

Abstract
The ascomycete fungus Gibberella zeae is an important plant pathogen that causes fusarium head blight on small grains. Molecular studies of this fungus have been performed extensively to uncover the biological mechanisms related to pathogenicity, toxin production, and sexual reproduction. Molecular methods, such as targeted gene deletion, gene overexpression, and gene fusion to green fluorescent protein (GFP), are relatively easy to perform with this fungus; however, conditional expression systems have not been developed. The purpose of this study was to identify a promoter that could be induced by zearalenone (ZEA) for the development of a conditional expression system in G. zeae. Through microarray analysis, we isolated one zearalenone response gene (ZEAR) whose expression was increased more than 50 times after ZEA treatment. Northern blot analysis showed that the ZEAR transcript dramatically increased after 1 h of ZEA treatment. To determine the utility of the ZEAR promoter, called Pzear, in a conditional expression system, we transformed a Pzear::GFP fusion construct into G. zeae. Our data showed a ZEA concentration-dependent increase in GFP expression. We also replaced the promoter of G. zeae metE (GzmetE), an essential gene for methionine biosynthesis, with the Pzear promoter. The growth of the Pzear-GzmetE mutant on minimal medium was dependent on the ZEA concentration supplemented in the medium and showed that GzMetE expression was induced by ZEA. This study is the first report of an inducible promoter in G. zeae. Our system will be useful for the characterization of essential gene functions in this fungus through differential and ZEA-dependent gene expression. In addition, the Pzear promoter may be applicable as a biosensor for the detection of ZEA contamination in agricultural products.
The ascomycete fungus Gibberella zeae (anamorph: Fusarium graminearum) is an important plant pathogen that causes fusarium ear rot disease on maize and head blight on barley, wheat, and rice (17). This fungus produces mycotoxins, such as trichothecenes and zearalenone (ZEA), which are harmful to humans and animals. Trichothecenes are sesquiterpenoids that are potent inhibitors of eukaryotic protein biosynthesis and cause food refusal, diarrhea, emesis, alimentary hemorrhaging, and contact dermatitis in animals (3). In addition, trichothecenes are virulence factors in plants (26). ZEA [6-(10-hydroxy-6-oxo-trans-1-undecenyl)-β-resorcyclic acid lactone] is a polyketide metabolite (3) that causes estrogenic disorders in laboratory rats, mice, and farm-raised swine that have ingested fusarium-contaminated maize, wheat, and barley (22). ZEA biosynthesis genes in G. zeae are located in a gene cluster that contains two polyketide synthase genes, one putative transcription factor, and one putative isoamyl alcohol oxidase gene (4, 12, 19). ZEA-nonproducing mutants generated from G. zeae field strains do not have any noticeable phenotypic changes except for a loss of ZEA production (4, 12, 19). The biological functions of ZEA in G. zeae have not been characterized to date.
Molecular studies of G. zeae have been widely performed by many research groups since the genome was sequenced by the Broad Institute (http://www.broadinstitute.org). Molecular manipulations, including targeted gene deletion, gene overexpression, and gene fusion to green fluorescent protein (GFP), are relatively easy to perform with this fungus. In addition, mutant collections of G. zeae generated through restriction enzyme-mediated integration mutagenesis are currently available (6, 29), and the functions of genes related to toxin biosynthesis, sexual reproduction, pigmentation, and virulence in the fungus have been well characterized (3, 9, 11, 14, 37). However, a conditional expression system that uses a conditional promoter for the study of essential genes and induction of transgenes is currently not available in G. zeae.
Regulation of gene expression by conditional promoters, including copper-, cadmium-, thiamine-, and alcohol-responsive promoters, has been developed in various organisms (23, 25, 30, 34-35). These systems have been used as tools for studying the molecular regulation of target gene expression and protein production of genes in industrial fungi (20, 39). Further, recombinant bacterial and fungal strains that possess a metal-inducible promoter have been used as biosensors for monitoring heavy metal contamination (25, 33). A wound-inducible promoter was also recently used to generate transgenic plants that can recognize fungal infections (36).
Previously, we performed a microarray analysis to characterize the biological functions of ZEA in G. zeae and found that the transcripts of certain genes, including ZEA biosynthetic genes, were highly elevated after ZEA treatment (S. Lee, J. Lee, and Y.-W. Lee, unpublished findings). We hypothesized that the promoters of genes upregulated by ZEA treatment could be used as inducible promoters for the conditional expression of target genes in response to ZEA treatment. From our analysis of genes upregulated by exogenous treatment of ZEA, one ZEA response gene (Broad Institute; FGSG_04581.3) was selected for further study based on an expression level that was 50 times higher than that of the control in cultures containing ZEA. The gene was designated a ZEA response gene (ZEAR). In this study, we have successfully identified a ZEA-inducible promoter and applied it as a tool for regulating gene expression in G. zeae.
MATERIALS AND METHODS
Fungal strains and media.
