

Abstract
GWAS have been successful in identifying disease susceptibility loci, but it remains a challenge to pinpoint the causal variants in subsequent fine-mapping studies. A conventional fine-mapping effort starts by sequencing dozens of randomly selected samples at susceptibility loci to discover candidate variants, which are then placed on custom arrays or used in imputation algorithms to find the causal variants. We propose that one or several rare or low-frequency causal variants can hitchhike the same common tag SNP, so causal variants may not be easily unveiled by conventional efforts. Here, we first demonstrate that the true effect size and proportion of variance explained by a collection of rare causal variants can be underestimated by a common tag SNP, thereby accounting for some of the “missing heritability” in GWAS. We then describe a case-selection approach based on phasing long-range haplotypes and sequencing cases predicted to harbor causal variants. We compare this approach with conventional strategies on a simulated data set, and we demonstrate its advantages when multiple causal variants are present. We also evaluate this approach in a GWAS on hearing loss, where the most common causal variant has a minor allele frequency (MAF) of 1.3% in the general population and 8.2% in 329 cases. With our case-selection approach, it is present in 88% of the 32 selected cases (MAF = 66%), so sequencing a subset of these cases can readily reveal the causal allele. Our results suggest that thinking beyond common variants is essential in interpreting GWAS signals and identifying causal variants.
Introduction
GWAS have been very successful in identifying and replicating disease-susceptibility loci for common and complex human diseases.1–3 A commonly held view is that the success of GWAS depends on the validity of the common disease/common variant (CD/CV) hypothesis, which specifies that most of the genetic risk for common diseases is due to disease loci where there is one common variant (minor allele frequency [MAF] > 5%) with small effect sizes.4,5 There has been a large volume of literature debating over whether the CD/CV hypothesis describes a large portion of the genetic susceptibility to common and complex diseases.6–15 However, it is generally assumed that association signals detected from GWAS represent linkage disequilibrium (LD) between a common tag SNP and a common causal variant with a small effect size and therefore explain only a minor proportion of disease heritability. If a common causal variant is responsible for an association signal in GWAS, it should be straightforward to zoom into the candidate region and identify the variant in subsequent fine-mapping studies with small sample sizes, although common variants with subtle effects could be difficult to recognize as causal even once identified.
We have recently demonstrated that rare variants can create synthetic association signals in GWAS, by occurring more often in association with one of the alleles of a common tag SNP and therefore resulting in a scenario in which common SNPs seem to confer risk for common diseases.16 The term “synthetic” does not imply that the association is spurious, but rather that it has different properties from what is commonly assumed (i.e., that one common causal variant underlies an association signal). An illustration of this concept is given in Figure 1, in which two causal variants emerge recently at the same haplotype background of a tag SNP, so the tag SNP represents the combined effects of both causal variants in present human populations. A third, very rare causal variant emerges in cis with the alternative allele of the tag SNP, so it may have minor antagonistic effects on the association signal. In short, the nature of genealogies presents multiple chances to partition rare variants such that an imbalance of allele frequencies can exist between cases and controls, and given the abundance of ancestral common SNPs in the genome, these differences can usually be picked by common tag SNPs. We note that the “tag” measure traditionally uses r2, which favors SNPs with similar allele frequencies (due largely to the relationship between power, sample size, and r2), whereas we focused on the D′ measure in the current study so as to better assess the relationship between a common tag SNP and rare causal variants. Extensive coalescence simulations show that such synthetic associations are not only possible but also inevitable; furthermore, if an association can be accounted for by multiple causal variants, each of which is likely to be rare, then some true causal variants could lie outside of the LD block containing the most significant tag SNPs, and multiple seemingly independent association signals may be present at the same locus.16 As a corollary, resequencing a narrow LD block at susceptibility loci in a small number of randomly selected control samples may not reveal the causal variants. Whole-genome imputation, even after the completion of the 1000 Genomes Project, may not find the causal variants either, unless rare variants (usually ethnicity-specific) are present in the haplotype data and can be imputed accurately.
Figure 1.
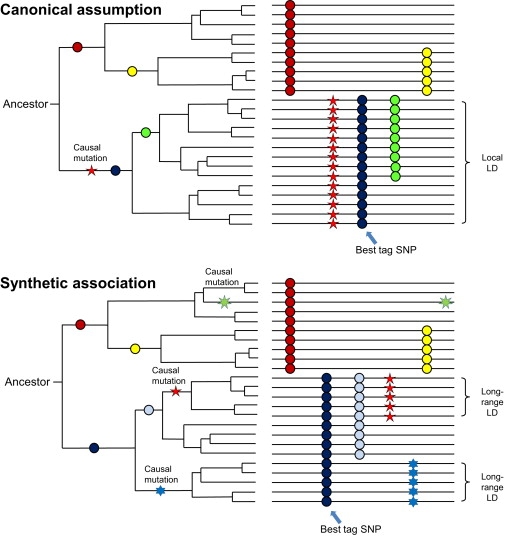
An Illustration Comparing the Canonical Common Disease/Common Variants Assumption and the “Synthetic Association” Theory
The left panel represents a genealogy tree showing the emergence of causal mutations and tag SNPs over human evolutionary history, while the right panel illustrates the catalog of variants within present human populations. Under synthetic association, the best tag SNP captures the combined effects of causal mutations; additionally, since causal variants arise recently, the tag SNP is in long-range LD with either causal variant.
