

Abstract
Retinitis pigmentosa is a genetically heterogeneous group of inherited ocular disorders characterized by progressive photoreceptor cell loss, night blindness, constriction of the visual field, and progressive visual disability. Homozygosity mapping and gene expression studies identified a 2 exon gene, C2ORF71. The encoded protein has no homologs and is highly expressed in the eye, where it is specifically expressed in photoreceptor cells. Two mutations were found in C2ORF71 in human RP patients: A nonsense mutation (p.W253X) in the first exon is likely to be a null allele; the second, a missense mutation (p.I201F) within a highly conserved region of the protein, leads to proteosomal degradation. Bioinformatic and functional studies identified and validated sites of lipid modification within the first three amino acids of the C2ORF71 protein. Using morpholino oligonucleotides to knockdown c2orf71 expression in zebrafish results in visual defects, confirming that C2ORF71 plays an important role in the development of normal vision. Finally, localization of C2ORF71 to primary cilia in cultured cells suggests that the protein is likely to localize to the connecting cilium or outer segment of photoreceptor cells.
Introduction
Retinitis pigmentosa (RP [MIM 268000]) is a group of inherited ocular disorders involving a progressive loss of photoreceptor cells and loss of vision. It is characterized by night blindness, bone spicule-like pigmentation of the retina, narrowed retinal arterioles, and constricted visual fields. RP exhibits a high level of genetic heterogeneity and can segregate in autosomal-dominant, autosomal-recessive, X-linked, and digenic patterns. The clinical presentation of RP is highly variable in age of onset and disease progression. Numerous genes have been shown to cause isolated RP (RetNet and OMIM), and it has been previously observed that approximately half of those genes known to cause inherited retinal disease are preferentially or specifically expressed in the photoreceptor cells of the retina.1 We identified genes that are preferentially expressed in photoreceptor cells by performing microarray expression studies comparing wild-type eye tissue to that collected from a mouse model, Bbs4−/−, that exhibits specific photoreceptor loss. The screen is based on the premise that genes that are predominantly or exclusively expressed in photoreceptor cells will show a large decrease in expression because these cells die as the retina degenerates. Expression data from such studies can then be applied to the genes in RP candidate intervals so that mutation-screening studies can be prioritized.2–5
In this study, we have utilized a combination of homozygosity mapping and gene expression studies to identify an RP gene, C2ORF71. It is highly expressed in the eye and shows specific localization to retinal photoreceptor cells. The encoded protein has no homologs. We identified both missense and nonsense pathogenic variants in C2ORF71 in families with autosomal-recessive RP and used expression knockdown studies in zebrafish to demonstrate that c2orf71 plays an important role in the development of normal vision. We used bioinformatic and functional studies to identify and validate sites of lipid modifications within the N terminus of the human C2ORF71 protein. Finally, we have demonstrated localization to the primary cilium, suggesting that C2ORF71 is localized to the photoreceptor outer segment or to the modified cilium of the photoreceptor.
Material and Methods
Homozygosity Mapping
Signed informed consent forms were obtained from all individuals at the time of their entry into the study. The control samples used for allele-frequency estimation in the general population included a subset of the DNA Polymorphism Discovery Resource (Coriell Cell Repository), which is a panel designed to reflect the diversity present in the human population.6 Genomic DNA was used as a template to generate probes for use on the XbaI (family 1) or HindIII (family 2) chip of the Affyemtrix Human Mapping 100K Set (Affymetrix). Sample processing and labeling were performed in accordance with the manufacturer's specifications (Affymetrix). The arrays were hybridized, washed, and scanned at either the Patterson Institute Microarray Service (Manchester) for family 1 or the University of Iowa DNA Core Facility for family 2. Homozygosity analysis was performed as described previously.3 Parametric linkage analysis for family 1 was carried out with Merlin software.7
Mutation Detection
PCR products for sequencing were amplified in a 20 μl reaction volume and were separated on 2% agarose gels as described elsewhere.8 Bands corresponding to the desired product were excised and purified with the QIAquick gel extraction kit (QIAGEN). A 4.5 μl volume of the purified PCR product was used as a template for sequencing reactions with dye-terminator chemistry (Applied Biosystems). These reactions were then analyzed on an ABI 3730XL DNA Sequencer. Primer sequences used for screening the human C2ORF71 gene are shown in Table S1.
RT-PCR
RNA was extracted from a pool of wild-type zebrafish embryos at the following stages: 8–10 somites; 24, 42, 48, and 72 hpf; 5 dpf; and adult retina. cDNA was synthesized with oligo dT primers, and gene expression was evaluated by PCR with primers specific for c2orf71 and β-actin (Table S2).
Morpholino Knockdown
Antisense morpholinos (MOs) were designed and purchased from Gene Tools. MOs were injected via air pressure into the one- to four-cell embryos. Zebrafish were maintained at standard conditions,9 and embryos were staged according to Kimmel et al., 1995.10 Morpholino sequences are shown in Table S3.
Vision Startle-Response Assay
The vision-evoked startle-response behavioral assay was performed as previously described (modified from Easter and Nicola11). In brief, 5 dpf zebrafish larvae were adapted to light for 1 hr, and the visual stimulus of a rapid change in the intensity of white light, via a 1 s block in light, was delivered under a standard dissecting microscope. A positive response was recorded if the animal made an abrupt movement (change in behavior) within 1 s of the change in light intensity. Five trials of visual stimulation were performed 30 s apart, followed by the mechanical stimulus of probing embryos with the tip of a blunt needle. Rarely, embryos did not respond to the mechanical stimulus, and these were not included in the analysis.
