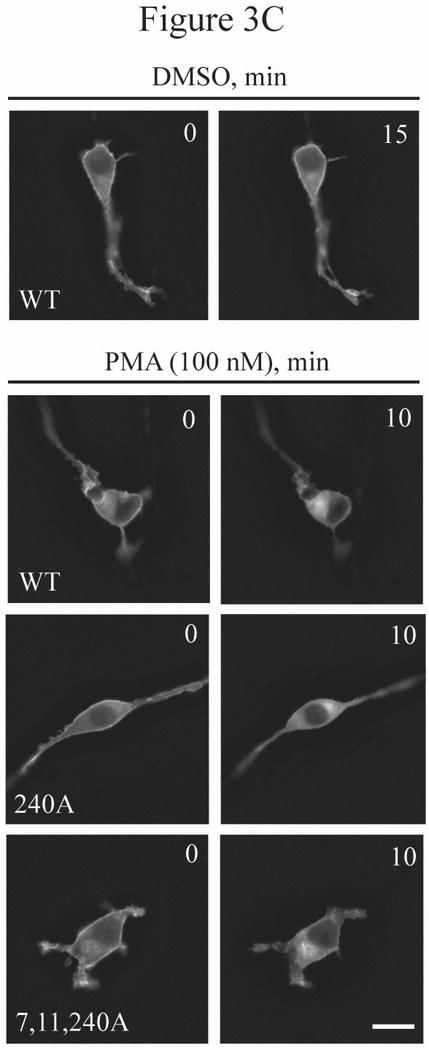
Multiple residues of Rnd3 are involved in the PKC-dependent translocation and gel mobility shift. (A) The phosphodeficient S240A mutant still displays a mobility shift. NIH 3T3 cells transiently expressing HA-Rnd3 proteins were treated with PMA (100 nM) for 10 min. Cell lysates were resolved on SDS-PAGE and immunoblotted with anti-HA antibody. The phosphodeficient Rnd3 S240A mutant still displayed a mobility shift (arrow), indicating that the shift is not caused by phosphorylation at S240. (B) An N-terminal peptide polyclonal antibody (aa 1-15) does not recognize the mobility-shifted form of Rnd3. NIH 3T3 cells transiently expressing HA-Rnd3 or empty vector were treated with either DMSO vehicle or PMA (100 nM) for 10 min -/+ CIP treatment. Cell lysates were resolved on SDS-PAGE and immunoblotted initially with anti-HA antibody. The blot was then stripped and blotted sequentially with a specific anti-Rnd3/RhoE antibody and anti-Rnd3 antiserum. The lack of a mobility-shifted form of HA-Rnd3 when using the N-terminal Rnd3 peptide polyclonal antibody indicates that the shifted form of Rnd3 (arrow) is dependent on the sequence found in the first 15 residues. (C) GFP-Rnd3 multiple phosphorylation mutants still translocate from the plasma membrane after PKC activation. NIH 3T3 cells were transiently transfected with either GFP-Rnd3-WT or GFP-Rnd3 phosphomutants and treated with either vehicle or PMA (100 nM) for the indicated times. Live images were taken on a confocal microscope. No differences in plasma membrane localization were noted between WT Rnd3 and the Rnd3 phosphorylation mutants after treatment with PMA. Scale bar is 20 μm.
Additional sites of PKC-mediated phosphorylation in Rnd3
In optimizing the SDS-PAGE gel mobility-shift experiments using HA-tagged Rnd3, we employed a rabbit antiserum produced against the N-terminus of Rnd3 [16]. Surprisingly, this antiserum did not detect the mobility shift of HA-Rnd3 in lysates from NIH 3T3 cells that had been treated with PMA. Yet, this shift was seen reproducibly when immunoblotting with either an antibody directed against the HA epitope tag or a mouse monoclonal antibody whose immunogen was the entire Rnd3/RhoE protein (Figure 3B). We therefore postulated that the site or sites of phosphorylation responsible for the shifted form of Rnd3 must be located in the first 15 amino acids that were used in producing the Rnd3 rabbit antiserum. Visual inspection of the Rnd3 sequence, and the phosphorylation prediction program NetPhos2.0, revealed two additional consensus PKC phosphorylation sites at serines 7 and 11, previously shown to be sites for phosphorylation by ROCK1 [15], which has a similar consensus motif. Together with the previous data demonstrating a correlation between alteration of Rnd3 subcellular localization and the presence of a mobility-shifted form of Rnd3 on SDS-PAGE, we hypothesized that phosphorylation of Rnd3 within this unique N-terminal extension, which would introduce negative charges, could be responsible for the effects on Rnd3 seen after treatment with PKC agonist. We reasoned that phosphorylation of Rnd3 at serines 7 and 11 in the N-terminal extension, along with phosphorylation at serine 240, may disrupt the polar interactions of Rnd3 with the plasma membrane. Site-directed mutagenesis was then used to generate GFP-tagged versions of Rnd3 that contained alanine substitutions at serines 7 and 11 along with serine 240, and this mutant GFP-Rnd3 construct was expressed transiently in NIH 3T3 cells. However, as with GFP-Rnd3-S240A, the subcellular localization of the triple serine mutant, GFP-Rnd3-S7,11,240A, was also indistinguishable from that of GFP-Rnd3-WT after treatment with PMA (Figure 3C). One possible explanation for lack of effect of the triple mutant S7A, S11A, S240A is that phosphorylation at one or more other site(s) is the primary target of PKC. To explore this possibility, six serine residues and a threonine (S7, S11, S210, T214, S220, S222 and S240) in Rnd3 were mutated to the corresponding phospho-deficient alanines to generate a nonphosphorylatable form of Rnd3 (Rnd3-All A) [15] and this nonphosphorylatable mutant was evaluated in a PMA-treatment translocation assay. As seen in Figure 4A, stimulation of NIH 3T3 cells with PMA caused the loss of GFP-tagged Rnd3 WT from the plasma membrane. Consistent with a requirement for Rnd3 to become phosphorylated in order for it to translocate upon PKC activation, PMA stimulation did not cause the loss of the nonphosphorylatable Rnd3-All A mutant from the plasma membrane. Furthermore, a FLAG-tagged version of Rnd3-All A did not display a gel mobility shift when expressed in NIH 3T3 cells stimulated with PMA (Figure 4B), as was seen with FLAG-Rnd3-WT. Our extensive efforts to identify the relevant specific Rnd3 phosphorylation site(s) through proteomic means have been unsuccessful. Thus, while further work will be needed to identify the minimal number of PKC phosphorylation sites needed for membrane translocation, it is clear that both the mobility shift and the translocation seen upon PKC activation require that Rnd3 itself be able to become phosphorylated.
