

Abstract
Cenani-Lenz syndrome (CLS) is an autosomal-recessive congenital disorder affecting distal limb development. It is characterized mainly by syndactyly and/or oligodactyly and is now shown to be commonly associated with kidney anomalies. We used a homozygosity-mapping approach to map the CLS1 locus to chromosome 11p11.2-q13.1. By sequencing candidate genes, we identified recessive LRP4 mutations in 12 families with CLS. LRP4 belongs to the low-density lipoprotein (LDL) receptor-related proteins (LRPs), which are essential for various developmental processes. LRP4 is known to antagonize LRP6-mediated activation of canonical Wnt signaling, a function that is lost by the identified mutations. Our findings increase the spectrum of congenital anomalies associated with abnormal lipoprotein receptor-dependent signaling.
Introduction
Spatial and temporal activation of canonical Wnt/β-catenin signaling is an essential developmental process during organogenesis and tissue regeneration.1 Wnt ligands bind to their specific coreceptors, such as frizzled and low-density lipoprotein-related proteins 5 and 6 (LRP5 [MIM 603506], LRP6 [MIM 603507]), leading to a stabilization of β-catenin and transcriptional activation.2 Alteration of the LRP5/6 signaling pathway has been described in cancer development and human diseases.1,3,4 LRP4 (MIM 604270) is another member of the low-density lipoprotein receptor family, but has an antagonistic effect on LRP5/6 signaling. Recent results of GWAS in bone mineral density5 and the finding that Lrp4 serves as a receptor for sclerostin regulating bone metabolism in mice6 highlight the importance of LRP4 in the regulation of bone mineral density and the development of osteoporosis. In mice, Lrp4 dysfunction also causes syndactyly.7
The genetic identification of factors regulating limb formation provided important insights into the role of major signaling pathways, such as sonic-hedgehog (SHH) and fibroblast growth factor (FGF), during limb development.8,9 Cenani-Lenz syndrome (CLS [MIM 212780]) is an autosomal-recessive congenital anomaly affecting mainly distal limb development. CLS is characterized by fusion and disorganization of metacarpal and phalangeal bones, radius and ulnar shortening, radioulnar synostosis, and severe syndactyly of hands and feet.10,11 Kidney hypoplasia has been described in one patient with CLS,12 but is not yet regarded as an associated trait.
Here, we map the CLS1 locus to chromosome 11p11.2-q13.1 and identify mutations in the LRP4 gene in 12 CLS families. We show that LRP4 function is required for the physiological regulation of Wnt signaling, and we identify mutations that cause loss of LRP4 function, which is important for normal limb and kidney development. Therefore, loss of human LRP4 function causes syndactyly, synostosis, and renal agenesis in Cenani-Lenz syndrome.
Material and Methods
Subjects
All subjects or their legal representatives gave written informed consent for participation in the study. The study was performed in accordance to the Declaration of Helsinki protocols and approved by the local institutional review boards. We collected peripheral blood samples from the affected children and parents, after informed consent was obtained, according to the protocols approved by the participating institutions. All of the research procedures followed were in accordance with the ethical standards of the responsible national and institutional committees on human subject research. Fourteen families with the clinical diagnosis of CLS were included in the study. In 12 of them, mutations were identified in LRP4. Clinical features of some of the families have already been published; see families CL-1,12 CL-2,13 CL-3,14 CL-6,15 CL-7.16 DNA from participating family members was extracted from peripheral blood lymphocytes by standard extraction procedures.
Linkage Analysis
We performed genome-wide linkage analysis in six families (CL-1 to CL-6; not all family members could be initially included into the genome scan), using the Affymetrix GeneChip Human Mapping 10K Array (version 2.0). This version of the 10K Chip Array comprises a total of 10,204 SNPs with a mean intermarker distance of 258 kb, equivalent to 0.36 cM. Genotypes were called by the GeneChip DNA Analysis Software (GDAS version 2.0, Affymetrix). We verified sample genders by counting heterozygous SNPs on the X chromosome. Relationship errors were evaluated with the help of the program Graphical Relationship Representation.17 The program PedCheck was applied to detect Mendelian errors,18 and data for SNPs with such errors were removed from the data set. Non-Mendelian errors were identified by use of the program MERLIN,19 and unlikely genotypes for related samples were deleted. Nonparametric linkage analysis using all genotypes of a chromosome simultaneously was carried out with MERLIN. Parametric linkage analysis was performed by a modified version of the program GENEHUNTER 2.120 through stepwise use of a sliding window with sets of 150 or 300 SNPs. Haplotypes were reconstructed with GENEHUNTER 2.1 and presented graphically with HaploPainter.21 This program also reveals informative SNP markers as points of recombination between parental haplotypes. All data handling was performed with the use of the graphical user interface ALOHOMORA,22 developed at the Berlin Gene Mapping Center to facilitate linkage analysis with chip data.