There are a few known examples supporting the hypothesis that multiple rare variants may collectively be responsible for an association signal. First, three rare variants in NOD2 (MIM 605956) (rs2066844, rs2066845 and rs2066847, MAF range from 1% to 5%) were found to be associated with susceptibility to Crohn disease (MIM 266600) in 200117,18 and, on the basis of functional assays, were potentially causal.19 Although there are no known common causal variants in NOD2, the Wellcome Trust Case Control Consortium (WTCCC) study in 2007 implicates a common tag SNP within NOD2 (rs17221417, MAF = 29%).20 In the HapMap CEU population (Utah residents with ancestry from northern and western Europe), two rare variants (rs2066844 and rs2066845) are in complete LD (D′ = 1) with the tag SNP, suggesting that the tag SNP may represent the combined effects of at least two rare variants (information for the third rare variant is not available from HapMap). Second, with the use of population-wide resequencing, multiple rare and ethnicity-specific mutations in PCSK9 (MIM 607786) have been associated with low-density lipoprotein (LDL) cholesterol levels in both European Americans and African Americans,21 multiple rare mutations in ANPGTL family members are associated with plasma triglyceride levels,22 and multiple rare mutations in ABCA1 (MIM 600046), APOA1 (MIM 107680), and ANPGTL4 are associated with high-density lipoprotein (HDL) cholesterol levels.23,24 Recent GWAS on dyslipidemia convincingly identified association between these genes and the corresponding lipid traits, through the use of common tag SNPs (rs11206510 for PCSK9, MAF = 19%; rs10889353 for ANPGTL3, MAF = 33%; rs964184 for APOA1, MAF = 14%; rs1883025 for ABCA1, MAF = 26%; rs2967605 for ANPGTL4, MAF = 16%).25 Third, Zhu et al. sequenced the angiotensinogen gene and found that multiple rare variants contribute to variation in angiotensinogen levels; interestingly, most of these variants sit on the same haplotype background created by three common SNPs.26 Fourth, three common tag SNPs encompassing MC1R (MIM 155555) were “independently” associated with melanoma in a recent GWAS.27 However, resequencing of the candidate gene, MC1R, indicates that these signals can be completely explained by the combined effects of several rare nonsynonymous mutations, suggesting that “ignoring rare variants can lead to incorrect inferences on the potential role of candidate genes carrying common SNPs identified by GWAS” (F. Demenais at al. [2009]. Importance of sequencing rare variants after a genome-wide association study (GWAS): the MC1R gene, 16q24 region and melanoma story, paper presented at American Society of Human Genetics 59th Annual Meeting, Honolulu, HI, USA). Fifth, many hundreds of human leukocyte antigen (HLA) alleles and haplotypes exist in human populations, and some of them cause autoimmune diseases.28 Although some SNPs can tag individual HLA alleles well,29,30 many association signals in GWAS by diallelic SNP markers potentially represent synthetic association, whereby one SNP allele tags multiple HLA alleles. Sixth, we have shown that even a single rare causal allele (MAF = 3.6%) can create significant association signals in a GWAS for sickle cell anemia (p = 1.1 × 10−136 for rs7120391, MAF = 11%) and that genome-wide significant (p < 5 × 10−8) signals can extend over 2.5 Mb across many dozens of r2-based LD blocks visually discernable in the HapMap population.16 The causal allele is under strong positive selection to protect against malaria, representing a classic example of heterozygote advantage,31 but it also represents an example that recently emerged causal alleles can leave a trace of long-range haplotypes surrounding the allele. Finally, we have shown that hearing loss, with hundreds of known causal mutations at the GJB2 ([MIM 121011])-GJB6 ([MIM 604418]) loci but without common causal variants,32 is associated with several seemingly independent common tag SNPs around GJB2.16 In summary, none of these association signals on common tag SNPs discussed above are spurious signals; instead, they may represent scenarios whereby multiple causal variants work together to create genome-wide significant association signals, which are being accredited to one common tag SNP.
The purpose of the current study is not to speculate on the fraction of association signals that can be attributed to the presence of multiple causal alleles. Rather, our aim is to accept the possibility that some signals in GWAS emerge from rare causal variants, and to use this possibility to leverage the extensive GWAS data in the search for causal variants. On the basis of the observation of differential LD between tag SNPs in cases compared to controls, together with the observation of long-range haplotypes surrounding tag SNPs, we present an approach for selecting cases to maximize the chance of finding causal variants. We evaluated this approach on a simulated data set and compared it with conventional fine-mapping approaches to identify causal variants. We also tested the approach on a GWAS on hearing loss, in which we know the identity of the causal variants, and we have sequenced all available cases for the presence of causal variants. We believe that our theoretic framework and our case-selection approach will have significant implications for the design of follow-up studies after a successful GWAS in order to facilitate success in finding the causal genes and their causal variants.
Material and Methods
Definition of a Synthetic Causal Marker
When more than one rare variant is present in the same gene in a population, in order to facilitate modeling of their joint effect, we create a synthetic marker that represents the combined effect of several rare variants. To simplify the description below, suppose that there are two rare causal variants with minor (causal) allele frequencies of pM and pN, respectively. Given that both variants are rare and recent, and that they are physically close (within the same gene), it is reasonable to assume that they are in complete LD with opposite direction (that is, that M and N alleles never occur in cis, or that only three haplotypes are present in population: Mn, mN and mn). Therefore, for the synthetic marker, the genotype is heterozygous when the two homologous chromosomes are in configuration of Mn/mn or mN/mn, and it is homozygous when the two homologous chromosomes are in configuration of Mn/mN, Mn/Mn, or mN/mN.
Let the two alleles for the synthetic marker be A as risk allele and a as non-risk allele. The allele frequency of the synthetic marker is the sum of M and N alleles. If the two causal variants are not in perfect LD, that is, if the MN haplotype exists in population, we can assume that that penetrance of an MN haplotype is identical to that of Mn or mN, whichever is larger. If we suppose that Mn is more penetrant than mN, then effectively we could consider a modified second causal variant with allele frequency of pN−pMN and pn+pMN, which has complete LD with the first causal variants. A synthetic causal marker can then be built from these two causal variants. Similarly, multiple causal variants can be modeled in a stepwise fashion. In cases where causal variants are protective, the allele frequency of the synthetic marker needs to be subtracted by that of the protective allele.
Relationship of Allelic Odds Ratio between the Synthetic Causal Marker and the Tag SNP
A general formula describing the allelic odds ratio (OR) for the tag SNP (with alleles B and b), based on the OR estimated from the synthetic causal variant (with alleles A and a), is
| (Equation 1) |
in which the allele frequency is pA < pB and D is the LD coefficient. The formula has been previously described.33
Assuming that the causal marker and the tag SNP have complete LD with identical direction, we have D = pA(1-pB). The above relationship can therefore be simplified as:
| (Equation 2) |
Relationship of the Locus-Specific Sibling Recurrence Risk Ratio between the Synthetic Causal Marker and the Tag SNP
The locus-specific sibling recurrence risk ratio, or λS, can be calculated as
| (Equation 3) |
in which VA is the additive genetic variance, VD is the dominance genetic variance, and K is the population prevalence of the disease.34 Let Va = VA /f02 and Vd = VD /f02, in which f0 is the penetrance of the wild-type genotype, as shown before,35 and the formula can be rewritten as
For the causal marker with A and a alleles in which PAR is the population attributable risk, we have:
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
In the study, we plot the relationships between λS for the synthetic causal marker and the tag SNP, given prespecified values of genotype relative risk (GRR) and a ladder of allele frequencies.
Simulation of Sequencing Data on a Susceptibility Locus
We simulated sequencing data that mimic the scenario described in Table 1 in order to evaluate different resequencing strategies for identifying causal variants from a susceptibility locus detected in GWAS when multiple causal variants are present. In the simulation, we assumed that three causal variants with MAF of ∼1%, 2%, and 4% are present at the same locus with a GRR of ∼3. Genealogical trees were simulated with GENOME,36 with an effective population size of 10,000 and a mutation rate of 10−8 used. A random gene genealogy was drawn, with mutations distributed along the genealogy, and disease-causing mutations were assigned at random from those variants that were within the allowed frequency range. Two simulated haplotypes were randomly selected with replacement for each individual, and 1000 individuals were generated, including an equal number of cases and controls. Case or control status was designated on the basis of the assigned risk. The simulation data sets can be downloaded from the website listed in the Web Resources section.
Table 1.
Effect Sizes of Three Causal Variants and the Tag SNP at the NOD2 Locus for Crohn Disease
| SNP | Function | MAF | GRR (Het) | GRR (Hom) | PAR | λS | Proportion of Genetic Risk Explaineda |
|---|---|---|---|---|---|---|---|
| Causal Variants | |||||||
| rs2066844 | Arg702Trp | 4.1% | 1.71 | 2.73 | 5.55% | 1.018 | 0.54% |
| rs2066845 | Gly908Arg | 1.5% | 2.53 | 12.13 | 4.55% | 1.040 | 1.2% |
| rs2066847 | Leu1007fsinsC | 1.9% | 3.64 | 12.06 | 9.29% | 1.118 | 3.4% |
| Three-SNP Combination | 18.2% | 1.184 | 5.1% | ||||
| Tag SNP | |||||||
| rs17221417 | Tag SNPb | 28.7% | 1.29 | 1.93 | 16.4% | 1.023 | 0.69% |
The three causal variants have a combined λS similar to that observed in linkage studies, explaining the heritability at the locus. The λS estimate based on the tag SNP creates a false impression of “missing heritability.” MAF, minor allele frequency; GRR, genotype relative risk; Het, heterozygotes; Hom, homozygotes; PAR, population attributable risk.
