

Abstract
The identification of genes to be deleted or amplified is an essential step in metabolic engineering for strain improvement toward the enhanced production of desired bioproducts. In the past, several methods based on flux analysis of genome-scale metabolic models have been developed for identifying gene targets for deletion. Genome-wide identification of gene targets for amplification, on the other hand, has been rather difficult. Here, we report a strategy called flux scanning based on enforced objective flux (FSEOF) to identify gene amplification targets. FSEOF scans all the metabolic fluxes in the metabolic model and selects fluxes that increase when the flux toward product formation is enforced as an additional constraint during flux analysis. This strategy was successfully employed for the identification of gene amplification targets for the enhanced production of the red-colored antioxidant lycopene. Additional metabolic engineering based on gene knockout simulation resulted in further synergistic enhancement of lycopene production. Thus, FSEOF can be used as a general strategy for selecting genome-wide gene amplification targets in silico.
One of the ultimate objectives of industrial biotechnology is the improvement of the yields and productivities of bioproducts. Toward this end, metabolic engineering has been successfully employed in improving industrial strains and has recently become more powerful due to the integration of omics and computational methods at the systems level (19, 27). Metabolic engineering improves the metabolic phenotype toward the overproduction of a desired product by the amplification and/or deletion of certain metabolic genes, together with the rewiring of regulatory circuits (10, 14).
Recent systemic approaches employing genome-scale metabolic models (7) have enabled researchers to identify gene deletion targets for improving microbial strains (1, 4, 30, 31) by utilizing various algorithms. For example, the nested optimization framework using mixed-integer linear programming (OptKnock) (4, 25) and a sequential gene deletion strategy based on the formalism of minimization of metabolic adjustment (MOMA) (1, 31) allowed successful identification of the genes to be deleted among a large number of gene candidates. Using this strategy in combination with regulatory engineering and omics analysis, systems-level metabolic engineering was performed for developing l-valine- and l-threonine-overproducing Escherichia coli strains (16, 24).
However, it has been difficult to identify gene amplification targets by similar approaches. This is because predicting the metabolic phenotypes after gene deletion is much easier than predicting the metabolic phenotype after gene amplification; gene deletion causes the corresponding flux to become zero, while gene amplification does not necessarily increase the corresponding metabolic fluxes, due to complex regulation of the metabolic network. Additionally, it is even more difficult to predict how much the flux will increase upon gene amplification. A creative algorithm, OptReg, has been developed (26), but its use in actual strain development has not been reported. 13C-based metabolic flux analysis (3) can also be used to identify gene amplification targets, but it is difficult to make predictions in a genome-scale metabolic network. Recently, Fowler and coworkers suggested a strategy for identifying gene overexpression targets based on the necessity ratio, defined as the ratio of a pathway flux under two different objective functions (9). The approach is simple and useful when the relationship between the perturbed intracellular flux and the product formation rate is monotonic. However, the relationship between the intracellular flux and the product formation rate has various patterns which are nonmonotonic and can result in erroneous predictions when using the necessity ratio. Here, we report the development of a strategy for identifying gene amplification targets and its application in strain development to enhance the production of a target product, using the red-colored antioxidant lycopene as an example. Also, the results of the combined application of both gene amplification and gene knockout simulation for lycopene overproduction are reported.
MATERIALS AND METHODS
Strains and media.