Mutation Screening
We identified candidate genes in the critical region by using the ENSEMBL and UCSC human genome databases. We amplified the 38 exons of the LRP4 gene (primers are listed in Table S1, available online) from DNA of index patients from all 14 families and sequenced the PCR products via the BigDye Terminator method on an ABI 3100 sequencer. We resequenced all identified mutations in independent experiments, tested for cosegregation within the families, and screened at least 200 healthy control individuals from Turkey, 150 from Pakistan, 50 from Germany, and 50 from Egypt for each mutation by PCR and/or restriction digestion or direct sequencing. We analyzed all identified alterations by using the server PolyPhen. The LRP4 protein structure was analyzed with the server Pfam in order to determine different protein domains of LRP4.
cDNA Analysis
RNA was extracted from fresh whole-cell blood through use of the Paxgene Blood RNA system. After cDNA transcription, nested PCR was used to amplify LRP4 cDNA (primers are listed in Table S1). Primers were designed according to the reference sequence.
Generation of Lrp4 Constructs
Five Lrp4 mutant constructs were generated by site-directed mutagenesis with the use of wild-type mouse Lrp4 in the pcDNA3.1/V5-His-TOPO vector (Invitrogen, Karlsruhe, Germany) as template. The correct sequence of all PCR amplicons and constructs was confirmed by direct sequencing from both sides with the use of the ABI BigDye Terminator v1.1 Cycle Sequencing Kit and the ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Cell Culture and Transfections
Human embryonic kidney (HEK)293T cells were cultured in Dulbecco's Modified Eagle Media (DMEM) containing 10% fetal bovine serum (FBS), amphothericin B, streptomycin, and penicillin. Cells were transfected with the use of Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions.
Luciferase Assay
One day before transfection, approx. 400.000 HEK293T cells were plated out in 12-well plates and grown up to 50% confluency in 10% FBS and DMEM. Transfections were performed in triplicate with the use of the TOP-Flash reporter system and the indicated expression plasmids with the following concentrations: 500 ng wild-type (WT) Lrp4 or 500 ng mutants, 250 ng Lrp6, 250 ng Wnt1, 100 ng Topflash Vector, 5 ng Renilla (p-RL-TK). Cells were transfected with the use of Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. Two days after transfection, cells were lysed and Luciferase activity was measured with the use of the Dual-Luciferase Reporter Assay Kit and a Glomex 96-microplate luminometer (Promega, Mannheim, Germany). Each transfection was also measured in triplicate.
Immunoblot
Immunoblot analysis was performed according to standard protocols. Detection of LRP4 was conducted with a C-terminal Lrp4 mouse monoclonal antibody (1:1000).
Cell-Surface Biotin-Labeling Assay
One day before the experiment, 50% confluent HEK293T cells were cotransfected with 1.5 μg LRP4, WT or mutant, and 1.5 μg insulin receptor (IR) in T75 cm2 flasks. Biotinylation was carried out with the use of the Cell Surface Protein Isolation Kit (Pierce, Bonn, Germany) according to the manufacturer's instructions. Protein concentrations were measured with the use of the BCA Protein Assay Kit (Pierce). Immunoblot analysis was performed according to standard protocols. Detection of LRP4 and IR was conducted with a C-terminal LRP4 mouse monoclonal antibody (1:1000) and an insulin-receptor rabbit monoclonal antibody (Abcam, Berlin, Germany) (1:1000).
Results
Clinical Findings in CLS Families
We have examined 14 CLS families presenting with a variable expression of clinical symptoms. In twelve of them we identified the molecular basis of the disease (Figure 1A, Table 1). We observed mild facial dysmorphism in the majority of CLS cases, with prominent forehead, hypertelorism, downslanting palpebral fissures, and micrognathia. Typical limb malformations included total to partial syndactyly of hands and feet, as well as distal bone malformations affecting the radius and ulna as well as the metacarpal and phalangeal bones (Figure 1A, Table 1). Interestingly, we also found kidney anomalies, including renal agenesis and hypoplasia, in over 50% of CLS families.
Figure 1.