Figure 4.
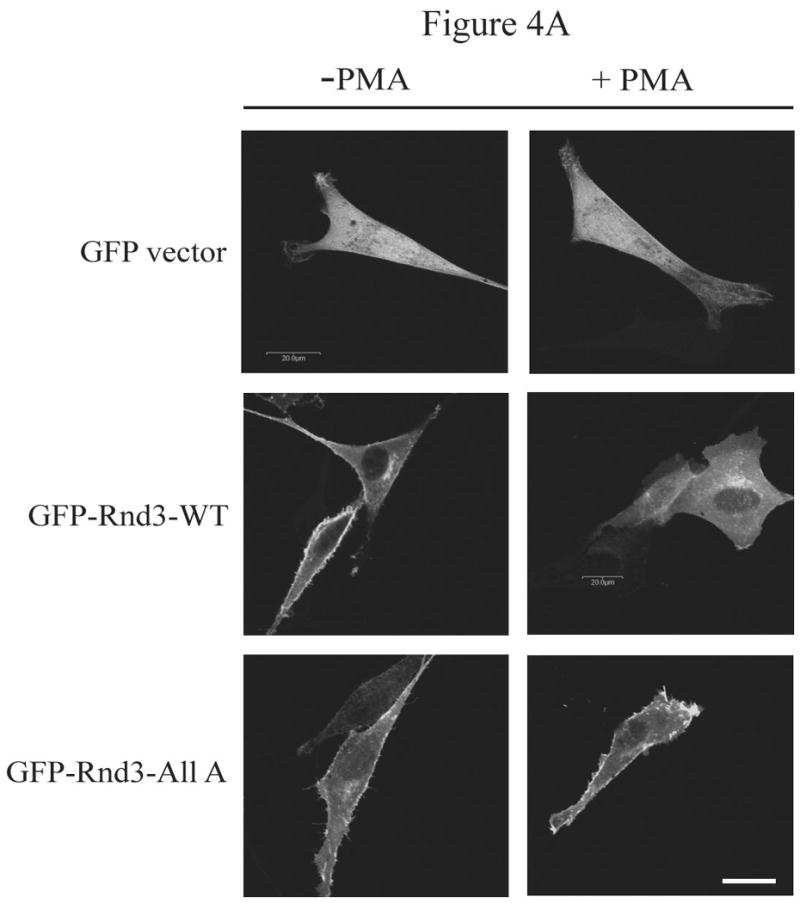
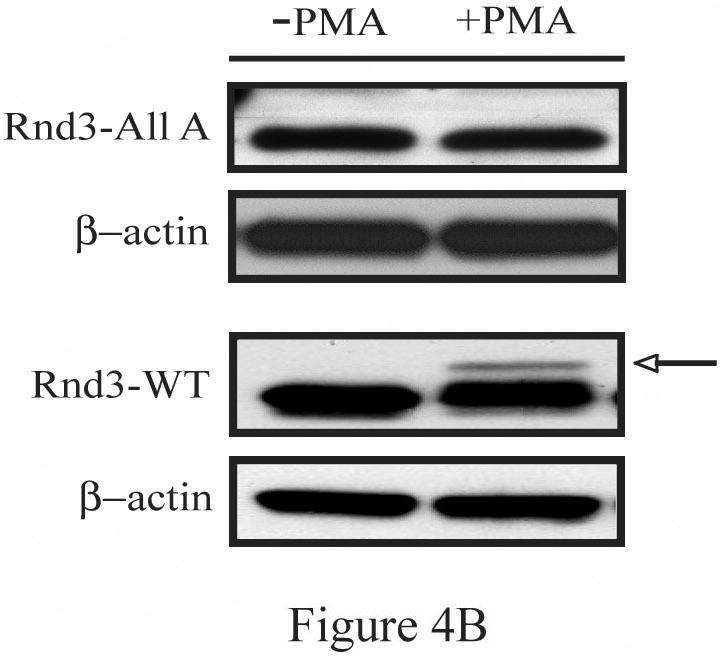
GFP-Rnd3-WT, but not GFP-Rnd3-All A, translocate from the plasma membrane after PKC activation. (A) NIH 3T3 cells were transiently transfected with either GFP-vector, GFP-Rnd3-WT or GFP-Rnd3-All A. Cells were treated with either vehicle or PMA (100 nM) for 10 min. Live cell images were taken on a confocal microscope. PMA treatment causes loss of GFP-WT-Rnd3 from the plasma membrane along with a corresponding flattened phenotype. A similar event was not seen in GFP-Rnd3-All A expressing cells. Scale bar is 20 μm. (B) FLAG-Rnd3-WT, but not FLAG-Rnd3-All A, displays a gel mobility shift on SDS-PAGE after PKC activation. NIH 3T3 cells were transiently transfected with either FLAG-Rnd3-WT or FLAG-Rnd3-All A and treated with either vehicle or PMA (100 nM) for 10 min. Lysates were resolved on SDS-PAGE and probed with anti-FLAG antibody to visualize FLAG-tagged Rnd3 protein. Only FLAG-Rnd3-WT was shifted in the presence of PMA (arrow).
