

Abstract
We present an autosomal-recessive frontonasal dysplasia (FND) characterized by bilateral extreme microphthalmia, bilateral oblique facial cleft, complete cleft palate, hypertelorism, wide nasal bridge with hypoplasia of the ala nasi, and low-set, posteriorly rotated ears in two distinct families. Using Affymetrix 250K SNP array genotyping and homozygosity mapping, we mapped this clinical entity to chromosome 12q21. In one of the families, three siblings were affected, and CNV analysis of the critical region showed a homozygous 3.7 Mb deletion containing the ALX1 (CART1) gene, which encodes the aristaless-like homeobox 1 transcription factor. In the second family we identified a homozygous donor-splice-site mutation (c.531+1G > A) in the ALX1 gene, providing evidence that complete loss of function of ALX1 protein causes severe disruption of early craniofacial development. Unlike loss of its murine ortholog, loss of human ALX1 does not result in neural-tube defects; however, it does severely affect the orchestrated fusion between frontonasal, nasomedial, nasolateral, and maxillary processes during early-stage embryogenesis. This study further expands the spectrum of the recently recognized autosomal-recessive ALX-related FND phenotype in humans.
Main Text
Frontonasal dysplasia (FND [MIM 136760]), or median facial cleft, is a remarkable outcome of developmental failure of the facial prominences surrounding the primitive mouth. Incomplete migration of the orbits into their proper positions results in various degrees of hypertelorism and causes a spectrum of malformations. Hypertelorism is frequently associated with cranial, cerebral, limb, and other abnormalities. Facial findings range from mild hypertelorism to severe midline clefts of the nose, lip, palate, and forehead. The nosology of this group of malformations remains unclear. Differentiation of the phenotypes of this spectrum is usually based on the presence of associated organ involvement rather than on overlapping facial findings.1–9 Autosomal-recessive frontofacionasal dysplasia (FFND [MIM 229400]), reported and named by Gollop, is the most severe form of frontonasal dysplasia.10 Cardinal clinical characteristics are midline facial defects, telecanthus, and eyelid malformation;10 however, extreme variability in the severity of abnormalities and associated findings, including anophthalmia and severe bilateral clefting, has been described.11
Genetic aspects of FND are not well defined. The mode of inheritance is unclear in the majority of cases presented in the literature. Until recently, mutation in the gene encoding the ephrin receptor tyrosine kinase 1 (EFNB1 [MIM 300035]), responsible for X-linked craniofrontonasal syndrome (CFNS [MIM 304110]), was the only known molecular cause.12,13 Two recent studies have drawn attention to the critical role of aristaless-related homeobox transcription factors (ALX3 [MIM 606014]) and (ALX4 [MIM 605420]) during craniofacial development, particularly in the molecular pathogenesis of frontonasal dysplasia in humans.14,15 Aristaless-like homeobox genes Alx1 (Cart1), Alx3, and Alx4 encode paired-type homeodomain proteins, which are thought to play an important role in the development of structures derived from craniofacial mesenchyme, the first branchial arch, and the limb bud.16 The fourth member of the vertebrate Alx gene family, which should be named as “Alx2,” has not been reported (P. Holland and T. Takahashi, personal communication).17 Although both heterozygous and homozygous Alx3 knockout mice do not show an obvious phenotype, homozygous ALX3 mutations in humans cause a distinctive presentation of frontonasal dysplasia, namely frontorhiny (FND [MIM 136760]).14 A large case-control study of facial clefts further suggested that common variants in ALX3 might have a role in isolated clefting.18 We recently demonstrated that homozygous mutations in the ALX4 gene cause a new frontonasal dysplasia phenotype associated with alopecia and genital abnormalities.15 Heterozygous ALX4 mutations have been described in patients with parietal foramina of the skull (FPM [MIM 168500]), without facial involvement. The nasal configurations of recessive ALX3- and ALX4-related frontonasal dysplasias are strikingly similar, despite the diversity of associated findings. Human mutations in aristaless-like homeobox gene 1 (CART1) (ALX1 [MIM 601527]) have not yet been reported.