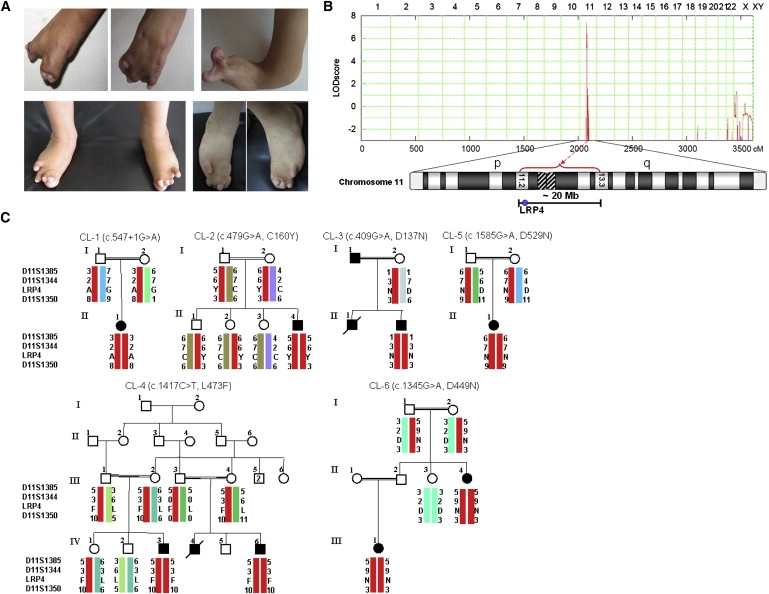
Clinical Findings in Families with CLS and Mapping of the CLS1 Locus
(A) Typical hand and feet anomalies seen in CLS patients.
(B) Graphical view of additive LOD-score calculations of genome-wide SNP mapping in families CL-1 to CL-6. Ideogram of chromosome 11 showing the localization of linked region.
(C) Haplotypes of CLS families included in the initial linkage analysis.
Table 1.
Clinical Findings in CLS Families Carrying LRP4 Mutations
| Family Data | CL-1 | CL-2 | CL-3 | CL-4 | CL-5 | CL-6 | CL-7 | CL-8 | CL-9 | CL-10 | CL-11 | CL-12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Consanguinity | + | + | + | + | + | + | + | + | + | + | - | + |
| No. of affected individuals | 1 | 1 | 3 | 3 | 1 | 2 | 1 | 6 | 2 | 2 | 1 | 1 |
| Mutation | c.547+1G>A | C160Y | D137N | L473F | D529N | D449N | T461P | C1017R | D529N | D529N | c.200-9G>A, c.4959G>C | D137N |
| Origin | Pakistan | Turkey | Egypt | Egypt | Turkey | Turkey | Pakistan | Jordan | Turkey | Turkey | Turkey | Egypt |
| Facial dysmorphism | ||||||||||||
| Prominent forehead | + | - | + | + | - | - | + | + | + | + | + | + |
| Hypertelorism | + | - | + | + | - | - | + | - | + | - | + | + |
| Downsl. palpebral fissures | - | - | + | + | - | - | - | + | - | - | - | + |
| Micro-, retrognathia | + | - | + | + | - | - | - | + | - | - | + | + |
| Teeth findings | ||||||||||||
| Hypodontia | - | - | - | - | - | - | - | - | + | - | ? | ? |
| Malar hypoplasia | + | - | + | + | - | - | + | + | - | - | + | + |
| Enamel hypoplasia | - | - | + | + | - | - | - | - | - | - | ? | ? |
| Early loss of permanent teeth | - | - | + | + | - | ? | - | - | ? | ? | ? | ? |
| Upper limb findings | ||||||||||||
| Typical syndactyly | + | +/− | + | + | + | + | +/− | + | + | - | + | + |
| Short forearms | + | - | + | + | + | + | - | - | - | - | + | + |
| Radius-ulnar synostosis | + | - | + | + | + | + | - | - | ? | ? | + | + |
| Disorganized / missing metacarpals and phalanges | + | + | + | + | + | + | + | + | + | + | + | + |
| Fused metacarpals | + | + | + | + | + | + | - | + | + | + | + | + |
| Nail aplasia | + | +/− | + | + | + | + | + | + | + | - | + | + |
| Lower limb findings | ||||||||||||
| Syndactyly | 2/3 | 2/3 | + | + | 2/3–2/5 | + | + | 1/2/3 | R 2/3 | 2/3 | 1/2 | + |
| Tibia-fibula synostosis | - | - | - | - | - | - | - | - | - | - | - | - |
| Disorganized / missing metatarsals and phalanges | - | - | + | + | +/− | - | + | + | + | - | -/+ | + |
| Nail aplasia /partial | - | - | + | + | +/− | + | + | + | -/+ | - | + | + |
| Kidney anomalies | ||||||||||||
| Agenesis | - | - | - | - | - | - | unilateral | - | bilateral | bilateral | bilateral | - |
| Hypoplasia | bilateral | - | - | - | - | unilateral | - | unilateral | - | - | - | - |
| Ectopic localization | - | - | - | - | - | + | - | + | - | - | - | - |
| Additional findings | ||||||||||||
| Developmental delay | - | - | + | - | - | - | - | - | - | mild gross motor delay | ? | ? |
| Other | bilateral broad hallux valgus anomaly | hypoplastic scrotum | scoliosis, hemivertebrae, mixed-type hearing loss | duplicated distal phalanges of the first and second toe, congenital cataract | pulmonary stenosis, congenital hip dislocation | hypothyroidism | G1: died at first day of life due to bilateral renal agenesis | G2: medical abortion (20. GW), bilateral renal agenesis | medical abortion (20. GW) | pectus excavatum |
Mapping of the CLS1 Locus and Identification of LRP4 Mutations