Histological Analysis
Embryos were maintained in 0.003% phenylthiourea (PTU) beginning at 80% epiboly so that pigmentation was minimized, and they were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). Gross morphology of the retinas was evaluated with standard hematoxylin and eosin staining.
DNA Constructs
A fragment encoding the full-length wild-type C2ORF71 of 1,288 amino acids (NCBI reference sequence: NM_001029883.1) was amplified from Human Retina Marathon-Ready cDNA (Clontech) and cloned into the pSTBlue-1 AccepTor vector (Novagen). We verified the entire coding sequence of this clone (pSTBlue-C2ORF71-WT) to ensure that the sequence matched the published NCBI reference sequence for C2ORF71. We observed no sequence changes. We created the I201F mutant by introducing the appropriate nucleotide change into the pSTBlue-C2ORF71-WT clone by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
To create tagged constructs, we designed PCR primers so that the 3′ end included a restriction site that was present in the destination vector and that would place the C2ORF71 coding sequence in the correct translational reading frame with the tag. These primers were used for PCR amplification off of the pSTBlue-C2ORF71 templates to create new fragments for cloning into the target vectors. C-terminal tagged C2ORF71 constructs were created with the following tags: pEGFP-N1 (Clontech) and pCDNA3.1-Myc (Invitrogen) modified to incorporate a FLAG tag downstream of the Myc tag.
Cell Culture and Immunoblot
hTERT-RPE and ARPE-19 cells were grown at 37°C and 5% CO2 in DMEM/F12 containing 10% (v/v) fetal bovine serum (Atlanta Biologicals). DNA was transfected into cells with Arrest-In (Open Biosystems) in growth medium containing serum according to the manufacturer's instructions. To induce primary cilia formation, we transferred cells to serum-free media 8 hr after transfection and starved them of serum for 24 hr. For proteosome inhibition studies, cells were transfected, and the peptide aldehydes MG115 (proteasome only) and MG132 (proteasome and cathepsin K) were added at 10 μM to the cells 24 hr after transfection. After incubation for an additional 12 hr, the cells were harvested in RIPA buffer. Whole-cell lysates (20 μg) were separated by SDS-PAGE and immunoblotted with mouse monoclonal FLAG (1:2,000; Sigma), rabbit polyclonal Myc (1:1,000; Abcam), and mouse monoclonal actin (1:10,000; Sigma) antibodies.
IHC
Cells were grown under the conditions described above with the exception that they were grown on poly-L-lysine-coated coverslips in 12-well culture plates. After transfection, the coverslips were washed with PBS, fixed with −20°C methanol for 10 min, washed four times for 5min. with PBS, incubated with primary antibody in Abdil (5% BSA, 5% NGS, and 0.01% Triton X-100 in PBS) for 1 hr, washed four times for 5 min with PBS, and then incubated with secondary antibody in Abdil for 1 hr. The second of the final four wash steps with PBS included DAPI in the wash buffer. The coverslips were mounted onto microscope slides with VectaShield anti-fade reagent (20 μl), visualized on an Olympus IX-71 microscope, and imaged with an Olympus DP71 digital camera. Primary antibodies included mouse monoclonal anti-γ-tubulin (1:1,000; Santa Cruz Biotechnology), mouse monoclonal anti-acetylated-tubulin (1:150,000; Sigma), and rabbit polyclonal TGN46 (1:500; Abcam). Secondary antibodies were goat anti-mouse Alexa 568 (1:500; Santa Cruz Biotechnology), goat anti-rabbit Alexa 568 (1:500; Santa Cruz Biotechnology), and goat anti-rabbit Alexa 488 (1:500; Santa Cruz Biotechnology).
Results
Mutations in C2ORF71 Are Associated with Isolated Retinal Degeneration
Homozygosity mapping via SNP genotyping was carried out on six affected individuals from two families with nonsyndromic autosomal-recessive RP (arRP). Homozygosity mapping in five affected members of family 1 revealed an ∼8 Mb region of shared homozygosity at 2p24.1-p23.1 between the SNPs rs2339705 and rs2303324 (22.9–31.0 Mb). Parametric linkage analysis using a fully penetrant recessive model produced a maximum LOD score of 4.5 for this region. In the second family, the mapping identified a single 21.8 Mb region of homozygosity (18.4–40.2 Mb) that contained the same chromosome 2 region that was identified in the first family. The common 8 Mb region encompasses 90 genes, none of which are known to cause isolated retinitis pigmentosa. Within this region are the hydroxyacyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA [MIM 600890]) and beta subunit (HADHB [MIM 143450]) genes that are mutated in long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (LCHAD [MIM 609016]), a recessive inborn error of metabolism that can be associated with retinal degeneration. Sequencing of the coding sequence of HADHA and HADHB in family 1 found no pathogenic variants.
In order to prioritize candidate gene screening, whole-eye gene expression data were generated for the Bardet-Biedl syndrome 4 (Bbs4 [MIM 600374]) knockout mouse model2 and were utilized to identify genes that were from the homozygous region on chromosome 2 and were likely to be preferentially expressed in the retina. This analysis identified two genes (glucokinase regulator (Gckr [MIM 600842] and C2orf71) that had significantly decreased expression in the Bbs4 knockout eyes as compared with wild-type control eyes. One gene, C2orf71, was found to exhibit an almost 26-fold decrease in expression in the 8-month-old knockout eyes and furthermore demonstrated a much greater loss in gene expression in 8-month-old versus five-month-old animals. A similar pattern of expression has been found for genes, such as that for rhodopsin, that are specific to the photoreceptor layer of the eye.