All strains used in this study are listed in Table 1. E. coli XL1-Blue was used as a host strain for recombinant DNA work. It was routinely cultured in Luria-Bertani (LB) medium (10 g Bacto tryptone, 5 g yeast extract, and 10 g NaCl per liter; Difco) at 37°C. E. coli DH5α and WL3110 (lacI mutant of W3110) (16) were used as host strains for lycopene production. Cells were cultured at 30°C for 48 h in a 250-ml flask containing 50 ml of 2×YT medium (16 g Bacto tryptone, 10 g yeast extract, 5 g NaCl per liter; Difco) (6). All mutant strains were constructed from E. coli W3110 by the one-step inactivation method using the RED recombinase system (5). For the batch fermentation, recombinant E. coli W3110 strains were cultivated at 30°C in a 6.6-liter Bioflo 3000 fermentor (New Brunswick Scientific) containing 2 liters of R/2 medium plus 20 g liter−1 glucose and 2 g liter−1 yeast extract. R/2 medium (pH 6.8) contains, per liter, 2 g (NH4)2HPO4, 6.75 g KH2PO4, 0.85 g citric acid, 0.7 g MgSO4·7H2O, and 5 ml of a trace metal solution [10 g FeSO4·7H2O, 2.25 g ZnSO4·7H2O, 1 g CuSO4·5H2O, 0.5 g MnSO4·5H2O, 0.23 g Na2B4O7·10H2O, 2g CaCl2·2H2O, and 0.1 g (NH4)6MO7O24 per liter of 5 M HCl]. Ampicillin (50 μg liter−1), chloramphenicol (34 μg liter−1), kanamycin (25 μg liter−1), and/or tetracycline (5 μg liter−1) was added depending on the antibiotic markers. Cell growth was monitored by measuring the absorbance at 600 nm (Ultraspec3000 spectrophotometer; Pharmacia Biotech). The dry cell weight (DCW) was determined by measuring the weight of dried cells after the culture broth was filtered through an ultrafine filter (0.7-μm-pore-size GF/F; Whatman).
TABLE 1.
Strains used in this study
| E. coli strain | Relevant properties | Source or reference |
|---|---|---|
| W3110 | F−mcrA mcrB IN(ΔrrnD rrnE)1λ− | Stratagene |
| WL3110 | W3110 (ΔlacI) | 16 |
| WLG | W3110 (ΔlacI ΔgdhA) | This study |
| WLA | W3110 (ΔlacI ΔgpmA) | This study |
| WLB | W3110 (ΔlacI ΔgpmB) | This study |
| WLGA | W3110 (ΔlacI ΔgdhA ΔgpmA) | This study |
| WLGB | W3110 (ΔlacI ΔgdhA ΔgpmB) | This study |
| WLAB | W3110 (ΔlacI ΔgpmA ΔgpmB) | This study |
| WLGB-RP | W3110 (ΔlacI ΔgdhA ΔgpmB ptrc dxs idi ispA) | This study |
| WLGB-RPP | W3110 (ΔlacI ΔgdhA ΔgpmB ptrc dxs idi ispA pps) | This study |
| DH5α | F− φ80dlacZΔM15 Δ(lacZYAI argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA ΔsupE44 thi-1 gyrA96 relA1 | New England Biolabs |
| XL1-Blue | recA1 endAI gyrA96 thi hsdR17(rK− mK+) supE44 relA1 lac [F′ proAB+lacIqZΔM15::Tn10(Tetr)] | Stratagene |
Plasmids.
The plasmids constructed and used in this study are listed in Table 2. All DNA manipulations were carried out using the standard protocols (29). Restriction enzymes were purchased from New England Biolabs. PCR was performed using a PTC-200 DNA Engine (PharmaTech) with the expand high fidelity PCR system (Roche Molecular Biochemicals). E. coli W3110 genomic DNA was used as a template for the PCR amplification of the dxs, fbaA, icdA, idi, mdh, pfkA, pgi, and tpiA genes using the primers listed in Table S1 in the supplemental material. The crt operon (crtE, crtX, crtY, crtI, crtB, and crtZ) was amplified by PCR using the genomic DNA of Erwinia uredovora as a template and the primers Car-F and Car-R and was cloned into the EcoRI site of pACYC184 to construct pCar184. The crtXY genes were removed from pCar184 to construct pLyc184. The crtEIB genes in pLyc184 were constitutively expressed from its own promoter. The dxs gene was cloned into the EcoRI and KpnI sites of pTrc99A under the trc promoter to construct pTrcDx (6.5 kb). The amplification target genes identified by an algorithm called flux scanning based on enforced objective flux (FSEOF) (see “FSEOF” below) were cloned into pTrcDx to construct pTrcDx-pfkA, pTrcDx-pgi, pTrcDx-fbaA, pTrcDx-tpiA, pTrcDx-icdA, pTrcDx-idi, pTrcDx-mdh, pTrcDx-idi-fbaA, pTrcDx-idi-tpiA, and pTrcDx-idi-mdh (Table 2). All genes under the trc promoter were expressed constitutively in E. coli DH5α because it is not a lacIq strain. The sequences of the cloned genes were confirmed by using an ABI PRISM 7700 sequencer (Applied Biosystems).
TABLE 2.
