

Abstract
Tissue-specific alternative splicing is an important mechanism for providing spatiotemporal protein diversity. Here we show that an in-frame splice mutation in BBS8, one of the genes involved in pleiotropic Bardet-Biedl syndrome (BBS), is sufficient to cause nonsyndromic retinitis pigmentosa (RP). A genome-wide scan of a consanguineous RP pedigree mapped the trait to a 5.6 Mb region; subsequent systematic sequencing of candidate transcripts identified a homozygous splice-site mutation in a previously unknown BBS8 exon. The allele segregated with the disorder, was absent from controls, was completely invariant across evolution, and was predicted to lead to the elimination of a 10 amino acid sequence from the protein. Subsequent studies showed the exon to be expressed exclusively in the retina and enriched significantly in the photoreceptor layer. Importantly, we found this exon to represent the major BBS8 mRNA species in the mammalian photoreceptor, suggesting that the encoded 10 amino acids play a pivotal role in the function of BBS8 in this organ. Understanding the role of this additional sequence might therefore inform the mechanism of retinal degeneration in patients with syndromic BBS or other related ciliopathies.
Main Text
Disorders of photoreceptor degeneration contribute a significant burden to human genetic disease and can manifest in syndromic and nonsyndromic forms under a variety of inheritance models.1–4 At present, mutations in excess of 150 genes have been causally linked with retinal degeneration, many of which (35) have been shown to be necessary and sufficient to cause isolated retinitis pigmentosa (RP [MIM 268000]). Despite this remarkable progress, however, some 40% of the genetic burden of RP remains elusive,3 highlighting both the poignant genetic heterogeneity of the disorder and the presence of a likely significant number of private mutations. Gene discovery in RP has also highlighted an important mechanism found in numerous other traits, where ubiquitously expressed genes of apparently broad functionality cause a spatiotemporally restricted phenotype by harboring disease-causing mutations in tissue-specific (alternatively spliced) isoforms.5–7
At the severe end of the phenotypic spectrum, the classical RP phenotype of bone spicule pigmentation in the fundus, initially in the midperiphery and, in advanced stages, in the macula and fovea,8 is also found in a number of syndromic traits, most prominently in Usher syndrome (MIM 276900), Bardet-Biedl syndrome (BBS [MIM 209900]), and other related ciliopathies.9 Recent studies have suggested that some genes that can cause syndromic retinal degeneration can also contribute to the development of isolated disease. For example, an intronic mutation that creates a splice-donor site and inserts a cryptic exon in NPHP6 (MIM 610142), the gene mutated in several ciliopathies including Joubert syndrome (JBTS5 [MIM 610188]), BBS, and Meckel syndrome (MKS4 [MIM 611134]),10–12 is sufficient to cause Leber congenital amaurosis (LCA10 [MIM 611755]), although the mechanism of phenotypic specificity is unclear.13
Here we report a splice-site mutation in a previously unknown exon of BBS8 (MIM 608132), one of the 14 genes that can cause BBS,14 that appears to be necessary and sufficient to cause nonsyndromic recessive RP in a consanguineous Pakistani family. In contrast to most known BBS8 mutations, which are predicted to significantly impact message stability and/or protein function,15 this mutation results in the skipping of 30 bp of an alternatively spliced exon (henceforth termed exon 2a), generating a shorter transcript that lacks 10 amino acids. Tissue-specific real-time PCR amplification suggests that exon 2a is expressed exclusively in the retina, whereas further studies of retinal tissue subsequent to laser capture microdissection indicate that this exon is expressed primarily in photoreceptors. These findings provide a potential mechanistic explanation for the restricted phenotype in the autosomal recessive RP (arRP) family and suggest that the role of this 10 amino acid sequence is critical for the retinal function of BBS8.
As part of a larger study to identify new disease-causing genes for inherited visual diseases, we recruited a large consanguineous Pakistani family PKRP179 with four affected and seven unaffected siblings (Figure 1A). Approval was obtained from the Institutional Review Board, and all participating subjects gave informed written consent consistent with the tenets of the Declaration of Helsinki. PKRP179 is from the southern region of the Punjab province of Pakistan, and detailed medical history was obtained by interviewing all family members. Patients were interviewed regarding age of first diagnosis, initial symptoms, progression, and any other ocular or systemic abnormalities. Ophthalmological examination with indirect dilated funduscopy was performed at Layton Rahmatullah Benevolent Trust Hospital, Lahore, Pakistan. Fundus photographs were acquired with a camera manufactured by Fuji. Electroretinography (ERG) measurements were recorded with equipment manufactured by LKC according to the standards of the International Society for Clinical Electrophysiology. Scotopic responses were recorded under dark-adapted conditions with a single bright flash stimulus, whereas the photopic responses were recorded under light-adapted conditions with a 30 Hz flicker stimulus.