Mutation screening of the complete coding sequence and splice sites of C2ORF71 was carried out on the two families. In family 1, a homozygous G-to-A transition that causes a nonsense mutation at residue 253 (p.W253X) of the 1,288 amino acid C2ORF71 protein was identified. This mutation is in the first exon of the C2ORF71 gene and was found to segregate with the disease in all affected family members (Figure 1A). The mutation was not detected in 100 patients of the same ethnic origin.
Figure 1.

Pedigrees of RP Families with C2ORF71 Mutations
Pedigrees of the two RP families with mutations in C2ORF71. For both panels, the presence of the mutant allele is indicated by a minus (-) symbol, whereas the presence of a wild-type allele is shown by a plus (+) symbol.
(A) A p.W253X mutation in C2ORF71 segregates with autosomal-recessive retinitis pigmentosa (arRP) in family 1. The p.W253X mutation was found in the homozygous state in all affected family members.
(B) A p.I201F mutation in C2ORF71 segregates with arRP in family 2. The proband (III-2) is homozygous for the C2ORF71 mutation. Patient II-4 is affected with Bardet-Biedl syndrome and is homozygous for the common BBS1 p.M390R mutation.
Sequencing of the RP patient from family 2 revealed an A-to-T transversion that was present at position 601 in the homozygous state. This missense variant results in the replacement of isoleucine by phenylalanine at position 201 (p.I201F) of the C2ORF71 protein. The p.I201F variant was detected in the heterozygous state in all unaffected members of this family, as is shown in Figure 1B. One member of the extended family has been diagnosed with Bardet-Biedl Syndrome (BBS [MIM 209900]), a syndromic form of retinal degeneration. This individual was found to be heterozygous for the C2ORF71 p.I201F mutation but homozygous for the common BBS1 p.M390R mutation. The RP patient did not have any pathogenic mutations in the BBS1 gene. The p.I201F mutation in C2ORF71 occurs in an evolutionarily conserved region of the protein, as is shown in Figure 2. To determine whether the p.I201F mutation was a common variant, we screened control samples from different ethnic populations. These samples included 90 from the NIH Diversity Panel (consisting of individuals of Caucasian, Asian and African descent) and 102 ethnically matched controls. We did not detect any evidence for this mutation. Thus, C2ORF71 p.I201F is not a common variant.
Figure 2.
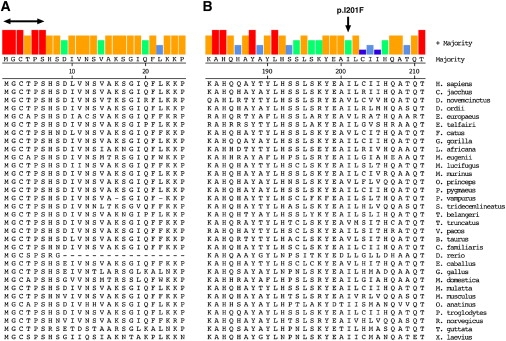
Evolutionary Comparison of Selected Regions of C2ORF71
Examination of evolutionary conservation of selected regions of C2ORF71 from 32 species.
(A) Comparison of the first 25 amino acids of the C2ORF71 protein sequence. Five of the first six amino acids are completely conserved among all organisms examined. Both the glycine at position 2 (G2) and the cysteine at position 3 (C3) are predicted to undergo post-translational acylation.
(B) Multiple alignment of C2ORF71 for a region surrounding the p.I201F missense mutation; the site of the mutation is indicated. For both panels, the consensus sequence is displayed at the top of the figure along with a color-coded indication of the level of conservation for each residue.
C2ORF71 p.I201F Causes Reduced Protein Stability
To examine the effect of p.I201F on protein expression, we expressed wild-type and mutant C-terminal Myc-FLAG-tagged C2ORF71 proteins (wild-type, C2ORF71-WT-MF; and p.I201F mutant, C2ORF71-I201F-MF) in ARPE-19 cells. By immunoblot analysis, C2ORF71-I201F-MF was found to exhibit a much lower level of expression than C2ORF71-WT-MF (Figure 3A). We then used two inhibitors of protein degradation, the peptide aldehydes MG115 (proteasome only) and MG132 (proteasome and cathepsin K), to inhibit specific pathways of protein degradation. Both inhibitors rescued expression of the mutant protein as shown in Figure 3B. It is notable that a higher level of protein expression was also observed for the wild-type C2ORF71-WT-MF protein in the presence of the inhibitors. At 1,288 amino acids, the C2ORF71 protein is relatively large, and it is likely that increased expression levels after transient transfection induce protein-degradation pathways. These findings support the proposition that one of the functional consequences of the p.I201F mutant is to induce proteasomal degradation of C2ORF71.
Figure 3.

C2ORF71 p.I201F Mutation Affects Protein Stability
Protein expression levels of wild-type and pI201F mutant protein were examined by transient transfection in human ARPE-19 cells.
(A) Immunoblot analysis of protein expression levels of C2ORF71 expression constructs with a C-terminal dual Myc-FLAG tag. Expression constructs were run in duplicate in adjacent lanes and detected with either Myc or FLAG antibodies, as indicated.