Plasmids used in this study
| Plasmid | Relevant propertiesa | Source |
|---|---|---|
| pACYC184 | Tcr Cmr, p15A ori, 4.2 kb | New England Biolabs |
| pCar184 | Tcr, Erwinia uredovora crt operon (crtEXYIB) under gntT104 promoter cloned between EcoRI of site of pACYC184, 11.1 kb | This study |
| pLyc184 | Tcr, deletion of crtXY genes from pCar184, 8.7 kb | This study |
| pTrc99A | Apr, trc promoter, ColE1 ori, 4.1 kb | Amersham Pharmacia |
| pTrcDx | Apr, trc promoter, dxs gene cloned at KpnI and EcoRI sites of pTrc99A, 4.7 kb | This study |
| pTrcDx-idi | Apr, trc promoter, idi gene cloned at KpnI and XbaI sites of pTrcDx under its own RBS, 6.6 kb | This study |
| pTrcDx-fbaA | Apr, trc promoter, fbaA gene cloned at KpnI and XbaI sites of pTrcDx under its own RBS, 7.1 kb | This study |
| pTrcDx-tpiA | Apr, trc promoter, tpiA gene cloned at KpnI and XbaI sites of pTrcDx under its own RBS, 6.8 kb | This study |
| pTrcDx-pfkA | Apr, trc promoter, pfkA gene cloned at XbaI and PstI sites of pTrcDx under its own RBS, 7.1 kb | This study |
| pTrcDx-pgi | Apr, trc promoter, pgi gene cloned at XbaI and PstI sites of pTrcDx under its own RBS, 7.7 kb | This study |
| pTrcDx-icdA | Apr, trc promoter, icdA gene cloned at XbaI site of pTrcDx under its own RBS, 7.3 kb | This study |
| pTrcDx-mdh | Apr, trc promoter, mdh gene cloned at XbaI site of pTrcDx under its own RBS, 7.0 kb | This study |
| pTrcDx-idi-fbaA | Apr, trc promoter, fbaA gene cloned at XbaI site of pTrcDx-idi under its own RBS, 7.7 kb | This study |
| pTrcDx-idi-tpiA | Apr, trc promoter, tpiA gene cloned at XbaI site of pTrcDx-idi under its own RBS, 7.4 kb | This study |
| pTrcDx-idi-mdh | Apr, trc promoter, mdh gene cloned at XbaI site of pTrcDx-idi under its own RBS, 7.9 kb | This study |
RBS, ribosome binding site.
Batch-fed fermentation.
For the production of lycopene using the final strains, batch-fed cultures were grown in a 6.6-liter jar fermenter (Bioflo 3000; New Brunswick Scientific Co.) containing 2 liters of R/2 medium supplemented with 20 g liter−1 glucose, 10 mg liter−1 tetracycline, and 50 mg liter−1 ampicillin. The R/2 medium (pH 6.8) contains, per liter, (NH4)2HPO4, 2 g; KH2PO4, 6.75 g; citric acid, 0.85 g; MgSO4·7H2O, 0.7 g; and trace metal solution [per liter of 5 M HCl, FeSO4·7H2O, 10 g; ZnSO4·7H2O, 2.25 g; CuSO4·5H2O, 1 g; MnSO4·5H2O, 0.5 g; Na2B4O7·10H2O, 0.23 g; CaCl2·2H2O, 2 g; and (NH4)6MO7O24, 0.1 g], 5 ml. The seed culture (200 ml) was prepared in the same medium. The culture pH was controlled at 6.8 by the addition of 14% (vol/vol) ammonia water. The dissolved oxygen (DO) concentration was controlled at 40% air saturation by automatically increasing the agitation speed up to 1,000 rpm and by changing the pure oxygen percentage. The nutrient feeding solution used for the batch-fed culture contained 600 g of glucose and 15 g of MgSO4·7H2O per liter. The feeding solution was added by the DO-stat feeding strategy (18).
Quantification of lycopene.
The lycopene concentration was determined by the cold acetone extraction method (23) using high-performance liquid chromatography (1100 series; Agilent Technology) equipped with a Spherisorb S5 ODS2 C18 column (4.6 by 250 mm, 5-μm particle size; Waters) and a diode array detector (474 nm; Agilent Technology). Lycopene purchased from Sigma-Aldrich was used to generate a standard curve. The mobile phase consisted of 30% (vol/vol) acetonitrile and 70% (vol/vol) methanol and was flown at 1.5 ml min−1.