We identified a Turkish patient who had severe frontofacionasal dysplasia and was born to consanguineous parents (family 1) (Figures 1A–1H). The affected male patient is currently 11 years old. The craniofacial features noted at birth included hypertelorism, bilateral extreme microphtalmia, upper eyelid colobomata, sparse eyelashes, absence of eyebrows, wide nasal base, hypoplasia of the ala nasi, bilateral nonmidline cleft lip with a prominent glabella, complete cleft palate, and low-set posteriorly rotated ears (Figure 1A, individual P1). The only extracranial finding was the presence of a caudal appendage in the sacral region (Figure 1B). Mild dilatation of the collecting system was observed with renal ultrasound. Orbital and brain MRI showed brachycephaly, bilateral extreme microphtalmia, and a large midline bone defect of the cranium (Figures 1G–1H). Karyotype analysis was normal. At present, the patient has mild mental retardation, necessitating special education. He follows commands very well, and his hearing is normal; however, his facial anatomy currently prevents sufficient expressive speech, and he is only able to say mama. Motor developmental milestones were surprisingly normal; he began to walk at 12 months. The mother had two consecutive pregnancies with affected fetuses that had an identical phenotype and were diagnosed prenatally (Figure 1, fetuses P2 and P3). The second pregnancy was terminated at the 25th week of gestation after prenatal ultrasound detected the craniofacial phenotype. Fetal autopsy revealed identical craniofacial findings, including bilateral extreme microphtalmia, bilateral nonmidline cleft lip, prominent glabella, and complete cleft palate (Figure 1C). A caudal appendage was also present in the sacral region, but there was no vertebral defect (Figure 1D). Examination of the central nervous system showed asymmetric optic nerves, agenesis of the corpus callosum, and mild cerebellar hypoplasia. Intra-abdominal organs were morphologically normal. Histological examination detected small cystic dilatations in the renal collecting system. The third pregnancy was terminated at the 23rd week of gestation (Figure 1, fetus P3). Fetal autopsy revealed similar craniofacial findings (Figures 1E–1F). Examination of the central nervous system showed corpus callosum agenesis, bilateral hypoplastic optic nerves, and neuronal heterotopia of the pons. Histological evaluation of the renal system revealed a significant delay in maturation, despite macroscopically normal kidneys. The study protocol was approved by the Hacettepe University Medical Faculty Ethics Committee (TBK 09/4-42). DNA samples were obtained after written consent was secured from the parents.
Figure 1.
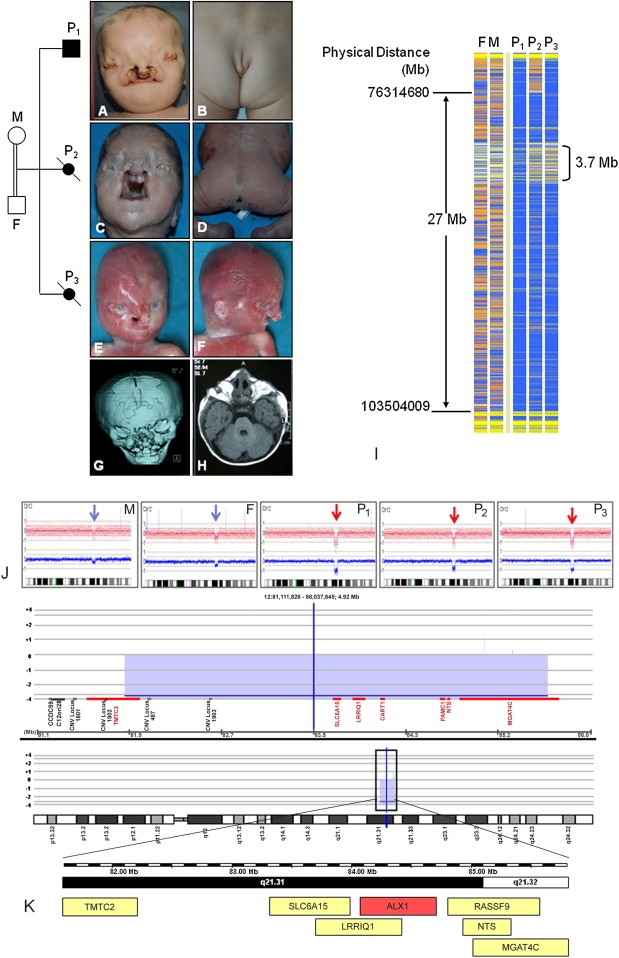
Clinical Presentation and Molecular Findings of Family 1
Pedigree of Family 1
(A–F) Identical facial appearance and caudal appendage in affected siblings.