PKCα is the isoform responsible for Rnd3 phosphorylation
While the specific target residue(s) have not yet been determined, the exact identity of the PKC isoform(s) responsible for the phosphorylation and altered localization of Rnd3 upon PKC activation also remained to be uncovered. Based on the PKC inhibitor data shown previously (Figures 1C and 2A) and the fact that NIH 3T3 cells express only the alpha isoform of conventional PKCs [20], we hypothesized that PKCα was the isoform involved. Therefore, we performed additional studies in mouse embryo fibroblasts (MEFs) in which PKCα had been genetically ablated [18]. We first tested whether PKCα is required for the electrophoretic mobility shift of Rnd3 seen upon stimulation with PKC agonists. PKCα -/- MEFs and WT control cells transiently expressing HA-Rnd3 were treated with the PKC agonist PMA. Lysates from these cells were resolved on SDS-PAGE and immunoblotted with anti-HA antibody. Furthermore, lysates were blotted with a PKCα-specific antibody demonstrating the absence of PKCα protein in PKCα -/- MEFs (Figure 5A). The slower migrating band of Rnd3 was seen only in the WT MEF cells and not in the PKCα -/- MEF cells, demonstrating that the PKCα isoform is required for the mobility shifted form of Rnd3, that we showed previously represents PKC-mediated phosphorylated Rnd3.
Figure 5.

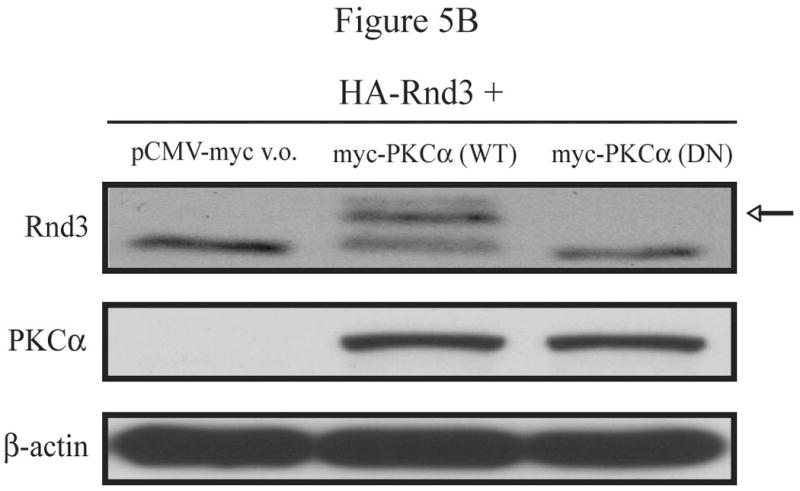
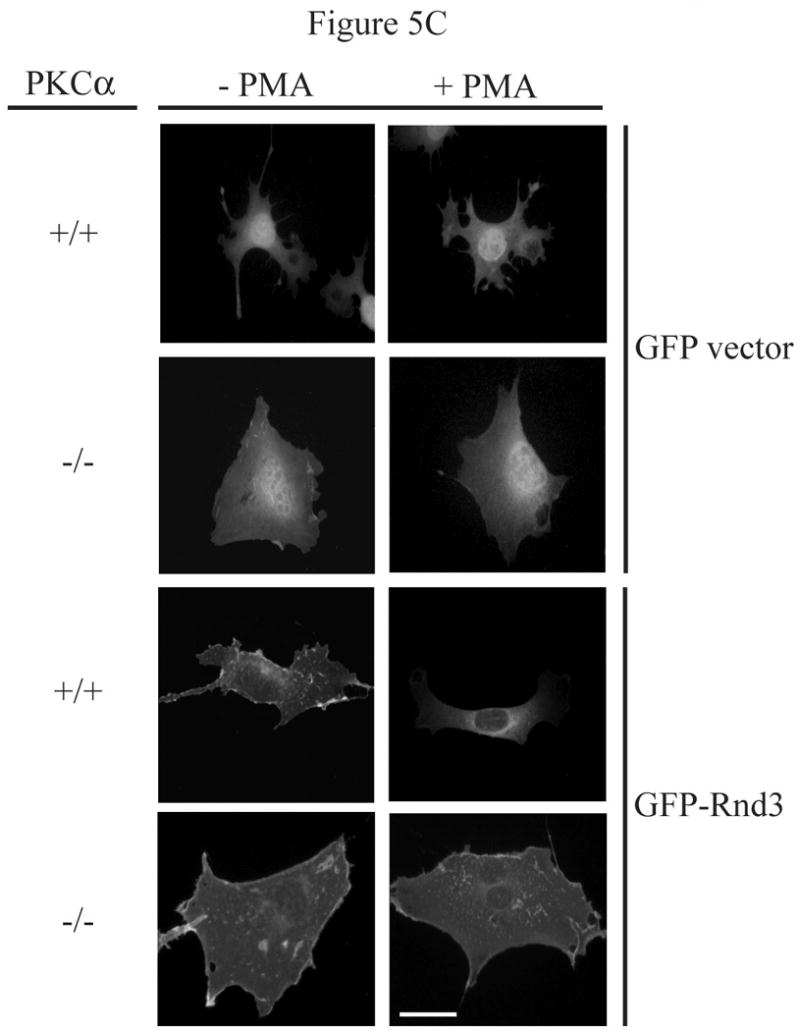
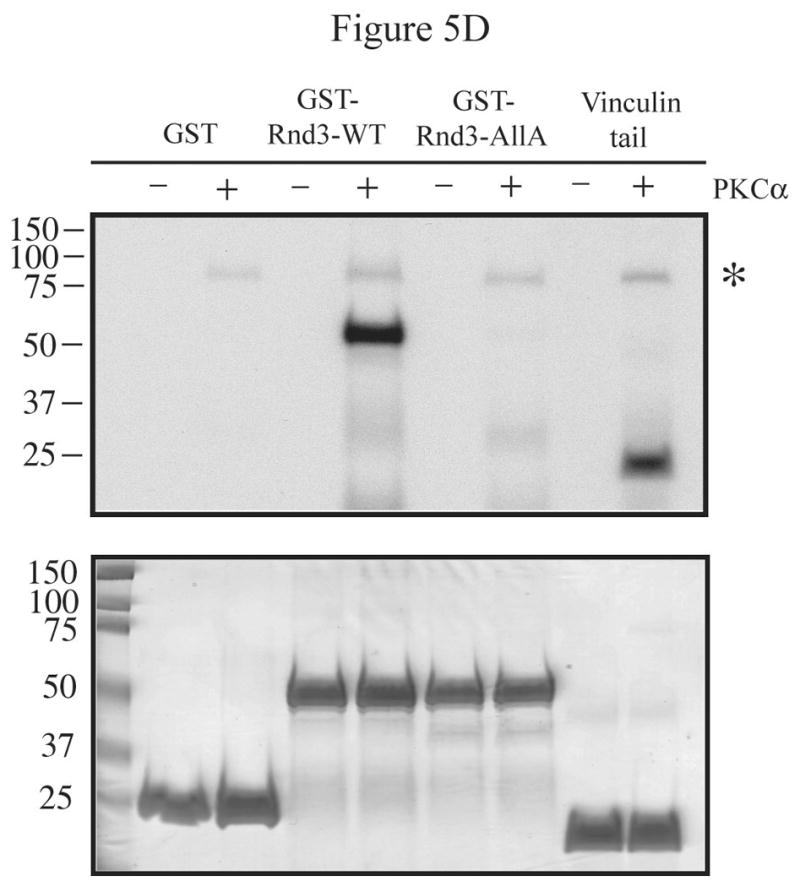