FSEOF.
An algorithm called flux scanning based on enforced objective flux (FSEOF) was developed for the selection of gene targets to be amplified (Fig. 1). The intracellular flux distributions were calculated by constraint-based flux analysis using MetaFluxNet (15) (http://mbel.kaist.ac.kr/lab/mfn) installed on a personal computer (2.40 GHz Pentium 4 CPU and Windows XP platform). The genome-scale metabolic network of E. coli EcoMBEL979 (see Table S3 in the supplemental material), which was constructed based on two excellent models reported by Reed et al. (28) and Ma and Zeng. (20), was used. For the simulation of recombinant E. coli producing lycopene, the EcoMBEL979 model was expanded with the heterologous lycopene biosynthetic pathways. The FSEOF procedure applied to lycopene production is described below.
FIG. 1.

Concept of FSEOF framework to select the gene amplification targets for enhanced product formation. FSEOF searches for the candidate fluxes to be amplified through scanning for those fluxes that increase with enforced objective (product formation) flux under the objective function of maximizing biomass formation flux. During the FSEOF implementation, three types of intracellular flux profiles are typically identified, increased, decreased, and unchanged; oscillating flux profiles can be found in rare cases. Among them, FSEOF identifies the fluxes showing the increased profile as the primary amplification targets.
First, the initial fluxes (vjinitial) were calculated by constraint-based flux analysis using the objective function of maximizing biomass formation, as follows.
Maximize vbiomass subject to ∑j=1NSijvjinitial=0(where ∀i ∈ M and ∀j ∈ N), vATPM ≥ vatp_maint, vZLYC = vlycopene, and vjα≤vj≤vjβ(where vj ∈ R) where Sij is the stoichiometric coefficient of metabolite i (belonging to a set of whole metabolites, M) in the jth reaction (belonging to a set of whole reactions, N) and vjinitialis the initial calculated flux of the jth reaction. vbiomass, vATPM, and vZLYC represent the biomass formation rate, the ATP consumption rate for maintenance, and the lycopene production rate, respectively. vatp_maint is the ATP flux required for non-growth-associated maintenance. vinitiallycopeneis the lycopene production rate measured from the initial experiment, which was 0.0002 mmol g DCW−1 h−1; it was measured by cultivating recombinant E. coli DH5α harboring pLyc184 and pTrcDx in 2×YT medium at 30°C. It should be noted that, in general, this can be provided as a measured constraint or calculated constraint if no experimental result is available. vjαand vjβare the lower and upper bounds of the flux of the jth reaction, respectively: for reversible reactions, −∞ ≤ vj ≤ ∞, and for irreversible reactions, 0 ≤ vj ≤ ∞.
Then, the theoretical maximum product (lycopene) formation rate was calculated; this was done by setting the objective function as maximizing the product formation flux during the constraint-based flux analysis, which can be formulated as follows.
Maximize vproduct subject to ∑j=1NSijvj=0(where ∀i ∈ M and ∀j ∈ N), vATPM ≥ vatp_maint, and vjα≤vj≤vjβ(where vj ∈ R) where vj is the flux of the jth reaction.
Finally, FSEOF was performed during the constraint-based flux analysis under the objective function of maximizing cell growth while the lycopene production rate (our actual objective) is gradually increased (enforced) from the initial flux value to a value adjacent to the theoretical maximum value for product formation (typically 90% of the maximum theoretical or higher value). This is because the flux distributions obtained by constraint-based flux analysis with the theoretical maximum production rate as an additional constraint are usually unrealistic; the biomass formation rate often becomes zero when the product formation objective function is set to its maximization during the simulation. Subsequently, the calculated intracellular fluxes under enforced product formation constraints were scanned for the selection of targets meeting the preset criteria for primary gene amplification. The targets were selected by identifying fluxes that increased upon the application of the enforced objective flux without changing the reaction's direction. This is mathematically formulated as follows.
Select vj that satisfies |vj|max>|vjinitial|and vjmax×vjmin≥0after the following calculations.