(G–H) Brachycephaly, bilateral extreme microphtalmia, and a large midline bone defect of the cranium.
(I) Schematic representation of homozygosity data of the chromosome 12q21.3 region. Genotype files (CHP files) were generated with Affymetrix GTYPE software and were transferred to the VIGENOS (Visual Genome Studio) program (Hemosoft, Ankara), which facilitates visualization of a large amount of genome data in comprehensible visual screens.15 Homozygous genotypes identical to the genotype data obtained from the index case (affected individual P1, family 1) are shown in blue. Contrasting homozygote genotypes are shown in white, whereas heterozygous SNPs appear in orange. Noninformativeness as a result of heterozygous genotypes in parent-child trios is indicated in yellow. Gray: no call; F: father; M: mother; P1-3: patients 1–3. The overlapping homozygous stretches of three patients are approximately 27 Mb in size. The Mendelian segregation error spanned a 3.7 Mb region (between 81.924.095 and 85.661.618 bases) and is marked in brackets.
(J) Diagrams obtained with CNAG software (blue and red arrows show single- and double-copy deletions observed in parents and affected siblings, respectively).
(K) Results of array-CGH analysis of chromosome 12 obtained from the Agilent Platform (Agilent Technologies, Santa Clara, CA, USA). The raw data (tiff images) were extracted with Feature Extraction software v.10.5.1.1. (Agilent Technologies, Santa Clara, CA, USA) and were analyzed with CGH Analytics software v.3.5.14 (Agilent Technologies, Santa Clara, CA, USA). In the chromosome view the region of interest showing a copy-number change on 12q21.31 is indicated above the ideogram by a black rectangle. The observed deletion spanning 81.9–85.6 Mb is marked by the blue block. Seven genes (TMTC2, SLC6A15, LRRIQ1, CART1 (= ALX1), PAMCI, NTS, and MGAT4C) are located within this 3.7 Mb region.
Genome-wide homozygosity mapping of both parents and three affected siblings was carried out with GeneChip mapping 250K SNP arrays from Affymetrix and revealed a single large homozygous stretch spanning approximately 27 Mb on chromosome 12q21.3 (Figure 1I). Interestingly, a Mendelian segregation error spanning between 81.9 and 85.6 Mb (approximaely 3.7 Mb) was detected within this homozygous stretch (Figure 1I). Analysis of the SNP array data by CNAG (Copy number analyzer for GeneChip v. 2.0, updated for the analysis of 500K SNP arrays19) showed a homozygous deletion of the entire region in all affected offspring (Figure 1J). The healthy parents were heterozygous carriers of this deletion. Cranial CT scans of both parents did not detect any abnormalities. The chromosomal imbalance was further confirmed by an independent method involving high-resolution Agilent array CGH (microarray-based comparative genomic hybridization) (Figure 1K). An array-CGH experiment was performed with the Human Genome Microarray Kit 105A (Agilent Technologies, Santa Clara, CA, USA). Seven genes (TMTC2, SLC6A15 [MIM 607971]), LRRIQ1, ALX1 (CART1), PAMCI [MIM 610383], NTS [MIM 162650], and MGAT4C [MIM 607385]) were located in the deleted region. The human ortholog of mouse aristaless-like homeobox 1 gene, ALX1 (CART1), was a highly relevant candidate gene potentially responsible for the craniofacial phenotype; homozygous Alx1 knockout mice exhibit large cranial defects and a lack of brain tissue.20,21
We expanded our study to include another Turkish patient with an almost identical phenotype from a family unrelated to that of the first patient. The affected girl was the first child of a nonconsanguineous couple with a negative family history. However, both parents originated from nearby villages in the same city, which suggested a recessive etiology. Her primary manifestations were hypertelorism, extreme microphtalmia on the right side, microphthalmia of the left eye, upper eyelid colobomata, lack of eyelashes and eyebrows, and palpable midline cranial cleft with a soft tissue mass in the left frontal area (Figure 2A). A wide nasal bridge with hypoplasia of the ala nasi, bilateral nonmidline cleft lip, very prominent glabella, complete cleft palate, and low-set posteriorly rotated ears were noted. Brain MRI at 4 months of age showed severely delayed myelinization, a thin corpus callosum, and asymmetric cerebellar vermis configuration on the left side. Three-dimensional cranial CT imaging (Figures 2B and 2C) demonstrated cranium bifidum and facial cleft, right occipital plagiocephaly, and right extreme microphthalmia. The left bulbus oculi was normal in size; however, both eyes had millimetric cystic lesions in the anterior chamber and microcalcification of the sclerae. The soft tissue mass in the left frontal area suggested a dermoid cyst or a fibroma. Abdominal, renal ultrasound, and transthoracic echocardiography were normal. A peripheral blood karyotype was normal. Motor development was delayed. She underwent reconstructive surgery but died in infancy because of a pulmonary infection.