PKCα is the isozyme responsible for Rnd3 phosphorylation. (A) PMA stimulation causes a gel mobility-shift of Rnd3 in WT MEF cells, but not in PKCα knock-out MEFs. PKCα knock-out MEFs and matched WT MEFs transiently expressing HA-Rnd3 were treated with PKC agonist PMA (100 nM) for 10 min. Cell lysates were resolved on SDS-PAGE and blotted with anti-HA antibody. Cell lysates were further probed with an anti-PKCα antibody to confirm absence of PKCα protein expression in knock-out MEF cells. The slower migrating band of Rnd3 was seen only in WT MEF cells (arrow). (B) Reintroduction of PKCα-WT, but not of PKCα-DN, into PKCα knock-out MEFS causes a mobility shift of Rnd3 when cells are treated with PKC agonist PMA. PKCα knock-out MEFs transiently expressing HA-Rnd3 along with either pCMV-vector, Myc- PKCα-WT or Myc-PKCα-DN were treated with PKC agonist PMA (100 nM) for 10 min. Cell lysates were resolved on SDS-PAGE and probed with anti-HA antibody. A gel mobility shift of Rnd3 (arrow) was seen only in lysates from PKCα knock-out cells when WT PKCα was reintroduced. (C) GFP-Rnd3 translocates from the plasma membrane in WT MEF cell, but not PKCα knock-out MEFs, after PKC stimulation. WT MEFs and PKCα knock-out MEFs were transiently transfected with either GFP vector or GFP-Rnd3. MEFs were treated with PKC agaonist PMA (100nM) for 10 min and live cell images were taken. Scale bar is 20 μm. (D) Rnd3 is phosphorylated by PKCα in vitro. GST alone, GST-Rnd3-WT, GST-Rnd3-All A and a tail fragment of vinculin (aa 881-1135) were used as substrates in a PKCα in vitro kinase assay. Rnd3-WT, but not Rnd3-All A, incorporates 32P-labelled phosphate. * autophosphorylated PKCα.
