

Abstract
Pyrococcus furiosus is a model organism for analyses of molecular biology and biochemistry of archaea, but so far no useful genetic tools for this species have been described. We report here a genetic transformation system for P. furiosus based on the shuttle vector system pYS2 from Pyrococcus abyssi. In the redesigned vector, the pyrE gene from Sulfolobus was replaced as a selectable marker by the 3-hydroxy-3-methylglutaryl coenzyme A reductase gene (HMG-CoA) conferring resistance of transformants to the antibiotic simvastatin. Use of this modified plasmid resulted in the overexpression of the HMG-CoA reductase in P. furiosus, allowing the selection of strains by growth in the presence of simvastatin. The modified shuttle vector replicated in P. furiosus, but the copy number was only one to two per chromosome. This system was used for overexpression of His6-tagged subunit D of the RNA polymerase (RNAP) in Pyrococcus cells. Functional RNAP was purified from transformed cells in two steps by Ni-NTA and gel filtration chromatography. Our data provide evidence that expression of transformed genes can be controlled from a regulated gluconeogenetic promoter.
Several reports addressed the initial establishment of genetic techniques for the Thermococcales, a major group of hyperthermophilic archaea including the genera Thermococcus and Pyrococcus. The first experiments described used the plasmid pGT5 from Pyrococcus abyssi. This plasmid is only 3,440 bp in size and replicates via a rolling circle mechanism (7). The archaeal plasmid was fused with a pUC19 vector to create a potential shuttle vector between Escherichia coli and Pyrococcus furiosus (1). This construct could be transformed in both organisms by CaCl2 treatment. Later, this construct was modified by introducing the alcohol dehydrogenase gene from Sulfolobus solfataricus as a selectable marker (3). The resulting plasmids pAG1 and pAG2 were maintained for several generations in E. coli, in the euryarchaeote P. furiosus, and also in the crenarchaeote Sulfolobus acidocaldarius. The presence of these plasmids in the two archaea conferred resistance to butanol and benzyl alcohol.
As the attempts to use this selection system for P. abyssi failed, a new shuttle vector, pYS2, was created (17). This construct is also based on the archaeal pGT5 plasmid and a bacterial vector, pLitmus38. It contains the pyrE gene of S. acidocaldarius, a key enzyme of the pyrimidine biosynthetic pathway, as a selectable marker. For the transformation procedure, a Pyrococcus strain was used containing a pyrE mutation which led to a uracil-auxotrophic phenotype. Using the shuttle vector pYS2 in combination with a polyethylene glycol-spheroplast method, it was possible to transform the pyrE mutant of P. abyssi to uracil prototrophy. Although the transformation frequency was very low, the shuttle vector was stably maintained at high copy number under selective conditions in both E. coli and P. abyssi (17).
A major breakthrough in the establishment of genetic tools for hyperthermophilic euryarchaeota was the development of a targeted gene disruption system by homologous recombination in Thermococcus kodakaraensis KOD1 (23). A uracil-auxotrophic strain was converted with a disruption vector harboring the pyrF marker within the trpE gene to a uracil-prototrophic and a tryptophan-auxotrophic strain by double-crossover recombination. Due to the natural competence for DNA uptake, the high transformation efficiency, and the high incorporation rate of DNA into its genome by homologous recombination, the system led to the identification of novel biochemical pathways, discovery of new enzyme functions, and further elucidation of proteins involved in the basic process of transcription (4, 11, 20, 22). A further improvement of this genetic system was the discovery that overexpression of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase gene is connected with the resistance against the antibiotic simvastatin (18). This selection system was first described in halophiles (15) and has the great advantage that there is no need for a certain host strain with a particular defect or auxotrophy toward an amino acid (18).
These new findings with respect to antibiotic resistance and the fact that the published shuttle vectors from Pyrococcus were never used in further investigations prompted us to redesign the pYS2 vector (17). In this article we describe the construction of a modified shuttle vector which allows overexpression of the HMG-CoA reductase in transformed Pyrococcus cells and leads to stable simvastatin-resistant cells. Furthermore, we also demonstrate overexpression of subunit D of the RNA polymerase (RNAP) by introducing an additional copy of subunit D with a C-terminal His tag under the control of a regulated promoter.
MATERIALS AND METHODS
Strains and growth conditions.
P. furiosus was cultivated under anaerobic conditions at 85°C in nutrient-rich medium based on SME medium (8) and supplemented with different organic substrates. SME-starch medium contained 0.1% each starch, yeast extract, and peptone. For SME-pyruvate medium, the starch was replaced with 40 mM Na-pyruvate. Gelrite (1%) was added for solidification of medium. The antibiotic simvastatin (Toronto Research Inc., Toronto, Canada) was dissolved in ethanol and sterilized by filtration.
General DNA manipulation.