The G. zeae strain GZ03639 was provided by Robert L. Bowden (U.S. Department of Agriculture-Agricultural Research Service, Manhattan, KS). The Fusarium oxysporum strain FO0901 and F. verticillioides strain FV0201 were isolated from South Korea and used as wild-type strains. The G. zeae methionine ΔGzmetE auxotroph strain, which was previously generated (6), was used as a negative control for the promoter replacement experiments. All strains used in this study were stored as frozen conidial suspensions in 15% glycerol at −70°C. For DNA and RNA extractions, strains were grown in 25 ml of complete medium (CM) or minimal medium (MM) (17). Conidia were produced on yeast malt agar (YMA) (7). The growth rates of strains were tested on CM, MM, and potato dextrose agar (PDA) (17), and strains were grown on rice medium for toxin production (15). To test for activity of an inducible promoter, strains were inoculated on CM and MM supplemented with 0, 3, 15, and 30 μM ZEA or 0.25 mM methionine (Sigma-Aldrich, St. Louis, MO).
Nucleic acid manipulations, primers, and PCR conditions.
Fungal genomic DNA and total RNA were prepared using a cetyl trimethyl ammonium bromide protocol (17) and the Trizol reagent (Invitrogen, Carlsbad, CA), respectively. Standard procedures were used for restriction endonuclease digestion, agarose gel electrophoresis, and Southern and Northern blot hybridization (28). One hundred ng of PCR product was labeled with [α-32P]dCTP by random priming and was used as a probe in the Southern and Northern blot analyses. Hybridization was carried out at 65°C for 18 h. The general PCR was performed as previously described (14). The PCR primers (Table 1) used in this study were synthesized by the Bioneer oligonucleotide synthesis facility (Bioneer, Daejon, South Korea).
TABLE 1.
Primers used in this study
| Name | Sequence (5′-3′) |
|---|---|
| gfp-f1-zear | GAGAGAACGAAAGTAACCATGGTGAGCAAGGGCGAGGAGC |
| hph-f1 | GGCTTGGCTGGAGCTAGTGGAGG |
| hph-r2 | AACCCGCGGTCGGCATCTACTCTA |
| hph-f3 | GATGTAGGAGGGCGTGGATATGT |
| hph-r4 | GAACCCGCTCGTCTGGCTAAG |
| zear-f1 | GAGCGTGTCACCTACCGAGAGC |
| zear-f1-nt | CATGCCCTGGCGTTGAAGTT |
| zear-r2-gfp | CCTCGCCCTTGCTCACCATGGTTACTTTCGTTCTCTCTGG |
| zear-f3-hph | CCTCCACTAGCTCCAGCCAAGCCTAATTTGGCGTTGGTTGGTCTT |
| zear-r4 | ACGATCCCGGTGCTTGGTT |
| zear-r4-nt | TGGTTCCGTGCCGTTTGTGATA |
| zear-orf1 | ATGGCTTCTGATCAGCAACGCC |
| zear-orf2 | TCATGCCTCCATCTTCTCTCTTTC |
| Pzear-f1-hph | TAGAGTAGATGCCGACCGCGGGTTCATGCCCTGGCGTTGAAGTT |
| Pzear-r2 | GGTTACTTTCGTTCTCTCTGGTCT |
| gzmetE-f1 | GCGGTGGTACGTAAGTCGGTTTCT |
| gzmetE-f1-nt | CGTGCTGCCCGAGTGAAGTTT |
| gzmetE-r2-hph | TCCACTAGCTCCAGCCAAGCCGGTATGGCGTAATTCTGCTTGG |
| gzmetE-f3-Pzear | GAGAGAACGAAAGTAACCATGTCAGAAATCAACACTACAAACGG |
| gzmetE-r4 | ATGGCCGTCGTTGTGGATTTG |
| gzmetE-r4-nt | ACTTGGGGTCGGCGTAGATGC |
| gzmetE-orf1 | TGTTGTCAAGCGCACCTCCATA |
| gzmetE-orf2 | CTGCGCGATCTCCTCTTGTTC |
| Fzear-orf1 | ATGGCACCCGAACAACAACGAC |
| Fzear-orf2 | CCTGATGCCTCTCATCAGGACTC |
| Fozear-f1 | TCGTCGTGGGCTTCATGTTGTAGA |
| Fozear-f1-nt | TGGTCGATGTTGGGCGGTAAGT |
| Fozear-r2-gfp | CGCCCTTGCTCACCATGGTTTGTGTTATACGATAAGCCACTAGCCG |
| Fozear-f3-hph | CCTCCACTAGCTCCAGCCAAGCCATGCGCTTGCTACGGGGACAG |
| Fozear-r4 | ACCAAAAGACGATGAGGGTGCTATC |
| Fozear-r4-nt | AGCCTCTCCTGAAATTGCTAAACGA |
| Fvzear-f1 | CCTCAGCGGTGTAGATAGTTTCTCCC |
| Fvzear-f1-nt | GTTTCTCCCCTGTGATGCTTGTGC |
| Fvzear-r2-gfp | CGCCCTTGCTCACCATGGTTTATGCTTATACGATATGCGCACTAGC |
| Fvzear-f3-hph | CCTCCACTAGCTCCAGCCAAGCCGGGTATTGGGTTATTGGGGAGTTCAT |
| Fvzear-r4 | TCCCCAGAATTTCCCCAACAATG |
| Fvzear-r4-nt | TAGGTCCAGGCCGCCATACTG |
Targeted gene deletion using GFP reporter constructs.