Initially, we genotyped DNA samples from six CLS families (CL-1 to CL-6, Figure 1) by using the Affymetrix GeneChip Human Mapping 10K Array. Affected individuals were born to consanguineous parents in all families. A combined parametric LOD score of 7.46 was obtained for a single region located on chromosome 11p11.2-q13.1 between SNPs rs1346671 and rs490192 (Figure 1B), defining a shared critical interval of about 19.7 Mb. Subsequent analysis of microsatellite markers and inclusion of additional family members confirmed homozygous haplotypes for the linked region in all affected individuals (Figure 1C). We considered LRP4 as a highly relevant positional and functional candidate gene. No additional gene from the critical region was tested. Sequencing of the 38 coding exons of LRP4 (Table S1) revealed different homozygous mutations in affected individuals in each of the six families. The mutations cosegregated with the disease in the families and were not found in at least 250 healthy control individuals. We found one donor splice-site mutation, c.547+1G>A (intron 6, CL-1), and five missense mutations: c.479G>A (p.C160Y, exon 5, CL-2), c.409G>A (p.D137N, exon 4, CL-3), c.1417C>T (p.L473F, exon 12, CL-4), c.1585G>A (p.D529N, exon 13, CL-5), c.1345G>A (p.D449N, exon 12, CL-6) (Figure 2A). All missense mutations are located in the extracellular domains of LRP4 within highly conserved regions, as shown by LRP4 protein alignments of various species (Figure S1). p.C160Y and p.D137N mutations lie within the ligand-binding (class A) repeat-containing domain of the receptor, p.D449N and p.L473F are located within calcium-binding epidermal growth factor (EGF) repeats, and D529 is located within the YWTD domain (Figure 2C).
Figure 2.
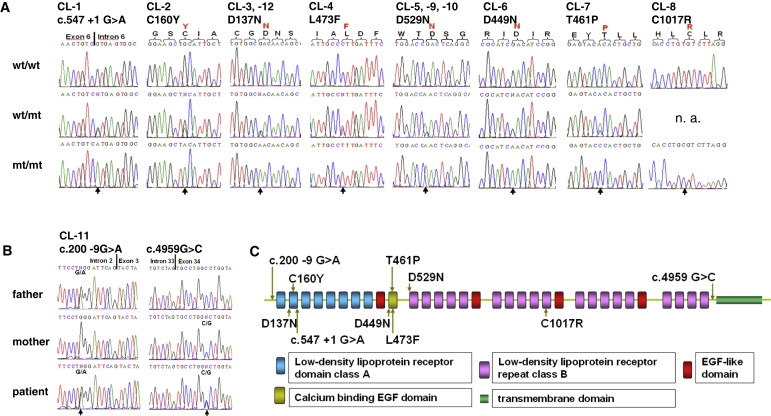
Mutations Identified in LRP4
(A) Electropherograms of identified homozygous LRP4 mutations compared with heterozygous carrier and WT sequences (n.a., not available).
(B) Electropherograms of identified compound-heterozygous splice-site mutations in LRP4 causing aberrant splicing and premature protein truncation.
(C) Schematic view of LRP4 receptor domains and localization of identified CLS mutations.
We continued the molecular analysis of LRP4 in eight additional CLS families (Figures 3A and 3C). In families CL-9 and CL-10, we found the same homozygous p.D529N mutation as identified before in the CL-5 family. All three families originated from Turkey, and haplotype analysis confirmed that p.D529N is a common founder mutation in Turkish CLS patients (Figure 3C). It is also interesting to note that the p.D137N mutation is repeatedly found in CLS families from Egypt, as we could identify a second family, CL-12, carrying this mutation. p.D137N in both families was located on identical haplotypes (Figure 3D), suggesting that p.D137N is a founder mutation. Two additional missense mutations were found: c.1382A>C (p.T461P, exon 11, CL-7) in a Jordanian patient and c.3049T>C (p.C1017R, exon 22, CL-8) in a large CLS family from Pakistan with six affected family members (Figure 2B, Figure 3A). These mutations also cosegregated with the disease, were not found in matched controls, and were located in highly conserved regions (Figure S1). Furthermore, no LRP4 mutation was found in two other consanguineous CLS families, and haplotype analysis did not show homozygosity of the LRP4 region in affected individuals from both families, supporting the idea of further locus heterogeneity (Figure 3B).