Figure 1.
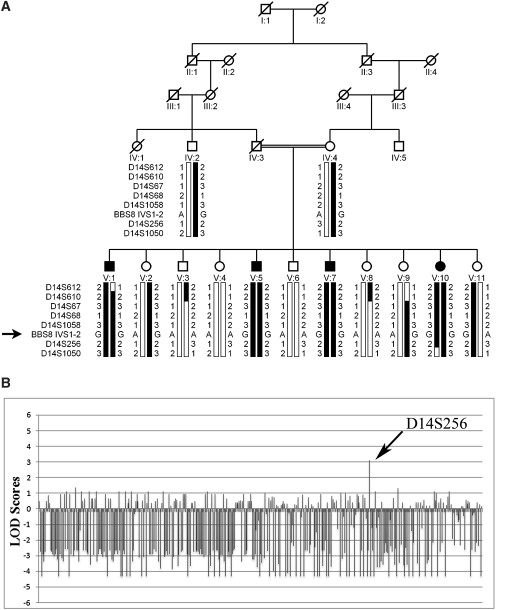
Pedigree Drawing and Two LOD Scores Attained for Family PKRP179 during the Genome-wide Linkage Scan
(A) Haplotypes for seven consecutive chromosome 14q microsatellite markers and the BBS8 splice-site mutation. The risk haplotype is shaded black, whereas alleles not cosegregating with risk haplotype are white.
(B) A graphical illustration of the two-point LOD scores obtained during the genome-wide scan (note: a two-point LOD score of −4.34 corresponds to −∞). We identified significant linkage with a single marker, D14S256, which yielded a two-point LOD score of 3.12 at θ = 0.
All affected individuals examined fulfilled the diagnostic criteria for diagnosis of RP, whereas retrospective analysis of available medical records was suggestive of early-onset RP in all four affected individuals (ages 2–4 at first diagnosis). We could detect no extraocular anomalies that typify syndromic RP. Fundus photographs of affected individuals showed typical changes of RP, including attenuation of retinal arteries and bone spicule pigment deposits in the midperiphery of the retina (Figure 2A). This diagnosis was further confirmed by ERG; all four affected individuals, but none of the unaffected siblings or the unaffected mother, had typical RP changes on ERG, including loss of both rod and cone responses (representative data in Figure 2B).
Figure 2.
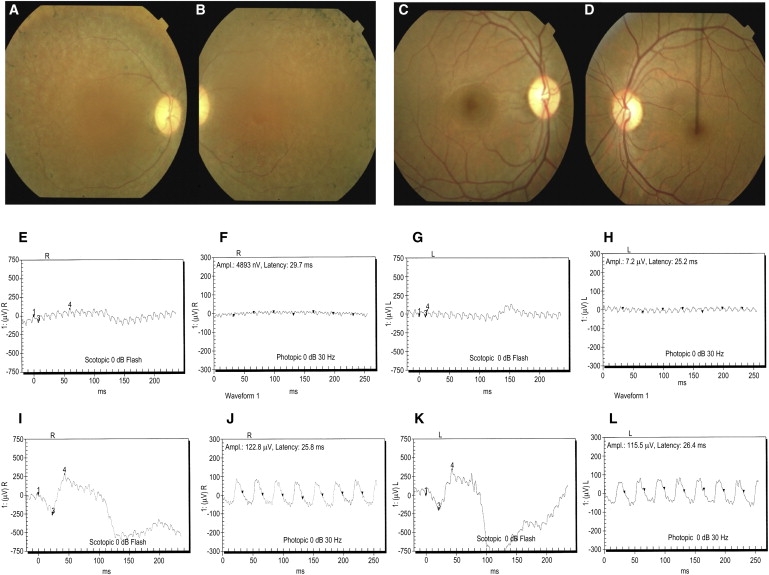
Fundus Photographs and Electroretinography Responses Illustrating the Retinal Phenotype of Family PKRP179
(A–D) Fundus photographs of individual V:5 Oculus Dexter (OD) (A) and Oculus Sinister (OS) (B) and individual V:6 OD (C) and OS (D). Individual V:5 shows several features associated with RP, including a waxy pallor of the optic disc, attenuated arterioles, atrophy of RPE, and peripheral bone spicules, which are absent in unaffected individual V:6.