Escherichia coli strain DH5α, used for vector construction and propagation, was cultivated at 37°C in Luria-Bertani (LB) medium. When needed, 100 μg/ml ampicillin was added to the medium. The vector pYS2 was provided by Gaël Erauso (Université de la Méditerranée, Marseille, France). Restriction and modification enzymes were purchased from NEB (Ipswich, MA). Plasmid DNA and DNA fragments from agarose gels were isolated using a plasmid mini-kit or gel extraction kit from Qiagen (Hilden, Germany). Phusion High-Fidelity DNA polymerase from Finnzymes (Keilaranta, Finland) was used as a polymerase for PCR. DNA sequencing was performed by Geneart (Regensburg, Germany). Genomic DNA from P. furiosus wild-type and transformed strains was isolated using a DNeasy Blood & Tissue kit from Qiagen.
Construction of the shuttle vectors pYS3 and pYS4.
The overexpression cassette for the HMG-CoA reductase gene from P. furiosus was constructed by replacing the native promoter with the strong promoter region (−250 to −1) of the glutamate dehydrogenase gene (gdh [10]). The fusion of this promoter to the coding region of the HMG-CoA reductase (primers: PF1848F 5′-ATGGAAATAGAGGAGATTATAGAG-3′ and PF1848BamHI 5′-ATCATCGGATCCTCATCTCCCAAGCATTTTATGAGC-3′) was done by PCR with overhanging ends at the reverse primer for the gdh promoter region gdhPromR-PF1848 (5′-CTCCTCTATTTCCATGTTCATCCCTCCAAATTAGGTG-3′). As a forward primer for the amplification, gdhPromFBamHI (5′-GGAACCGGATCCTTGAAAATGGAGTGAGCTGAG-3′) was used. The cassette was inserted into the pYS2 vector by replacing the BamHI fragment containing pyrE with the HMG-CoA reductase gene. The created vector pYS3 was sequenced and used later for transformation and further modification.
To obtain shuttle vector pYS4, RNA polymerase subunit D (rpoD) was linked to the fructose-1,6 bisphosphatase (fbp) promoter and inserted into pYS3. A His6 tag was attached at the C terminus of subunit D in addition. The promoter sequence of the fbp was amplified from genomic DNA using the primers EcoRV-PF0613Pr-F (5′-CTATTAGATATCTCCTTAACATTTCTCCAAA-3′) and PF0613Prom-R (5′-CTGAACTTCAATTCCGGCCATTTTTTCACCTCCAGAAT-3′). Via the PF0613Prom-R primer, the promoter sequence had a 3′ overhang for the fusion with the coding region of rpoD. For the amplification of subunit D, the primers PF1647-F (5′-AAATGGCCGGAATTGAAGTTCAGATTCTTGA-3′) and PF1647-His-R (5′-GTGATGGTGATGGTGATGAGAGGTCAATTTTTGAAGTTCAC-3′) were used. This step introduced the incorporation of the sequence for the His6 tag at the C terminus of rpoD. The RpoD-His6 gene was fused with the terminating region of the histone A1 gene of P. furiosus (24). The primer pair His-PF1831Term-F (5′-CATCACCATCACCATCACTGAAATCTTTTTTAGCACTT-3′) and PF1831T-EcoRV-R (5′-TCAATTGATATCACCCTAGAAAAAGATAAGC-3′) created the terminating region of the histone A1 gene with a part overlapping the RpoD-His6 gene at the 5′-end that was used to fuse the fragments by PCR. Finally, the construct was integrated into the pYS3 vector next to the HMG-CoA gene reductase cassette using the flanking EcoRV sites. The construction of the plasmid was verified by DNA sequencing.
Transformation of P. furiosus.
P. furiosus cultures grown at 75°C to a cell density between 0.8 × 108 and 1.0 × 108 per ml were used for transformation. For a transformation reaction, the cells of 3 ml grown culture were collected anaerobically by centrifugation (10 min at 6,000 × g) and resuspended in a total volume of 100 μl transformation solution containing SME (without KH2PO4), 40 mM Na-pyruvate, 4.7 mM NH4Cl, and 80 mM CaCl2. The pH was adjusted to 7.0 with HCl. Cells were incubated at 4°C for 90 min under anaerobic conditions. After 30 min, 0.5 pmol pYS3 or pYS4 was added. After a heat shock at 80°C for 3 min, the cells were again incubated for 10 min at 4°C and then cultivated in SME-starch liquid medium in the presence of 10 μM simvastatin at 85°C for 48 h. Later, the cells were plated on SME medium with starch as a substrate and containing 10 μM simvastatin. The plates were incubated at 85°C for 48 h.
Growth properties of P. furiosus and P. furiosus pYS3 and pYS4 transformants.
To analyze resistance toward simvastatin, pYS3-transformed cells were cultivated in SME-starch medium supplemented with 1, 5, 10, or 20 μM simvastatin at 85°C. Wild-type P. furiosus cells were also cultivated in SME-starch medium at 85°C but without simvastatin. Cell densities were measured at appropriate intervals. Cell counts were analyzed with a Thoma counting chamber (0.02-mm depth; Marienfeld, Lauda-Königshofen, Germany) under a phase-contrast microscope. To determine the expression of subunit D under glycolytic or gluconeogenetic conditions, P. furiosus pYS4 cells were grown in either SME-starch or SME-pyruvate medium in the presence of 10 μM simvastatin at 85°C.