(B) Rescue of C2ORF71 protein expression by inhibition of proteasome activity by the peptide aldehydes MG115 and MG132. C2ORF71 was detected with FLAG antibody. Full-length wild-type (WT) and p.I201F mutant (MT) C2ORF71-Myc or -FLAG expression constructs were used.
Retina-Specific Expression of C2orf71
The initial prioritization of the candidate genes by gene expression studies indicated that C2orf71 was likely to be expressed specifically in the mouse retina. In 5-month-old Bbs4 knockout mice, expression of C2orf71 in whole eyes was about 3-fold lower than in wild-type controls. However, by 8 months of age, there was almost complete loss of photoreceptors in the Bbs4 knockout mouse retina, and at this stage, C2orf71 expression was nearly 26-fold lower in the knockout eyes. These results are consistent with a gene for which expression in the eye is specifically confined to photoreceptor cells. Evaluation of human and mouse EST sequences is also consistent with the idea that C2ORF71 is a gene whose expression is largely confined to the eye. Of the 19 unique human ESTs listed for C2ORF71 as part of UniGene cluster Hs.354243, 14 (73%) are annotated as being from libraries derived from the eye. Furthermore, of the 18 unique ESTs that are documented as part of the mouse UniGene cluster Mm.134151, 17 (94%) are annotated as being derived from eye cDNA libraries. Microarray experiments of tissue expression at the GeneNetwork website12 confirms eye-specific expression of C2orf71. In Figure 4, the mRNA tissue expression levels of C2orf71 are plotted against the expression levels of rhodopsin. It is observed that both genes have higher expression levels in the eye than in any other tissues examined. Specific expression of c2orf71 in the zebrafish retina was demonstrated by RT-PCR, as shown in Figure S1.
Figure 4.
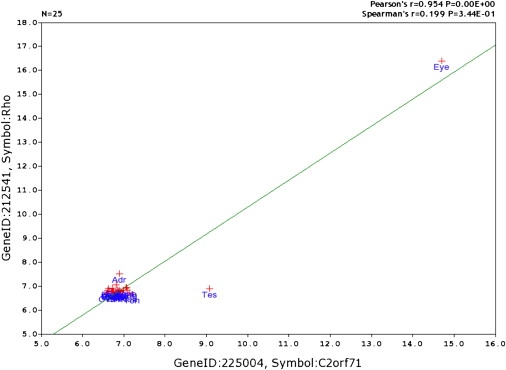
Tissue-Expression Correlation of C2orf71 and Rhodopsin
Tissue-expression correlation between the known retina-specific gene rhodopsin (Rho) and C2orf71. Both Rho and C2orf71 are highly expressed in the eye, whereas C2orf71 is also expressed at a low level in the testis.
Knockdown of c2orf71 in Zebrafish Causes Vision Defects
Having established a genetic link between C2ORF71 mutations and retinitis pigmentosa, we next examined the in vivo role of c2orf71 in zebrafish vision. Zebrafish are an ideal model system for examining visual responsiveness because of their rapid development, the opportunity to knock down specific gene expression, and their amenability to a vision-based behavioral assay. The zebrafish retina is fully laminated and light responsive by three days post fertilization (dpf).11,13,14 We first examined when the zebrafish ortholog of C2ORF71 is expressed by RT-PCR analysis. Using a developmental time course of whole-embryo cDNA, we determined that the c2orf71 transcript is present beginning at 48 hpf (Figure 5A). This time point corresponds developmentally with the initiation of photoreceptor differentiation.15 Furthermore, the c2orf71 transcript is present in the zebrafish adult retina (Figure 5A).
Figure 5.
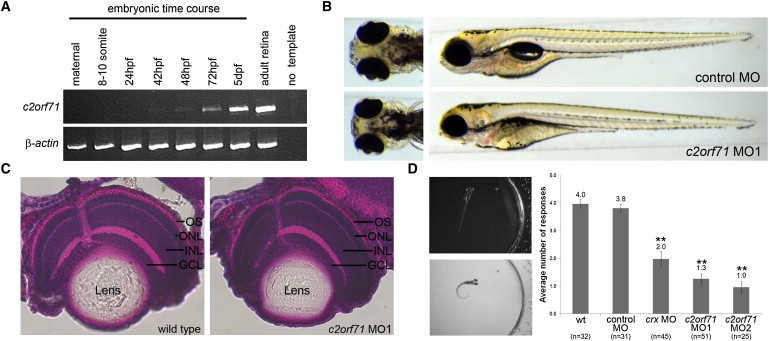
Characterization of c2orf71 in the Zebrafish Model System
Functional characterization of zebrafish c2orf71 by RT-PCR and morpholino knockdown.
(A) RT-PCR of c2orf71 expression throughout a zebrafish developmental time course and in adult retinal tissue. β-actin was used as a control. The c2orf71 transcript is expressed by 48 hpf, a time point correlating with photoreceptor development, and is expressed in the adult retina.
(B) Ventral and side views of 5 dpf embryos that were injected with either 15 ng of control MO or 5 ng of c2orf71 MO1.
(C) H&E histological staining of transverse sections through 5 dpf zebrafish retinas of both wild-type and c2orf71 MO-injected embryos. Pigment in the retinal pigment epithelium (RPE) apical to the photoreceptors was chemically inhibited so that it did not prevent visualization of the zebrafish outer segment. Abbreviations are as follows: GCL, ganglion cell layer; INL inner nuclear layer; ONL, outer nuclear layer; and OS, outer segment of the photoreceptor.
