

Abstract
Micrognathia, glossoptosis, and cleft palate comprise one of the most common malformation sequences, Robin sequence. It is a component of the TARP syndrome, talipes equinovarus, atrial septal defect, Robin sequence, and persistent left superior vena cava. This disorder is X-linked and severe, with apparently 100% pre- or postnatal lethality in affected males. Here we characterize a second family with TARP syndrome, confirm linkage to Xp11.23-q13.3, perform massively parallel sequencing of X chromosome exons, filter the results via a number of criteria including the linkage region, use a unique algorithm to characterize sequence changes, and show that TARP syndrome is caused by mutations in the RBM10 gene, which encodes RNA binding motif 10. We further show that this previously uncharacterized gene is expressed in midgestation mouse embryos in the branchial arches and limbs, consistent with the human phenotype. We conclude that massively parallel sequencing is useful to characterize large candidate linkage intervals and that it can be used successfully to allow identification of disease-causing gene mutations.
Introduction
Advances in genomic technologies can markedly speed the analysis of genetic contributions to disease and make once-intractable questions tractable. The technique of massively parallel sequencing with exon capture allows rapid assessment of mutations that cause human diseases.1,2 The syndrome we have chosen to study with this new technology is an X-linked pleiotropic developmental anomaly syndrome (MIM 311900) comprising micrognathia, glossoptosis, and cleft palate (currently described as Robin sequence), persistent left superior vena cava, atrial septal defect, and talipes equinovarus, which was originally called “Robin's syndrome” by Gorlin et al.3 Subsequently, this disorder was designated as TARP syndrome based on the acronym formed by talipes equinovarus, atrial septal defect, Robin sequence, and persistence of the left superior vena cava (to avoid confusion of Robin sequence and “Robin's syndrome”). Linkage analysis of the family originally evaluated by Gorlin et al.3 confirmed the X-linked inheritance by mapping the locus to Xp11.23-q13.3.4 Although this mapping confirmed the inheritance pattern and narrowed the candidate gene list, the region was nearly 28 Mb (from 46.42 Mb to 74.04 Mb; genome build 36) and contained more than 200 genes or transcripts, including several complex gene families of G antigen transcripts. Thus, the task of interrogating all of these genes for sequence variants and validating the variants was daunting.
Here we report the identification of the second family manifesting this syndrome, describe the phenotype in three affected males, show that the second family has linkage that is consistent with that of the initial family, describe our use of chromosome X exon target capture and massively parallel (so-called “next-gen”) sequencing to identify RBM10 (MIM 300080) mutations in the two families, and show that the murine Rbm10 gene is expressed in midgestation embryos in a pattern consistent with the human phenotype.
Materials and Methods
Target Selection and Sequencing
DNA isolation, genotyping, and haplotype analysis were performed as previously described.4 To sequence genes, we used solution hybridization selection (SureSelect, Agilent) to generate a single-end sequencing library according to the manufacturer's directions (Illumina). For the purpose of this project, the exon targets were defined as all coding DNA sequence (UCSC) gene exons between positions 2,710,679 bp and 154,500,000 bp, which is from XG (MIM 314700) to TMLHE (MIM 300777), inclusive, human genome build 36 (hg18). The target sequence was 2,784,426 bp, and the oligonucleotide library was designed to target 2,264,175 bp of this (81.3%). Briefly, 3 μg of genomic DNA was sheared via acoustic fragmentation (model S2, Covaris) with the following parameters: duty cycle, 10%; intensity, 5; cycle burst, 200; time, 180 s; set mode frequency, sweeping; temperature, 4°C. The DNA was subjected to end repair and adaptor ligation (NEBNext DNA Sample Prep Reagent Set 1, New England Biolabs). Size selection of 200–300 bp fragments was performed by using electrophoresis (4% NuSieve GTG gel, Lonza Group). The prepped library was amplified with primers 1.1 and 2.1 (Illumina). The amplified library was hybridized to biotinylated RNA library baits (Agilent), and targeted sequences were purified with magnetic beads (Dynal M-280 streptavidin, Invitrogen). Purified target sequences were amplified with proprietary PCR primers (Agilent) and DNA polymerase (Herculase II fusion, Stratagene).