Detection of RpoD and RpoD-His6 by Western blot analysis.
For the preparation of cell extracts, 10 g of P. furiosus wild-type or P. furiosus cells transformed with pYS4 was resuspended in 30 ml buffer (40 mM HEPES, 500 mM NaCl, 10 mM imidazole, 15% glycerol, pH 7.5), sonicated on ice, and treated with glass beads using a FastPrep-24 (M. P. Biomedicals, Irvine, CA) for complete cell lysis. After centrifugation (100,000 × g for 1 h at 4°C), the protein concentrations of the clarified supernatants were determined by Bradford assays. For quantification of the expression levels of RpoD or RpoD-His6, Western blot experiments were done as previously described using polyclonal antibodies raised against recombinant subunits A" or D from P. furiosus (9). The signals were visualized using a Cy5-labeled secondary anti-rabbit antibody from Thermoscientific (Waltham, MA) and a fluorescence image analyzer (FLA-5000, Fuji, Japan).
Purification of RpoD-His6 and RNAP-His6.
The cell extracts prepared as described in the previous section were applied onto 1-ml Ni2+-charged HisTrap HP columns (GE Healthcare). Bound proteins were eluted in one step using an elution buffer containing 300 mM imidazole instead of 10 mM. To separate free RpoD-His6 from the fraction incorporated into the RNAP, the eluate was loaded onto a Superdex 200 gel filtration column (GE Healthcare) equilibrated with 40 mM HEPES, pH 7.3, 250 mM KCl, 2.5 mM MgCl2, 0.5 mM EDTA, and 20% glycerol. Aliquots of the fractions were analyzed for RNAP activity using a specific in vitro transcription assay (10) and SDS-PAGE analysis.
Southern blot analysis.
Total genomic DNA was digested with EcoRV, and the resulting restriction fragments were separated on a 1% agarose gel. After electrophoresis, the DNA was transferred to a nylon membrane (Roche Applied Science, Mannheim, Germany) by capillary blot. A part of the rpoD gene was amplified by PCR using the primer pair RpoD500-F (5′-CCAACATTTGCAGTTGATGAAG-3′) and RpoD500-R (5′-CTCTTCGAAATCCTTTGGTATGTAG-3′). This segment was used as a probe to detect the RNAP subunit D gene in genomic and in plasmid DNA. The labeled probe was generated by the random primed method using the NEBlot kit (NEB, Ipswich, MA) in the presence of digoxigenin-11-dUTP (Roche Applied Science, Mannheim, Germany). After hybridization, the signals were detected using anti-digoxigenin antibodies conjugated with alkaline phosphatase according to the instructions of the producer (Roche Applied Science, Mannheim, Germany).
RESULTS
Transformation in P. furiosus with a redesigned shuttle vector of pYS2.
The selection mechanism of the shuttle vector pYS2 is based on a uracil auxotrophic strain of P. abyssi which has a mutation in the pyrE gene (17). The plasmid contains a wild-type copy of the pyrE gene of S. acidocaldarius, and successful transformation complements the uracil auxotrophy. As our attempts to construct a uracil auxotrophic strain of P. furiosus were not successful (data not shown), we redesigned the vector pYS2. In the new construct pYS3, the pyrE gene was substituted by the HMG-CoA reductase gene and for an efficient expression this gene was fused with the strong gdh promoter from P. furiosus (10) (Fig. 1).
FIG. 1.

Schematic diagram of the shuttle vectors pYS2, pYS3 and pYS4. To create pYS3, the pyrE marker of pYS2 was replaced with a simvastatin resistance cassette via BamHI restriction sites. This enables the overexpression of the HMG-CoA reductase under the control of the gdh promoter. The plasmid pYS4 contains an additional fragment for the expression of a His6-tagged version of RNAP subunit D. This further copy is under the control of the fbp promoter and followed by the hpyA1 terminator sequence. The fragment was inserted into the EcoRV restriction site of pYS3. dso, double-stranded origin of replication; sso, single-stranded origin of replication; ori, origin of replication.
As overexpression of the HMG-CoA reductase led to the resistance against simvastatin in T. kodakaraensis (18), we also analyzed the effect of various concentrations of simvastatin on the growth of P. furiosus. In SME-pyruvate medium supplemented with 5, 10, or 20 μM simvastatin, growth was inhibited for only one day if the cells were incubated at 95°C. In contrast, incubation at 85°C with similar concentrations prevented growth for three days. This indicates that the stability of simvastatin is dramatically decreased at 95°C, but 85°C seems to be an appropriate temperature for selection of transformants. This reduced incubation temperature still allows growth of P. furiosus in a reasonable time in liquid as well as in solidified medium.