(E–L) Electroretinography responses of individual V:5 are OD combined rod and cone response (E), OD cone response (F), OS combined rod and cone response (G), and OS cone response (H); and of individual V:6 are OD combined rod and cone response (I), OD cone response (J), OS combined rod and cone response (K) and OS cone response (L). Individual V:5 has typical RP changes, including loss of both rod and cone responses (E–H), whereas ERG readings of unaffected individual V:6 show no changes in the rod and cone response (I–L).
The size of the pedigree suggested sufficient power to achieve genome-wide statistical significance under a fully penetrant autosomal recessive mode of RP transmission (two-point LODmax = 3.44). We therefore conducted a genome-wide scan with 382 polymorphic short tandem repeat (STR) markers spaced equally across the genome. We performed two-point linkage analyses with the FASTLINK version of MLINK from the LINKAGE Program Package16 and calculated maximum LOD scores with ILINK. Autosomal recessive RP was analyzed as a fully penetrant trait with a disease allele frequency of 0.001. The marker order and distances between the markers were obtained from the Marshfield database and the National Center for Biotechnology Information chromosome 14 sequence maps. Equal allele frequencies were assumed for the initial genome scan, whereas for fine mapping, allele frequencies were derived from 96 unrelated and unaffected individuals.
We identified significant linkage with a single marker, D14S256, which yielded a two-point LOD score of 3.12 at θ = 0 (Figure 1B; Table 1). Additional STRs were selected to confirm this finding and to refine the critical interval; we observed two-point LOD scores of 3.05, 3.09, 3.03, and 3.32 at θ = 0 with D14S610, D14S67, D14S68, and D14S1058, respectively.
Table 1.
Two-Point LOD Scores of Family PKRP179
| Marker | cM | Mb | 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | θmax |
|---|---|---|---|---|---|---|---|---|---|---|
| D14S612 | 93.76 | 87.27 | −∞ | −2.57 | −1.15 | −0.67 | −0.59 | −0.17 | 0.08 | 0.40 |
| D14S610 | 95.89 | 88.20 | 3.05 | 2.97 | 2.70 | 2.36 | 1.78 | 1.09 | 3.05 | 0.00 |
| D14S67 | 95.89 | 88.38 | 3.09 | 2.99 | 2.74 | 2.38 | 1.79 | 1.14 | 3.09 | 0.00 |
| D14S68 | 95.89 | 88.62 | 3.03 | 2.96 | 2.71 | 2.38 | 1.71 | 1.04 | 3.03 | 0.00 |
| D14S1058 | 95.89 | 88.64 | 3.32 | 3.25 | 3.06 | 2.68 | 1.90 | 1.30 | 3.32 | 0.00 |
| D14S256∗ | 96.42 | 89.21 | 3.12 | 3.06 | 2.81 | 2.49 | 1.83 | 1.14 | 3.12 | 0.00 |
| D14S1050∗ | 107.13 | 92.91 | −∞ | −2.37 | −1.04 | −0.61 | −0.54 | −0.16 | 0.10 | 0.40 |
STR markers included in the genome-wide scan.
Reconstruction of the haplotypes supports the linkage analysis, localizing the pathogenic locus to chromosome 14q (Figure 1), an area not associated previously with mapped or cloned isolated RP loci. We found recombination events in affected individual V:1 at D14S612, defining the proximal boundary. Similarly, there was distal recombination in unaffected individual V:10 at D14S1050, placing the critical interval in a 13.4 cM (5.6 Mb) region between D14S612 and D14S1050. Notably, alleles for all STRs within the interval were homozygous in all affected individuals, supporting a homozygosity by descent (Figure 1); retrospective examination of genome-wide genotypes showed this region to be the sole genomic segment uniquely homozygous for all four patients, but not for any of their seven unaffected siblings, confirming that we have mapped another arRP locus, RP51.