The total λS for Crohn disease is estimated to be 27.2, following 50.
rs17221417 is the tag SNP reported in Table 2 of the WTCCC paper.20 The MAF and GRR were estimated with the use of WTCCC data.
The simulation data set contains a total of 4116 segregating sites within a 2 Mb region. To simulate genotyping data on these subjects in a GWAS, we randomly selected SNPs so that their MAF distribution followed a uniform distribution between 0 and 0.5. In total, 504 SNPs were selected as if they were genotyped by a GWAS, with an average intermarker distance of ∼4 kb. We tested all variants in the hypothetical genotyping array for association with disease status, by allelic association tests in PLINK.37
Genome-wide Search for Long-Range Haplotypes in Cases
Because rare causal variants arose recently, they often exist on long-range haplotypes spanning multiple blocks of high LD (as observed in control populations), which recombination has not yet had a chance to further fragment. The concept of the long-range haplotype has been widely used in human genetics research. For example, it has been used for inferring positive selection from the human genome,38,39 used in population-based linkage analysis for identifying disease-susceptibility loci,37–40 and used in inferring the population origin of private alleles.41 In addition, previous resequencing studies on rare variants clearly demonstrated the presence of long-range haplotypes, suggesting that rare causal alleles were generally recent in origin.42 Furthermore, contrasting LD patterns in cases versus controls offers improved power to localize some association signals.43,44 Therefore, long-range haplotypes that are preferentially observed in cases tend to be those that harbor rare causal alleles, and such loci will display differences in LD structure between cases and controls.
We performed genome-wide scanning of the SNP genotype data to identify regions likely to harbor long-range haplotypes in cases, by contrasting LD patterns between cases and controls with the use of index SNPs. This analysis was facilitated with the use of the “clumping” function in the PLINK software.37 The clumping procedure takes all significant SNPs (by default p < 1 × 10−4) as index SNPs and forms clumps of all nearby SNPs (by default p < 0.01), using the “--clump-r2 0” argument to include all SNPs regardless of r2 with the index SNP. Next, we compared the D′ values between the index SNP and all nearby clumped SNPs in cases versus controls. Our rationale is that even if two SNPs are not in LD in the control population, if two alleles in the two SNPs happen to be in the same long-range haplotypes that were carried through the genealogy, then greater LD should be detected in cases that enrich for such long-range haplotypes. Thus, for a given index SNP, we calculated the D′ with all nearby SNPs in cases (D′case) and in controls (D′control), as well as the ratio of the D′ measure in cases versus in controls (D′case / D′control). For each index SNP, summary statistics can be calculated as the median of these ratios, and a higher value indicates better correlation of adjacent SNPs in cases than in controls.
Phasing Long-Range Haplotypes at Susceptibility Loci
Assuming that rare causal variants can be tagged by specific long-range haplotypes, we attempted to identify a subset of tag SNPs that are maximally informative for long-range haplotype construction, because these tag SNPs need to differentiate the effect of long-range haplotypes and ancestral short-range LD blocks in the human genome. Therefore, from the “clump” of SNPs at a susceptibility locus, we specifically chose SNPs that appear to be tightly linked to the index SNP in cases versus in controls, on the basis of D′case / D′control. In the hearing-loss data, possibly due to the presence of one major risk allele, the ratio tends to be relatively large, so we arbitrarily picked a threshold of > 2 to select SNPs for haplotype phasing. In the simulation data, we used a threshold of > 1.2 to select six SNPs.
We relied on the fastPHASE program45 for building long-range haplotypes for cases and controls together, using the SNPs selected from the previous step. All default parameters were used, and the “-i” option was used to minimize individual error as opposed to switch error.
Identify Subset of Cases for Sequencing
After haplotype phasing, we aimed to identify a subset of cases to enrich for samples who are more likely to harbor causal variants. The best-guess haplotypes for each sample are used to assess the frequency of each haplotype in cases and controls, and Fisher's exact test is used to assess whether a long-range haplotype is associated with disease status. We acknowledge that fastPHASE does not account for haplotype-phasing uncertainty, but it offers greatly improved speed. Because our purpose is to enrich a subset of cases carrying specific causal variants, some phasing errors can be tolerated. On the basis of the hypothesis that the first few long-range haplotypes that are overrepresented in cases versus controls could tag the major classes of rare causal variants, we can select cases that carry these specific haplotypes for resequencing studies.
Results
Effect Sizes May Be Underestimated in GWAS
One of the most consistent themes from analysis of GWAS data is that, with rare exceptions, the effect sizes of the susceptibility loci tend to be modest, with an OR typically less than 1.3 2. Because the power of association studies depends on effect sizes, it likely that most additional undetected susceptibility loci have even smaller effect sizes. However, if an association signal is due to the presence of multiple rare variants, then the estimated effect size merely reflects that assigned to the tag SNPs, which could be very different from those of the causal variants. This possibility has been recognized, but mainly in the context of the assumption that the causal site is imperfectly tagged by the common variant. The possibility that much of the signal comes from multiple rare variants has not been systematically addressed.
To investigate the relationships of effect sizes between common tag SNPs and rare causal variants in a quantitative manner, we need to create an artificial “synthetic causal marker,” which represents the combined effect of one or more causal variants. Assuming that causal variants do not occur in cis (one variant masks the effect of another if they do), the allele frequency of the synthetic marker is the sum of all causal variants, whereas the effect sizes (odds ratio and GRR) can be expressed as a weighted sum from those of the causal variants. In the discussion below, we treat the susceptibility locus as if there is only one synthetic causal marker (with A as risk allele and a as non-risk allele), and we examine how well the tag SNP (with B as risk allele and b as non-risk allele) represents the effects of this synthetic causal marker. It is well known that differences in odds ratio estimates could exist if allele frequencies of a tag SNP differ much from a causal variant in LD.33 This relationship is illustrated in Figure 2, by assuming that the tag SNP has an MAF of 30% whereas the causal marker has an MAF ranging from 1% to 10%. In all scenarios, the tag SNP underestimates the true effect sizes of the causal marker. For example, when the MAF = 1% for the causal marker, the OR of 1.2, as suggested by the tag SNP, actually reflects a true OR of 7 for the causal marker.
Figure 2.