In the first experiments, the new construct pYS3 was used to transform P. furiosus according to the published CaCl2 procedure for T. kodakaraensis with some minor modifications (23): the heat shock was performed for 3 min at 80°C instead of 45 s at 85°C, and cells were incubated in the cold at 4°C instead of at 0°C. Transformants were selected by growing cells for 48 h in liquid medium in the presence of 10 μM simvastatin. Growth was observed only when cells were transformed with plasmid pYS3 and not when cells were treated in control reactions with transformation solution not containing the plasmid. The transformation efficiency in liquid medium was approximately 5 × 102 transformants per μg pYS3 plasmid DNA. For the isolation of single transformants, cells grown in liquid cultures were plated on culture medium containing 10 μM simvastatin. The plating efficiency of the transformants in the presence of 10 μM simvastatin was ∼15% (the plating efficiency of wild-type cells on media not containing the antibiotics was ∼78%).
A few simvastatin-resistant colonies were selected and further analyzed for the presence of the plasmid by PCR amplification. To provide evidence that the plasmid was stably replicated in Pyrococcus, the plasmid was isolated again from Pyrococcus after several transfers (four to five times) of cells in fresh culture medium. Using this isolated plasmid DNA it was possible to successfully retransform E. coli (data not shown). This clearly demonstrates that this redesigned shuttle vector, including the plasmid pGT5 from P. abyssi, was also stably replicated as an external DNA element in P. furiosus.
Induced expression of subunit D of the RNAP.
As the next step, we analyzed whether the shuttle vector could be converted into an expression vector which allows the expression of proteins under the control of a regulated promoter. Subunit D of the archaeal RNAP was used as a model protein, and an additional copy of this gene was inserted into the shuttle vector under the control of the fructose-1-6 bisphosphatase (PF0613) promoter (Fig. 1, pYS4). To allow a simple and rapid purification of the protein, a His6 tag at the C terminus was introduced, and for efficient termination of transcription, the terminator from the histone gene hpyA1 was linked to the 3′-end of the gene (24). The PF0613 promoter is repressed under glycolytic and induced under gluconeogenetic conditions (13, 16).
The new construct pYS4 was transformed into Pyrococcus, and a single colony was transferred into liquid medium and first cultivated under glycolytic conditions in the presence of starch. Later, the same culture was transferred to gluconeogenetic conditions using a medium containing pyruvate as the energy source. In each case the expression of subunit D was analyzed in crude extracts and compared with that of the wild type by a Western blot assay using antibodies against RNAP subunit D (Fig. 2). Identical amounts of RNAP were applied to gels used for Western blots as shown by immunostaining using the antibody raised against RNAP subunit A". Analysis of the crude extracts of the wild-type strain revealed only one signal corresponding to subunit D (Fig. 2, lanes 7 to 9). In contrast, the crude extracts of the transformants grown with starch (lanes 1 to 3) or pyruvate (lanes 4 to 6) contained an additional polypeptide migrating slightly more slowly than wild-type subunit D. This signal corresponding to the additional copy of subunit D encoded on the plasmid differed in size due to the existence of the His6 tag at the C terminus. The additional signal found in transformants was rather weak in cells grown with starch and much stronger in cells grown with pyruvate (Fig. 2, compare lanes 1 to 3 with 4 to 6). This clearly demonstrates that the promoter of the additional subunit D copy on the plasmid is strongly induced under gluconeogenetic conditions and therefore this system is useful for regulated expression of proteins in P. furiosus.
FIG. 2.
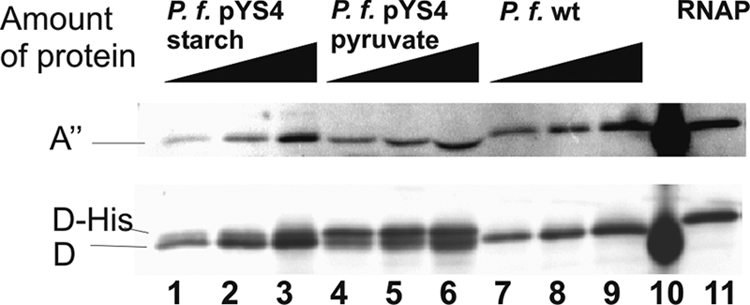
Comparison of the expression of wild-type and His6-tagged subunit D encoded on plasmid pYS4 by a Western blot analysis. To analyze the relative amount of subunit D in the transformant grown with starch or pyruvate and in wild-type cells grown with starch, we used comparable amounts of RNAP in each extract. For titration of equal amounts of RNAP, we used an anti-subunit A" antibody (upper signal). The corresponding amounts of proteins in crude extracts from the transformant grown with starch were 218 ng, 436 ng, and 872 ng (lanes 1 to 3). Lanes 4 to 6 contained the transformant grown with pyruvate with protein amounts of 1,122 ng, 2,244 ng, and 4,488 ng. Wild-type crude extracts were 166 ng, 332 ng, and 664 ng protein (lanes 7 to 9). For identification of subunit D, we used an anti-subunit D antibody (lower signals). Lane 10 contains a prestained size marker (Pager Ruler; Fermentas) and lane 11 contains 50 ng of purified RNAP (with a His tag at subunit D). P. f., P. furiosus.