Interpretation of Coding Variations
To predict possible deleterious effects of coding changes on the native state of the protein, we developed novel software to predict the deviation from the “normal” function relative to the human reference sequence and associated UCSC known gene models (hg18). This software computes the consequence of the change to the sequence and predicts whether the nucleotide variation leads to a silent, missense, nonsense, canonical splice-site, or frameshift change. CDPred, a module within the coding analysis software, is an algorithm that scores amino acid variants on the basis of evolutionary conservation in conserved protein coding domains. CDPred assigns all nonsense, splice-site, and frameshift changes in a protein the most damaging score (−30), under the hypothesis that these variants lead to truncated protein products or nonsense-mediated mRNA decay, both of which can be equivalent to a null protein. This algorithm also predicts whether a missense change would be deleterious to the function of the protein, based on a log-likelihood score computed from a position-specific scoring matrix of the best-aligning protein-conserved domain model to the query protein. For a given missense change (relative to the reference codon), the scores are in a range of +20 to −20.
The CDPred algorithm is initialized with a reference protein sequence and the variant (missense or nonsense) relative to the reference. An alignment of the reference protein sequence to a database of protein domain models (position-specific scoring matrices) was computed with RPS-BLAST (parameters: –e 0.00001 and –F T). Sequence alignments with greater than 80% overlap of the length of the model with an E value less than 1e-5 were considered to be significant. The delta log-likelihood scores were computed as the difference in position-specific scores of the variant amino acid and the reference amino acid. The lower or more negative the score, the more deleterious was the predicted change. Positive scores above 3 may also be potentially damaging, but a strongly positive score generally results from a situation in which the normal human amino acid at a position is different from many or all other aligned species over a domain and in which the variant allele is closer to the ancestral form. For example, if an individual had the ancestral form of the FOXP2 gene (MIM 605317), that individual would potentially exhibit loss of speech; however, the delta score for the amino acid positions that are unique to all other humans would be positive because that individual more closely resembles the ancestral form. The CDPred software is available for evaluation and implementation at the website listed in the Web Resources section of the paper.
Mutation Confirmation and Segregation Studies
PCR amplification and Sanger sequencing of DNA samples to confirm the mutations and cosegregation were performed as previously described.5 PCR primer sequences are available upon request. Mutation numbering was performed according to Human Gene Variation Society nomenclature with reference sequence NM_005676.3. Human studies were performed according to an approved human subjects research protocol of the NHGRI institutional review board, and informed consent was obtained.
In Situ Analysis
C57BL/6J mouse embryos were fixed overnight in 4% paraformaldehyde in PBS. Reverse-transcribed digoxigenin-conjugated probes were made from PCR products with polymerase binding site linkers. The following DNA source was used for probe synthesis: Rbm10, reverse transcription PCR from C57BL/6J skin total RNA (Rbm10-F-T3- GCGCGAATTAACCCTCACTAAAGGGCGGGATGGATTAGGCAGTGAC; Rbm10-R-T7-GCGCGTAATACGACTCACTATAGGGACAAAAGGAACATGATTTGAG). In situ hybridizations were performed by using published protocols6 with the following modifications. After probe hybridization, ribonuclease A digestion was omitted, and tris-buffered saline was used in place of PBS. BM-purple substrate (Roche, Molecular Biochemicals) was used in place of 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium.7 Multiple embryos were evaluated at each stage of development analyzed: E9.5 (n = 6), E10.5 (n = 5), and E11.5 (n = 4). Murine procedures were performed in accordance with NIH guidelines and under NHGRI mouse protocol G94-7.