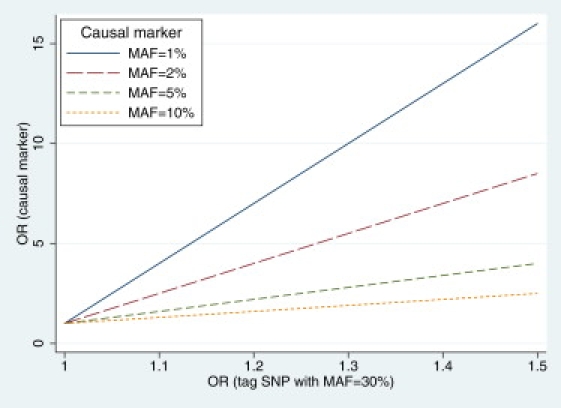
Illustration of an Odds Ratio for a Tag SNP and a Synthetic Causal Marker
The relationship between the odds ratio (OR) observed on a tag SNP with MAF = 30% and the true OR for a synthetic causal marker is shown, with MAF ranging from 1% to 10%. In all scenarios, the tag SNP underestimate the true effect size, especially when the causal marker is relatively rare.
To demonstrate the underestimation of genetic effects with the use of real data, we next examined the NOD2 locus for Crohn disease as an example. Three nonsynonymous or frameshift mutations in NOD2 were previously associated with Crohn disease17,18 and were subsequently confirmed by many replication studies and meta-analysis.46 The three mutations have been studied by in vitro biochemical assays and by mouse models with introduced mutations,19 therefore confirming that they play causal roles rather than are in LD with a causal variant. A large-scale meta-analysis on NOD2 reported estimates on allelic OR for the three causal variants (ranging from 2.2 to 4.1),46 and a recent NOD2 genetic study has estimated the GRR (ranging from 1.7 to 3.6 for heterozygotes)47 (Table 1). In the WTCCC study,20 the NOD2 locus was convincingly associated with Crohn disease, through the use of the common tag SNP rs17221417 (MAF = 29%, OR = 1.37).20 Comparing the true effect size of causal variants with that inferred from rs17221417 (Table 1), it is clear that the tag SNP severely underestimates the true effect size of any one of the three causal variants.
Heritability Due to Identified GWAS Loci May Be Underestimated
A question that often arises in GWAS is “Where is the missing heritability?”48,49. This question refers to the fact that the collection of variants discovered in GWAS explains only a minor fraction of the heritability, even for diseases or traits that are highly heritable. Multiple reasons have been proposed to explain the missing heritability.48 However, the presence of multiple rare causal variants offers additional explanations: First, some rare causal variants may tag the non-risk allele of a common tag SNP (see example in Figure 1), antagonizing the effect size of the tag SNP; similarly, a rare protective variant may tag the risk allele of a common tag SNP, antagonizing the observed effect size. Second, even if a tag SNP represents the combined effects of all causal variants perfectly (D′ = 1), its heritability measure may dramatically underestimate the true contribution of the susceptibility locus.
The theoretic foundation of differing familial risk between a common and a rare variant, which are in complete LD (D′ = 1) but have different allele frequencies, has been previously described in an excellent article by Hemminki et al.35 Although this article serves the purpose of explaining the discordance between population attributable risk (PAR) and familial risk, it turns out that the statistical derivation can also be applied to an investigation of our hypothesis. In our analysis, we considered a tag SNP with an MAF of 30% and a causal marker with an MAF ranging from 1% to 10% (with a 1% ladder of increase). We then investigated the relationships of the locus-specific λS estimates based on the tag SNP or the causal marker. The λS represents the relative risk of siblings of patients divided by the population prevalence of the disease, and it is used to calculate the proportion of heritability explained by a locus for binary phenotypes.34 We tested several effect sizes for the causal marker by assuming multiplicative models (Figure 3). In all cases, it is clear that the λS estimates based on the tag SNP severely underestimate that based on the causal marker, supporting previous speculations that rare functional alleles could explain a much larger proportion of familial aggregation of cases than common tag SNPs.35
Figure 3.
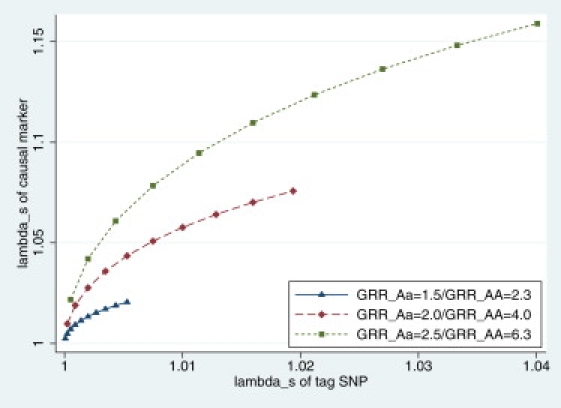
Illustration of λS for a Tag SNP and a Synthetic Causal Marker
The relationship between λS for a tag SNP with MAF = 30% and the true λS for a synthetic causal marker is shown, with MAF ranging from 1% to 10% (each dot represents 1% increase), under three effect sizes with multiplicative genetic models. In all cases, the tag SNP underestimates the familial aggregation explained by the true causal variant.
To investigate the missing heritability in real data, we again examined the NOD2 locus in the Crohn disease data set (Table 1). First, the PAR for the tag SNP is almost identical to the PAR for the three causal variants combined together (16.4% versus 18.2%), demonstrating that the tag SNP indeed captures information from all three causal variants together. Next, using the GRR estimates,47 we calculated the locus-specific λS for the three causal variants. Their combined effects result in a λS estimate of 1.184, which is quite close to the estimate of λS = 1.3 for NOD2 observed in linkage study.18 Therefore, two-thirds of the familial risk at NOD2 can be explained by these three causal variants. In fact, assuming a total λS of 27.2 for Crohn disease (weighted estimate from multiple studies50), the three causal variants explain > 5% of the genetic risk of Crohn disease, suggesting that NOD2 is a major disease locus. Examination of the tag SNP rs17221417 reported by WTCCC20 reveals a locus-specific λS estimate of merely 1.023, which is not even remotely close to the expected value of 1.3. Therefore, the tag SNP creates the false impression that the association signal explains a tiny fraction of the linkage signal at NOD2 and that some major-effect causal variants cannot be tagged by rs17221417. In other words, the tag SNP creates a scenario of “missing heritability,” in which the strongest Crohn disease-susceptibility locus, NOD2, explains only ∼1% of the total genetic risk for Crohn disease, if we simply take the GWAS results at face value. Altogether, extending previous studies, our analysis suggests that the “missing heritability” observed in GWAS may not be as severe as it appeared to be, if multiple rare variants are responsible for at least a proportion of the discovered susceptibility loci.
Comparative Strategies for Identifying Causal Variants in a Simulated Data Set
After a successful GWAS, the next natural extension is to fine-map the discovered susceptibility loci to identify causal variants. Researchers may sequence a few dozen subjects on the entire HapMap r2-based LD block harboring the most significantly associated SNPs, identify all common and rare variants in these subjects, then design a custom fine-mapping panel with all of these variants to examine a large number of cases and controls. We note that these approaches have already been adopted for multiple diseases by many groups (for example, see P. Deloukas and WTCCC [2008], High throughput approaches to fine mapping in regions of confirmed association, paper presented at 58th Annual meeting of American Society of Human Genetics, Philadelphia, PA, USA). However, as of today, these types of efforts have not identified “smoking gun” mutations (O. Harismendy et al. [2009], Population resequencing and functional annotation of the 9p21 interval associated with coronary artery disease and type 2 diabetes, paper presented at The Biology of Genomes, Cold Spring Harbor, NY, USA; G. McVean and WTCCC [2009], Targeted resequencing and fine-mapping of variants in association studies, paper presented at The Biology of Genomes. Cold Spring Harbor, NY, USA).51 This could be due to the insufficient power for identifying the “needle” from a haystack of common variants with very similar association statistics, or it could be due to the properties of synthetic associations. If multiple rare variants exist, then we would expect the following: (1) causal variants may fall outside of the r2-based LD block naturally observed in control subjects; (2) some causal variants may not be observed in selected subjects and therefore may not be designed on the custom arrays; and (3) some causal variants may be on the custom arrays, but their test statistics (p values) may not be as significant as common tag SNPs that represent multiple causal variants.