To confirm that the kinase activity of PKCα is required for its effects on Rnd3 phosphorylation, we reintroduced either WT or kinase-deficient (KD) PKCα (K368R) into PKCα -/- MEFs and looked for restoration of the appearance of the slower migrating form. The PKCα-K368R mutant is considered kinase-deficient as it abolishes ATP binding ability. PKCα -/- MEFs, transiently expressing HA-Rnd3 along with either empty Myc-vector only, Myc-PKCα-WT or Myc-PKCα-KD were treated with the PKC agonist PMA, cell lysates were resolved on SDS-PAGE and immunoblotted with anti-HA antibody. As shown in Figure 5B, a gel mobility-shift of HA-Rnd3 was seen only upon reintroduction of PKCα-WT but not kinase-deficient PKCα. Thus, the kinase activity of PKCα is required for Rnd3 phosphorylation. Next, we transiently expressed GFP vector only and GFP-Rnd3 in both PKCα -/- and matched control WT MEFs. Cells were treated with the PKC agonist PMA and images were taken before and after treatment. As expected, PMA treatment had no effect on the cellular localization of GFP alone in either PKCα -/- or the matched control WT MEFs (Figure 5C). Consistent with a requirement for the PKCα isoform, PMA treatment caused the loss of GFP-Rnd3 from the plasma membrane in WT MEFs but not in PKCα -/- MEFs.
Finally, an in vitro kinase assay was used to determine whether PKCα directly phosphorylates Rnd3. As seen in Figure 5D, GST-Rnd3-WT, but neither the GST tag alone nor GST-Rnd3-All A, became phosphorylated in the presence of recombinant PKCα. As a positive control, a truncated tail of vinculin (aa 881-1135) was also phosphorylated in the presence of PKCα. Furthermore, kinase activity was visualized by the presence of autophosphorylated PKCα. A lack of incorporated 32P in the Rnd3-All A mutant suggests that one or more of the Ser/Thr residues mutated in this construct is phosphorylated by PKCα. Thus, the data presented, through the use of a conventional PKC-specific inhibitor and PKCα -/- MEFs, along with the direct phosphorylation Rnd3 by PKCα, give strong evidence that PKCα is likely to be the kinase responsible for Rnd3 phosphorylation in the present study.
PKCα-dependent Rnd3 phosphorylation downregulates Rnd3 inhibitory activity and leads to increased signaling through the Rho-ROCK pathway
As mentioned earlier, Rnd3 exerts its biological activity by counteracting the effects of RhoA signaling. Because we predicted that PKC-mediated phosphorylation decreases Rnd3 activity, we investigated whether Rnd3 phosphorylation leads to an increase in signaling through the Rho-ROCK pathway. To this end, NIH 3T3 cells were transiently transfected with either GFP only, GFP-Rnd3-WT or GFP-Rnd3-All A expression constructs. Transfected cells were treated with either DMSO vehicle or PMA and then fixed and stained with Rhodamine-conjugated phalloidin to mark actin. As seen in Figure 6A, PMA treatment caused not only translocation of GFP-Rnd3-WT from the plasma membrane but also the restoration of stress fibers, along with greater spreading and a flattened appearance of the cells. None of these changes were seen when cells were treated with only DMSO vehicle. In direct opposition to the results seen with GFP-Rnd3-WT, PMA treatment had no effect on the plasma membrane localization of the PKC-insensitive mutant GFP-Rnd3-All A; stress fibers were not restored, and the cells did not flatten and spread. To uncover a possible molecular mechanism for the restoration of stress fibers and cell spreading in PMA-treated cells expressing Rnd3-WT but not Rnd3-All A, lysates from PMA-treated cells were resolved on SDS-PAGE and blotted for phospho-myosin light chain phosphatase (P-MYPT1), a downstream target of the Rho-ROCK pathway [24]. As seen in Figure 6B, in Rnd3-WT expressing cells, the levels of P-MYPT1 were higher after treatment with PMA, as compared to treatment with DMSO vehicle only. In contrast, Rnd3-All A effectively acted as a dominant negative in this pathway, abrogating the ability of PKC activation to increase P-MYPT1. The data are thus consistent with a model in which Rnd3 located at the plasma membrane is able to disrupt signals from the Rho-ROCK pathway that are involved in stress fiber formation/maintenance. We envision that, when Rnd3 becomes phosphorylated after PKC activation and is translocated from the plasma membrane, it is no longer able to disrupt the Rho-ROCK signaling pathway. The Rnd3-All A mutant, which is no longer subject to PKC-mediated phosphorylation, remains located on the plasma membrane, even after PKC activation. Hence, in Rnd3-All A expressing cells, signals coming from the Rho-ROCK pathway can still be disrupted by this PKC-insensitive mutant Rnd3. Previous work has demonstrated that the Rnd3-All A mutant is still able to interact with ROCK, as demonstrated by in vitro binding and pulldown assays [15]. These results demonstrate that PKCα phosphorylation of Rnd3 represents an important negative feedback loop that may be critical to restoration of signaling through the Rho-ROCK pathway following transient activation of the GTPase-insensitive Rho family protein Rnd3.