A green fluorescent protein (GFP) reporter construct was created by double-joint PCR (38). A gfp-hph fragment (2.7 kb) that carries a GFP open reading frame (ORF) and hygromycin phosphotransferase cassette (hph) was amplified from pIGPAPA (8) with gfp-f1-zear/hph-f1 primers (Table 1). The 5′ and 3′ flanking regions of ZEAR (Broad Institute; FGSG_04581.3) were amplified by PCR from the wild-type strain GZ03639 with the zear-f1/zear-r2-gfp and zear-f3-hph/zear-r4 primers, respectively. The PCR amplification conditions were 2 min at 94°C, followed by 30 cycles of 30 s at 94°C, 1 min at 55°C, and 2 min at 72°C, followed by a final extension for 10 min at 72°C. The PCR products were purified with the DNA purification system (Promega, Madison, WI) using the manufacturer's instructions. The three amplicons were fused by PCR in a 25-μl reaction mixture containing 2 μl of the 5′-flanking amplicon (50 ng/μl), 2 μl of the 3′-flanking amplicon (50 ng/μl), 3 μl of the hph amplicon (100 ng/μl), 2 μl of deoxynucleoside triphosphates (dNTPs) (2.5 mM each), 2.5 μl of 10× PCR buffer including MgCl2, 1 U of ExTaq polymerase (Takara Bio Inc., Japan), and 13.25 μl of water. The PCR amplification conditions were 2 min at 94°C, followed by 10 cycles of 30 s at 94°C, 20 min at 58°C, and 5 min at 72°C, followed by a final extension of 10 min at 72°C. One μl of this amplification mixture was reamplified as a template in PCRs with zear-f1-nt/hph-f3 and hph-r4/zear-r4-nt primer sets and a 50-μl reaction volume. The PCR conditions were 2 min at 94°C, followed by 30 cycles of 30 s at 94°C, 1 min at 60°C, and 3 min at 72°C, followed by a final extension for 10 min at 72°C. Constructs for F. oxysporum and F. verticillioides were created using the same approach. These amplification products were combined and used to directly transform fungal protoplasts using a polyethylene glycol (PEG)-mediated method (15).
Promoter replacement with an inducible promoter.
The 5′ flanking region of the GzmetE (Broad Institute; FGSG_05658.3) gene, which is essential for methionine biosynthesis in G. zeae and deletion of which causes methionine auxotrophy (6), was replaced with the 5′ flanking region of ZEAR. The hph (1.4 kb) gene was amplified from pIGPAPA with the hph-f1/hph-r2 primer pairs. An 838-bp fragment corresponding to the −838 to −1 bp region of the ZEAR translational start site was amplified from the wild-type strain GZ03639 with the Pzear-f1-hph/Pzear-r2 primers. The 5′ flanking region and ORF of GzmetE were amplified from the wild-type strain with the gzmetE-f1/gzmetE-r2-hph and gzmetE-f3-Pzear/gzmetE-r4 primer sets, respectively. After PCR purification, the four PCR products were fused by PCR, and 1 μl of this amplification mixture was reamplified as a template in PCRs with the gzmetE-f1-nt/hph-f3 and hph-r4/gzmetE-r4-nt primer sets. These amplification products were transformed into protoplasts of the wild-type strain GZ03639.
Expression profiles of genes in response to ZEA treatment.
Strains were incubated in 25 ml CM for 72 h at 24°C on a rotary shaker (150 rpm), and mycelia harvested through filtration subsequently were washed twice with 50 ml of sterile, distilled water. The mycelia were reincubated in 25 ml CM or MM supplemented with 0, 3, 15, and 30 μM ZEA, and total RNA was extracted from the cultures at 1, 2, 4, and 8 h after ZEA treatment for Northern blot analysis. PCR fragments amplified from the wild-type strains with the zear-orf1/zear-orf2, gzmetE-orf1/gzmetE-orf2, and Fzear-orf1/Fzear-orf2 primer sets were used as probes for analyzing transcript levels of ZEAR, GzmetE, and ZEAR homologs, respectively.
Microscopic observation.
Conidia (105) produced on YMA were reincubated in 3 ml of CM supplemented with 0, 3, 6, 9, 12, 15, 30, 300, 1,500, and 3,000 μM ZEA and observed at 1, 2, 4, and 8 h after inoculation with a DE/Axio Imager A1 microscope (Carl Zeiss, Oberkochen, Germany) with excitation and emission wavelengths of 488 and 515/530 nm, respectively.
RESULTS AND DISCUSSION
Identification of ZEA-inducible genes.
We previously performed a microarray analysis to characterize the biological functions of ZEA in G. zeae. To synthesize cDNA for the microarray analysis, the wild-type strain GZ03639 of G. zeae was grown in 25 ml of an SG medium (1), which is used for ZEA production by G. zeae, and supplemented with 0 or 15 μM ZEA for 3 days at 24°C (Lee et al., unpublished). Interestingly, the expression level of ZEAR (Broad Institute; FGSG_04581.3) in the SG culture supplemented with 15 μM ZEA was 50 times higher than that in the SG culture without ZEA. We hypothesized that this gene was induced by ZEA treatment and that the promoter of the gene could be developed as an inducible promoter in G. zeae. Since G. zeae does not produce ZEA when grown in CM, we confirmed our hypothesis by performing Northern blot analysis of RNA extracted from the CM culture treated with exogenous ZEA. The expression of ZEAR markedly increased in the CM culture after 1 h of ZEA treatment and subsequently decreased over time. The transcript was not detected in the culture without ZEA treatment. In addition, the level of ZEAR expression was dependent on the ZEA concentration used (Fig. 1). This result supported the hypothesis that expression of ZEAR was induced by ZEA. Moreover, the expression profile of the gene satisfied the requirements for an inducible promoter since the gene expression rapidly increased upon ZEA treatment and was not detected in the control. We designated the promoter of ZEAR Pzear.