To examine the effectiveness of conventional approaches for pinpointing causal variants when multiple causal variants exist, we analyzed a simulated data set with genotypes for a disease locus in 1000 subjects (500 cases and 500 controls). The locus contains three causal variants with a MAF of ∼1%, ∼2%, and ∼4% and a GRR of ∼3. We simulated genotypes for 4116 segregating sites in a 2 Mb region by a coalescence model, and we then randomly selected 504 SNPs with MAF following a uniform distribution between 0 and 0.5, as if there are 504 SNPs in a hypothetical genotyping array for this region. The most significantly associated SNP (SNP2276) is a common tag SNP with p = 1.1 × 10−10 and a MAF of 28% in cases and 16% in controls.
From the simulation data, we evaluated a conventional strategy of selecting a random set of 30 controls for the identification of variants by sequencing. We performed 1000 replicate experiments, and we found that 397, 500, and 73 of these experiments were able to include one, two, and three causal variants from the 30 subjects, respectively. Therefore, this experimental design is unlikely to lead to the discovery of all three causal variants to be placed in custom fine-mapping arrays. Furthermore, even under the assumption that all three causal variants are indeed on the fine-mapping arrays, we then examined their association statistics by comparing cases versus controls: their p values are 2.3 × 10−4, 2.0 × 10−8 and 1.0 × 10−10, respectively. However, in the entire simulation data set, there is also a common variant, SNP 2034, with p = 8.8 × 10−12, which completely tags all three causal variants (D′ = 1). Because most researchers assume the existence of one common causal variant, they would usually consider only the most significant p value in a region in fine-mapping studies, so these causal SNPs would be missed. Finally, we examined the role of conditional regression analysis, by including SNP 2034 as covariate in a logistic regression model and assessing association of all other variants. Although all three causal variants show some levels of residual association, only one of them ranks at the top of the list. Therefore, even by conditional regression analysis, we still cannot identify all three true causal variants. On the basis of our experiments above, if multiple causal variants do exist at a susceptibility locus, the traditional fine-mapping approaches are unlikely to identify all of them.
To address this issue and to improve the likelihood of success, we propose a case-selection approach based on examining long-range haplotypes, which are haplotypes expected to contain recently emerged causal alleles. The rationale is that resequencing studies must be focused on subjects who are more likely to carry causal alleles, in order to ensure a reasonably good chance of identifying these variants for the design of fine-mapping panels. We treated SNP2276 (the most significant SNP in the hypothetical genotyping array) as an index SNP and identified five additional SNPs, which have higher D′ with the index SNP in cases versus controls. Interestingly, these SNPs range from 241 kb to the left of to 207 kb to the right of the index SNP. We then phased all cases and controls and identified a few haplotypes that appear to have higher frequency in cases versus controls. Strikingly, 96/100, 59/112, and 21/36 subjects carrying the first, second, and third haplotypes do carry each of the three causal variants, respectively. Therefore, the presence of long-range haplotypes improves the prediction of the carrier status of the causal variants, although it is not perfect. In any case, the selection procedure resulted in high enrichment of subjects for each of the three causal variants, potentially facilitating the discovery of these variants and subsequent fine-mapping efforts by custom arrays.
To further examine how the long-range haplotype-based method works, we performed additional experiments by constructing local haplotypes. We used PLINK37 to estimate haplotype blocks for SNPs on the hypothetical genotyping array, and we identified an LD block with four SNPs encompassing SNP2276. We then followed the same procedure described above to identify haplotypes associated with disease, but only one haplotype showed higher frequency in cases than in controls. This haplotype is carried by 235 cases, including 98, 23, and 68 subjects with the three causal variants, respectively. Therefore, compared to long-range haplotypes, SNPs within a local LD block performed less well in enriching subjects with specific causal variants.
Case-Selection Approach Identifies Causal Variants in a Real Data Set
Next, we used a real GWAS data set on hearing loss to test the case-selection method based on long-range haplotypes. Hearing loss represents an extreme example because of the existence of a major-effect locus near GJB2-GJB652 (similar to the MHC region for type 1 diabetes [MIM 222100]), making it different from many other complex diseases (such as type 2 diabetes [MIM 125853]). Nevertheless, because hundreds of causal variants have been documented in many studies sequencing this region (Connexin-deafness database), the knowledge regarding the known causal variants helps us test our method of identifying them by sequencing. We previously performed a GWAS on 418 cases and 6892 control subjects, and we identified genome-wide significant associations within the GJB2-GJB6 locus, the most significant tag SNP being rs870729 (MAF = 19%, p = 3.4 × 10−11, OR = 1.7).16 Here, we surveyed the whole-genome genotype data to search for loci with evidence of long-range haplotypes in cases, using a simple summary statistic called the median D′case / D′control ratio. In essence, the summary statistic evaluates differences in LD patterns in cases compared to controls in a given locus surrounding an index SNP (the most significant SNP at a locus). A locus on 10q25.1 showed the strongest evidence of long-range haplotypes (with rs7085286 as index SNP, association p = 2.1 × 10−5, median D′case / D′control ratio = 3.3). The GJB2-GJB6 locus (with rs870729 as index SNP) showed the second strongest evidence of long-range haplotypes (median D′case / D′control ratio = 1.8). At the GJB2-GJB6 locus, for 23/23 (100%) of the SNPs, we observed a higher D′ value in cases than in controls (Table 2), and some of the strongest D′ ratios were observed 850 kb away from the index SNP across multiple LD blocks, supporting the assumption that a long-range haplotype is indeed present in cases.
Table 2.