The critical interval harbors more than 100 annotated genes according to the USCS database. Subsequent to prioritizing genes on the basis of their known function and available expression data in the retina, we systematically started sequencing each candidate in one affected and one unaffected individual. Primer pairs for individual exons in the critical interval were designed with the Primer3 program (sequences and amplification conditions are available upon request). The PCR primers for each exon were used for bidirectional sequencing with BigDye Terminator Ready reaction mix, and sequencing was performed on an ABI PRISM 3100 automated sequencer (Applied Biosystems). Sequencing results were assembled with ABI PRISM sequencing analysis software version 3.7 and analyzed with SeqScape (Applied Biosystems).
The third transcript in our list was BBS8/TTC8, rendered a strong candidate because of its causal involvement in BBS, one of the major hallmarks of which is RP. Analysis of the BBS8 transcript with the UCSC Genome Browser and the expressed sequence tag (EST) databases indicated the possibility that, in addition to the known, previously reported exons, this gene might also contain an additional 30 bp exon adjacent to its canonical second exon. We therefore included this putative expressed fragment in our sequence analysis of the entire transcript and found a homozygous A to G substitution in intron 1: IVS1-2A>G (Figure 3A). The substitution maps to the splice acceptor site of the new exon, labeled exon 2a, and is predicted to result in skipping this exon, causing a 10 amino acid deletion of the protein (Figure 3B). Segregation of this change showed it to be transmitted with the disease phenotype; all affected individuals were homozygous, whereas unaffected individuals were either heterozygous or homozygous for the wild-type allele. Moreover, we did not find this change in 384 Pakistani control chromosomes or 384 chromosomes of northern European descent. Finally, alignment of the genomic sequence of this region with all available genomic sequences in the databases showed the splice acceptor site to be completely invariant in 33 species (Figure 3C), adding further evidence to the pathogenic potential of the mutation.
Figure 3.
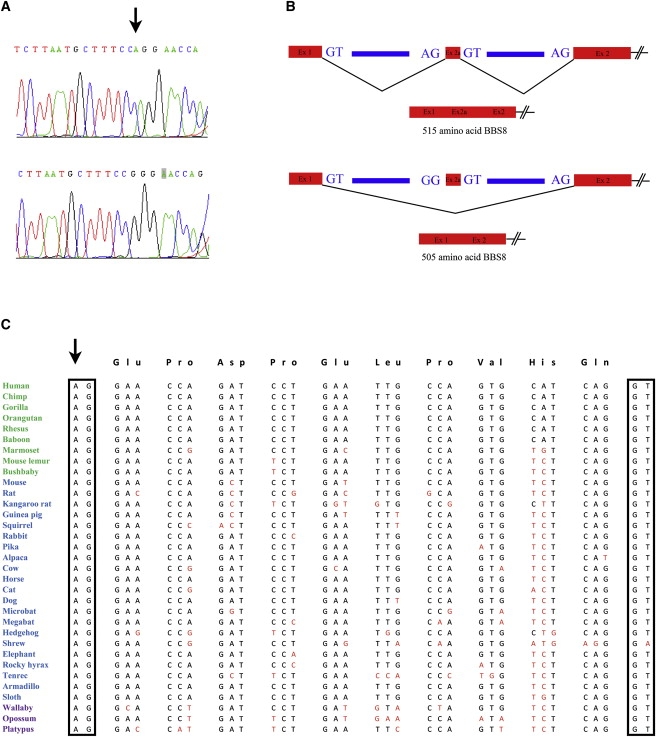
Sequence Chromatogram of the Splice-Site Mutation Identified in Family PKRP179, and the Predicted Effect of this Conserved Splice-Acceptor Site
(A) Sequence chromatograms of a wild-type (top) and homozygote (bottom) for the IVS1-2A>G splice-site mutation.
(B) The predicted effect of the mutation on the splicing of alternative exon 2a. The arrow points to the adenine residue mutated in family PKRP179.
(C) Sequence conservation of the acceptor splice site and of exon 2a in BBS8 orthologs (primates are colored green; placental mammals are blue; vertebrates are purple). The acceptor site is conserved in 33 of 33 species with available genomic sequence for this locus, whereas the donor site at the 3′ end of the exon is conserved in 32 of 33 species. Arrow points to the splice acceptor site mutated in family PKRP179.