FIG. 1.
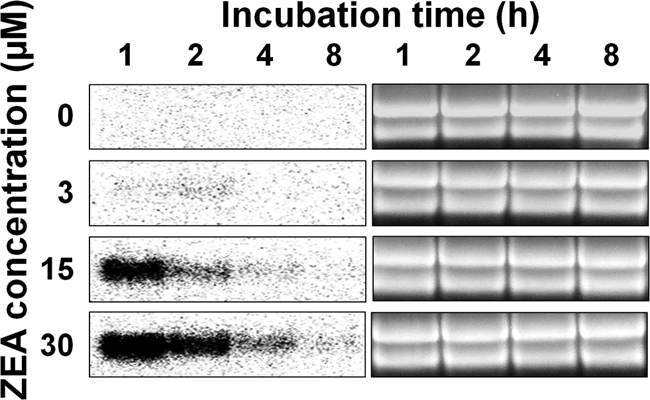
Analysis of ZEAR mRNA expression level. Total RNA of the wild-type strain GZ03639 of G. zeae was extracted from CM cultures grown for 1, 2, 4, and 8 h after ZEA treatment. Twenty micrograms was subjected to Northern analysis, with the PCR product of ZEAR, amplified from GZ03639 with zear-orf1/zear-orf2 primers, serving as a probe. An ethidium-bromide-stained gel is shown as a loading control.
ZEAR is a putative 114-amino-acid fungus-specific C6 transcription factor that contains one intron and an N-terminal fungal transcriptional regulatory domain (amino acid residues 1 to 54). It also contains a nuclear localization signal (KRSRRKP; amino acid residues 48 to 54) located at the end of the transcriptional regulatory domain. ZEAR is not conserved in plants and animals but is highly conserved in the genus Fusarium. However, the functions of the gene have not been characterized for any of the fungal species.
In addition to ZEAR, we also selected another putative transcription factor, called ZEB2 (Broad Institute; FGSG_02398.3), which controlled the ZEA gene cluster (12). Expression of ZEB2 was five times higher in SG cultures supplemented with 15 μM ZEA than in the SG culture without ZEA. Our previous study showed that ZEB2 expression gradually increased over time in wild-type G. zeae cultured in SG medium but was low in the ZEA-nonproducing mutants (12). However, in contrast to that of ZEAR, the expression of ZEB2 was not rapidly induced by ZEA treatment, suggesting that the ZEB2 promoter would not be suitable for a conditional system that used a ZEA-responsive inducible promoter.
Targeted gene deletion using GFP reporter constructs.
The ZEAR ORF was replaced with GFP and hph by targeted deletion. Positive mutant clones were confirmed by the presence of a 6.3-kb band on a Southern blot (Fig. 2). The wild-type construct migrated as a 4.0-kb band. The Δzear-gfp deletion mutant did not show any noticeable phenotypic changes in the following characteristics: growth rate, mycelial morphology on MM, CM, and PDA, conidiation and conidial morphology on YMA, sexual reproduction on carrot agar, trichothecene and ZEA production on rice culture, and pathogenicity in wheat. Further, the mutant had no phenotypic changes under the following stress conditions: NaCl, KCl, sorbitol, FeSO4, menadione, fludioxonil, iprodione, sodium dodecyl sulfate (SDS), Congo red, benomyl, and acidic (pH 4) and basic (pH 11) conditions (data not shown).
FIG. 2.

Strategy for targeted gene fusion with GFP. The promoter and terminator regions of ZEAR were fused with gfp::hph to generate the Δzear-gfp mutant, where GFP expression was controlled by the promoter of ZEAR. In the Southern blot, lane 1 is GZ03639, lanes 2 and 3 are mutants that carry multiple integration events, and lanes 4 and 5 are positive mutants where the target gene was replaced with Pzear::gfp::hph.
Microscopic observation showed that GFP in the Δzear-gfp mutant was not expressed in the control CM culture without ZEA but was detected at the 1-h time point in cultures treated with ZEA (Fig. 3). The level of GFP expression was dependent on the ZEA concentration used: it was gradually elevated in response to 3 to 30 μM ZEA but decreased at ZEA concentrations higher than 30 μM. Further, expression of GFP in these cultures decreased over time.
FIG. 3.

GFP expression of the Δzear-gfp mutant. Conidia were incubated for 1, 2, 4, and 8 h in CM supplemented with ZEA. First, third, and fifth lines, bright-field microscopy; second, fourth, and sixth lines, fluorescence microscopy.
Promoter replacement.