Figure 2.
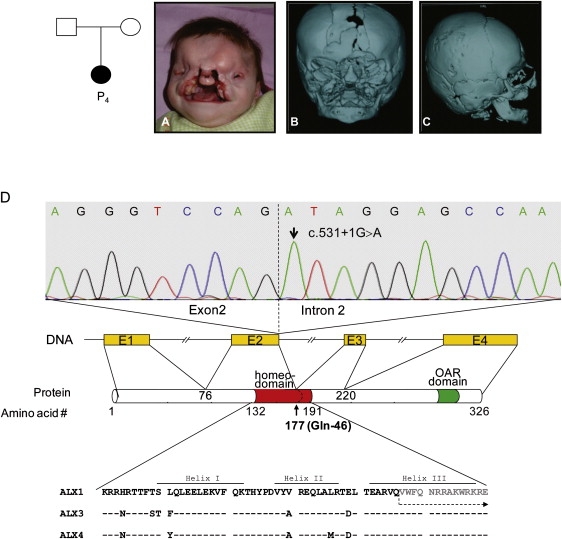
Clinical and Molecular Findings of Family 2
(A–C) Pedigree and clinical and radiological presentation of the patient in family 2.
(D) Identification of the homozygous donor splice-site mutation. The upper panel shows the sequence chromatogram of the index patient carrying the homozygous c.531+1G > A mutation. The dotted line represents the border between exon 2 and intron 2 of the ALX1 gene in humans. The position of domains and motifs of human ALX1 was obtained from the Uniprot database (Uniprot accession number Q15699). The amino acid sequence alignment of the homeodomains of ALX family proteins are shown in the lower panel. At the protein level, the splice site mutation targets the glutamine residue at position 177 (marked in bold) in helix III of the ALX1 homeodomain. The residues 76–177 containing a gross part of the homeodomain are encoded by exon 2. The amino acid sequence following the splice site mutation is shown in gray and is marked with a dotted arrow. The highly conserved homeodomain as well as the subsequent C-terminal parts of the protein will be affected by the splice-site mutation through different mechanisms.
In addition to other homozygous stretches throughout the genome, 250K Affymetrix SNP array analysis of the affected child identified homozygosity at the ALX1 region on chromosome 12q21. Analysis of 250K array data indicated that the affected child inherited regions identical by descent in approximately 5% (1/20) of her genome through both parents, which supports consanguinity between parents. CNV analysis did not show any structural chromosomal anomalies. Considering the relatively low-resolution profile of the 250K array, we have performed a higher-resolution analysis by using an Affymetrix 6.0 SNP array; nevertheless, we did not detect any significant chromosomal imbalance throughout the genome, including the chromosome 12q21 region. Sequencing of ALX1 (Ensembl accession number ENST00000316824) showed a homozygous G > A substitution at the invariant +1 position of the donor splice site of intron 2, c.531+1G > A (Figure 2D). The homozygous mutation was confirmed by an independent method—restriction-digestion analysis with Hpy188III. The parents were heterozygous carriers, and the mutation was not detected in 171 healthy Turkish controls. The c.531+1G > A mutation is predicted to disrupt the donor site of ALX1 intron 2. RNA of the patient was not available for further investigation of aberrant ALX1 transcripts caused by the splice-site mutation. Therefore, we can only speculate that the disruption of the donor site could lead to complete skipping of exon 2, encoding the majority of the homeodomain; complete intron retention, causing frameshift and premature truncation of the protein; or activation of a cryptic splice site with an unknown effect on the protein. In any case, there is a likely structural change of the mutant protein near the glutamine residue at position 177 (Glu46) within the DNA recognition helix (helix III) of the ALX1 homeodomain (Figure 2D). The recognition helix is known to be crucial for optimal DNA interactions with the major groove of the target DNA.22 Thus, alterations or complete deletion of this region most likely disrupts the DNA binding ability of the protein. The molecular findings in this patient further support the idea that likely loss of ALX1 function is the underlying pathogenic mechanism for the new FND phenotype presented in this study.