Clinical Reports
Individual III-3 (Figure 1) was born at 32 weeks gestation to a 22-year-old gravida 4 para 2–3 abortus 1 mother. The pregnancy was complicated by oligohydramnios and premature rupture of the membranes. Delivery was breech. Although a fetal heartbeat was noted on examination within 2 min prior to delivery, APGAR scores were 0 at 1, 5, and 10 min. The baby boy died at 5 min of age. Upon postmortem examination, his weight was 1335 g and his crown heel length was 41 cm (both appropriate for gestational age). External examination showed apparently low-set ears, micrognathia, and a large cleft of the hard and soft palate. The hands were broad, with ulnar deviation. Bilateral talipes equinovarus and rocker bottom feet were present. The right testicle was undescended. Upon internal examination, he was found to have a large atrial septal defect, and his lung showed an underdeveloped third lobe with overall marked underdevelopment of alveoli. A Meckel diverticulum was present. Radiography showed a severely underdeveloped mandible. Microscopic description showed extramedullary hematopoiesis of the liver. Standard resolution karyotype was reported as normal, 46,XY.
Figure 1.
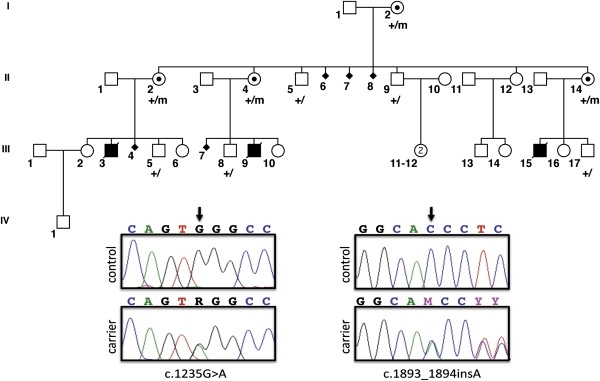
Pedigree of TARP Syndrome Family 2
The three affected male individuals are shown with darkened symbols. Obligate female carriers have a dot within their symbol. Nine family members were genotyped for the c.1235G>A mutation; their status is indicated by the + (wild-type) or m (mutant) designations below each pedigree symbol. Also shown are Sanger electropherograms of the two mutations; on the left is the nonsense mutation c.1235G>A found in family 2 below the control sequence, and on the right is the insertion mutation c.1893_1894insA found in family 1, again below the control sequence.
Individual III-9 was born at 37.5 weeks gestation to a 26-year-old gravida 1 para 1 mother. The prenatal course was notable for intrauterine growth retardation late in pregnancy. At birth, the baby boy weighed 2090 g (∼tenth centile) and his length was 46 cm (appropriate for gestational age). He was apneic, hypotonic, and cyanotic post delivery. He had micrognathia, cleft palate, glossoptosis, and hyaline membrane disease. He developed seizures at 24 hr. Head ultrasound showed basal ganglia hemorrhages and subdural hematoma. He was intubated, but he died at 8 days of age with liver failure, kidney failure, hyaline membrane disease, athetoid movements, and seizures. Autopsy was declined.
Individual III-15 was born at 28 weeks gestation to a 23-year-old gravida 2 para 1–2 mother. Few clinical details are available about this affected baby boy. At birth, he was found to have multiple anomalies including cleft palate (Robin sequence), possible cardiac defects, and bilateral talipes equinovarus. He died of his multiple congenital anomalies.
Results
Linkage analysis of the original family identified an 11 cM region in Xp11.23-q13.3 with a peak LOD score of 2.75 at marker DXS1039.4 The borders of the region were defined by recombinant markers at DXS1003 and DXS8092. Haplotype analysis was performed in family 2 with these three markers and additional markers both within and outside of the region (DXS8054, DXS1208, DXS7132, and DXS6800). Haplotypes were consistent with affection or carrier status in all individuals tested, and there were no recombinations in family 2 that narrowed the region defined by family 1 (data not shown). The linked short tandem repeat polymorphism haplotype in the two families was distinct, suggesting that the mutations in the two families were likely to be distinct.
The target-selected DNA libraries from one female heterozygote from each of the two families (females were used for optimal DNA quantity and quality) were sequenced on one lane each of a sequencing instrument (Illumina GAII) in single-end 36 bp configuration, which yielded 20,262,045; 18,775,942 reads (family 1; family 2) or 729,433,620; 675,933,912 bp of total sequence. Of this sequence, 43.8%; 45.2% could be uniquely aligned to the targeted exons. This aligned sequence yielded a gross overall coverage of 115×; 110× of the target. The capture efficiency varied across the targets with 2,239,228; 2,234,963 bp (80.5%) of the target with ≥1× coverage, 2,136,202; 2,128,057 bp (76.5%) with ≥10× coverage, and 2,071,297; 2,059,012 bp (74.1%) with ≥20× coverage. The most probable genotype variant-calling software (J.K.T., N.F.H., J.C.M., and L.G.B. et al., unpublished data) was able to make high-confidence genotype calls on 1,956,070; 1,941,688 bp of this sequence (70.0%).