Purification of archaeal RNAP by immobilized metal ion affinity chromatography.
To analyze whether this modified subunit D containing a His6 tag at the C terminus also assembles into the archaeal RNAP, the crude extract of cells transformed with pYS4 was applied onto Ni-NTA columns. Specific bound proteins were eluted with a buffer containing 300 mM imidazole. To separate subunit D assembled into the archaeal RNAP from the free polypeptide, the pooled subunit D-containing fractions from the Ni-NTA column were further purified by gel filtration chromatography. The RNAP-containing fraction isolated by this two-step procedure from the transformant grown with pyruvate was compared with conventionally purified native RNAP (19) by gradient SDS-PAGE and silver staining (Fig. 3A, lanes 2 and 3). Both RNAPs showed an almost identical pattern. This indicates that the overexpressed subunit D with the His6 tag assembles into the RNAP, and the whole enzyme can be isolated by immobilized metal ion affinity chromatography.
FIG. 3.
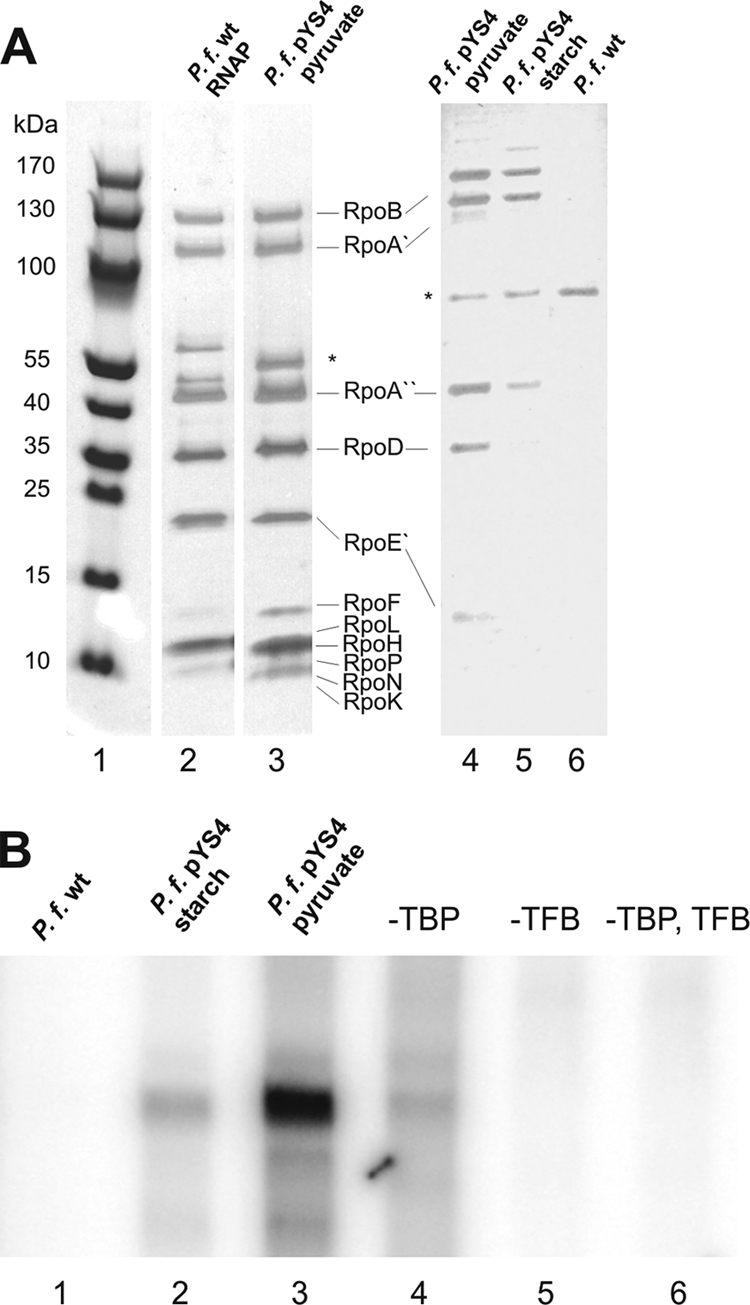
Purification and functional analysis of the RNAP. (A) Silver-stained gradient SDS gel (4 to 20%) of 2 μg purified RNAP from P. furiosus (P. f.). Lane 2 contains a wild-type RNAP, purified as described previously (10). Lane 3 contains the RNAP from the transformant grown with starch and purified with Ni-NTA and gel filtration chromatography. The corresponding fractions (identical volumes) of similar amounts of two-step-purified cell extracts from wild-type cells (lane 4, without RNAP activity) and transformants grown with starch (lane 5) and pyruvate (lane 6) were analyzed on a 10% SDS gel. The band labeled with an asterisk is an unknown protein which was copurified in all three extracts. The corresponding subunits of the RNAP are indicated. Lane 1 contains a molecular size marker. (B) In vitro transcription with the purified RNAP fractions. The RNAP fractions from the transformants grown with starch (lane 2) or pyruvate (lane 3) and from the wild type (lane 1) were used to transcribe the gdh promoter in the presence of the archaeal transcription factors TBP and TFB. Lanes 4 to 6 contain control experiments without transcription factor TBP, transcription factor TFB, or both. The transcription assays were performed as described previously (19).