Alx1 (Cart1) plays a crucial role in the early phase of chondrocyte development.23 Its expression is restricted to forebrain mesenchyme, branchial arches, limb buds, and cartilage.20,21 Zhao et al. reported that mice homozygous for Alx1 null mutations have neural tube closure defects that lead to anencephaly and acrania.21 Nasal cartilage was also severely affected, in addition to other facial structures. The cranial neural-tube defect observed in Alx1 mutant mice was prevented by prenatal administration of folic acid. Consequently, the authors suggested that ALX1 is a candidate gene leading to defects in neural-tube closure in humans.21
Neither cranial nor vertebral neural-tube closure defects (such as spina bifida and/or anencephaly) were observed in our affected patients. Although the caudal appendage could be a sign of a hidden neural-tube closure defect,24 it was considered a vestigial human tail because there were no bony structures inside the appendage and an autopsy showed no evidence of spinal-cord anomalies. The human embryo has a tail until the 6th–7th week of gestation.25 This tail regresses, and caudal vertebrae fuse to form the coccyx at the 7th–8th week of embryogenesis. The nonvertebral apex remains temporarily and eventually disappears. All true vestigial human tails reported in the literature lack bone and cartilage and do not represent abnormalities of the spinal cord.26 As such, we propose that, unlike Alx1-homozygous mutant mice, disruption of ALX1 in humans does not lead to defects in neural-tube closure.
The literature contains descriptions of at least 16 syndromes characterized by microphthalmia associated with cleft lip and/or cleft palate.27 Because of the severity of its expression and its recessive mode of inheritance, the ALX1-related phenotype can be considered to be borderline between autosomal-recessive frontofacionasal dysplasia (FFND [MIM 229400]) and Fryns microphthalmia syndrome (anophthalmia plus syndrome [MIM 600776]). Anophthalmia plus syndrome was first reported in a pair of siblings with bilateral anophthalmia, facial clefting, microtia, large sacral neural-tube defects, and uterus unicornis.28 Additional associated findings, such as primary congenital hypothyroidism (CHNG1 [MIM 275200] and CHNG2 [MIM 218700]) and adrenal hypoplasia congenita (AHC [MIM 300200]), were subsequently included in the anophthalmia plus spectrum.29,30 Absence of the above listed anomalies, as well as the absence of neural-tube defects in our cases, clinically distinguishes the presented phenotype from anophthalmia plus syndrome and suggests a previously unrecognized phenotype, namely ALX1-related FND, within the FFND malformation spectrum.
The new spectrum of ALX-related frontonasal dysplasias (ALX-related FNDs) that involve loss-of-function mutations in the ALX1, ALX3, and ALX4 genes provides insight into the molecular basis of recessively inherited complex frontonasal dysplasias. Figure 3 illustrates various stages of facial development in relation to ALX-related FND phenotypes. The human face originates from several primordia in the 4th week of embryonic development.31 These primordia consist of a single frontonasal prominence and paired nasomedial and nasolateral processes—components of the nasal (olfactory) primordia and paired maxillary and mandibular prominences—both of which are components of the first branchial arch.31 Morphological evaluation of homozygous ALX3 and ALX4 loss-of-function mutant phenotypes revealed embryonic disruption of the fusion of the frontal and medial nasal prominences, which eventually led to bifid nose and hypertelorism (Figure 3). A long philtrum and prominent philtral ridges are characteristic of ALX3-related FND (also known as frontorhiny), but not of the ALX4-related FND facial phenotype.14,15 ALX4-related FND represents a more severe manifestation, with remarkable hypertelorism, additional eye involvement, blepharophimosis, and total alopecia. Palate development seems to be normal in both ALX3- and ALX4-related FNDs. Although facial appearances in both ALX3- and ALX4-related FNDs resemble those seen at approximately the tenth week of human embryonic development, studies on Alx3/Alx4 double mutant mice demonstrated that apoptosis in the frontonasal area could start much earlier than this, i.e., at E10.0, which is equivalent to the 28th day in human embryonic development.32 No palatal or tongue involvement was observed in Alx3/Alx4 double mutant mice.32
Figure 3.