We filtered the results for variants in genes on the basis of several attributes that we reasoned were appropriate for this disorder (Table 1). Heterozygosity was used because the test subjects were both unaffected female carriers for an X-linked trait. We reasoned that the variant should be severe because it caused a lethal phenotype in hemizygous males, so we filtered for nonsynonymous and stop alleles. Similarly, we also scanned the sequences for insertion or deletion variants, reasoning that most of these would cause frameshifting, null alleles. The criterion of novelty was applied in two ways. First, we filtered for variants not present in dbSNP, reasoning that a variant causing a rare phenotype should not be common. Second, we filtered for the absence of a sequence variant in three control DNA samples. The controls included two samples from males with syndromic microphthalmia and a single sample from a female parent of a child with a previously uncharacterized X-linked lethal disorder. We also applied a filter that bounded the variants genomically within the defined linkage interval from the original family, which was confirmed in family 2.
Table 1.
The Number of Genes with One or More Variants Following Each Filtering Criterion
|
All X Exons1 |
Linkage Region |
|||
|---|---|---|---|---|
| Family 1 | Family 2 | Family 1 | Family 2 | |
| Total substitutions | 360 | 330 | 85 | 76 |
| Heterozygous | 271 | 229 | 54 | 54 |
| Nonsynonymous | 71 | 65 | 14 | 14 |
| Not in dbSNP | 14 | 16 | 5 | 4 |
| Not in three controls | 11 | 11 | 3 | 3 |
| Nonsense | 0 | 1 | 0 | 1 |
| Total indels | 53 | 47 | 9 | 7 |
| Nonsynonymous | 8 | 7 | 2 | 1 |
| Not in dbSNP | 3 | 2 | 1 | 0 |
| Not in three controls | 1 | 1 | 1 | 0 |
| Frameshifting | 1 | 0 | 1 | 0 |
Refers to targetable exons between XG and TMLHE.
The initial analysis focused on single base pair substitutions, which, with the application of the heterozygosity + nonsynonymous + nonsense + novelty filters (Table 1), showed a single nonsense mutation, c.1235G>A in RBM10 (RNA binding motif 10), which predicts p.Trp412X in family 2 (Figure 1, lower left panels; Figure 2). No other nonsense mutations were identified in either family with this alignment and these filters. A second analysis for small deletions and insertions was performed with the following filters: nonsynonymous + novelty + frameshifting. This allowed identification of a single mutation in family 1, which was c.1893_1894insA in RBM10, which predicts p.Pro632ThrfsX41 (Figure 1, lower right panels; Figure 2). Each RBM10 variant was confirmed by PCR amplification and Sanger sequencing (Figure 1, bottom) in multiple individuals within the respective family, and mutation status segregated with known carrier status in all individuals (cosegregation is shown for family 2 in Figure 1; cosegregation for family 1 is not shown). We concluded that these were causative variants on the basis of the co-occurrence of a nonsense and a frameshift mutation in the same gene in two families with a highly similar and extremely rare phenotype.
Figure 2.
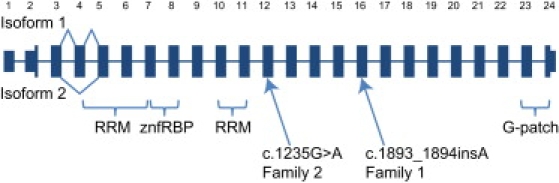
Cartoon of the RBM10 Gene Structure, Alternative Splicing, Conserved Domains, and Mutations Found in Two Families with TARP Syndrome
For clarity, the width of the rectangles is the same for all exons and is therefore not proportional to the actual length of the exons. The 5′ and 3′ UTRs are shown as rectangles with reduced vertical height. The gene has two mRNA isoforms: variant 1, which includes exon 4, and variant 2, which does not. Note that all other exons are believed to be constitutively spliced into both isoforms, so splicing lines are not shown for those exons. The portions of the gene that encode for the four recognized conserved domains are shown (two RRM, RNA recognition motif znfRBP, a zinc finger Ran binding protein, and a G-patch domain). Finally, the two independent mutations found in families 1 and 2 are shown.