Figure 3.
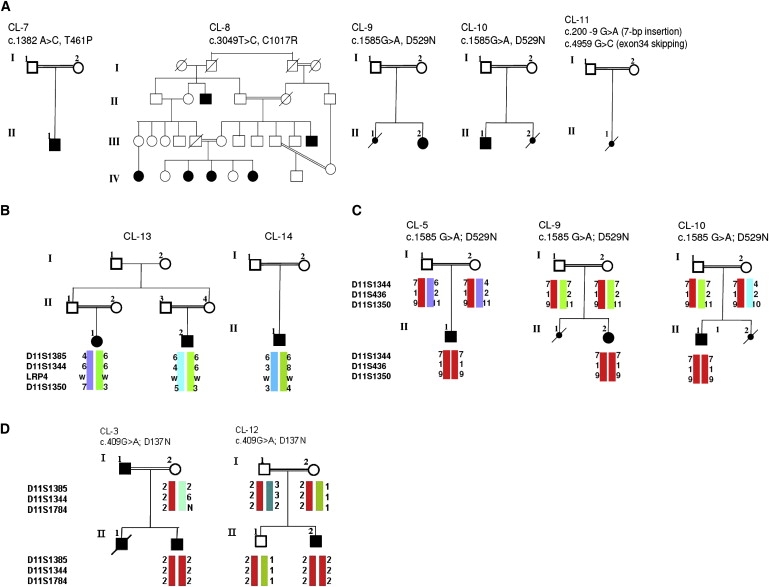
Additional CLS Families and Genetic Heterogeneity
(A) Pedigrees of additional CLS families.
(B) The LRP4 locus was excluded in families CL-13 and CL-14 by haplotype analysis.
(C and D) Identification of an LRP4 founder mutation, p.D529N, in three Turkish CLS families (C) and of p.D137N in two families from Egypt (D). Identical haplotypes are shown in red.
We found initial evidence for an impairment of LRP4 function as the underlying pathomechanism of CLS by identifying compound-heterozygous splice-site mutations in a typically affected fetus with CLS. Both mutations, c.200-9G>A and c.4959G>C (Figure 2C), caused aberrantly spliced LRP4 transcripts (r.199_200insGATTCAG and r.4952_4987del, respectively), and both mutations lead to a truncated protein (Figures 4A and 4B).
Figure 4.
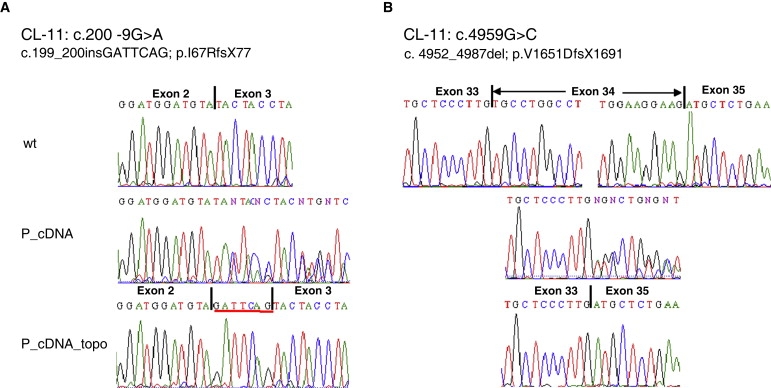
Splicing Effects of Mutations in Family CL-11
(A) Analysis of the heterozygous c.200-9G>A mutation. cDNA sequences of the exon 2-exon 3 boundary of wild-type (wt) and patient cDNA are shown (without [P_cDNA] and after subcloning via the TOPO-Vector system [P_cDNA_topo]).
(B) Analysis of the heterozygous c.4959G>C mutation. Electropherograms show the cDNA sequences of LRP4 transcripts encoded by exon 33 to exon 35 of wild-type (wt), and skipping of exon 34 in the patient cDNA.
LRP4 Mutations Cause Loss of Protein Function
To investigate whether the identified missense mutations confer loss of function or whether they are functionally hypomorphic with biochemically detectable residual protein activity, we analyzed the effect of five missense mutations (p.D137N, p.C160Y, p.L473F, p.D449N, and p.D529N) on the transduction and activation of canonical Wnt signaling by using a Dual Luciferase Reporter Assay in transiently transfected HEK293T cells. Consistent with earlier findings,5 we found that WNT1 was able to significantly activate LRP6-mediated β-catenin signaling and that additional coexpression of LRP4 potently antagonized this activation (Figure 5A). In contrast, coexpression of each of the five missense mutations abolished the observed antagonistic LRP4 effect on LRP6-mediated activation of Wnt/β-catenin signaling. Moreover, mutant LRP4 receptors failed to be efficiently transported to the plasma membrane, as shown by cell-surface biotinylation (Figure 5B).