Starting with similar amounts of cells for purification, five times more RNAP was isolated from transformant cells grown with pyruvate than from cells grown with starch (Fig, 3A, compare lanes 4 and 5). As expected, RNAP was not enriched in similar fractions purified from extracts of the wild-type strain by the same procedure (Fig. 3A, lane 6). The band labeled with an asterisk was purified from wild-type cells as well as from the transformants after Ni-NTA affinity chromatography and Superdex 200 gel filtration. This multimeric polypeptide was not characterized in any detail here.
To check whether the affinity-purified RNAP fractions (Fig. 3A, lanes 4 to 6) are functionally active, these fractions were analyzed by in vitro transcription experiments. The affinity-purified fractions were able to synthesize runoff RNA products from the gdh promoter in the presence of both archaeal transcription factors, TBP and TFB (10) (Fig. 3B, lanes 2 and 3). When one transcription factor or both were omitted, transcription was abolished (Fig. 3B, lanes 4 to 6). Taken together, these data indicate that subunit D with a C-terminal His6 tag assembles into the RNAP. As expected, the amount of RNAP containing His6-tagged D that can be purified from a given amount of cells was higher when subunit D was overexpressed from the gluconeogenetic promoter. Furthermore, it is possible to specifically purify this fraction of the RNAP by Ni-NTA and size exclusion chromatography from the crude extract. The purified fraction is functionally active and not contaminated with TBP and TFB. As expected, this procedure did not allow the purification of RNAP without the C-terminal His6 tag at subunit D (Fig. 3B, lane 1).
Copy number of pYS4 in P. furiosus.
To determine the copy number of this shuttle vector in P. furiosus, EcoRV-digested total DNA was analyzed by Southern blot experiments. The DNA sequence of subunit D was used as a probe. When wild-type P. furiosus DNA was analyzed, a 4.3-kb signal was identified. This exactly corresponds to the predicted size of a fragment containing subunit D in chromosomal DNA restricted with EcoRV (Fig. 4, lanes 2 to 6). When transformants were analyzed beside the genomic fragment, an additional band with a size of 1.1 kb was observed (Fig. 4, lanes 7 to 11). As this signal was also present in the control lane with EcoRV-hydrolyzed plasmid DNA (lane 1), these results clearly demonstrate the presence of plasmid pYS4 in transformed cells. The finding that both signals arising from the genomic fragment and from the plasmid are in a similar strength range indicates that the copy number of the plasmid is approximately in the same ratio as the number of chromosomes of one cell. To exclude the possibility that the ratio between plasmid and chromosomal DNA was changed during the DNA purification procedure, the ratio between plasmid and chromosomal DNA was analyzed in crude extracts in addition. This experiment confirmed the results that the copy number of the plasmid pYS4 in P. furiosus was between one and two (data not shown).
FIG. 4.

Analysis of the copy number of pYS4 in P. furiosus (P. f.) by Southern blotting. Identical amounts from 25 ng to 400 ng of EcoRV-digested genomic DNA of P. furiosus wild-type (lanes 2 to 6) and pYS4 transformant (lanes 7 to 11) were used and analyzed with the rpoD probe. Lane 1 contained the EcoRV-digested vector pYS4 as a positive control.
DISCUSSION
In this paper we describe a shuttle expression vector system for P. furiosus and E. coli allowing the regulated expression of proteins in Pyrococcus. We have redesigned a published shuttle vector of P. abyssi using the overexpression of the HMG-CoA reductase as a selection marker which confers resistance to the antibiotic simvastatin, as described earlier for T. kodakaraensis (18). The copy numbers of the new vectors pYS3 and pYS4 were dramatically reduced in comparison to that of the pYS2 shuttle vector used in P. abyssi. The copy number of the shuttle vector pYS2 was 20 to 30 copies per chromosome and was therefore in the same range as described for the wild-type pGT5 plasmid from P. abyssi (6, 17). At present we have no explanation for the dramatic reduction of the copy number to one or two per chromosome in P. furiosus. Since in our construct the transcription of the HMG-CoA reductase gene occurs in a direction opposite to that of the replication of the plasmid, we have also analyzed whether insertion of the HMG-CoA reductase gene in the opposite direction affects the copy number. First experiments indicate that the transcriptional orientation of the HMG-CoA reductase gene does not influence the copy number of the plasmid (data not shown). We assume that the maintenance of this plasmid in P. furiosus is mainly driven by the antibiotic resistance and the mechanism responsible to maintain a certain copy number in P. abyssi is absent in P. furiosus. A reduced copy number in a different host was also observed when plasmids pAG1 and pAG2 from P. abyssi were transferred to P. furiosus (1) or when a plasmid from Thermococcus nautilus was transferred to T. kodakaraensis (21).