Because the RBM10 gene product was poorly characterized and mutations had not been described with any abnormal phenotype in humans or in animal models, we set out to characterize the expression of this gene in midgestation mouse embryos. The orthologous murine Rbm10 isoform 1 gene product is 96% identical to the human protein. We isolated an Rbm10 antisense probe from the mouse sequence via RT-PCR of whole mouse RNA isolated from embryonic day E16.5 skin. Whole-mount in situ expression analysis of the murine Rbm10 gene at E9.5 and E10.5 of development showed a similar pattern of expression, with the most robust staining observed in the first branchial arch (which gives rise to the mandible), second branchial arch, developing limb buds, and tailbud (Figure 3). Robust expression remained for E11.5 embryos in both the limb and tail bud regions, whereas expression in branchial arches one and two decreased at this stage (data not shown). This pattern of expression correlated well with the human malformations observed in TARP syndrome, which include severe micrognathia and limb defects.
Figure 3.
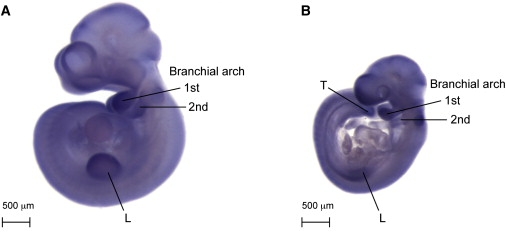
Expression of the Murine Ortholog Rbm10 in Midgestation Embryos
(A) In situ hybridization of a probe to murine Rbm10 in a wild-type E10.5 mouse embryo. There is expression of the transcript primarily in branchial arches 1 and 2. There is some expression in the limb (L), in a region that partially overlaps the apical ectodermal ridge.
(B) Expression of Rbm10 in an E9.5 mouse embryo. At this stage, the expression is slightly less strong in the second branchial arch and limb but is strong in the first branchial arch. Expression was also noted in the tail (T) at both stages.
Discussion
Here we show that the use of targeted exon capture with filters that included a genomic delimitation defined by a linkage region, the zygosity state, novelty, a severe deleterious mutation prediction, and a common mutation among two affected families can identify a gene mutated in a rare disorder. This approach was efficient and less time consuming compared with what would be required for PCR amplification and Sanger sequencing of the approximately 200 genes within the ∼28 Mb candidate interval. These data show two rare variants in RBM10, one in each of two families with a highly similar, rare, pleiotropic multiple congenital anomaly syndrome. We suggest that the delineation of nonsense or frameshift mutations in each of two families with a rare disorder is unlikely by chance alone. A simple estimate of the probability of finding these variants in the same gene in two patients in a candidate gene region of 200 genes is 1/200, or 0.005. Notably, nonsense or stop variants in RBM10 were not found in the recent report of 208 patients with mental retardation.8 In addition, the mouse expression data showed a striking correlation with the phenotypic manifestations in the human disorder. Finally, the variants cosegregated with the carrier status of the females in both families. We conclude that these data show that nonsense or null mutations in RBM10 cause syndromic Robin sequence, or TARP syndrome.
The only previous report of RBM10 and human phenotypes that we identified was a case report of a girl with an X;15 translocation with hypertelorism, a small face, high forehead, small, low-set ears, ulnar deviation of the hands, agenesis of the corpus callosum, and a hypoplastic fifth toe and metatarsal.9 This X;15 translocation breakpoint was near RBM10, although it was not determined whether the expression of RBM10 was altered by this translocation. This girl and the boys from families 1 and 2 with TARP did not have extensive phenotypic similarity; thus, it is unlikely that this translocation mediated its phenotypic effects primarily through disruption or dysregulation of RBM10.