These results prompted us to revisit the family for a thorough clinical examination. We found no evidence for syndromic disease (Table 2): none of the four affected individuals had renal problems; their body mass index was in the normal range; there were no signs of polydactyly (or scarring from a possible early excision of extra digits); and we found no evidence of developmental delay, appreciable cognitive impairment, or inappropriate social behavior. Finally, we observed no evidence of midline defects (high arched palates) or facies reminiscent of BBS. Although we were unable to examine for primary or secondary sexual characteristics, the absence of all other features of BBS strongly suggested that this family does not manifest the syndrome.
Table 2.
Summary of the Phenotypic Finding of Affected Individuals in Family PKRP179: Individuals V:1, V:5, V:7, and V:10
| Individual ID | V:1 | V:5 | V:7 | V:10 |
|---|---|---|---|---|
| RP (age at the time of first detection) | 4 yrs | 3 yrs | 2 yrs | 3 yrs |
| Developmental delay | No | No | No | No |
| Tongue morphology | Normal | Normal | Normal | Normal |
| Dental architecture | Normal | Normal | Normal | Normal |
| Learning disability | No | No | No | No |
| Spastic paraplegia | None | None | None | None |
| Kidney defects | None | None | None | None |
| Behavior: speech | Normal | Normal | Normal | Normal |
| Behavior: walk | Normal | Normal | Normal | Normal |
| Polydactyly | None | None | None | None |
| Colobomas | None | None | None | None |
| Type II diabetes | None | None | None | None |
| Deafness | None | None | None | None |
| Weight (lb) | 158 | 113 | 125 | 90 |
| Height (in) | 75 | 68 | 70 | 61 |
| Body mass index | 19.7 | 17.2 | 17.9 | 17.0 |
Note: The weight and height of the affected individuals V:1, V: 5, V:7, and V:10 were recorded at ages 29, 24, 21, and 16, respectively.
Our clinical and genetic findings raised the conundrum of a ubiquitously expressed gene giving rise to an isolated phenotype. One possible explanation is that the splice mutation is an in-frame event, and therefore a hypomorph. However, we and others have found a number of hypomorphic alleles in BBS8 that appear to be genetically sufficient to cause BBS. Therefore, although we could not formally exclude this possibility, we noticed that exon 2a was present in five ESTs present in the human EST database: notably, the originating tissue in all five clones was the eye. Because 26 of 30 bases (and both splice sites) of this exon are conserved in the mouse, we amplified both predicted Bbs8 splice isoforms from a panel of 12 mouse tissues known to express Bbs8 (eye, cortex, cerebellum, heart, liver, lung, kidney, testes, muscle, pancreas, stomach, and thymus). We found the shorter isoform (verified, upon sequencing, to lack exon 2a) in all tissues tested; by contrast, a longer isoform containing the predicted 30 bp exon could only be detected in the eye (data not shown). To verify these data, we sought to perform real-time quantitative RT-PCR (qRT-PCR). We amplified total RNA with the RNeasy micro kit (QIAGEN), and cDNAs were reverse transcribed with superscript III (Invitrogen) with random hexamers. PCR amplification was carried out with 10 ng of cDNA, and all reactions were performed in an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). Reactions consisted of a 3 min denaturation at 95°C followed by 45 cycles of denaturation for 10 s at 95°C and 30 s annealing at 60°C, as well as 30 s elongation at 72°C. Data were analyzed with Bio-Rad iQ5 Standard Edition V 2.1 program. The amount of each splice isoform was normalized against GAPDH (MIM 138400). By using this protocol on a subset of the original tissues tested, we were able to replicate these data (Figures 4A and 4B).
Figure 4.
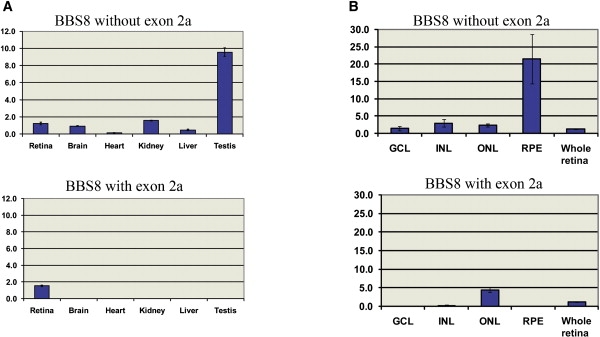
Expression Analysis of the Short and Long BBS8 mRNA Isoforms
Expression levels of the short and long BBS8 mRNA isoforms (with or without exon 2a) in mouse tissue and retinal layers.