Figure 5.
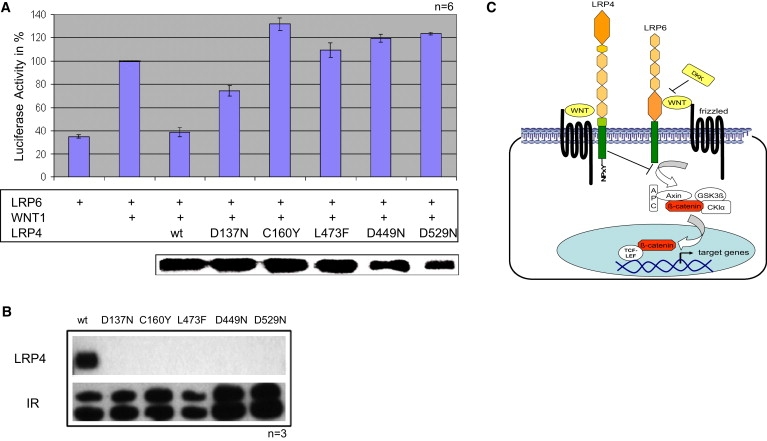
Functional Analysis of LRP4 Missense Mutations
(A) Results from the Dual Luciferase Reporter Assay after coexpression of LRP6, WNT1, and wild-type and mutant LRP4 in different combinations. Graph depicting the relative Luciferase activities (mean and standard deviations of six experiments, in triplicate each time). Comparable steady-state expression levels of wild-type and mutant LRP4 proteins are shown by immunoblotting of total lysates after measurement of luciferase activity.
(B) Compared to wild-type LRP4 and wild-type insulin receptor (IR), mutant LRP4 proteins are not detectable by cell-surface biotinylation. Results from three independent experiments are shown.
(C) Schematic representation of LRP4 function in LRP6-mediated activation of Wnt/β-catenin signaling.
Discussion
We report the mapping of the CLS1 locus to chromosome 11p11.2-q13.1 and present convincing evidence that mutations in the LRP4 gene cause CLS. Clinical findings in our CLS patients showed that in addition to the well-described distal limb malformations (ranging from total to partial syndactyly and bone malformations of both hands and feet), patients presented with facial features such as prominent forehead, hypertelorism, downslanting palpebral fissures, and micrognathia. Previously, renal hypoplasia has been reported in only one case16 and was therefore not regarded as an associated trait of CLS. Our finding that over 50% of CLS families present with renal agenesis and/or hypoplasia adds kidney anomalies to the clinical spectrum of CLS. In this context, it is interesting to note that a subpenetrant phenotype of kidney agenesis was observed in Lrp4 homozygous null mice. In the Lrp4−/− homozygous kidneys the ureteric budding is often delayed, resulting in insufficient stimulation of the mesenchyme. This results in destruction of prenephric mesenchymal structures, and no kidneys are formed (J. Herz, personal communication). These findings clearly show that Lrp4 has an important function for kidney development in mice and humans.
Recently, murine LRP4 was shown to serve as a coreceptor for agrin in the formation of the neuromuscular junction.23 Mutations in genes encoding other members of this complex have been associated with akinetic and myasthenic syndromes in humans (MIM 288150 and MIM 254300).24,25 Given that we did not observe a clinically detectable neuromuscular phenotype in our CLS patients, the role of LRP4 in the development and function of the neuromuscular junction in humans (as opposed to mice) will require further investigation. In the case that the LRP4 mutations found in CLS patients are not complete loss-of-function mutations and do have minor residual functionality in vivo, there could be a different but overlapping phenotype caused by, for example, homozygous nonsense mutations or deletions in LRP4, and this could include neuromuscular symptoms.
The LRP4 mutations identified in our study are frequently missense mutations, which are located in the large extracellular domain of LRP4 within the ligand-binding (class A) repeat-containing domain, calcium-binding EGF repeats, and the YWTD domain of the receptor (Figure 2C). These changes might cause structural alterations of the extracellular LRP4 domain that interfere with normal folding and thus prevent the efficient export of the protein through the secretory pathway, but this hypothesis has to be proven in future experimental studies. Our functional analysis of five of the missense mutations clearly demonstrated a functional impairment of LRP4 mutant proteins. LRP4 is important for control and modification of Wnt signaling by its antagonistic effect on LRP6-mediated activation of WNT signaling (Figure 5C). This antagonistic function is completely lost in four out of five LRP4 mutants, as shown in our in a Dual Luciferase Reporter Assay. The p.D137N mutant seems to show some residual antagonistic function in the Reporter Assay experiment, and the biotinylation experiments clearly demonstrated that p.D137N mutant protein is not getting to the cell surface. Whether a yet unknown function of LRP4, which is not dependent on its membrane integration, could be responsible for this residual function remains to be elucidated.