The RBM10 gene and its 930 amino acid protein product is a member of the RNA binding motif (RBM) gene family. The RBM gene family is large, but mutations in only a few of the family members cause recognized human disorders, including dilated cardiomyopathy10 (MIM 613172), which is caused by mutations in RBM20 (MIM 613171), and alopecia, neurologic defects, and endocrinopathy syndrome11 (MIM 612079), which are caused by mutations in RBM28 (MIM 612074). A number of RBM genes have been shown to be important for RNA processing, RNA splicing, apoptosis, and other diverse biologic roles.12 RBM10 has been shown to undergo typical X chromosome inactivation.13–15 RBM10 is predicted to include a zinc finger motif, a G patch, and two RNA recognition motif (RRM) domains (Figure 2). This architecture is found in a variety of RNA binding proteins, including different heterogeneous nuclear ribonucleoproteins (RNPs), protein components of small nuclear RNPs, and those implicated in the regulation of alternative splicing. A typical RRM domain has two RNA or DNA binding sites and one RRM dimerization site.
The RBM10 gene has two known alternative splice forms. Variant 1 codes for a 930 amino acid protein (NM_005676, 23 coding exons), and variant 2 codes for an 852 amino acid protein (NM_152856, 22 coding exons). The nonsense change (p.Trp412X in the long form) would truncate the protein just after the end of the second RRM motif, which may destabilize the overall structure of the RBM10 protein. The frameshift (p.Pro632ThrfsX41) would also affect the last third of the protein. Alternatively, these truncating mutations may induce nonsense-mediated mRNA decay. In either case, the loss of function of RBM10 in TARP syndrome demonstrates that this gene is critical for normal mammalian development. Further elucidation of the role of this gene in early embryogenesis will lead to a better understanding of congenital anomalies that affect the face, heart, and limbs.
The filtering of the sequence data shows that the RBM10 gene could have been identified without linkage data, because the two RBM10 mutations were the only variants that met all of the filtering criteria for the entire targeted set of exons in these two families. We recognize that the determination of appropriate filtering criteria is subjective and that the outcome may be difficult to predict. For example, our criterion that the mutations should be frameshifting or nonsense would not hold for all X-linked male lethal disorders. Our overall approach was to begin with stringent or conservative filters and relax them in successive analyses in an attempt to identify the mutated gene. We were fortunate that the first filtering screen was successful, but we recognize that this will not always be the case. As noted in the methods section, this sequence experiment included five samples. The others were three patients who, among them, had two disorders distinct from TARP. We believe that we have identified the causative mutations in one of these two disorders by use of the same filtering used to identify the TARP mutations, but the other has failed to identify a causative gene mutation (data not shown). Much more data on similar projects will need to be generated to develop optimal approaches to filtering.
Another potential cause of a failure to identify causative mutations by exon capture is oligonucleotide design and sequencing coverage. This implementation of solution hybridization exon targeting included oligonucleotide designs for just over 80% of the exonic base pairs. After generating >100× of aligned sequence coverage for each sample, we had adequate coverage (≥10 quality base score depth) for detection of heterozygous changes in 80% of the base pairs of RBM10. This would suggest that for any pair of samples from patients with TARP syndrome, where each sample has one causative mutation in each of two distinct targets, there is about a 60%–65% chance that the mutations in both will be detected. In addition, nonexonic variants, synonymous variants that may affect splice enhancers, and other types of causative mutations are likely to be challenging to detect by exon targeting. Therefore, early successes in gene identification by target selection will not be representative of all disorders, and it will be important to develop new approaches to address these challenges.
Acknowledgments
This research was supported by Intramural Funds of the National Human Genome Research Institute, National Institutes of Health. The authors thank Joyce T. Turner for the initial consenting and records gathering for the family and Emma J. Spaulding for technical support. This paper is in memory of Robert J. Gorlin, the lead author of the paper describing family 1 and a generous, good-humored supporter of the work of L.G.B.