(D) Panels on the left are an example of a positive behavioral response (change in swimming direction) to an abrupt change in light intensity. The graph shows the quantification of the vision startle response. Cone-rod homeobox (crx) gene knockdown (15 ng) was used as a control for vision impairment. There was a statistically significant reduction in the number of responses in crx and c2orf71 (MO1 and MO2) morphant embryos compared to wild-type and control MO-injected embryos, indicating visual impairment in the crx and c2orf71 morphant embryos. n = sample size, ∗∗p < 0.01, ANOVA with Tukey's test. The error bars represent the standard error of the mean.
To examine the functional role of c2orf71 in development and vision, we knocked down c2orf71 protein expression by using two different translational-start-site-targeted antisense morpholino oligonucleotides. Morphological examination of the morpholino-injected embryos (morphants) revealed no gross defects in body axis, consistent with a gene expressed late in development (Figure 5B). Histological examination by H&E staining revealed that, although overall eye size was reduced, the retina was fully laminated. Notably, the photoreceptor outer segments appeared shorter in length in the morphant retinas than in the wild-type retinas (Figure 5C).
Assessment of visual function was performed on 5 dpf embryos with a visual-startle-response assay paired with a touch-sensitivity assay for motility (modified from Easter and Nicola11). Zebrafish embryos elicit a characteristic escape response to rapid changes in light intensity. An immediate positive escape response or no response was monitored over a series of five short blocks in light spaced 30 s apart (Figure 5D, Movies S1 and S2). Morphant embryos were also evaluated for motility and the ability to respond to stimuli. In this assay, embryos were probed with a blunt needle on the flank, which invokes the same escape response as that observed in the vision assay (Movie S2). Uninjected wild-type and control morpholino-injected embryos positively responded on average in four out of five trials of the vision assay. Knockdown of the cone-rod homeobox (crx [MIM 602225])) gene, a gene necessary for photoreceptor formation in the zebrafish, was used as a control for visual impairment.16,17 The crx morphant embryos respond on average in only two out of the five trials (p < 0.01). Similarly, knockdown of c2orf71 causes visual impairment comparable to that of crx knockdown (Figure 5D). This data strongly supports a functional role for c2orf71 in vision.
Lipid Modification of the N-Terminal Region of C2ORF71
The C2ORF71 protein is annotated on the ENSEMBLE web site as having a proline-rich domain at the C-terminal region of the protein; however, it is not clear what role this motif plays in C2ORF71 function. To identify other potential functional motifs, we screened C2ORF71 ortholog sequences from different species for the presence of potential functional domains with the PSORT II suite of programs.18 Consistent among the various C2ORF71 orthologs was the prediction of a palmitoylation modification at the C3 residue. Protein sequence alignment of the first 25 amino acids of the C2ORF71 orthologs demonstrates complete conservation of the first three amino acids (methionine, glycine, and cysteine) as well as the proline at position 5 and the serine at position 6, as is illustrated in Figure 2. The complete conservation of glycine at position 2 (G2) suggests that this position may be myristoylated, a modification that is found to occur at the G2 position of many proteins. Myristoylation at the G2 position of C2ORF71 is predicted by both the MYRISTOYLATOR (score = 0.153) and NMT (score = 0.451) programs.19,20 In addition, the TERMINATOR program predicts both N-myristoylation at G2 and S-palmitoylation at C3 with a likelihood of 87%.21 It is thought that the myristoylation at G2 provides a protein with the capability to transiently interact with membranes and that C3 palmitoylation provides additional stability to the membrane binding.22–24
The X-linked human retinitis pigmentosa 2 (RP2 [MIM 300757]) gene is an RP gene with a dual G2/C3 motif that is subject to lipid modification. The RP2 protein is predominantly targeted to the plasma membrane, and it has been shown that its first seven amino acids are sufficient to localize a C-terminal GFP-tagged construct to the plasma membrane in a variety of cells.25 We therefore created constructs that contained the first 20 amino acids of the human C2ORF71 protein fused to a C-terminal GFP reporter construct. When the wild-type version of the construct was expressed in hTERT-RPE cells, an intense signal was detected in a region surrounding the centrosome, as shown in Figure 6A. The C2ORF71 expression colocalized with the trans-Golgi network (Figure 6B). We next created mutant versions of the construct at the G2 (G2A) and the C3 (C3S) positions to examine how the prevention of myristoylation and/or palmitoylation affects localization. For the G2A construct, the distinct localization pattern observed for the wild-type construct was lost, and only a diffuse cytoplasmic pattern was observed (Figure 6C). Similar to what was found for the wild-type construct, the C3S construct was found to localize to a region surrounding the centrosome, but the pattern was more diffuse (Figure 6D). Therefore, we conclude that these two lipid modification sites are functional in the C2ORF71 protein because disruption of these motifs affects the localization of the reporter constructs in a manner that is consistent with their previously proposed functions of membrane targeting and stabilization.
Figure 6.
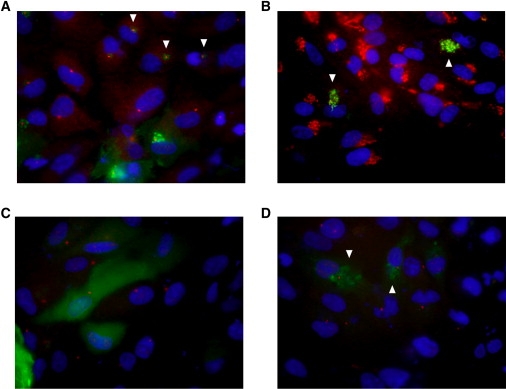
Cellular Localization of N-terminal C2ORF71 Fragments
The first 20 amino acids of the human C2ORF71 protein were fused to a C-terminal green fluorescent protein reporter tag and transfected into hTERT-RPE cells.