Although the copy number of the shuttle vector described in this paper is low, the possibility to transform P. furiosus and to use this shuttle vector for the regulated expression of plasmid-borne genes now allows the development of a genetic system. Our recent results suggest that it will be also possible to mutate the chromosome of P. furiosus using the overexpression of the HMG-CoA reductase as a selection marker against simvastatin (data not shown). But also the shuttle vector-based regulated expression system offers novel intriguing possibilities for future developments. As we could successfully demonstrate that this system allows overexpression of a His6-tagged subunit D under the control of a gluconeogenetic promoter whose expression was dependent upon the substrate in the growth medium, the system described here can be used for production of recombinant proteins in P. furiosus. It could be an alternative system for expression of proteins which are difficult to produce in E. coli, especially for proteins of hyperthermophiles which might have a low propensity to fold properly at low temperature, like the Sor (sulfur oxygenase/reductase) protein from Acidianus ambivalens, which was produced with higher efficiency in a hyperthermophilic Sulfolobus expression system than in E. coli (2).
We provide also evidence that this system can be used to isolate an active fraction of RNAP in a two-step purification procedure when a His6-tagged additional copy of subunit D was overexpressed in Pyrococcus cells. This system allows overexpression of mutant subunits in Pyrococcus and isolation of the RNAP-containing mutations for structure-function analyses as described for T. kodakaraensis (11, 21). Using a low level of expression of a particular subunit from the PF0613 promoter (growth on starch), it should be also possible to introduce point mutations into functional important regions of this subunit. Therefore, this system is a perfect complementation of our previously described system for the reconstitution of the 11-subunit RNAP from individual subunits in vitro (19). Furthermore, this system will be useful for the construction of a reporter gene assay, which should allow a rapid in vivo analysis of promoter sequences or of regulatory DNA elements in P. furiosus. Taken together, the presented shuttle vector-based transformation system for P. furiosus is an important first step to establish a complete genetic toolbar for one of the hyperthermophilic key organisms in archaeal research for analysis of recombination (26), replication (5, 14), transcription (25), and metabolism (12).
Acknowledgments
We thank Renate Richau for technical assistance and Gaël Erauso, Université de la Méditerranée, Marseille, France, for providing plasmid pYS2.
Footnotes
Published ahead of print on 2 April 2010.
REFERENCES
- 1.Aagaard, C., I. Leviev, R. N. Aravalli, P. Forterre, D. Prieur, and R. A. Garrett. 1996. General vectors for archaeal hyperthermophiles: strategies based on a mobile intron and a plasmid. FEMS Microbiol. Rev. 18:93-104. [DOI] [PubMed] [Google Scholar]
- 2.Albers, S. V., M. Jonuscheit, S. Dinkelaker, T. Urich, A. Kletzin, R. Tampe, A. J. Driessen, and C. Schleper. 2006. Production of recombinant and tagged proteins in the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 72:102-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aravalli, R. N., and R. A. Garrett. 1997. Shuttle vectors for hyperthermophilic archaea. Extremophiles 1:183-191. [DOI] [PubMed] [Google Scholar]
- 4.Atomi, H., and T. Imanaka. 2008. Targeted gene disruption as a tool for establishing gene function in hyperthermophilic Archaea, p. 213-223. In F. Robb, G. Antranikian, D. Grogan, and A. Driessen (ed.), Thermophiles: biology and technology at high temperatures, CRC Press, Boca Raton, FL.