Maximize vbiomass subject to 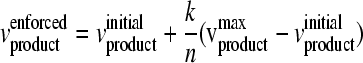 and K = {k | k = 1, 2, …, n − 1} (where n ≥ 10) subject to ∑j=1NSijvj=0(where ∀i ∈ M and ∀j ∈ N), vATPM ≥ vatp_maint, and vjα≤vj≤vjβ(where vj ∈ R) where vjmaxand vjminare the maximum and minimum fluxes of the jth reaction calculated during the implementation of FSEOF. vproductenforcedis the additional constraint provided during the constraint-based flux analysis; it starts with the initial value vproductinitialplus one nth of the difference between the vproductmaxand vproductinitialand is increased to a value adjacent to vproductmaxin k steps.
and K = {k | k = 1, 2, …, n − 1} (where n ≥ 10) subject to ∑j=1NSijvj=0(where ∀i ∈ M and ∀j ∈ N), vATPM ≥ vatp_maint, and vjα≤vj≤vjβ(where vj ∈ R) where vjmaxand vjminare the maximum and minimum fluxes of the jth reaction calculated during the implementation of FSEOF. vproductenforcedis the additional constraint provided during the constraint-based flux analysis; it starts with the initial value vproductinitialplus one nth of the difference between the vproductmaxand vproductinitialand is increased to a value adjacent to vproductmaxin k steps.
RESULTS AND DISCUSSION
Flux scanning based on enforced objective flux.
FSEOF starts with the constraint-based flux analysis of a genome-scale metabolic model, which calculates the internal metabolic fluxes by linear optimization of an objective function (7). A set of mass balances around metabolites yields the stoichiometric model Sij·vj = 0 with an assumption of pseudo steady state, in which Sij is a stoichiometric coefficient of a metabolite i in the jth reaction and vj is the flux of the jth reaction given in mmol g DCW−1 h−1. The metabolic model constructed in this study is an underdetermined system, having infinite sets of solutions. Thus, linear programming (LP) was performed, with maximizing the biomass formation rate, represented by the overall macromolecule composition of the cell, as the objective function, with constraints of mass conservation, reaction thermodynamics, and metabolic capacity in order to calculate intracellular fluxes. We introduced the enforced flux toward product formation as an additional constraint during the linear optimization of the objective function given as the maximum biomass formation.
In general, the yield of metabolite production is lower than the theoretical yield. This is because the objective of microbial metabolism is different from our desired objective. Thus, the magnitude of the enforced objective constraint employed during FSEOF simulation can be a key point in that the objective function is intentionally pushed in the direction of our objective. In this study, the enforced objective constraint was formulated by setting the product formation rate at the value currently attained by the strains and increasing it to 90% of the theoretical maximum. Usually, there is a direct inverse relationship between the biomass formation and product formation rates. This is why the biomass formation rate often becomes zero when the product formation rate achieves the theoretical maximum value; in practice, genome-scale simulations can provide unrealistic flux values to achieve the theoretical maximum due to the limitation of cell growth. Since the gene amplification targets are identified by observing the tendencies of the flux changes, we designed the FSEOF algorithm in such a way that it scans for the flux changes when the enforced objective flux is applied.
During this process, the internal metabolic fluxes that increase with increasing flux toward product formation were scanned for; these fluxes are potential targets to be amplified for increased yield of the desired product (Fig. 1). Although the flux patterns rather than the actual flux values were utilized for selecting the amplification targets by FSEOF simulation, the problem of multiple optima in linear programming calculation still exists. Thus, flux variability analysis was also carried out (21) for the selected targets, as well as for some nonselected targets, to evaluate the performance of FSEOF simulation (Fig. 2B to E; also see below).
FIG. 2.