Figure 6.
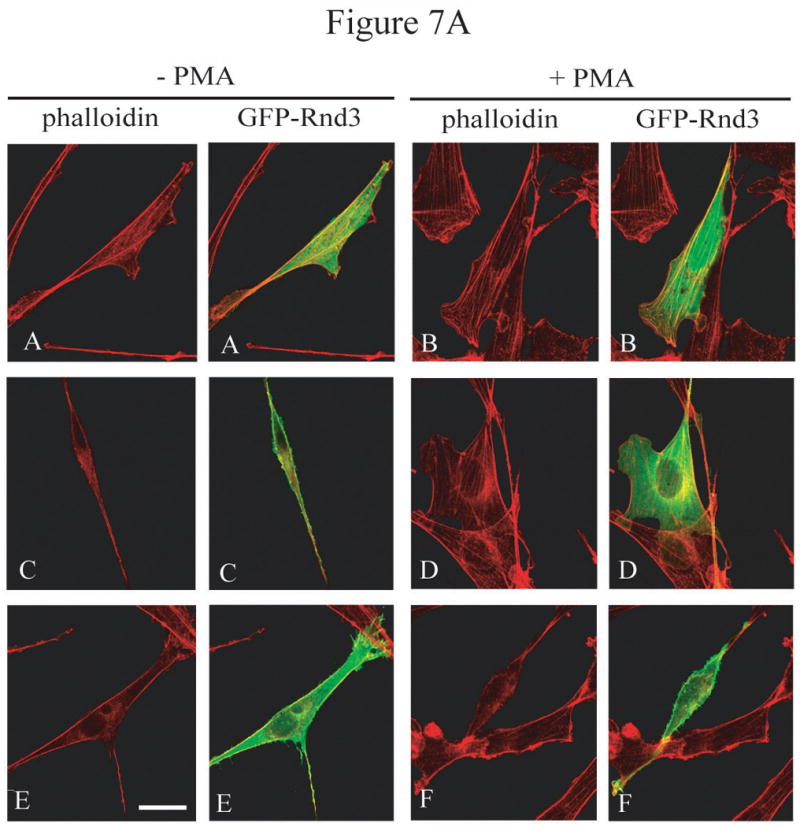
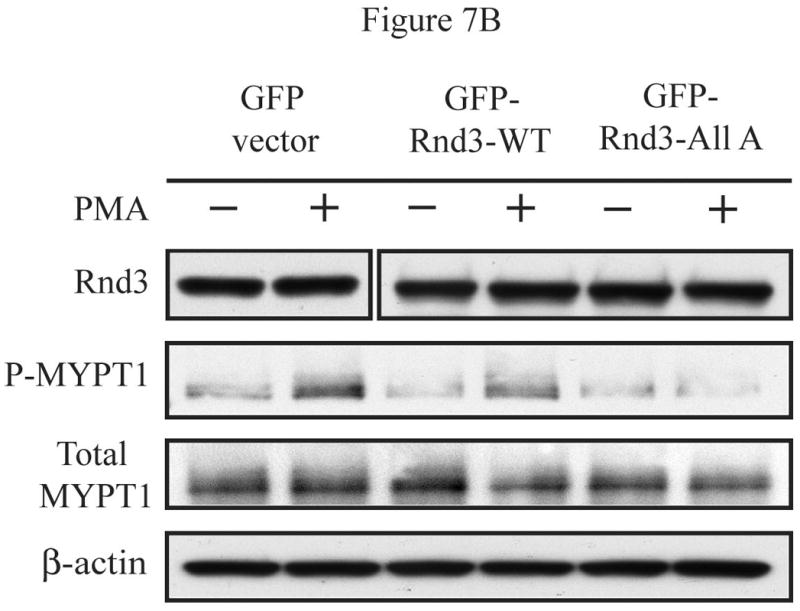
PKCα-dependent Rnd3 phosphorylation downregulates Rnd3 inhibitory activity and leads to increased signaling through the Rho-ROCK pathway. (A) Treatment with PMA causes reformation of stress fibers in GFP-Rnd3-WT but not in GFP-Rnd3-All A expressing cells. NIH 3T3 cells were transiently transfected with either GFP-vector (A and B), GFP-Rnd3-WT (C and D) or GFP-Rnd3-All A phosphorylation mutant (E and F) and treated with either DMSO vehicle (A,C,E) or PMA (100 nM) for minutes (B,D,F). Live images were taken on a confocal microscope. Actin structures were visualized with phalloidin. PMA treatment caused loss of GFP-WT-Rnd3 from the plasma membrane along with a corresponding flattened phenotype. Furthermore, there was a reappearance of stress fibers seen in GFP-Rnd3-WT expressing cells when treated with PMA. Similar events were not seen in GFP-Rnd3-All A expressing cells. Scale bar is 20 μm. (B) PKCα-dependent phosphorylation leads to increased signaling through the Rho-ROCK pathway. NIH 3T3 cells transientlty expressing GFP-vector, GFP-Rnd3-WT or GFP-Rnd3-All A were treated similarly as above. Lysates were separated by SDS-PAGE and probed with anti-GFP-antibody to visualize equal expression of the GFP-fusion proteins and GFP protein alone. Additionally, lysates were probed with anti-P-MYPT1 antibody to detect signaling from the Rho-ROCK pathway. An increase in P-MYPT1 signal was seen only in GFP-Rnd3-WT- and not in GFP-Rnd3-All A-expressing cells, indicating increased signaling through the Rho-ROCK pathway only in the presence of phosphorylatable Rnd3.