A List of SNPs that Are Associated with Hearing Loss with p < 0.01 and Are within 1 Mb from the Index SNP rs870729
| SNPa | Distance to rs870729 | p Value | r2caseb | D′caseb | r2controlb | D′controlb | D′ Ratioc |
|---|---|---|---|---|---|---|---|
| rs2992950 | −853628 | 0.002274 | 0.001 | 0.106 | 0 | 0.014 | 7.57 |
| rs17080523 | −849341 | 0.002406 | 0 | 0.092 | 0 | 0.002 | 46.00 |
| rs7329467 | −62457 | 6.87E-08 | 0.117 | 0.495 | 0.003 | 0.071 | 6.97 |
| rs7984378 | −47459 | 0.003742 | 0.015 | 0.234 | 0.002 | 0.084 | 2.79 |
| rs2313475 | −31103 | 0.00413 | 0.056 | 0.257 | 0 | 0.027 | 9.52 |
| rs3751385 | −18923 | 1.50E-09 | 0.744 | 0.889 | 0.673 | 0.843 | 1.05 |
| rs6490527 | 2550 | 0.006606 | 0.211 | 1 | 0.265 | 1 | 1.00 |
| rs7319601 | 56340 | 0.007069 | 0.026 | 0.237 | 0.009 | 0.166 | 1.43 |
| rs9509124 | 59891 | 0.005835 | 0.031 | 0.256 | 0.01 | 0.172 | 1.49 |
| rs9315439 | 63701 | 0.000129 | 0.051 | 0.235 | 0 | 0.007 | 33.57 |
| rs7992230 | 66438 | 0.001725 | 0.081 | 0.663 | 0.036 | 0.5 | 1.33 |
| rs1537788 | 82693 | 4.71E-05 | 0.075 | 0.373 | 0.033 | 0.277 | 1.35 |
| rs9285110 | 85313 | 9.38E-05 | 0.057 | 0.369 | 0.03 | 0.305 | 1.21 |
| rs1547721 | 86181 | 0.004168 | 0.075 | 0.639 | 0.037 | 0.51 | 1.25 |
| rs7327522 | 89416 | 0.007661 | 0.035 | 0.261 | 0.001 | 0.048 | 5.44 |
| rs4769989 | 93031 | 0.002192 | 0.04 | 0.313 | 0.001 | 0.048 | 6.52 |
| rs7323752 | 95958 | 0.002151 | 0.04 | 0.323 | 0.002 | 0.084 | 3.85 |
| rs7323769 | 95987 | 0.00054 | 0.043 | 0.409 | 0.008 | 0.202 | 2.02 |
| rs1953516 | 99267 | 0.00039 | 0.049 | 0.447 | 0.01 | 0.236 | 1.89 |
| rs7337231 | 114739 | 0.002022 | 0.028 | 0.309 | 0.009 | 0.204 | 1.51 |
| rs9509167 | 125622 | 0.001591 | 0.016 | 0.215 | 0.008 | 0.173 | 1.24 |
| rs12868032 | 128169 | 0.003955 | 0.017 | 0.211 | 0.006 | 0.151 | 1.40 |
| rs9550637 | 130648 | 0.007389 | 0.029 | 0.221 | 0.002 | 0.06 | 3.68 |
SNPs selected for constructing long-range haplotypes are marked in bold font.
r2 and D′ measure correlation between each SNP and the index SNP rs870729.
D′ ratio is defined as D′case / D′control.
Next, we obtained sequencing data on the GJB2 locus for a set of 329 cases, whose DNA were available for sequencing. On the basis of well-documented annotations of mutations, a total of 19 unique causal variants were observed. Not surprisingly, we found that 37 cases carry the 35delG mutation, which has an MAF of 8.2% in cases, far higher than the known frequency (1.25%) in controls of European ancestry.53 Several less frequently observed causal variants in cases include M34T (MAF = 1.4%), 167delT (MAF = 1.2%), L90P (MAF = 1.1%), V37I (MAF = 0.5%), and a GJB6 deletion (MAF = 0.3%), and they collectively account for 86% of the causal mutations at the GJB2-GJB6 locus in our data. Interestingly, 35delG is in high LD with the tag SNP rs870729 (D′ = 0.95). The other five causal variants (M34T, 167 delT, L90P, V37I, GJB6 deletion) are in complete LD with rs870729 (D′ = 1), though we acknowledge that LD calculation is unstable when the MAF for one variant is very low. However, it is clear that the MAF of rs870729 is far higher than that of any causal variants, so if we had sequenced only a handful of control subjects for variant discovery, we would not find many of these rare causal variants. Because we cannot put these causal variants into custom arrays or use them in imputation, we would not identify them by traditional fine-mapping efforts.
We then focused on whether we can confidently identify a subset of cases carrying the 35delG mutation, using SNP genotype data alone. We set a D′case / D′con > 2 threshold, and we selected 11 SNPs (bold font in Table 2) for phasing long-range haplotypes by fastPHASE45 and comparing the best-guess haplotype frequencies in cases versus controls (Table 3). The most striking difference was observed for a long-range haplotype that has an allele frequency of 0.8% in controls but 5.3% in cases, suggesting that it may tag a causal allele with major effects. By comparison with the resequencing data, we found that 28/32 (88%) of the cases carrying this haplotype also have the 35delG mutation, indicating a high positive predictive value of this haplotype for the presence of 35delG. The allele frequency of 35delG is 66% in these 32 cases, suggesting that sequencing a few of these cases can easily identify the causal variants. In addition, the case-selection method has high specificity, because only 0.8% of the controls carry this long-range haplotype, and some of them probably do carry the 35delG mutation. Our results thus provide strong evidence supporting the validity and effectiveness of the case-selection approach.
Table 3.
A List of Haplotypes and Their Estimated Frequencies in Hearing Loss Cases and Controls
| Haplotypea | Frequency (Cases) | Frequency (Controls) | Odds Ratio | p Valueb | Cases with 35delG | Cases with 167delT |
|---|---|---|---|---|---|---|
| 212121121112 | 0.053 | 0.008 | 6.8 | 0 | 88% (28/32) | |
| 212121121111 | 0.010 | 0.001 | 12.7 | 4.73E-05 | 67% (4/6) | |
| 211111121112 | 0.010 | 0.002 | 4.08 | 0.00608 | ||
| 211121121112 | 0.015 | 0.006 | 2.55 | 0.00967 | ||
| 121212212221 | 0.018 | 0.009 | 2.08 | 0.0212 | ||
| 211111221112 | 0.009 | 0.003 | 2.81 | 0.0276 | ||
| 121112212221 | 0.014 | 0.008 | 1.77 | 0.111 | ||
| 211112221112 | 0.021 | 0.016 | 1.35 | 0.267 | 38% (5/13) | |
| 211122121112 | 0.015 | 0.011 | 1.37 | 0.342 | ||
| 211122212211 | 0.009 | 0.007 | 1.27 | 0.482 | ||
| 211112222111 | 0.011 | 0.009 | 1.23 | 0.519 |
Best-guess haplotypes with > 1% frequency in cases and with an OR > 1 are shown; 1 and 2 in the haplotype refers to the A and B alleles for SNPs listed in Table 2, per Illumina's TOP/BOT allele designation.
p value is calculated by Fisher's exact test.
Several additional haplotypes also show enrichment as being overrepresented in cases versus controls, so we investigated whether they were tagging additional rare variants. We found that the second haplotype, despite being rare, actually also tagged the 35delG causal variant, with 4/6 (67%) of the cases predicted to harbor this haplotype carrying the 35delG variant. Additionally, we also found that another haplotype, with a frequency of 1.6% in controls and 2.1% in cases, predicts the presence of the 167delT mutation, as 5/13 (38%) of the cases with this haplotype also carry a 167delT mutation. No other haplotypes seem to be tagging other causal variants, such as M34T, L90P, or V37I. In practice, given limited sequencing resources, some researchers may choose to sequence one best-candidate haplotype first, but other haplotypes should also be surveyed to find a more comprehensive ensemble of causal variants. Altogether, if we had sequenced a dozen cases carrying a few specific long-range haplotypes, we could have easily identified at least one causal mutation for hearing loss in the GWAS data. It is likely that diseases that are more complex than hearing loss may not be as straightforward to analyze, but as an extreme example, the hearing loss data suggest the potential utility of enriching cases to discover causal variants.