The promoter of the GzmetE gene was replaced with Pzear, which corresponded to a −838 to −1 bp region of the ZEAR translational start site, and confirmed by Southern blotting. Positive mutants (Pzear-GzmetE) that contained Pzear fused to GzmetE showed a 3.3-kb band, in contrast to the wild type, which showed a 1.5-kb band (Fig. 4). The GzmetE gene was constitutively transcribed in the wild-type strain regardless of the ZEA concentration, while the expression of GzmetE from the Pzear-GzmetE mutant exhibited a rapid increase after ZEA treatment followed by a decrease over time (Fig. 5). Expression of GzmetE was dependent on the concentration of ZEA in a manner similar to that of ZEAR expression.
FIG. 4.

Strategy for promoter replacement. The GzmetE promoter (PgzmetE) was replaced with the zearalenone-inducible promoter, Pzear. In the Southern blot, lane 1 is GZ03639, lanes 2 and 3 are mutants that carry multiple integration events, and lanes 4 and 5 are positive mutants that carry the Pzear promoter.
FIG. 5.

Analysis of GzmetE mRNA expression. Total RNAs from wild-type strain GZ03639 of G. zeae and the Pzear-GzmetE mutant were extracted from MM cultures grown for 1, 2, 4, and 8 h after ZEA treatment. Twenty micrograms was subjected to Northern blot analysis, with the PCR product of GzmetE, amplified from GZ03639 with gzmetE-orf1/gzmetE-orf2 primers, serving as a probe. An ethidium-bromide-stained gel is shown as a loading control.
To confirm that the Pzear promoter fused to GzmetE regulated the expression of GzmetE in Pzear-GzmetE mutants, we inoculated those strains on MM supplemented with ZEA. The Pzear-GzmetE and ΔgzmetE mutants did not grow on MM without ZEA compared to the wild-type control. The mutants did grow on MM supplemented with 3 μM ZEA; however, radial growth was not completely recovered. In contrast, radial growth of the mutants did recover to a level comparable to that of the wild-type strain when grown on MM supplemented with 15 and 30 μM ZEA (Fig. 6). These data suggested that GzmetE expression in the mutants was fully induced by ZEA treatment. However, Pzear-GzmetE mutants produced more pigment and less aerial mycelia on MM supplemented with ZEA than the wild-type strain, despite complete restoration of the growth rate (Fig. 6). The observed side effects may have been caused by differences in expression profiles of the gene between the mutant and wild-type strains, rather than ZEA treatment. Since gene expression in the mutants exhibited a rapid increase followed by a gradual decrease after ZEA treatment, as opposed to constitutive expression in the wild-type strain, it is possible that the old mycelia in the mutants experienced nutrient starvation. Several inducible promoters developed in other systems rapidly trigger expression of a target gene in response to an inducer with a subsequent decrease in gene expression over time. For example, a copper-inducible promoter in Histoplasma capsulatum (5) showed the same regulation pattern as the ZEA-inducible promoter analyzed in this study. In order to overcome these types of side effects in conditional expression systems, other promoters need to be identified that can be differentially or constitutively induced. Our next study will focus on the identification and characterization of new and novel inducible promoters in G. zeae.
FIG. 6.

Mycelial growth of the strains on MM supplemented with ZEA. Spot 1, GZ03639; spot 2, ΔgzmetE; spots 3 and 4, Pzear-GzmetE mutants. The ZEA concentration is indicated next to each panel. Photographs were taken 3 days after inoculation.
Identification of ZEAR homologs in F. oxysporum and F. verticillioides.
Homologs of ZEAR were identified from the Fusarium Comparative Database (http://www.broadinstitute.org/annotation/genome/fusarium_group/MultiHome.html) by a BlastP search. Protein sequences of FOXG_13654.2 and FVEG_11090.3 from F. oxysporum and F. verticillioides, respectively, which share 96% identity, had 74% identity with ZEAR. Expression of FOXG_13654.2 and FVEG_11090.3 increased in the CM culture after 1 h of ZEA treatment and decreased over time, similar to ZEAR expression in G. zeae. The transcripts were not detected in the culture without ZEA treatment but increased in a ZEA dose-dependent manner (Fig. 7). FOXG_13654.2 and FVEG_11090.3 were then replaced with GFP and hph, and positive mutant clones were confirmed by Southern blotting. FOXG_13654.2 and FVEG_11090.3 deletion mutants designated the Δfozear-gfp and Δfvzear-gfp mutants, respectively, did not show any noticeable phenotypic changes (data not shown). Microscopic observation showed that GFP in the Δfozear-gfp and Δfvzear-gfp mutants was not expressed in the control CM culture without ZEA but was detected at the 1-h time point in cultures treated with ZEA (Fig. 8), showing that the level of GFP expression in the mutants was similar to that in the Δzear-gfp mutant. This result suggested that expression of FOXG_13654.2 and FVEG_11090.3 is induced by ZEA and that the promoters of these genes are useful in a conditional expression system in F. oxysporum and F. verticillioides.
FIG. 7.

Expression profiles of ZEAR homolog in F. oxysporum and F. verticillioides. Total RNA of each strain was extracted from CM cultures grown for 1, 2, 4, and 8 h after ZEA treatment. Twenty micrograms of total RNA was subjected to Northern analysis, with the PCR product of FOXG_13654.2 and FVEG_11090.3, amplified from F. oxysporum strain FO0901 and F. verticillioides strain FV0201, respectively, with Fzear-orf1/Fzear-orf2 primers, serving as a probe. An ethidium-bromide-stained gel is shown as a loading control.