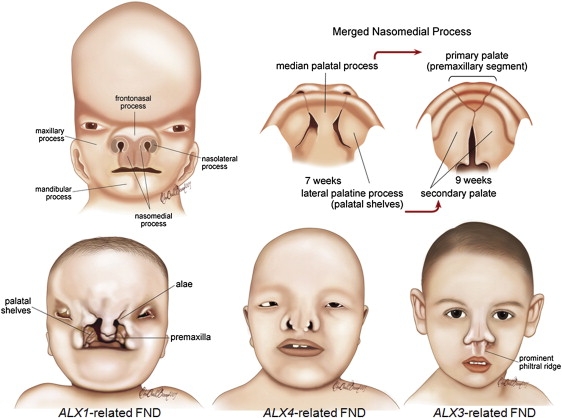
Morphological Comparison of ALX-Related FNDs
Facial and palate development are illustrated in the upper part of the figure. Regions of the human face that originate from various prominences and palate development in two different stages are shown on the upper left and right, respectively. Palate development occurs between the sixth and seventh week of embryonic development and originates from the median and paired lateral palatine processes (upper right).31 During facial and palate development, the frontonasal prominence contributes to the formation of the bridge of the nose; the lateral processes form the sides of the nose (alae). The merged nasomedial process is crucial to both nasal and palatal development; it forms the intermaxillary segment, which is a precursor of the philtrum of the lip, the pre-maxillary component of the upper jaw, and the primary palate. ALX1 expression is essential not only for building the oral and nasal cavities but also for proper eye development during early embryogenesis. It is not possible to compensate for complete loss of ALX1 function, and frontonasal and palatal development is likely to be disrupted in early stages of development. ALX4 and ALX3 expression are related more to regulating the formation of the final shape of the nose. The ALX4-related FND phenotype is more pronounced than that of ALX3-related FND in terms of the severity of hypertelorism and eye involvement. Alopecia is also associated only with ALX4-related FND. A prominent philtral ridge is characteristic of ALX3-related FNDs (frontorhiny).
Obviously, the ALX1-related FND phenotype presents the extreme end of developmental disruption and clinical severity among the recessive ALX-related FNDs. Normal development of structures originating from the frontal and nasomedial prominences, such as the bridge of the nose and the premaxillary component of the upper jaw, respectively, is observed; however, bilateral oblique clefts between premature medial and lateral structures indicate disruption of the fusion of these structures, namely nasomedial and nasolateral prominences. The absence of an upper lip and macrostomia indicates that the nasomedial and maxillary processes failed to fuse. If the medial and lateral palatine processes do not fuse, it can result in complete cleft palate. A lack of fusion of the apices of the lateral palatine process (palatal shelves) in the midline suggests that embryonic development might be disrupted before the seventh week of gestation. However, studies on homozygous Alx1(Cart1) mutant mice indicated that disruption in facial development could be placed even earlier.21 Histological defects in head mesenchyme are first observed at E9.0 (equivalent to day 24 in human development) in Alx1(Cart1) mutant embyos.21 Alx1(Cart1) expression in palate mesenchyme is clearly shown during rat embryogenesis, although a potential role of this gene in palate differentiation is still not well understood.20 Mandibular processes originating from the first branchial arch are not affected in the ALX-related FND spectrum.
In conclusion, the present study expands the spectrum of the newly recognized autosomal-recessive ALX-related FND phenotypes. The craniofacial findings in our patients with ALX1-related FND indicate that ALX1 plays an essential role in the orchestrated fusion of frontonasal, nasomedial, nasolateral, and maxillary processes during early-phase embryogenesis. Future research will probably unravel the interaction or cascade of events that occur between different ALX genes.
Acknowledgments
We are grateful to the families for their participation in the study. We thank Han Brunner for critical reading and comments, Ebru Oralli Bircan for illustrations, Hacettepe University Craniofacial Surgery Study Group members Yucel Erk, Emin Mavili, Aycan Kayikcioglu (Plastic and Reconstructive Surgery), Kemal Benli (Neurosurgery), Aysenur Cila (Radiology), Tulin Taner, and Ilken Kocadereli (Orthodonty) for evaluating the frontonasal malformation cases in the registry. This work was supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK) (grant numbers 108S420 to N.A.A), and the overall consortium (CRANIRARE) was supported by the European Research Area Network (E-RARE) (project number R07197KS).