Discussion
It has long been appreciated that Rnd3/RhoE is constitutively GTP-bound [9], and therefore that its activity must be regulated by means other than GTP-GDP cycling. Rnd3 protein expression is known to be tightly regulated and responsive to both internal and external cues. However, modulation of expression is a relatively slow process compared to many signaling activities mediated by small GTPases. Rapid modulation of Rnd3 activity by other means might be required to regulate dynamic signaling processes. Rnd3/RhoE is known to interact with and be a direct target of the serine/threonine kinase ROCK1 [15, 21]. Here we have presented evidence that post-translational regulation of Rnd3 activity can also be accomplished via differential subcellular localization due to PKCα-mediated phosphorylation. These results add an additional mechanism of regulation to those documented previously, and provide clarification of how Rnd3 modulates Rho signaling to alter cytoskeletal organization. Several closely related proteins have been shown previously to be substrates for phosphorylation by PKC. In particular, K-Ras4B was shown to be a substrate for PKC phosphorylation in its C-terminal polybasic region [13], and we have shown that this phosphorylation influences both its subcellular localization and function [14]. The presence of similar serine residues in Rnd3 at sites homologous to those of phosphorylation in related proteins prompted us to investigate whether Rnd3 displayed a similar mode of regulation. Inspection of the Rnd3 amino acid sequence revealed a potential PKC phosphorylation site at residue S240, just upstream of the CAAX motif in the C-terminal hypervariable membrane targeting domain.
Here we demonstrate that Rnd3 is phosphorylated upon PKC activation, and that inhibition of conventional PKC isoforms abrogates this phosphorylation. However, we have determined that phosphorylation at S240 alone could not be responsible for the effects seen with Rnd3 due to PKC activation. Rather, multiple residues found in both the unique N-terminal and C-terminal extensions are necessary. Perhaps phosphorylation of residues located in both the N- and C-terminal extensions would reduce Rnd3 plasma membrane affinity. The exact phosphorylation sites contained within these extensions necessary for loss of Rnd3 from the plasma membrane and translocation to the cytosol still remain to be deciphered. We concluded that phosphorylation of Rnd3 at serine 240 alone is not sufficient to regulate Rnd3 subcellular localization or to produce the mobility-shifted form of Rnd3.
Given that the consensus sequences for PKC and ROCK1 are similar, several of the same Ser/Thr residues in Rnd3 could be phosphorylated by either kinase or by both, depending on the stimulus as well as the relative abundance of PKC and ROCK1. The relative abundance of these proteins in the HeLa cancer cells used in our previous study [15] versus the NIH 3T3 fibroblast cells used here is not known, but cell type differences may be important in phosphorylation of Rnd3 following PMA stimulation. It is also possible that PKC might act upstream of ROCK1, enhancing its ability to interact with and phosphorylate Rnd3. In this regard, we saw only partial inhibition of PMA-mediated phosphorylation of Ser11 by ROCK inhibitors in HeLa cells [15], and thus it is likely that there is both a direct phosphorylation of at least this site by PKC, as well as an indirect effect via stimulation of ROCK1-mediated phosphorylation of Ser11, i.e., as a result of an indirect pathway of PKC-ROCK1. However, it is important to note that, regardless of the relative involvement of PKC versus ROCK, in both of our studies we have observed the same functional consequence of PMA stimulation to induce a shift of Rnd3 from plasma membrane to cytoplasm.
Thus, we have also shown in this report that the phosphorylation state of Rnd3 has direct consequences on its cellular location, with phosphorylation causing loss of plasma membrane localization and translocation to the cytosol. Similar results have been documented for other proteins, such as the ARF nucleotide exchange factor ARNO and the small GTPase RalA, where phosphorylation events also result in loss of protein localization at the plasma membrane and translocation to the cytosol [25-29]. We have further presented evidence that PKCα-mediated phosphorylation of Rnd3 leads to increased signaling through the Rho-ROCK signaling pathway. We suggest that phosphorylation of Rnd3 leads to relocalization away from sites where it can antagonize signaling from the Rho-ROCK pathway, thus leading to remodeling of the actin cytoskeleton. We offer compelling evidence that Rnd3 may represent an important link directly connecting PKCα with the Rho-ROCK pathway and the myriad of cell responses they control through cytoskeletal organization via actomyosin contractility.
Acknowledgments
We thank Steen Hansen for anti-Rnd3 antibody, William Davis for rat PKCα cDNA, Yanping Zhang for the expression vector pHIT-FLAG3 and Keith Burridge, Adi Dubash and Marisa Menold for invaluable technical advice.
Funding: Our work was supported by US National Institutes of Health grants to ADC (CA063071, CA067771 and CA109550) and CJD (CA063071, CA67771 and CA92240). The authors have no declarations of interest.