FIG. 8.

GFP expression of Δfozear-gfp and Δfvzear-gfp mutants. FOXG_13654.2 and FVEG_11090.3 from F. oxysporum and F. verticillioides, respectively, were replaced with GFP and hph to generate the Δfozear-gfp and Δfvzear-gfp mutants. Conidia were incubated for 1 and 4 h in CM supplemented with ZEA. First and third lines, bright-field microscopy; Second and fourth lines, fluorescence microscopy.
We introduced the Pzear::GFP construct, which was used to generate the Δzear-gfp mutant from wild-type strain GZ03639 of G. zeae, into F. oxysporum and F. verticillioides. Although we screened more than 10 mutants from each species, we found that GFP was constitutively expressed in all of the mutants regardless of the ZEA concentration (data not shown). This suggests that the Pzear promoter from G. zeae is not suitable for a conditional expression system in other fungi. This unexpected result also suggests that the expression of ZEAR and that of its homologs are indirectly activated by ZEA. The binding of ZEA to a ZEA receptor could have a role in a signal transduction pathway, as previously discussed (18). We hypothesize that ZEAR and its homologs may be regulated by a signal pathway and that the factor responsible for regulating the expression of ZEAR and its homologs may not be conserved enough to be functional among G. zeae and other Fusarium spp.
Applications.
In this study, the promoter replacement method using the Pzear promoter successfully restored the growth of an auxotroph mutant when ZEA was supplied to MM lacking methionine, suggesting that this promoter may be useful in a conditional gene expression system exogenously treated with ZEA. In addition, our data suggest that this method can be used for studying essential genes in this fungus. For example, addition of ZEA during fungal transformation would allow mutants that carry the Pzear promoter fused to an essential target gene to survive. The cellular function of the target gene could also be characterized by adjusting the amount of ZEA supplemented in the phenotyping media.
To date, the Pzear promoter has been more useful than the promoters developed in other fungal systems for molecular biological studies. The most frequently used inducible promoters are the alcohol dehydrogenase promoter (27), amylase A promoter (32), and copper-inducible promoter (5). The alcohol dehydrogenase and amylase A promoters, however, have the disadvantage of having their expression repressed by carbon sources in the medium. In addition, the copper-inducible promoter may not be suitable for G. zeae, since copper has physiological and toxic effects on fungal growth and development. In contrast, ZEA did not affect mycelium morphology of G. zeae, and repression of the ZEAR gene did not occur in the presence of carbon sources such as glucose and sucrose.
The conditional system described in this study could also be used for characterizing the mechanism of self/nonself recognition in G. zeae. Fungi have a recognition process called vegetative incompatibility (VIC) during vegetative growth that plays an important role in both gene flow and genetic barriers within fungal species (16). To date, VIC studies have been performed in a few fungi, including Podospora anserina and Neurospora crassa (2, 21), but have not occurred in G. zeae. A genome database (http://www.broadinstitute.org/annotation/genome/fusarium_group/MultiHome.html), genetic map (10), and alignment between genetic and physical maps (13) in G. zeae are currently available and would allow for the identification of vic genes in this fungus. After identifying vic genes, coexpression of these genes from different vegetative compatibility groups is necessary for understanding the entire process that is triggered by vic. However, one major problem in the coexpression of vic genes in one strain is that this would immediately trigger cell death. Therefore, the time and level of expression of the target genes would need to be regulated. The inducible system that we have developed in this study will be an excellent strategy for overcoming this problem and is anticipated to play a key role in future VIC studies in G. zeae.
The approach developed in this study may also be applicable for the genetic engineering of plant systems to generate plants that can recognize a G. zeae attack. Expression of antimicrobial peptides and defense-related genes under the control of a constitutively active CaMV35S promoter has been performed in plants to protect them from disease (24). Organ-specific promoters in genetically modified plants have also been characterized (31). However, these promoters require a fitness cost, since they express the target gene even in the absence of pathogens. Therefore, specific promoters are needed that induce only the target gene when pathogens are introduced into the plant host. Promoters induced by ZEA are excellent candidates for developing this type of system because the promoter specifically responds to ZEA produced by G. zeae.
Another possible application for this system is as a biosensor for the detection of ZEA contamination. Recombinant yeast and bacterial strains have been developed for heavy metal detection using a metal-specific gene promoter (25, 33). The detection ranges for cadmium (Cd) in a recombinant yeast strain of Saccharomyces cerevisiae and Hansenula polymorpha were 1 to 50 μM and 1 to 900 μM, respectively (25). Further, a recombinant bacterium strain of Staphylococcus aureus had a higher sensitivity to metal but showed severe growth inhibition at 100 nM Cd (33). Similarly, a recombinant H. capsulatum strain containing lacZ that was controlled by a copper-inducible promoter showed β-galactosidase activity at 1 to 500 μM CuSO4 (5). The recombinant G. zeae strains developed in this study had a detection range of 3 to 1,500 μM ZEA.
In conclusion, we have developed a conditional expression system using ZEA in G. zeae. Northern blot analysis and expression studies using a GFP fusion construct in recombinant strains showed that the Pzear promoter could function as an inducible promoter that was successfully controlled by a wide concentration range of ZEA. One important advantage of this system is the observation that ZEA does not cause any phenotypic defects in G. zeae and allows the extensive study and functional characterization of many genes that are essential for fungal life. Temporal and spatial studies of G. zeae gene expression could also be performed in this system. In addition, the system that we have developed may be useful as a biosensor for the detection of ZEA contamination and may help us understand the biological functions of ZEA in G. zeae.