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Ensembl, http://www.ensembl.org
References
- 1.Sedano H.O., Cohen M.M., Jr., Jirasek J., Gorlin R.J. Frontonasal dysplasia. J. Pediatr. 1970;76:906–913. doi: 10.1016/s0022-3476(70)80374-2. [DOI] [PubMed] [Google Scholar]
- 2.Sedano H.O., Gorlin R.J. Frontonasal malformation as a field defect and in syndromic associations. Oral Surg. Oral Med. Oral Pathol. 1988;65:704–710. doi: 10.1016/0030-4220(88)90014-x. [DOI] [PubMed] [Google Scholar]
- 3.Hing A.V., Syed N., Cunningham M.L. Familial acromelic frontonasal dysostosis, autosomal dominant inheritance with reduced penetrance. Am. J. Med. Genet. A. 2004;128A:374–382. doi: 10.1002/ajmg.a.30091. [DOI] [PubMed] [Google Scholar]
- 4.Guion-Almeida M.L., Richieri-Costa A. Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation and CNS structural anomalies: Defining the phenotype. Clin. Dysmorphol. 2001;10:81–86. doi: 10.1097/00019605-200104000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Toriello H.V., Radecki L.L., Sharda J., Looyenga D., Mann R. Frontonasal “dysplasia,” cerebral anomalies, and polydactyly, report of a new syndrome and discussion from a developmental field perspective. Am. J. Med. Genet. Suppl. 1986;2:89–96. doi: 10.1002/ajmg.1320250613. [DOI] [PubMed] [Google Scholar]
- 6.Toriello H.V., Higgins J.V., Mann R. Oculoauriculofrontonasal syndrome: Report of another case and review of differential diagnosis. Clin. Dysmorphol. 1987;4:338–346. doi: 10.1097/00019605-199510000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Pai G.S., Levkoff A.H., Leithiser R.E., Jr. Median cleft of the upper lip associated with lipomas of the central nervous system and cutaneous polyps. Am. J. Med. Genet. 1987;26:921–924. doi: 10.1002/ajmg.1320260421. [DOI] [PubMed] [Google Scholar]
- 8.Lees M.M., Hodgkins P., Reardon W., Taylor D., Stanhope R., Jones B., Hayward R., Hockley A.D., Baraitser M., Winter R.M. Frontonasal dysplasia with optic disc anomalies and other midline craniofacial defects, a report of six cases. Clin. Dysmorphol. 1998;7:157–162. doi: 10.1097/00019605-199807000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Masuno M., Imaizumi K., Aida N., Nishimura G., Kimura J., Kuroki Y. Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation, and CNS structural anomalies, another observation. Clin. Dysmorphol. 2000;9:59–60. doi: 10.1097/00019605-200009010-00012. [DOI] [PubMed] [Google Scholar]
- 10.Gollop T.R., Kiota M.M., Martins R.M.M., Lucchesi E.A., Alvarenga E. Frontofacionasal dysplasia: Evidence for autosomal recessive inheritance. Am. J. Med. Genet. 1984;19:301–305. doi: 10.1002/ajmg.1320190212. [DOI] [PubMed] [Google Scholar]
- 11.Reardon W., Hockey A., Silberstein P., Kendall B., Farag T.I., Swash M., Stevenson R., Baraitser M. Autosomal recessive congenital intrauterine infection-like syndrome of microcephaly, intracranial calcification, and CNS disease. Am. J. Med. Genet. 1994;52:58–65. doi: 10.1002/ajmg.1320520112. [DOI] [PubMed] [Google Scholar]
- 12.Twigg S.R.F., Kan R., Babbs C., Bochukova E.G., Robertson S.P., Wall S.A., Morriss-Kay G.M., Wilkie A.O.M. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. USA. 2004;101:8652–8657. doi: 10.1073/pnas.0402819101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieland I., Jakubiczka S., Muschke P., Cohen M., Thiele H., Gerlach K.L., Adams R.H., Wieacker P. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am. J. Hum. Genet. 2004;74:1209–1215. doi: 10.1086/421532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Twigg S.R.F., Versnel S.L., Nürnberg G., Lees M.M., Bhat M., Hammond P., Hennekam R.C.M., Hoogeboom A.J.M., Hurst J.A., Johnson D. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am. J. Hum. Genet. 2009;84:698–705. doi: 10.1016/j.ajhg.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayserili H., Uz E., Niessen C., Vargel I., Alanay Y., Tuncbilek G., Yigit G., Uyguner O., Candan S., Okur H. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum. Mol. Genet. 2009;18:4357–4366. doi: 10.1093/hmg/ddp391. [DOI] [PubMed] [Google Scholar]