Results of FSEOF (gene amplification) simulation, flux variability analysis, and MOMA (gene knockout) simulation. (A) Overview of central metabolic pathways for lycopene production and the amplification target genes identified by FSEOF. Red, blue, and gray arrows indicate the fluxes that are increased, decreased and unchanged, respectively, with the enforced objective of increased lycopene production. Red-colored and blue-colored gene names indicate that these genes were amplified and deleted, respectively, in this study. Abbreviations of metabolites are as follows: 3C4MOP, 3-carboxy-4-methyl-2-oxopentanoate; AC_ex, acetate (extracellular); ACA, acetyl-CoA; ACGAM1P, N-acetyl-d-glucosamine 1-phosphate; ACGLU, N-acetyl-l-glutamate; ACGSSA, N-acetyl-l-glutamate-5-semialdehyde; ACSER, O-acetyl-l-serine; ACTP, acetyl phosphate; CYS-L, l-cysteine; DHAP, dihydroxyacetone phosphate; E4P, d-erythrose-4-phosphate; F6P, d-fructose 6-phosphate; FPP, trans,trans farnesyl pyrophosphate; FUM, fumarate; GLX, glyoxylate; GPP, geranyl diphosphate; l-Glu, l-glutamate; malACP, malonyl acyl carrier protein; OAA, oxaloacetate; P5P, alpha-d-ribose 5-phosphate; PEP, phosphoenolpyruvate; PGA, d-glycerate 2-phosphate; PYR, pyruvate; RL5P, d-ribulose 5-phosphate; S7P, sedoheptulose 7-phosphate; SER-L, l-serine; X5P, d-xylulose 5-phosphate. Other abbreviations which are not mentioned here are defined in the text and in Table 3. The detailed results of FSEOF are provided in Table S3B in the supplemental material. (B to E) Flux variability patterns of the targets selected by FSEOF calculation. The number above each plot indicates the number of fluxes out of the 35 fluxes identified by FSEOF that follow the corresponding pattern group. The remaining 5 fluxes resulted in an unbounded pattern and were not shown. The upper and bottom lines represent the maximum and minimum flux values, respectively. The various patterns are classified as fluxes increasing without variability (B), increasing in a narrow range in which the minimum of the upper line is lower than the maximum of the bottom line (C), increasing within a broad range (D), and showing no change or decreasing-increasing (E). (F) Results of MOMA simulation. The red circle represents the results for the E. coli wild-type strain. Red diamonds represent the results for single gene deletions. Green rectangles represent the results for the deletion of two genes.
Implementation of FSEOF on lycopene-producing E. coli.
FSEOF was employed to identify the amplification target genes for enhanced lycopene production in E. coli. Based on previous reports (6, 13, 17, 22, 23), a recombinant E. coli DH5α strain harboring the two plasmids pLyc184 and pTrcDx, which contain the Erwinia uredovora crtEIB genes and the E. coli dxs gene (23), respectively, was constructed as a parental strain for the production of lycopene and implementation of FSEOF. The genome-scale metabolic network of E. coli EcoMBEL979 was expanded with the heterologous lycopene biosynthetic pathways (see Tables S3A and B in the supplemental material) and was simulated using MetaFluxNet (15). The experimentally measured lycopene flux value in a parental recombinant strain was 0.0002 mmol g DCW−1 h−1. The maximum theoretical lycopene production flux value calculated using the objective function of its maximized formation was 0.5658 mmol g DCW−1 h−1. Thus, FSEOF was carried out with the enforced objective constraints of varying the lycopene production flux value at 9 steps from 0.0002 to 0.5092 mmol g DCW−1 h−1 (90% of 0.5658). Then, the selection criteria of |vj|max>|vjinitial|and vjmax×vjmin≥0were implemented during FSEOF to select fluxes showing at least one period of the increasing pattern without changing the reaction's direction. The newly predicted flux values using FSEOF should be higher than the initial flux values, vj. For predicted gene targets where the flux direction changed, they were not considered overexpression targets. This resulted in 35 out of 983 reactions being identified as initial gene amplification targets for increasing lycopene production (Fig. 2A and Table 3; also see Table S4A in the supplemental material). It is also important to set up the range of the enforced objective flux within a realistic range that can be achieved by the strain. In fact, the best strain developed in this study possessed a lycopene production rate that was much lower than the upper limit of the enforced objective flux. Obviously, there are many reasons for this, one being that lycopene is a product that becomes membrane associated. Thus, its production is limited by the capacity of the membrane and is also affected by its consequent alteration of the functions of proteins on the membrane. Therefore, we tested whether varying the enforced objective function within a more realistic range affected the FSEOF prediction results; the FSEOF results obtained within the enforced objective function in a smaller range (from 0.0002 mmol g DCW−1 h−1 to 0.01131 mmol g DCW−1 h−1) were compared with those of the original simulation. As shown in Table S4B in the supplemental material, the new simulation with the smaller objective range suggested the same 35 candidates as gene amplification targets. Thus, varying the range of the enforced objective function from the initial value to 90% of the theoretical maximum value was found to be valid.
TABLE 3.