- 5.Emptage, K., R. O'Neill, A. Solovyova, and B. A. Connolly. 2008. Interplay between DNA polymerase and proliferating cell nuclear antigen switches off base excision repair of uracil and hypoxanthine during replication in archaea. J. Mol. Biol. 383:762-771. [DOI] [PubMed] [Google Scholar]
- 6.Erauso, G., F. Charbonnier, T. Barbeyron, P. Forterre, and D. Prieur. 1992. Preliminary characterization of a hyperthermophilic archaebacterium with a plasmid, isolated from a north Fiji basin hydrothermal vent. C. R. Acad. Sci. 314:387-393. [Google Scholar]
- 7.Erauso, G., S. Marsin, N. Benbouzid-Rollet, M. F. Baucher, T. Barbeyron, Y. Zivanovic, D. Prieur, and P. Forterre. 1996. Sequence of plasmid pGT5 from the archaeon Pyrococcus abyssi: evidence for rolling-circle replication in a hyperthermophile. J. Bacteriol. 178:3232-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiala, G., and K. O. Stetter. 1986. Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C. Arch. Microbiol. 145:56-61. [Google Scholar]
- 9.Hausner, W., and M. Thomm. 1995. The translation product of the presumptive Thermococcus celer TATA-binding protein sequence is a transcription factor related in structure and function to Methanococcus transcription factor B. J. Biol. Chem. 270:17649-17651. [DOI] [PubMed] [Google Scholar]
- 10.Hethke, C., A. C. Geerling, W. Hausner, W. M. de Vos, and M. Thomm. 1996. A cell-free transcription system for the hyperthermophilic archaeon Pyrococcus furiosus. Nucleic Acids Res. 24:2369-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata, A., T. Kanai, T. J. Santangelo, M. Tajiri, K. Manabe, J. N. Reeve, T. Imanaka, and K. S. Murakami. 2008. Archaeal RNA polymerase subunits E and F are not required for transcription in vitro, but a Thermococcus kodakarensis mutant lacking subunit F is temperature-sensitive. Mol. Microbiol. 70:623-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenney, F. E., Jr., and M. W. Adams. 2008. Hydrogenases of the model hyperthermophiles. Ann. N. Y. Acad. Sci. 1125:252-266. [DOI] [PubMed] [Google Scholar]
- 13.Kanai, T., J. Akerboom, S. Takedomi, H. J. van de Werken, F. Blombach, J. van der Oost, T. Murakami, H. Atomi, and T. Imanaka. 2007. A global transcriptional regulator in Thermococcus kodakaraensis controls the expression levels of both glycolytic and gluconeogenic enzyme-encoding genes. J. Biol. Chem. 282:33659-33670. [DOI] [PubMed] [Google Scholar]
- 14.Kiyonari, S., S. Tahara, M. Uchimura, T. Shirai, S. Ishino, and Y. Ishino. 2009. Studies on the base excision repair (BER) complex in Pyrococcus furiosus. Biochem. Soc. Trans. 37:79-82. [DOI] [PubMed] [Google Scholar]
- 15.Lam, W. L., and W. F. Doolittle. 1992. Mevinolin-resistant mutations identify a promoter and the gene for a eukaryote-like 3-hydroxy-3-methylglutaryl-coenzyme A reductase in the archaebacterium Haloferax volcanii. J. Biol. Chem. 267:5829-5834. [PubMed] [Google Scholar]
- 16.Lee, S. J., M. Surma, W. Hausner, M. Thomm, and W. Boos. 2008. The role of TrmB and TrmB-like transcriptional regulators for sugar transport and metabolism in the hyperthermophilic archaeon Pyrococcus furiosus. Arch. Microbiol. 190:247-256. [DOI] [PubMed] [Google Scholar]
- 17.Lucas, S., L. Toffin, Y. Zivanovic, D. Charlier, H. Moussard, P. Forterre, D. Prieur, and G. Erauso. 2002. Construction of a shuttle vector for, and spheroplast transformation of, the hyperthermophilic archaeon Pyrococcus abyssi. Appl. Environ. Microbiol. 68:5528-5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumi, R., K. Manabe, T. Fukui, H. Atomi, and T. Imanaka. 2007. Disruption of a sugar transporter gene cluster in a hyperthermophilic archaeon using a host-marker system based on antibiotic resistance. J. Bacteriol. 189:2683-2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naji, S., S. Grunberg, and M. Thomm. 2007. The RPB7 orthologue E′ is required for transcriptional activity of a reconstituted archaeal core enzyme at low temperatures and stimulates open complex formation. J. Biol. Chem. 282:11047-11057. [DOI] [PubMed] [Google Scholar]
- 20.Santangelo, T. J., L. Cubonova, C. L. James, and J. N. Reeve. 2007. TFB1 or TFB2 is sufficient for Thermococcus kodakaraensis viability and for basal transcription in vitro. J. Mol. Biol. 367:344-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santangelo, T. J., L. Cubonova, and J. N. Reeve. 2008. Shuttle vector expression in Thermococcus kodakaraensis: contributions of cis elements to protein synthesis in a hyperthermophilic archaeon. Appl. Environ. Microbiol. 74:3099-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato, T., T. Fukui, H. Atomi, and T. Imanaka. 2005. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71:3889-3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato, T., T. Fukui, H. Atomi, and T. Imanaka. 2003. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185:210-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spitalny, P., and M. Thomm. 2008. A polymerase III-like reinitiation mechanism is operating in regulation of histone expression in archaea. Mol. Microbiol. 67:958-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomm, M., C. Reich, S. Grunberg, and S. Naji. 2009. Mutational studies of archaeal RNA polymerase and analysis of hybrid RNA polymerases. Biochem. Soc. Trans. 37:18-22. [DOI] [PubMed] [Google Scholar]
- 26.Williams, R. S., G. Moncalian, J. S. Williams, Y. Yamada, O. Limbo, D. S. Shin, L. M. Groocock, D. Cahill, C. Hitomi, G. Guenther, D. Moiani, J. P. Carney, P. Russell, and J. A. Tainer. 2008. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]