(A) The wild-type construct was found to localize to a region surrounding the centrosome (arrowheads).
(B) Colocalization of wild-type C2ORF71 expression with a trans-Golgi network marker (arrowheads).
(C) Changing the glycine at position 2 to an alanine (G2A) prevented myristoylation and resulted in the loss of specific localization.
(D) Changing the cysteine at position 3 to a serine (C3S) prevented palmitoylation and resulted in a slightly more diffuse localization pattern surrounding the centrosome (arrowheads) than was present in the wild-type. For all of the panels, DAPI nuclear staining is shown in blue, and C2ORF71 is in green. For (A), (C), and (D), γ-tubulin staining of the centrosome is in red, whereas in (B), TGN46 staining of the trans-Golgi network is shown in red.
Cellular Localization of C2ORF71 to the Primary Cilia
Finally, we sought to determine the cellular localization of full-length wild-type C2ORF71 protein tagged at the C terminus with a GFP tag (C2ORF71-GFP). In hTERT-RPE cells, C2ORF71-GFP was found to localize to a confined region near the nucleus, as is shown in Figure 7A. Colabeling with γ-tubulin indicates that this localization of C2ORF71 is pericentrosomal: the signal from C2ORF71 was often observed to be in close proximity to, but distinct from, γ-tubulin. We then examined the localization of the p.I201F missense mutant (C2ORF71-I201F-GFP). Consistent with the low level of protein expression observed for the p.I201F mutant, the number of cells that were found to express the mutant protein was much lower than the number of cells expressing the wild-type construct. In addition, the number of cells that demonstrated high levels of protein expression was greatly decreased. However, for those cells in which signal was present, localization of the mutant protein was similar to that seen for the wild-type protein (Figure 7B). This result implies that the p.I201F variant results in the production of a decreased amount of protein that can be correctly targeted.
Figure 7.
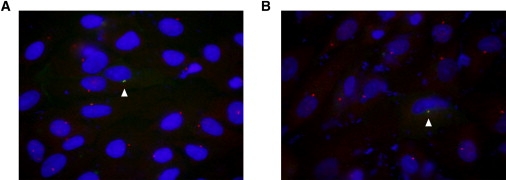
Cellular Localization of Full-Length C2ORF71
Full-length C2ORF71 was fused to a C-terminal green fluorescent protein (GFP) reporter tag and transfected into hTERT-RPE cells so that protein localization could be determined.
(A) The wild-type construct was found to localize to a region surrounding the centrosome (arrowhead).
(B) The p.I201F mutant was found to display a localization (arrowhead) similar to that of the wild-type C2ORF71 protein, although global expression levels were decreased. For both panels, C2ORF71 is in green, DAPI nuclear staining is shown in blue, and γ-tubulin staining of the centrosome is shown in red.
Because C2orf71 is highly expressed in photoreceptor cells, we hypothesized that it localizes to the outer segment, a highly modified primary cilium. Therefore, in order to determine whether C2ORF71 could be observed in primary cilia, we aimed to induce the formation of primary cilia by repeating the expression experiments with cells that were stressed by serum starvation. The C2ORF71-WT-GFP construct was found to be present within primary cilia (Figure 8). No cells expressed C2ORF71-I201F-GFP in primary cilia; however, lower expression levels of the mutant protein may have precluded its detection.
Figure 8.

Cellular Localization of C2ORF71 to the Primary Cilia
Full-length C2ORF71 was fused to a C-terminal green fluorescent protein (GFP) reporter tag and transfected into hTERT-RPE cells. After transfection, the cells were starved of serum for 24 hr to produce primary cilia.
(A) Expression of C2ORF71.
(B) Decoration of the basal body and ciliary axoneme by dual γ-tubulin/acetylated tubulin staining.
(C) DAPI nuclear staining.
(D) Merged image of (A)–(C). The location of the primary cilia is indicated by the arrowheads in (A), (B), and (D).
Discussion
Homozygosity mapping in small consanguineous families in combination with the prioritization of candidate genes on the basis of expression studies has been a powerful tool for the identification of new disease genes. We have previously used this approach to identify a gene, parathyroid hormone-responsive B1 gene (PTHB1 [MIM 607968]), that causes the syndromic retinal disorder known as Bardet-Biedl syndrome.3 We now extend this technique to the identification of a gene for retinitis pigmentosa, C2ORF71, which was one of two genes that were identified by positional cloning and that showed significantly decreased expression in Bbs4 mouse knockout eyes compared to wild-type control eyes. Further informatics analyses, as well as expression studies, have confirmed that the gene is selectively expressed in the retina. It has been observed that about half of the genes that cause retinal degeneration demonstrate a specific or elevated expression profile in the retina.1 We have previously found that approximately half of the genes with the highest levels of retinal specificity have already been documented as causing disorders of the retina.2 Thus, it is likely that large-scale mutational screening of genes whose expression is restricted or preferential to the retina will identify additional genes that play a role in retinal degeneration.