We also found that WNT1 was able to significantly activate LRP6-mediated β-catenin signaling, which is consistent with earlier findings.7 We demonstrated that the main reason for the loss-of-function effect is the failure of mutant LRP4 receptors to be efficiently transported to the plasma membrane. In addition, the heterozygous splice-site mutations identified in the CL-11 fetus, c.200-9G>A and c.4959G>C, caused aberrantly spliced LRP4 transcripts and premature protein truncations. Conclusively, we suggest complete or near-complete loss of LRP4 function as the underlying pathogenetic mechanism of CLS. As a result, developmental limb and kidney malformations in patients occur through a mechanism that likely also involves excessive LRP6-mediated Wnt/β-catenin activation (Figure 5C).
It has been previously shown that Lrp4 dysfunction also causes polysyndactyly in mice7 and syndactyly with variable penetrance in bovines, termed mulefoot disease.26 Lrp4 was shown to be expressed in the apical ectodermal ridge (AER) in the developing limb bud,7 a structure important for coordination of patterning and growth of the distal limb.8 Various signaling molecules are secreted from the AER, such as sonic hedgehog (Shh), bone morphogenic proteins (Bmps), fibroblast growth factors (Fgfs), and Wnts, and the complex interactions of these signaling pathways are essential for normal limb development.27,28 Extensive analysis of the limb phenotype in Lrp4−/− knockout mice showed that loss of Lrp4 causes structural AER alterations as well as ectopic expression of different key signaling molecules (e.g., Fgf8, Bmp4, and Shh).7 Whether Lrp6 expression was upregulated during limb development in the AER of Lrp4−/− mice was not analyzed. Given the facts that (1) Lrp4 was described as an integrator of Wnt and Bmp signaling,29 (2) Lrp4 has an antagonistic function on Lrp6-mediated Wnt/β-catenin activation, and (3) Lrp6 is critical for Wnt signaling during limb development in mice,30 it is a reasonable working hypothesis that loss of LRP4 during limb development in CLS patients could lead to an overactivation of LRP6, which then causes altered Wnt signaling. Future approaches are needed to show that knocking down LRP4 expression upregulates Wnt/β-catenin in vivo.
In two CLS families, we did not find LRP4 mutations, and haplotype analysis did now show homozygosity in affected individuals born to consanguineous parents. In the CL-13 family with only a single affected individual, homozygosity is only an assumption due to parental consanguinity, but lack of homozygosity does not completely exclude LRP4 as causative gene. In the CL-14 family, haplotype analysis of microsatellite markers as well as results from 250K arrray analysis excluded common haplotypes in both affected individuals, suggesting further locus heterogeneity in CLS. Future identification of a causative gene(s) in these families will highlight additional key proteins for distal limb development.
We conclude that LRP4 function is required for the physiological regulation of Wnt signaling, which is important for normal limb and kidney development. Homozygous loss of human LRP4 function causes syndactyly, synostosis, and renal agenesis in Cenani-Lenz syndrome.
Acknowledgments
We are thankful to all family members that participated in this study, to Bernhard Zabel for referral of patients, to Esther Milz for excellent technical assistance, and to Christian Kubisch, Brunhilde Wirth, and Karin Boss for critical reading of the manuscript. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant numbers 01GM0880 (SKELNET) and 01GM0801 (E-RARE network CRANIRARE) to B.W. J.H. is supported by grants from the National Institutes of Health, the American Health Assistance Foundation, the Perot Family Foundation, and the Wolfgang-Paul Program of the Alexander-von-Humboldt Foundation. C.N. was supported by the German Research Foundation (DFG), grant NE826/3-2.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ENSEMBL, http://www.ensembl.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Pfam Server, http://pfam.sanger.ac.uk/
PolyPhen, http://coot.embl.de/PolyPhen
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Gordon M.D., Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 3.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D., Osteoporosis-Pseudoglioma Syndrome Collaborative Group LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 4.Mani A., Radhakrishnan J., Wang H., Mani A., Mani M.A., Nelson-Williams C., Carew K.S., Mane S., Najmabadi H., Wu D., Lifton R.P. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Styrkarsdottir U., Halldorsson B.V., Gretarsdottir S., Gudbjartsson D.F., Walters G.B., Ingvarsson T., Jonsdottir T., Saemundsdottir J., Snorradóttir S., Center J.R. New sequence variants associated with bone mineral density. Nat. Genet. 2009;41:15–17. doi: 10.1038/ng.284. [DOI] [PubMed] [Google Scholar]