List of key genes and enzymes investigated based on the FSEOF simulation
| Gene | Enzyme | Functional category |
|---|---|---|
| acnAB | Aconitase | TCA cycle |
| gltA | Citrate (CIT) synthase | |
| fumAB | Fumarase | |
| icdAa | Isocitrate (ICIT) dehydrogenase (NADP) | |
| mdha,c | Malate (MAL) dehydrogenase | |
| sdhABCD | Succinate (SUC) dehydrogenase | |
| sucCD | Succinyl-CoA (SUCOAS) synthetase (ADP-forming) | |
| sucAB | 2-Oxoglutarate (AKG) dehydrogenase | |
| sdhABCD | Succinate dehydrogenase | |
| dxr | 1-Deoxy-d-xylulose 5-phosphate reductoisomerase | Methylerythritol phosphate pathwayb |
| dxsc | 1-Deoxyxylulose-5-phosphate synthase | |
| idia | Isopentenyl diphosphate (IPDP) isomerase | |
| ispAa | Geranyltranstransferase/dimethylallyltranstransferase | |
| ispD | 4-Diphosphocytidyl-2C-methyl-d-erythritol synthase | |
| ispE | 4-Diphosphocytidyl-2-C-methylerythritol kinase | |
| ispF | 2C-Methyl-d-erythritol 2,4-cyclodiphosphate synthase | |
| ispG | 1-Hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase | |
| ispH | 1-Hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase | |
| crtE | Geranylgeranyl pyrophosphate (GGPP) synthase | Heterogeneous genes for lycopene synthesis |
| crtB | Phytoene (PHYTO) synthetase | |
| crtI | Phytoene dehydrogenase | |
| fbaAa | Fructose-bisphosphate (FBP) aldolase | Glycolysis |
| pfkABa | Phosphofructokinase | |
| pgia | Glucose-6-phosphate (G6P) isomerase | |
| tpiAa,c | Triose-phosphate isomerase |
A gene that was overexpressed in this study.
Enhancing fluxes of the methylerythritol phosphate pathway has a positive effect on lycopene production. Among them, the products of dxs, idi, and ispA were previously shown to enhance lycopene production.
A key gene that was newly identified as an amplification target by using FSEOF.
The results of FSEOF suggest that the upper glycolytic fluxes to glyceraldehyde-3-phosphate (G3P) and the fluxes of methylerythritol phosphate (MEP) and the lycopene biosynthetic pathway should be increased to enhanced lycopene production. Also, it was found that the fluxes downstream of G3P should be decreased. It is interesting to note from a previous report (1) that lycopene production could be enhanced by employing the gpmAB and talB mutants, resulting in a decrease in pentose phosphate pathway fluxes, which is in agreement with the FSEOF prediction. These results suggest that the availability of a G3P pool is important for the synthesis of lycopene.
Another important pattern observed from FSEOF simulation involves the fluxes through the metabolite acetyl-coenzyme A (CoA). At the acetyl-CoA node, the incoming flux from pyruvate decreased and was found to be redirected toward 1-deoxy-d-xylulose-5-phosphate (DXYL5P), as seen in the increase in flux from pyruvate to DXYL5P. This would naturally result in the decrease which was observed in the outgoing fluxes from the acetyl-CoA node, with the exception of the flux through citrate synthase (TCA cycle). The reason for the increased flux into the TCA cycle seems to be for the generation of NADH, NADPH, and ATP, which are required for the synthesis of isopentenyl diphosphate (IPP) or dimethylallyl diphosphate (DMPP) via the MEP pathway (Fig. 2A). As 6 mol IPP and 2 mol DMPP are required for the synthesis of 1 mol lycopene, eight moles each of these cofactors are needed.
Flux variability analysis for the selected target.
Constraint-based flux analysis does not give a unique flux distribution, and thus, there exists a rather large degree of freedom within the network to reach the optimal flux value for the objective function (e.g., growth rate). Therefore, the flux values obtained in FSEOF might not be the optimal value and can result in a false-positive prediction. To confirm that the fluxes obtained from FSEOF are meaningful, the flux variability analysis was performed.