(A) The BBS8 mRNA without exon 2 was detected in all tissues tested, whereas exon 2a was only found in the retinas.
(B) Subsequent to laser capture microdissection and qRT-PCR, the BBS8 short message was detectable in all layers tested (GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; and RPE, retinal pigment epithelium), whereas the long isoform was found almost exclusively in the ONL. The y axis shows ratios of each transcript against GAPDH.
The RP phenotype is thought to be driven primarily by photoreceptor degeneration. We therefore interrogated the expression of the Bbs8 long message in different retinal layers by obtaining pertinent tissue through laser-capture microdissection. Briefly, adult C57BL/6 mice were euthanized, and eyes were enucleated. After removing the lens, the eye cups were incubated in calcium/magnesium-free Hanks' balanced salt solution at 37°C for 20 min to facilitate separation of the retina from the pigment epithelium. Eye cups were then immersed in 10%, 15%, and 25% sucrose for 30 min each and were snap frozen in OCT/25% sucrose. The eye cups were sectioned at 7 μm and mounted onto slides with charged PEN-foil membranes (Leica Microsystems). Sections were fixed in 70% ice-cold ethanol for 30 s, rinsed in RNase-free water, stained with hematoxylin for 10 s, and dehydrated in 70% ethanol and 100% ethanol for 1 min each. Retinal pigment epithelium (RPE), outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL) samples were taken independently with an LMD6000 laser capture microdissection microscope (Leica Microsystems).
Using the material obtained from this process, we isolated and purified mRNA from the retinal pigment epithelium, the outer nuclear layer, the inner nuclear layer, and the ganglion cell layer. qRT-PCR from each layer with probes detecting each of the two Bbs8 isoforms showed that, in contrast to the short Bbs8 mRNA species, which was detectable ubiquitously in the retina, the expression of the Bbs8 isoform affected by the patient mutation was almost exclusive to the photoreceptor layer, with a barely detectable message in the INL (Figures 4C and 4D). Taken together, these data suggest that the long BBS8 isoform is likely important to the function of this protein in the photoreceptors and suggest that loss of the ONL enriched, retina-specific exon can explain the isolated RP phenotype in humans.
Here we have reported the mapping of RP51 with the aid of a large, consanguineous Pakistani kindred and demonstrate that a homozygous splice-site mutation affecting the conserved splice-acceptor site of an alternative exon in BBS8, exon 2a, is sufficient to explain the disorder at this locus. Although we cannot exclude the formal possibility that another lesion under the linkage peak can explain the phenotype, sequencing of most visible (on the genomic sequence) exons in the region did not reveal plausible candidate changes. Moreover, the mutant allele affects a base pair conserved completely in all 33 species in which this genomic segment is detectable, segregates with the disorder, and is absent from >1000 controls of various ethnicities (including 384 Pakistani control chromosomes), HapMap, and all available human genomes. Analysis of RP cohorts from Pakistan (n = 192), northern Europe (n = 302), and China (n = 192) did not identify additional lesions in this residue (or elsewhere in exon 2a), consistent with the suggestion that, with the exception of a few major loci, private mutations will constitute the majority of the missing 40% load of arRP.3,4
Detailed clinical examination of this family could exclude extraocular signs of BBS, indicating that, in contrast to all other BBS8 mutations, this allele only affects the function of the BBS8 protein in the photoreceptor. Although we cannot formally exclude the possibility that a separate cis or trans allele might convey protection against the nonretinal BBS phenotypes, that model is difficult to reconcile with the presence of isolated RP in all four affected siblings. Rather, our finding of expression of the affected exon exclusively in the retina, and almost uniquely in the ONL, suggests that the most likely explanation for our data is that failure of inclusion of exon 2a specifically impedes the function of a photoreceptor-specific (or heavily enriched) BBS8 protein isoform. The absence of this isoform from accessible patient tissues naturally precludes the direct study of the mutant message, and, in that context, it might be important to model this allele in the mouse as a means of both confirming our observations and developing a mechanistic model.