Discussion
In the current study, we illustrated the potential effect-size distortion of causal variants in GWAS and the potential underestimation of heritability explained for loci detected by GWAS, through the analysis of a well-known locus for Crohn disease. We then proposed a case-selection approach that enriches samples likely to carry long-range haplotypes containing causal alleles, and we demonstrated the effectiveness of this approach on a simulated data set and on a real data set with resequencing data. There are several caveats related to the three major aspects of this study:
When an association signal on a common tag SNP was due to one or more rare variants, our simple theoretical calculation and real-data analysis confirmed that tag SNPs may underestimate the true effect sizes of causal variants in GWAS. However, we stress several issues here in the interpretation of effect sizes. First, the magnitude of underestimation depends on the allele frequency of the synthetic causal marker rather than each individual causal variant, because the tag SNP measures the combined effects of several causal variants. Second, given that rare mutations have emerged relatively recently, the casual variants in the same gene may vary between ethnicity groups (for example, causal variants in PCSK9 for low-density lipoprotein cholesterol levels differ completely between European Americans and African Americans21). Therefore, in the presence of synthetic association, a tag SNP may be associated with the same phenotype in different ethnicity groups, but the effect sizes could differ substantially or even be in opposite directions, because the tag SNP may tag different sets of causal variants. Third, synthetic association could result in distortion of disease models, whereas causal variants with dominant or recessive effects could manifest as if they increase disease risk in a somewhat multiplicative fashion. Finally, synthetic association does not exclude the possibility that rare variants with strong effects and common variants with modest effects coexist at the same locus. Many disease loci (for example, KCNJ11 for type 2 diabetes54) were discovered through study of a few families with extreme or Mendelian form of the disease, whereas common variants with lower penetrance were later discovered at the same loci (for example, the E23K mutation in KCNJ11 has an OR value less than 255 and is functionally validated as potentially causal56).
If a susceptibility locus is subject to synthetic association, how can we estimate the magnitude and recover the missing heritability? In the absence of resequencing data at the locus on many subjects, this question cannot be answered directly by association results from GWAS alone, because we do not know a priori the distribution of frequencies and effect sizes of causal variants at the locus. However, for many diseases, it is feasible to dig out old linkage results on the same locus: although the LOD scores may not reach stringent criteria for statistical significance, the identity-by-descent sharing statistics at the locus can be used to estimate heritability explained.34 If several linkage studies are examined and they show largely consistent results, one could in principle recover the true heritability explained by the locus from linkage statistics. Therefore, old linkage data sets could be very useful for interpreting new GWAS results. This idea remains to be tested, after large-scale sequencing data on many subjects is available for some disease loci.
Many current fine-mapping efforts aim at first sequencing a small group of subjects to discover variants, because we do not yet have a comprehensive catalog of genetic variants in humans in diverse ethnicity groups. Our study highlights the importance of the 1000 Genomes Project to catalog rare variants, but we caution that some ethnicity-specific variants that contribute to risk could be well below the threshold of detection in the 1000 Genomes Project. Additionally, our study also suggests the need to generate high-coverage sequencing data, so that we can have reasonably accurate estimates of allele frequencies of some rare variants in control populations. This is because most sequencing studies of candidate loci will generate high levels of coverage (30× or more) to ensure accurate genotyping, but if only 4–6× coverage is available on the 1000 Genomes Project data, we could not determine whether a given rare variant is overrepresented in cases, unless we sequence our own controls at high coverage as well. Furthermore, imputation algorithms may work less well for rare variants than for common ones for low-coverage sequencing data. Therefore, the availability of high-coverage data from the 1000 Genomes Project will reduce the need of individual investigators to sequence controls, so that valuable sequencing resources can be focused on more cases for improving the power of finding associated variants.
We also acknowledge that a case-only design for resequencing could potentially induce false positives (inflated type I error); that is, many of the discovered variants from sequencing could be spuriously associated with disease. This issue has already been extensively discussed before.57 Therefore, researchers should not utilize custom arrays only on the original GWAS data set to infer causal variants. Instead, just like a replication study on GWAS, a custom array with candidate causal variants should always be tested in independent sample sets for assessment of their true effects. Finally, functional validation is the ultimate answer to the causality of candidate causal variants, and it is required for the biological understanding of disease-locus relationships.
In conclusion, in those instances when the association signals detected in GWAS are due to the presence of multiple causal variants, researchers need to take caution in interpreting the true effect sizes and heritability explained by the causal variants. With the successful identification of hundreds of disease-susceptibility loci, many research groups have now started to apply fine-mapping experiments to identify causal alleles, mostly by designing custom fine-mapping arrays, which require large amounts of investments and human endeavor. These types of custom arrays, which in design may miss many rare variants (because of the way in which variants were ascertained), cannot interrogate the full spectrum of causal alleles. On the other hand, it is important to consider the possibility of “synthetic association” when designing fine-mapping experiments, in order to gain at least empirical data supporting the comparative effectiveness between custom SNP panels versus targeted long-range resequencing in finding causal variants. It is also a priority to develop methods for selecting extreme cases for resequencing studies and performing subsequent association tests on rare variants.57–59 Ultimately, resequencing a few well-phenotyped cases, supplemented with the deep-sequencing data from the 1000 Genomes Project, may turn out to be more cost efficient and may provide more insights than what could be gleaned from traditional fine-mapping approaches.
Acknowledgments
We gratefully thank all of the patients and their families who were enrolled in this study, as well as all of the control subjects who donated blood samples to Children's Hospital of Philadelphia (CHOP) for genetic research purposes. We thank the technical staff at the Center for Applied Genomics at CHOP for producing the genotypes used for analyses and the nursing, medical assistant, and medical staff for their invaluable help with recruitment of patient and control subjects for the study. We thank the Wellcome Trust Case Control Consortium (WTCCC) for making the Affymetrix data available for our analysis. The WTCCC is funded by Wellcome Trust award 076113, and a full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. The study was supported by a Methodological Study Award (K.W. and H.H.) from the National Institutes of Health and National Center for Research Resources (UL1 RR025774). All genome-wide genotyping was funded by an Institute Development Award from CHOP.
Contributor Information
Kai Wang, Email: kaichop@gmail.com.
Hakon Hakonarson, Email: hakonarson@email.chop.edu.