Abbreviations
- CIP
calf intestinal phosphatase
- DMSO
dimethyl sulfoxide
- GFP
green fluorescent protein
- HA
haemagglutinin
- MARCKS
myristoylated alanine-rich C kinase substrate
- MEF
mouse embryonic fibroblast
- MYPT1
myosin light chain phosphatase 1
- PKC
protein kinase C
- PMA
phorbol myristic acid
- ROCK
Rho-kinase
References
- 1.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 2.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 3.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 4.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 5.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–1952. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol. 2006;7:54–62. doi: 10.1038/nrm1788. [DOI] [PubMed] [Google Scholar]
- 7.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–197. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guasch RM, Scambler P, Jones GE, Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–4771. doi: 10.1128/mcb.18.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol. 1996;16:2689–2699. doi: 10.1128/mcb.16.6.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 11.Moores SL, Schaber MD, Mosser SD, Rands E, O'Hara MB, Garsky VM, Marshall MS, Pompliano DL, Gibbs JB. Sequence dependence of protein isoprenylation. J Biol Chem. 1991;266:14603–14610. [PubMed] [Google Scholar]
- 12.Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, Cox AD, Wilson O, Kirschmeier P, Der CJ. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem. 2008;283:25150–25163. doi: 10.1074/jbc.M800882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ballester R, Furth ME, Rosen OM. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem. 1987;262:2688–2695. [PubMed] [Google Scholar]
- 14.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, Yim D, Fein A, Perez de Castro I, Li C, Thompson CB, Cox AD, Philips MR. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005;24:1170–1180. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen SH, Zegers MM, Woodrow M, Rodriguez-Viciana P, Chardin P, Mostov KE, McMahon M. Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf-MEK-extracellular signal-regulated kinase pathway. Mol Cell Biol. 2000;20:9364–9375. doi: 10.1128/mcb.20.24.9364-9375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiordalisi JJ, Johnson RL, 2nd, Ulku AS, Der CJ, Cox AD. Mammalian expression vectors for Ras family proteins: generation and use of expression constructs to analyze Ras family function. Methods Enzymol. 2001;332:3–36. doi: 10.1016/s0076-6879(01)32189-4. [DOI] [PubMed] [Google Scholar]
- 18.Maly K, Strese K, Kampfer S, Ueberall F, Baier G, Ghaffari-Tabrizi N, Grunicke HH, Leitges M. Critical role of protein kinase C alpha and calcium in growth factor induced activation of the Na(+)/H(+) exchanger NHE1. FEBS Lett. 2002;521:205–210. doi: 10.1016/s0014-5793(02)02867-3. [DOI] [PubMed] [Google Scholar]
- 19.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 20.Formisano P, Oriente F, Miele C, Caruso M, Auricchio R, Vigliotta G, Condorelli G, Beguinot F. In NIH-3T3 fibroblasts, insulin receptor interaction with specific protein kinase C isoforms controls receptor intracellular routing. J Biol Chem. 1998;273:13197–13202. doi: 10.1074/jbc.273.21.13197. [DOI] [PubMed] [Google Scholar]
- 21.Komander D, Garg R, Wan PT, Ridley AJ, Barford D. Mechanism of multi-site phosphorylation from a ROCK-I:RhoE complex structure. EMBO J. 2008;27:3175–3185. doi: 10.1038/emboj.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang JH, Jiang Y, Toita R, Oishi J, Kawamura K, Han A, Mori T, Niidome T, Ishida M, Tatematsu K, Tanizawa K, Katayama Y. Phosphorylation of Rho-associated kinase (Rho-kinase/ROCK/ROK) substrates by protein kinases A and C. Biochimie. 2007;89:39–47. doi: 10.1016/j.biochi.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Tanabe A, Kamisuki Y, Hidaka H, Suzuki M, Negishi M, Takuwa Y. PKC phosphorylates MARCKS Ser159 not only directly but also through RhoA/ROCK. Biochem Biophys Res Commun. 2006;345:156–161. doi: 10.1016/j.bbrc.2006.04.082. [DOI] [PubMed] [Google Scholar]
- 24.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 25.Chardin P, Paris S, Antonny B, Robineau S, Beraud-Dufour S, Jackson CL, Chabre M. A human exchange factor for ARF contains Sec7- and pleckstrin-homology domains. Nature. 1996;384:481–484. doi: 10.1038/384481a0. [DOI] [PubMed] [Google Scholar]
- 26.Frank SR, Hatfield JC, Casanova JE. Remodeling of the actin cytoskeleton is coordinately regulated by protein kinase C and the ADP-ribosylation factor nucleotide exchange factor ARNO. Mol Biol Cell. 1998;9:3133–3146. doi: 10.1091/mbc.9.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagel W, Schilcher P, Zeitlmann L, Kolanus W. The PH domain and the polybasic c domain of cytohesin-1 cooperate specifically in plasma membrane association and cellular function. Mol Biol Cell. 1998;9:1981–1994. doi: 10.1091/mbc.9.8.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santy LC, Frank SR, Hatfield JC, Casanova JE. Regulation of ARNO nucleotide exchange by a PH domain electrostatic switch. Curr Biol. 1999;9:1173–1176. doi: 10.1016/S0960-9822(00)80019-6. [DOI] [PubMed] [Google Scholar]
- 29.Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, Tang MJ, Chou CK, Lin WJ, Yuan CJ, Huang CY. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005;280:9013–9022. doi: 10.1074/jbc.M411068200. [DOI] [PubMed] [Google Scholar]