Web Resources
The URLs for data presented herein are as follows:
Conserved Domain-Based Prediction (CDPred) Software, http://research.nhgri.nih.gov/software/CDPred/
NCBI Single Nucleotide Polymorphism Database (dbSNP), http://www.ncbi.nlm.nih.gov/projects/SNP/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorlin R.J., Cervenka J., Anderson R.C., Sauk J.J., Bevis W.D. Robin's syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am. J. Dis. Child. 1970;119:176–178. [PubMed] [Google Scholar]
- 4.Kurpinski K.T., Magyari P.A., Gorlin R.J., Ng D., Biesecker L.G. Designation of the TARP syndrome and linkage to Xp11.23-q13.3 without samples from affected patients. Am. J. Med. Genet. A. 2003;120A:1–4. doi: 10.1002/ajmg.a.10201. [DOI] [PubMed] [Google Scholar]
- 5.Johnston J.J., Olivos-Glander I., Killoran C., Elson E., Turner J.T., Peters K.F., Abbott M.H., Aughton D.J., Aylsworth A.S., Bamshad M.J. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 2005;76:609–622. doi: 10.1086/429346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilkinson D., Nieto M. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. In: Wassarman P., DePamphilis M., editors. Methods in Enzymology: Guide to Techniques in Mouse Development. Academic Press; San Diego, FL: 1993. pp. 361–373. [DOI] [PubMed] [Google Scholar]
- 7.Loftus S.K., Baxter L.L., Buac K., Watkins-Chow D.E., Larson D.M., Pavan W.J. Comparison of melanoblast expression patterns identifies distinct classes of genes. Pigment Cell Melanoma Res. 2009;22:611–622. doi: 10.1111/j.1755-148X.2009.00584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gläser B., Shirneshan K., Bink K., Wirth J., Kehrer-Sawatzki H., Bartz U., Zoll B., Bohlander S.K. Molecular cytogenetic analysis of a de novo balanced X; autosome translocation: Evidence for predominant inactivation of the derivative X chromosome in a girl with multiple malformations. Am. J. Med. Genet. A. 2004;126A:229–236. doi: 10.1002/ajmg.a.20584. [DOI] [PubMed] [Google Scholar]
- 10.Brauch K.M., Karst M.L., Herron K.J., de Andrade M., Pellikka P.A., Rodeheffer R.J., Michels V.V., Olson T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nousbeck J., Spiegel R., Ishida-Yamamoto A., Indelman M., Shani-Adir A., Adir N., Lipkin E., Bercovici S., Geiger D., van Steensel M.A. Alopecia, neurological defects, and endocrinopathy syndrome caused by decreased expression of RBM28, a nucleolar protein associated with ribosome biogenesis. Am. J. Hum. Genet. 2008;82:1114–1121. doi: 10.1016/j.ajhg.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutherland L.C., Rintala-Maki N.D., White R.D., Morin C.D. RNA binding motif (RBM) proteins: A novel family of apoptosis modulators? J. Cell. Biochem. 2005;94:5–24. doi: 10.1002/jcb.20204. [DOI] [PubMed] [Google Scholar]
- 13.Coleman M.P., Ambrose H.J., Carrel L., Németh A.H., Willard H.F., Davies K.E. A novel gene, DXS8237E, lies within 20 kb upstream of UBE1 in Xp11.23 and has a different X inactivation status. Genomics. 1996;31:135–138. doi: 10.1006/geno.1996.0022. [DOI] [PubMed] [Google Scholar]
- 14.Thiselton D.L., McDowall J., Brandau O., Ramser J., d'Esposito F., Bhattacharya S.S., Ross M.T., Hardcastle A.J., Meindl A. An integrated, functionally annotated gene map of the DXS8026-ELK1 interval on human Xp11.3-Xp11.23: Potential hotspot for neurogenetic disorders. Genomics. 2002;79:560–572. doi: 10.1006/geno.2002.6733. [DOI] [PubMed] [Google Scholar]
- 15.Goto Y., Kimura H. Inactive X chromosome-specific histone H3 modifications and CpG hypomethylation flank a chromatin boundary between an X-inactivated and an escape gene. Nucleic Acids Res. 2009;37:7416–7428. doi: 10.1093/nar/gkp860. [DOI] [PMC free article] [PubMed] [Google Scholar]