- 16.Meijlink F., Beverdam A., Brouwer A., Oosterveen T.C., Berge D.T. Vertebrate aristaless-related genes. Int. J. Dev. Biol. 1999;43:651–663. [PubMed] [Google Scholar]
- 17.Holland P.W., Booth H.A,F., Bruford E.A. Classification and nomenclature of al human homeobox genes. BMC Biol. 2007;5:47. doi: 10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jugessur A., Shi M., Gjessing H.K., Lie R.T., Wilcox A.J., Weinberg C.R., Christensen K., Boyles A.L., Daack-Hirsch S., Trung T.N. Genetic determinants of facial clefting: Analysis of 357 candidate genes using two national cleft studies from Scandinavia. PLoS ONE. 2009;4:e5385. doi: 10.1371/journal.pone.0005385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 20.Zhao G.Q., Eberspaecher H., Seldin M.F., de Crombrugghe B. The gene for the homeodomain-containing protein Cart-1 is expressed in cells that have a chondrogenic potential during embryonic development. Mech. Dev. 1994;48:245–254. doi: 10.1016/0925-4773(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Q., Behringer R.R., de Crombrugghe B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat. Genet. 1996;13:275–283. doi: 10.1038/ng0796-275. [DOI] [PubMed] [Google Scholar]
- 22.Chi Y.I. Homeodomain revisited: A lesson from disease-causing mutations. Hum. Genet. 2005;116:433–444. doi: 10.1007/s00439-004-1252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao G.Q., Zhou X., Eberspaecher H., Solursh M., de Crombrugghe B. Cartilage homeoprotein1, a homeoprotein selectively expressed in chondrocytes. Proc. Natl. Acad. Sci. USA. 1993;90:8633–8637. doi: 10.1073/pnas.90.18.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman B.A. Neural tube defects. Pediatr. Clin. North Am. 2004;51:389–419. doi: 10.1016/S0031-3955(03)00207-4. [DOI] [PubMed] [Google Scholar]
- 25.Spiegelmann R., Schinder E., Mintz M., Blakstein A. The human tail: A benign stigma. Case report. J. Neurosurg. 1985;63:461–462. doi: 10.3171/jns.1985.63.3.0461. [DOI] [PubMed] [Google Scholar]
- 26.Dao A.H., Netsky M.G. Human tails and pseudotails. Hum. Pathol. 1984;15:449–453. doi: 10.1016/s0046-8177(84)80079-9. [DOI] [PubMed] [Google Scholar]
- 27.Warburg M., Jensen H., Prause J.U., Bolund S., Skovby F., Miranda M.J. Anophthalmia-microphthalmia-oblique clefting syndrome: Confirmation of the Fryns anophthalmia syndrome. Am. J. Med. Genet. 1997;73:36–40. doi: 10.1002/(sici)1096-8628(19971128)73:1<36::aid-ajmg8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 28.Fryns J.P., Legius E., Moerman P., Vandenberghe K., Van den Berghe H. Apparently new ‘anophthalmia-plus’ syndrome in sibs. Am. J. Med. Genet. 1995;58:113–114. doi: 10.1002/ajmg.1320580204. [DOI] [PubMed] [Google Scholar]
- 29.Akalin I., Senses D.A., Ilgin-Ruhi H., Misirlioglu E., Yalciner M., Cetinkaya E., Fryns J.P., Tukun A. A novel Fryns ‘anophthalmia-plus’ syndrome associated with primary hypothyroidism. Genet. Couns. 2005;16:145–148. [PubMed] [Google Scholar]
- 30.Ozalp O., Ozcimen E.E., Yilmaz Z., Yanik F., Sahin F.I. Fryns ‘anophthalmia-plus’ syndrome with hypoplastic adrenal glands. Genet. Couns. 2008;19:43–46. [PubMed] [Google Scholar]
- 31.Carlson B.M. Mosby-Year Book Inc.; St Louis: 1994. Human Embryology and Developmental Biology. [Google Scholar]
- 32.Beverdam A., Brouwer A., Reijnen M., Korving J., Meijlink F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 2001;128:3975–3986. doi: 10.1242/dev.128.20.3975. [DOI] [PubMed] [Google Scholar]