Phenotypic restriction in broadly expressed genes driven by tissue-specific alternative splicing is not new. In the context of RP, a retina-specific exon of retinitis pigmentosa GTPase regulator (RPGR [MIM 312610]) harbors pathogenic mutations present in patients with isolated RP.5 Similarly, a frame-shifting mutation in an alternatively spliced exon of COL2A1 (MIM 120140) that is expressed specifically in vitreous collagen mRNA has been implicated in autosomal-dominant rhegmatogenous retinal detachment (MIM 609508), with the patients escaping the broader phenotypes associated with canonical COL2A1 mutations in Stickler syndrome (MIM 108300).6,7 Such examples add to the weight of evidence whereby variation of the mature mRNA message, through alternative splicing and editing, can specify organ and/or cell type pathology.
Computational modeling of the additional 10 amino acid sequence with a variety of protein structure/domain programs did not detect any structural domain; as such, there are no immediate clues as to why the inclusion of exon 2a conveys photoreceptor-specific signaling properties to the protein. Nonetheless, we speculate that an understanding of the additional properties conferred to BBS8 by this extra sequence will inform the pathomechanism of retinal degeneration not only for some forms of arRP but also for BBS and other ciliopathies, which will lead to better therapeutics and treatments.
Acknowledgments
We gratefully acknowledge the family members who donated samples to make this work possible. Financial support was provided by the Ministry of Science and Technology, Islamabad Pakistan, National Institutes of Health grants R01HD04260, R01DK072301, R01DK075972 (N.K.), and R01EY007142 (S.P.D.), the Macular Vision Research Foundation, and the Foundation Fighting Blindness (N.K.). N.K. is the George R. Brumley Professor.
Web Resources
The URLs for data presented herein are as follows:
International HapMap Project, http://www.hapmap.org/
Marshfield Clinic Research Foundation, http://www.marshfieldclinic.org/research/pages/index.aspx
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Primer3 Input (version 0.4.0), http://frodo.wi.mit.edu/primer3
Retinal Information Network (RetNet), http://www.retnet.org
Simple Modular Architecture Research Tool (SMART), http://smart.embl-heidelberg.de/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Inglehearn C.F. Molecular genetics of human retinal dystrophies. Eye (Lond) 1998;12:571–579. doi: 10.1038/eye.1998.147. [DOI] [PubMed] [Google Scholar]
- 2.Hims M.M., Diager S.P., Inglehearn C.F. Retinitis pigmentosa: Genes, proteins and prospects. Dev. Ophthalmol. 2003;37:109–125. doi: 10.1159/000072042. [DOI] [PubMed] [Google Scholar]
- 3.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Daiger S.P., Bowne S.J., Sullivan L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007;125:151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirschner R., Rosenberg T., Schultz-Heienbrok R., Lenzner S., Feil S., Roepman R., Cremers F.P., Ropers H.H., Berger W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Genet. 1999;8:1571–1578. doi: 10.1093/hmg/8.8.1571. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S.K., Leonard B.C., Damji K.F., Bulman D.E. A frame shift mutation in a tissue-specific alternatively spliced exon of collagen 2A1 in Wagner's vitreoretinal degeneration. Am. J. Ophthalmol. 2002;133:203–210. doi: 10.1016/s0002-9394(01)01339-3. [DOI] [PubMed] [Google Scholar]
- 7.McAlinden A., Majava M., Bishop P.N., Perveen R., Black G.C., Pierpont M.E., Ala-Kokko L., Männikkö M. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum. Mutat. 2008;29:83–90. doi: 10.1002/humu.20603. [DOI] [PubMed] [Google Scholar]
- 8.Heckenlively J.R., Yoser S.L., Friedman L.H., Oversier J.J. Clinical findings and common symptoms in retinitis pigmentosa. Am. J. Ophthalmol. 1988;105:504–511. doi: 10.1016/0002-9394(88)90242-5. [DOI] [PubMed] [Google Scholar]
- 9.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 10.Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 11.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Al-Fadhel M., Lewis R.A., Eyaid W. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 12.Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ansley S.J., Badano J.L., Blacque O.E., Hill J., Hoskins B.E., Leitch C.C., Kim J.C., Ross A.J., Eichers E.R., Teslovich T.M. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 16.Lathrop G.M., Lalouel J.M. Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 1984;36:460–465. [PMC free article] [PubMed] [Google Scholar]