Web Resources
The URLs for data presented herein are as follows:
Connexin-deafness database, http://davinci.crg.es/deafness/
fastPHASE, http://depts.washington.edu/ventures/UW_Technology/Express_Licenses/fastPHASE.php
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Simulation data, http://www.openbioinformatics.org/synassoc/
References
- 1.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 2.Manolio T.A., Brooks L.D., Collins F.S. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakravarti A. Population genetics—making sense out of sequence. Nat. Genet. 1999;21(1, Suppl):56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- 5.Lander E.S. The new genomics: global views of biology. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 6.Wright A.F., Hastie N.D. Complex genetic diseases: controversy over the Croesus code. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-8-comment2007. COMMENT2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schork N.J., Murray S.S., Frazer K.A., Topol E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pritchard J.K., Cox N.J. The allelic architecture of human disease genes: common disease-common variant…or not? Hum. Mol. Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 9.Reich D.E., Lander E.S. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 10.Iles M.M. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genet. 2008;4:e33. doi: 10.1371/journal.pgen.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bodmer W., Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng B., Kimmel M. Simulations provide support for the common disease-common variant hypothesis. Genetics. 2007;175:763–776. doi: 10.1534/genetics.106.058164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith D.J., Lusis A.J. The allelic structure of common disease. Hum. Mol. Genet. 2002;11:2455–2461. doi: 10.1093/hmg/11.20.2455. [DOI] [PubMed] [Google Scholar]
- 14.Iyengar S.K., Elston R.C. The genetic basis of complex traits: rare variants or “common gene, common disease”? Methods Mol. Biol. 2007;376:71–84. doi: 10.1007/978-1-59745-389-9_6. [DOI] [PubMed] [Google Scholar]
- 15.Weiss K.M., Terwilliger J.D. How many diseases does it take to map a gene with SNPs? Nat. Genet. 2000;26:151–157. doi: 10.1038/79866. [DOI] [PubMed] [Google Scholar]
- 16.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hugot J.P., Chamaillard M., Zouali H., Lesage S., Cézard J.P., Belaiche J., Almer S., Tysk C., O'Morain C.A., Gassull M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 18.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 19.Cho J.H., Weaver C.T. The genetics of inflammatory bowel disease. Gastroenterology. 2007;133:1327–1339. doi: 10.1053/j.gastro.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen J.C., Boerwinkle E., Mosley T.H., Jr., Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 22.Romeo S., Yin W., Kozlitina J., Pennacchio L.A., Boerwinkle E., Hobbs H.H., Cohen J.C. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 24.Romeo S., Pennacchio L.A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H.H., Cohen J.C. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kathiresan S., Willer C.J., Peloso G.M., Demissie S., Musunuru K., Schadt E.E., Kaplan L., Bennett D., Li Y., Tanaka T. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X., Fejerman L., Luke A., Adeyemo A., Cooper R.S. Haplotypes produced from rare variants in the promoter and coding regions of angiotensinogen contribute to variation in angiotensinogen levels. Hum. Mol. Genet. 2005;14:639–643. doi: 10.1093/hmg/ddi060. [DOI] [PubMed] [Google Scholar]
- 27.Bishop D.T., Demenais F., Iles M.M., Harland M., Taylor J.C., Corda E., Randerson-Moor J., Aitken J.F., Avril M.F., Azizi E. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009;41:920–925. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernando M.M., Stevens C.R., Walsh E.C., De Jager P.L., Goyette P., Plenge R.M., Vyse T.J., Rioux J.D. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Bakker P.I., McVean G., Sabeti P.C., Miretti M.M., Green T., Marchini J., Ke X., Monsuur A.J., Whittaker P., Delgado M. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miretti M.M., Walsh E.C., Ke X., Delgado M., Griffiths M., Hunt S., Morrison J., Whittaker P., Lander E.S., Cardon L.R. A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005;76:634–646. doi: 10.1086/429393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartl D.L., Clark A.G. Sinauer; Sunderland, MA: 2000. Principles of population genetics. [Google Scholar]
- 32.Marlin S., Feldmann D., Blons H., Loundon N., Rouillon I., Albert S., Chauvin P., Garabédian E.N., Couderc R., Odent S. GJB2 and GJB6 mutations: genotypic and phenotypic correlations in a large cohort of hearing-impaired patients. Arch. Otolaryngol. Head Neck Surg. 2005;131:481–487. doi: 10.1001/archotol.131.6.481. [DOI] [PubMed] [Google Scholar]
- 33.Zondervan K.T., Cardon L.R. The complex interplay among factors that influence allelic association. Nat. Rev. Genet. 2004;5:89–100. doi: 10.1038/nrg1270. [DOI] [PubMed] [Google Scholar]
- 34.Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am. J. Hum. Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- 35.Hemminki K., Försti A., Bermejo J.L. The ‘common disease-common variant’ hypothesis and familial risks. PLoS ONE. 2008;3:e2504. doi: 10.1371/journal.pone.0002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang L., Zöllner S., Abecasis G.R. GENOME: a rapid coalescent-based whole genome simulator. Bioinformatics. 2007;23:1565–1567. doi: 10.1093/bioinformatics/btm138. [DOI] [PubMed] [Google Scholar]
- 37.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabeti P.C., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald G.J. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 39.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francks C., Tozzi F., Farmer A., Vincent J.B., Rujescu D., St Clair D., Muglia P. Population-based linkage analysis of schizophrenia and bipolar case-control cohorts identifies a potential susceptibility locus on 19q13. Mol Psychiatry. 2008;15:19–25. doi: 10.1038/mp.2008.100. [DOI] [PubMed] [Google Scholar]
- 41.Schroeder K.B., Jakobsson M., Crawford M.H., Schurr T.G., Boca S.M., Conrad D.F., Tito R.Y., Osipova L.P., Tarskaia L.A., Zhadanov S.I. Haplotypic background of a private allele at high frequency in the Americas. Mol. Biol. Evol. 2009;26:995–1016. doi: 10.1093/molbev/msp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen J.C., Pertsemlidis A., Fahmi S., Esmail S., Vega G.L., Grundy S.M., Hobbs H.H. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl. Acac. Sci. U S A. 2006;103:1810–1815. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T., Zhu X., Elston R.C. Improving power in contrasting linkage-disequilibrium patterns between cases and controls. Am. J. Hum. Genet. 2007;80:911–920. doi: 10.1086/516794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaykin D.V., Meng Z., Ehm M.G. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am. J. Hum. Genet. 2006;78:737–746. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Economou M., Trikalinos T.A., Loizou K.T., Tsianos E.V., Ioannidis J.P. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. Am. J. Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 47.Pascoe L., Zouali H., Sahbatou M., Hugot J.P. Estimating the odds ratios of Crohn disease for the main CARD15/NOD2 mutations using a conditional maximum likelihood method in pedigrees collected via affected family members. Eur. J. Hum. Genet. 2007;15:864–871. doi: 10.1038/sj.ejhg.5201839. [DOI] [PubMed] [Google Scholar]
- 48.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 49.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis C.M., Whitwell S.C., Forbes A., Sanderson J., Mathew C.G., Marteau T.M. Estimating risks of common complex diseases across genetic and environmental factors: the example of Crohn disease. J. Med. Genet. 2007;44:689–694. doi: 10.1136/jmg.2007.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeager M., Xiao N., Hayes R.B., Bouffard P., Desany B., Burdett L., Orr N., Matthews C., Qi L., Crenshaw A. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum. Genet. 2008;124:161–170. doi: 10.1007/s00439-008-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kenneson A., Van Naarden Braun K., Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet. Med. 2002;4:258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 53.Green G.E., Scott D.A., McDonald J.M., Woodworth G.G., Sheffield V.C., Smith R.J. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211–2216. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- 54.Gloyn A.L., Pearson E.R., Antcliff J.F., Proks P., Bruining G.J., Slingerland A.S., Howard N., Srinivasan S., Silva J.M., Molnes J. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 55.Gloyn A.L., Weedon M.N., Owen K.R., Turner M.J., Knight B.A., Hitman G., Walker M., Levy J.C., Sampson M., Halford S. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 56.Schwanstecher C., Meyer U., Schwanstecher M. K(IR)6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta-cell ATP-sensitive K(+) channels. Diabetes. 2002;51:875–879. doi: 10.2337/diabetes.51.3.875. [DOI] [PubMed] [Google Scholar]
- 57.Li B., Leal S.M. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]