- 6.Choi H.Y., Dieckmann M., Herz J., Niemeier A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE. 2009;4:e7930. doi: 10.1371/journal.pone.0007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson E.B., Hammer R.E., Herz J. Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum. Mol. Genet. 2005;14:3521–3538. doi: 10.1093/hmg/ddi381. [DOI] [PubMed] [Google Scholar]
- 8.Niswander L. Pattern formation: old models out on a limb. Nat. Rev. Genet. 2003;4:133–143. doi: 10.1038/nrg1001. [DOI] [PubMed] [Google Scholar]
- 9.Ingham P.W., Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat. Rev. Genet. 2006;7:841–850. doi: 10.1038/nrg1969. [DOI] [PubMed] [Google Scholar]
- 10.Cenani A., Lenz W. Total syndactylia and total radioulnar synostosis in 2 brothers. A contribution on the genetics of syndactylia. Ztschr. Kinderheilk. 1967;101:181–190. [PubMed] [Google Scholar]
- 11.Elçioglu N., Atasu M., Cenani A. Dermatoglyphics in patients with Cenani-Lenz type syndactyly: studies in a new case. Am. J. Med. Genet. 1997;70:341–345. doi: 10.1002/(sici)1096-8628(19970627)70:4<341::aid-ajmg1>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 12.Bacchelli C., Goodman F.R., Scambler P.J., Winter R.M. Cenani-Lenz syndrome with renal hypoplasia is not linked to FORMIN or GREMLIN. Clin. Genet. 2001;59:203–205. doi: 10.1034/j.1399-0004.2001.590312.x. [DOI] [PubMed] [Google Scholar]
- 13.Seven M., Yüksel A., Ozkiliç A., Elçioğlu N. A variant of Cenani-Lenz type syndactyly. Genet. Couns. 2000;11:41–47. [PubMed] [Google Scholar]
- 14.Temtamy S.A., Ismail S., Nemat A. Mild facial dysmorphism and quasidominant inheritance in Cenani-Lenz syndrome. Clin. Dysmorphol. 2003;12:77–83. doi: 10.1097/00019605-200304000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Percin E.F., Percin S. Two unusual types of syndactyly in the same family; Cenani-Lenz type and “new” type versus severe type I syndactyly? Genet. Couns. 2003;14:313–319. [PubMed] [Google Scholar]
- 16.Jarbhou H., Hamamy H., Al-Hadidy A., Ajlouni K. Cenani-Lenz syndactyly with facial dysmorphism, hypothyroidism, and renal hypoplasia: a case report. Clin. Dysmorphol. 2008;17:269–270. doi: 10.1097/MCD.0b013e328306a6ed. [DOI] [PubMed] [Google Scholar]
- 17.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 18.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 20.Strauch K., Fimmers R., Kurz T., Deichmann K.A., Wienker T.F., Baur M.P. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am. J. Hum. Genet. 2000;66:1945–1957. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiele H., Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 22.Rüschendorf F., Nürnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 23.Kim N., Stiegler A.L., Cameron T.O., Hallock P.T., Gomez A.M., Huang J.H., Hubbard S.R., Dustin M.L., Burden S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huzé C., Bauché S., Richard P., Chevessier F., Goillot E., Gaudon K., Ben Ammar A., Chaboud A., Grosjean I., Lecuyer H.A. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009;85:155–167. doi: 10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michalk A., Stricker S., Becker J., Rupps R., Pantzar T., Miertus J., Botta G., Naretto V.G., Janetzki C., Yaqoob N. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am. J. Hum. Genet. 2008;82:464–476. doi: 10.1016/j.ajhg.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson E.B., Steffen D.J., Lynch K.W., Herz J. Defective splicing of Megf7/Lrp4, a regulator of distal limb development, in autosomal recessive mulefoot disease. Genomics. 2006;88:600–609. doi: 10.1016/j.ygeno.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Capdevila J., Izpisúa Belmonte J.C. Patterning mechanisms controlling vertebrate limb development. Annu. Rev. Cell Dev. Biol. 2001;17:87–132. doi: 10.1146/annurev.cellbio.17.1.87. [DOI] [PubMed] [Google Scholar]
- 28.Barrow J.R., Thomas K.R., Boussadia-Zahui O., Moore R., Kemler R., Capecchi M.R., McMahon A.P. Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17:394–409. doi: 10.1101/gad.1044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohazama A., Johnson E.B., Ota M.S., Choi H.Y., Choi H.J., Porntaveetus T., Oommen S., Itoh N., Eto K., Gritli-Linde A. Lrp4 modulates extracellular integration of cell signaling pathways in development. PLoS ONE. 2008;3:e4092. doi: 10.1371/journal.pone.0004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinson K.I., Brennan J., Monkley S., Avery B.J., Skarnes W.C. Nature. 2000;28:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.