The encoded protein C2ORF71 has no homologs and is specifically expressed in photoreceptors. We have identified two unique C2ORF71 mutations that are present in patients affected with RP. The first mutation is a p.W253X nonsense mutation that is likely to be subjected to NMD. The phenotype in the family is variable in that six of eight individuals have an adult-onset form of RP, whereas the other two individuals had a much earlier-onset and severe generalized dystrophy (<5 years) associated with nystagmus. The second mutation is a missense variant (p.I201F) found in a patient with adult-onset RP. The amino acid substitution occurs within an evolutionarily conserved region of the protein, and our study shows that this point mutation decreases C2ORF71 protein levels by impacting protein stability via proteosomal degradation.
The identification of potential functional domains in the C2ORF71 protein can provide important clues as to its function in the retina. There is strong evidence that C2ORF71 contains sites that undergo post-translational lipid modifications. The G2 and C3 residues are completely conserved in all known C2ORF71 orthologs. The presence of these motifs suggests that C2ORF71 interacts with membranes during some portion of its existence in the retina. One class of post-translational modification involves proteins that are covalently modified by a variety of lipids (myristate, palmitate, farnesyl, etc.).23,26,27 Palmitoylation enhances the surface hydrophobicity and membrane affinity of protein substrates and plays important roles in protein trafficking,28,29 stability,29 sorting,22 apoptosis,30,31 signaling,24,26,32 and other cellular processes.
With increasing knowledge of the mammalian transcriptome, the discovery of C2ORF71 is likely to represent the identification of one of a relatively small number of the remaining transcripts that are of unknown function, are highly (or exclusively) expressed in the retina, and are specific to photoreceptor cells. The precise location of the protein within the photoreceptor cell remains to be determined, although on the basis of our detection of C2ORF71-GFP-tagged protein in primary cilia, we hypothesize that it is localized within the outer segment of the retina and/or the connecting cilium. This observation is supported by the finding that the C2ORF71 protein has an expression signature similar to that of other proteins, such as rhodopsin, that have preferential expression in the photoreceptors and are localized to the outer segment. Furthermore, analyses of the protein and its interactions are now required for characterization of its specific function within the normal retina.
We have observed C2ORF71 mutations in human RP patients of varying phenotypes, including those of both adult and juvenile onset. Furthermore, the demonstration that loss of function of the zebrafish ortholog leads to early-onset alterations in visual behavior confirms that the protein is likely to play a role in the development of vision. This suggests that a comprehensive screen of C2ORF71 is required in patients not only with adult-onset but also with early-onset severe autosomal-recessive retinal dystrophies (including Leber Congenital Amaurosis) so that the full range of pathogenic mutations in the gene associated with human retinal disease can be examined.
Acknowledgments
We acknowledge the families who provided DNA samples and their consent for inclusion in these studies. The following individuals provided technical support: Gun-Hee Kim, Svetha Swaminathan, Pamela Pretorius, and John Beck. We also acknowledge the following sources of funding: Iowa Cardiovascular Center Institutional Research Fellowship (L.M.B.), Foundation Fighting Blindness (E.M.S. and V.C.S.), the National Institutes of Health grants REY110298 (V.C.S.), REY017168 (V.C.S.), R01CA112369 (D.C.S.), Fight for Sight (G.B. and F.M.), the Manchester Academic Health Sciences Centre (MAHSC), and NIHR Manchester Biomedical Research Centre. V.C.S. and E.M.S. are investigators of the Howard Hughes Medical Institute.
Supplemental Data
Wild-type embryo showing an immediate response to the change in light intensity. show real-time imaging of embryos during the startle response assay. The contrast of the dark frames was modified with Adobe Photoshop Software so that the embryo would be visible.
c2orf71 morphant embryo showing no response to the change in light intensity. A subsequent robust response to the mechanical stimulation with a blunt needle demonstrates embryo motility.
Web Resources
The URLs for data presented herein are as follows:
ENSEMBLE, http://www.ensembl.org/index.html
MYRISTOYLATOR, http://ca.expasy.org/tools/myristoylator/
NMT, http://mendel.imp.univie.ac.at/myristate/SUPLpredictor.htm
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PSORT II, http://psort.ims.u-tokyo.ac.jp/
Retinal Information Network (RetNet), http://www.sph.uth.tmc.edu/retnet/
TERMINATOR, http://www.isv.cnrs-gif.fr/terminator3/index.html
The GeneNetwork, http://www.genenetwork.org
References
- 1.Blackshaw S., Fraioli R.E., Furukawa T., Cepko C.L. Comprehensive analysis of photoreceptor gene expression and the identification of candidate retinal disease genes. Cell. 2001;107:579–589. doi: 10.1016/s0092-8674(01)00574-8. [DOI] [PubMed] [Google Scholar]
- 2.Swiderski R.E., Nishimura D.Y., Mullins R.F., Olvera M.A., Ross J.L., Huang J., Stone E.M., Sheffield V.C. Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest. Ophthalmol. Vis. Sci. 2007;48:3329–3340. doi: 10.1167/iovs.06-1477. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura D.Y., Swiderski R.E., Searby C.C., Berg E.M., Ferguson A.L., Hennekam R., Merin S., Weleber R.G., Biesecker L.G., Stone E.M. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet. 2005;77:1021–1033. doi: 10.1086/498323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demos C., Bandyopadhyay M., Rohrer B. Identification of candidate genes for human retinal degeneration loci using differentially expressed genes from mouse photoreceptor dystrophy models. Mol. Vis. 2008;14:1639–1649. [PMC free article] [PubMed] [Google Scholar]