Acknowledgments
This work was supported by a National Research Foundation (NRF) grant funded by the Korea government (MEST) (2009-0063350), by a grant (CG 1411) from the Crop Functional Genomics Center of the 21st Century Frontier Research Program funded by the Ministry of Education, Science and Technology, and by the Basic Science Research Program through the NRF funded by the Ministry of Education, Science and Technology (2009-0076168).
We thank Heesoo Lee and Injung Song for technical assistance.
Footnotes
Published ahead of print on 26 March 2010.
REFERENCES
- 1.Bacon, C. W., J. D. Robbins, and J. K. Porter. 1977. Media for identification of Gibberella zeae and production of F-2-(Zearalenone). Appl. Environ. Microbiol. 33:445-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevanne, D., E. Bastiaans, A. Debets, S. J. Saupe, C. Clave, and M. Paoletti. 2009. Identification of the het-r vegetative incompatibility gene of Podospora anserina as a member of the fast evolving HNWD gene family. Curr. Genet. 55:93-102. [DOI] [PubMed] [Google Scholar]
- 3.Desjardins, A. E. 2006. Fusarium mycotoxins: chemistry, genetics and biology. APS Press, St. Paul, MN.
- 4.Gaffoor, I., and F. Trail. 2006. Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl. Environ. Microbiol. 72:1793-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gebhart, D., A. K. Bahrami, and A. Sil. 2006. Identification of a copper-inducible promoter for use in ectopic expression in the fungal pathogen Histoplasma capsulatum. Eukaryot. Cell 5:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han, Y.-K., T. Lee, K.-H. Han, S.-H. Yun, and Y.-W. Lee. 2004. Functional analysis of the homoserine O-acetyltransferase gene and its identification as a selectable marker in Gibberella zeae. Curr. Genet. 46:205-212. [DOI] [PubMed] [Google Scholar]
- 7.Harris, S. D. 2005. Morphogenesis in germinating Fusarium graminearum macroconidia. Mycologia 97:880-887. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz, B. A., A. Sharon, S. W. Lu, V. Ritter, T. M. Sandrock, O. C. Yoder, and B. G. Turgeon. 1999. A G protein alpha subunit from Cochliobolus heterostrophus involved in mating and appressorium formation. Fungal Genet. Biol. 26:19-32. [DOI] [PubMed] [Google Scholar]
- 9.Hou, Z., C. Xue, Y. Peng, T. Katan, H. C. Kistler, and J.-R. Xu. 2002. A mitogen-activated protein kinase gene (MGV1) in Fusarium graminearum is required for female fertility, heterokaryon formation, and plant infection. Mol. Plant Microbe Interact. 15:1119-1127. [DOI] [PubMed] [Google Scholar]
- 10.Jurgenson, J. E., R. L. Bowden, K. A. Zeller, J. F. Leslie, N. J. Alexander, and R. D. Plattner. 2002. A genetic map of Gibberella zeae (Fusarium graminearum). Genetics 160:1451-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim, J.-E., J. Jin, H. Kim, J.-C. Kim, S.-H. Yun, and Y.-W. Lee. 2006. GIP2, a putative transcription factor that regulates the aurofusarin biosynthetic gene cluster in Gibberella zeae. Appl. Environ. Microbiol. 72:1645-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim, Y.-T., Y.-R. Lee, J. Jin, K.-H. Han, H. Kim, J.-C. Kim, T. Lee, S.-H. Yun, and Y.-W. Lee. 2005. Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. Mol. Microbiol. 58:1102-1113. [DOI] [PubMed] [Google Scholar]
- 13.Lee, J., J. E. Jurgenson, J. F. Leslie, and R. L. Bowden. 2008. Alignment of genetic and physical maps of Gibberella zeae. Appl. Environ. Microbiol. 74:2349-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee, J., J. F. Leslie, and R. L. Bowden. 2008. Expression and function of sex pheromones and receptors in the homothallic ascomycete Gibberella zeae. Eukaryot. Cell 7:1211-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, T., Y.-K. Han, K.-H. Kim, S.-H. Yun, and Y.-W. Lee. 2002. Tri13 and Tri7 determine deoxynivalenol- and nivalenol-producing chemotypes of Gibberella zeae. Appl. Environ. Microbiol. 68:2148-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leslie, J. F. 1993. Fungal vegetative compatibility. Annu. Rev. Phytopathol. 31:127-150. [DOI] [PubMed] [Google Scholar]
- 17.Leslie, J. F., and B. A. Summerell. 2006. The Fusarium laboratory manual. Blackwell Professional, Ames, IA.