- 5.Chowers I., Gunatilaka T.L., Farkas R.H., Qian J., Hackam A.S., Duh E., Kageyama M., Wang C., Vora A., Campochiaro P.A. Identification of novel genes preferentially expressed in the retina using a custom human retina cDNA microarray. Invest. Ophthalmol. Vis. Sci. 2003;44:3732–3741. doi: 10.1167/iovs.02-1080. [DOI] [PubMed] [Google Scholar]
- 6.Collins F.S., Brooks L.D., Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura D.Y., Swiderski R.E., Alward W.L., Searby C.C., Patil S.R., Bennet S.R., Kanis A.B., Gastier J.M., Stone E.M., Sheffield V.C. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 1998;19:140–147. doi: 10.1038/493. [DOI] [PubMed] [Google Scholar]
- 9.Westerfield M. University of Oregon Press; Eugene: 1995. The Zebrafish Book. [Google Scholar]
- 10.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 11.Easter S.S., Jr., Nicola G.N. The development of vision in the zebrafish (Danio rerio) Dev. Biol. 1996;180:646–663. doi: 10.1006/dbio.1996.0335. [DOI] [PubMed] [Google Scholar]
- 12.Geisert E.E., Lu L., Freeman-Anderson N.E., Templeton J.P., Nassr M., Wang X., Gu W., Jiao Y., Williams R.W. Gene expression in the mouse eye: An online resource for genetics using 103 strains of mice. Mol. Vis. 2009;15:1730–1763. [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt E.A., Dowling J.E. Early retinal development in the zebrafish, Danio rerio: Light and electron microscopic analyses. J. Comp. Neurol. 1999;404:515–536. [PubMed] [Google Scholar]
- 14.Branchek T. The development of photoreceptors in the zebrafish, brachydanio rerio. II. Function. J. Comp. Neurol. 1984;224:116–122. doi: 10.1002/cne.902240110. [DOI] [PubMed] [Google Scholar]
- 15.Hu M., Easter S.S. Retinal neurogenesis: The formation of the initial central patch of postmitotic cells. Dev. Biol. 1999;207:309–321. doi: 10.1006/dbio.1998.9031. [DOI] [PubMed] [Google Scholar]
- 16.Shen Y.C., Raymond P.A. Zebrafish cone-rod (crx) homeobox gene promotes retinogenesis. Dev. Biol. 2004;269:237–251. doi: 10.1016/j.ydbio.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Shen Y., Rest J.S., Raymond P.A., Zack D.J. Isolation and characterization of a zebrafish homologue of the cone rod homeobox gene. Invest. Ophthalmol. Vis. Sci. 2001;42:481–487. [PubMed] [Google Scholar]
- 18.Horton P., Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997;5:147–152. [PubMed] [Google Scholar]
- 19.Bologna G., Yvon C., Duvaud S., Veuthey A.L. N-Terminal myristoylation predictions by ensembles of neural networks. Proteomics. 2004;4:1626–1632. doi: 10.1002/pmic.200300783. [DOI] [PubMed] [Google Scholar]
- 20.Maurer-Stroh S., Eisenhaber B., Eisenhaber F. N-terminal N-myristoylation of proteins: Prediction of substrate proteins from amino acid sequence. J. Mol. Biol. 2002;317:541–557. doi: 10.1006/jmbi.2002.5426. [DOI] [PubMed] [Google Scholar]
- 21.Martinez A., Traverso J.A., Valot B., Ferro M., Espagne C., Ephritikhine G., Zivy M., Giglione C., Meinnel T. Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics. 2008;8:2809–2831. doi: 10.1002/pmic.200701191. [DOI] [PubMed] [Google Scholar]
- 22.Greaves J., Chamberlain L.H. Palmitoylation-dependent protein sorting. J. Cell Biol. 2007;176:249–254. doi: 10.1083/jcb.200610151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadolski M.J., Linder M.E. Protein lipidation. FEBS J. 2007;274:5202–5210. doi: 10.1111/j.1742-4658.2007.06056.x. [DOI] [PubMed] [Google Scholar]
- 24.Resh M.D. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2006;2:584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 25.Chapple J.P., Hardcastle A.J., Grayson C., Willison K.R., Cheetham M.E. Delineation of the plasma membrane targeting domain of the X-linked retinitis pigmentosa protein RP2. Invest. Ophthalmol. Vis. Sci. 2002;43:2015–2020. [PubMed] [Google Scholar]
- 26.Casey P.J. Protein lipidation in cell signaling. Science. 1995;268:221–225. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 27.Resh M.D. Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods. 2006;40:191–197. doi: 10.1016/j.ymeth.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Draper J.M., Xia Z., Smith C.D. Cellular palmitoylation and trafficking of lipidated peptides. J. Lipid Res. 2007;48:1873–1884. doi: 10.1194/jlr.M700179-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linder M.E., Deschenes R.J. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabandhu K., Herincs Z., Huault S., Dost B., Peng L., Conchonaud F., Marguet D., He H.T., Hueber A.O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 2007;26:209–220. doi: 10.1038/sj.emboj.7601456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feig C., Tchikov V., Schutze S., Peter M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2007;26:221–231. doi: 10.1038/sj.emboj.7601460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurayoshi M., Yamamoto H., Izumi S., Kikuchi A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem. J. 2007;402:515–523. doi: 10.1042/BJ20061476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wild-type embryo showing an immediate response to the change in light intensity. show real-time imaging of embryos during the startle response assay. The contrast of the dark frames was modified with Adobe Photoshop Software so that the embryo would be visible.
c2orf71 morphant embryo showing no response to the change in light intensity. A subsequent robust response to the mechanical stimulation with a blunt needle demonstrates embryo motility.