- 18.Lysøe, E., K. R. Bone, and S. S. Klemsdal. 2009. Real-time quantitative expression studies of the zearalenone biosynthetic gene cluster in Fusarium graminearum. Phytopathology 99:176-184. [DOI] [PubMed] [Google Scholar]
- 19.Lysøe, E., S. S. Klemsdal, K. R. Bone, R. J. Frandsen, T. Johansen, U. Thrane, and H. Giese. 2006. The PKS4 gene of Fusarium graminearum is essential for zearalenone production. Appl. Environ. Microbiol. 72:3924-3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mach, R. L., and S. Zeilinger. 2003. Regulation of gene expression in industrial fungi: Trichoderma. Appl. Microbiol. Biotechnol. 60:515-522. [DOI] [PubMed] [Google Scholar]
- 21.Micali, C. O., and M. L. Smith. 2006. A nonself recognition gene complex in Neurospora crassa. Genetics 173:1991-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mirocha, C. J., and C. M. Christensen. 1974. Oestrogenic mycotoxins synthesized by Fusarium sp., p. 129-148. In I. F. H. Purchase (ed.), Mycotoxins. Elsevier Scientific Publishing, Amsterdam, Netherlands.
- 23.Munson, G. P., D. L. Lam, F. W. Outten, and T. V. O'Halloran. 2000. Identification of a copper-responsive two-component system on the chromosome of Escherichia coli K-12. J. Bacteriol. 182:5864-5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osusky, M., G. Zhou, L. Osuska, R. E. Hancock, W. W. Kay, and S. Misra. 2000. Transgenic plants expressing cationic peptide chimeras exhibit broad-spectrum resistance to phytopathogens. Nat. Biotechnol. 18:1162-1166. [DOI] [PubMed] [Google Scholar]
- 25.Park, J. N., M. J. Sohn, D. B. Oh, O. Kwon, S. K. Rhee, C. G. Hur, S. Y. Lee, G. Gellissen, and H. A. Kang. 2007. Identification of the cadmium-inducible Hansenula polymorpha SEO1 gene promoter by transcriptome analysis and its application to whole-cell heavy-metal detection systems. Appl. Environ. Microbiol. 73:5990-6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proctor, R. H., T. M. Hohn, and S. P. McCormick. 1995. Reduced virulence of Gibberella zeae caused by disruption of a trichothecene toxin biosynthetic gene. Mol. Plant Microbe Interact. 8:593-601. [DOI] [PubMed] [Google Scholar]
- 27.Romero, B., G. Turner, I. Olivas, F. Laborda, and J. R. De Lucas. 2003. The Aspergillus nidulans alcA promoter drives tightly regulated conditional gene expression in Aspergillus fumigatus permitting validation of essential genes in this human pathogen. Fungal Genet. Biol. 40:103-114. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook, J. S., and D. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Seong, K., Z. Hou, M. Tracy, H. C. Kistler, and J.-R. Xu. 2005. Random insertional mutagenesis identifies genes associated with virulence in the wheat scab fungus Fusarium graminearum. Phytopathology 95:744-750. [DOI] [PubMed] [Google Scholar]
- 30.Shoji, J.-Y., J.-I. Maruyama, M. Arioka, and K. Kitamoto. 2005. Development of Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiol. Lett. 244:41-46. [DOI] [PubMed] [Google Scholar]
- 31.Skadsen, R. W., P. Sathish, M. L. Federico, T. Abebe, J. Fu, and H. F. Kaeppler. 2002. Cloning of the promoter for a novel barley gene, Lem1, and its organ-specific promotion of GFP expression in lemma and palea. Plant Mol. Biol. 49:545-555. [DOI] [PubMed] [Google Scholar]
- 32.Tada, S., K. Gomi, K. Kitamoto, K. Takahashi, G. Tamura, and S. Hara. 1991. Construction of a fusion gene comprising the Taka-amylase A promoter and the Escherichia coli beta-glucuronidase gene and analysis of its expression in Aspergillus oryzae. Mol. Gen. Genet. 229:301-306. [DOI] [PubMed] [Google Scholar]
- 33.Verma, N., and M. Singh. 2005. Biosensors for heavy metals. Biometals 18:121-129. [DOI] [PubMed] [Google Scholar]
- 34.Waring, R. B., G. S. May, and N. R. Morris. 1989. Characterization of an inducible expression system in Aspergillus nidulans using alcA and tubulin-coding genes. Gene 79:119-130. [DOI] [PubMed] [Google Scholar]
- 35.Willyerd, K. L., A. M. Kemp, and A. L. Dawe. 2009. Controlled gene expression in the plant pathogen Cryphonectria parasitica by use of a copper-responsive element. Appl. Environ. Microbiol. 75:5417-5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yevtushenko, D. P., V. A. Sidorov, R. Romero, W. W. Kay, and S. Misra. 2004. Wound-inducible promoter from poplar is responsive to fungal infection in transgenic potato. Plant Sci. 167:715-724. [Google Scholar]
- 37.Yu, H.-Y., J.-A. Seo, J.-E. Kim, K.-H. Han, W.-B. Shim, S.-H. Yun, and Y.-W. Lee. 2008. Functional analyses of heterotrimeric G protein G alpha and G beta subunits in Gibberella zeae. Microbiology 154:392-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu, J.-H., Z. Hamari, K.-H. Han, J.-A. Seo, Y. Reyes-Dominguez, and C. Scazzocchio. 2004. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 41:973-981. [DOI] [PubMed] [Google Scholar]
- 39.Zuo, J., P. D. Hare, and N. H. Chua. 2006. Applications of chemical-inducible expression systems in functional genomics and biotechnology. Methods Mol. Biol. 323:329-342. [DOI] [PubMed] [Google Scholar]