The flux variability analysis in response to the enforced product formation flux was performed for the 35 fluxes identified by FSEOF and 5 additional fluxes that were not gene amplification targets (as negative controls). As shown in Fig. 2B to E, there were 4 meaningful flux variability patterns (see Fig. S1 in the supplemental material for more information on the flux variability analysis). Eleven fluxes were found to have an increasing pattern without variability (Fig. 2B), the same as FSEOF predicted. Ten fluxes were also found to have an increasing pattern but within a narrow increasing range in which the minimum of the upper line was lower than the maximum of the bottom line (Fig. 2C). This suggests that these 10 fluxes should increase with the enforced objective flux, which is consistent with the results of FSEOF. Thus, these 21 fluxes (Fig. 2B and C) showing increasing trends are consistent with the results from FSEOF simulation. Of the remaining 14 fluxes, 5 showed an increasing pattern within a broad increasing range (Fig. 2D), 4 showed almost no change and/or a decreasing-increasing pattern (Fig. 2E), and the remaining 5 reactions were unbound (not shown). These results suggest that 26 out of 35 fluxes identified as amplification targets are consistent with the results of flux variability analysis. Among the other 9 fluxes, 5 were unbound when flux variability analysis was performed, providing no further information. The remaining 4 fluxes were the only ones which differed from the results from FSEOF by showing no increasing pattern (Fig. 2E). It should be stated that the 9 fluxes found to be either unbound or not changing were also experimentally verified in this study and were found to be good targets, with the exception of the Pfk amplification (Pgi and IcdA also resulted in marginal changes), contrary to the results of flux variability analysis. Thus, FSEOF provides a simple and powerful way of identifying the gene amplification targets.
To further examine whether flux variability analysis can suggest results different from those of FSEOF, five central metabolic fluxes that were not suggested as targets using FSEOF were tested (as negative controls). The variable ranges of the fluxes for the Eno, GapA, Pgl, Ppc, and Pyk reactions showed a decreasing pattern with increasing lycopene production, as expected for the negative controls (see Fig. S1E in the supplemental material). Thus, FSEOF simulation allows the prediction of gene amplification targets that are mostly consistent with (and even better than) the results from flux variability analysis.
Experimental validation of the targets identified by FSEOF simulation.
In order to validate the amplification target genes selected by FSEOF, the target genes were cloned into pTrcDx carrying the dxs gene and subsequently introduced into the recombinant E. coli DH5α harboring pLyc184 carrying the crtEIB genes (see Materials and Methods and Tables 1 to 3). First, all the target genes whose products are involved in glycolysis were amplified because it was expected that the reinforcement of these fluxes would increase G3P, an important precursor of lycopene. Among the TCA cycle genes, only the icdA and mdh genes were considered for actual experiments as a proof of concept; this was because other reactions in the TCA cycle are catalyzed by relatively large enzyme complexes which are rather difficult to properly amplify in the cell.
The lycopene concentration obtained with the control strain was 2.52 mg liter−1, with a content of 1.48 mg g DCW−1. When the dxs gene was overexpressed, the lycopene concentration increased to 4.95 mg liter−1 with a content of 3.09 mg g DCW−1. When the pfkA, pgi, fbaA, tpiA, icdA, and mdh genes were amplified together with the dxs gene, the lycopene concentrations obtained were 3.25, 5.32, 6.94, 6.84, 4.94, and 9.06 mg liter−1 and the lycopene contents were 1.63, 2.73, 4.62, 4.89, 2.98, and 4.83 mg g DCW−1, respectively (Fig. 3A; see Table S2 in the supplemental material). Amplification of the fbaA, tpiA, and mdh genes resulted in the highest enhancement of lycopene production. One of the in silico-predicted targets, pfkA, showed a negative effect on lycopene production upon its amplification. This is due to the imperfect nature of the in silico genome-scale metabolic model that cannot account for complex regulatory mechanisms. Nonetheless, the identification of at least three nonintuitive gene amplification targets (in particular, the mdh gene) proves the usefulness of FSEOF.
FIG. 3.

Identification of gene targets for enhanced lycopene production by FSEOF, MOMA, and their combination. (A) Production of lycopene by recombinant E. coli strains engineered based on the results of FSEOF, MOMA, and their combination. All strains harbor the plasmid pLyc184 in addition to the plasmid shown. The lycopene concentrations (mg liter−1) and contents (mg g DCW−1) are shown by black and white bars, respectively. Error bars show standard deviations. Strains and plasmids used are described in Tables 1 and 2. All experiments were carried out in triplicate. The products of genes shown in the plasmid names are phosphofructokinase (pfkA), phosphoglucose isomerase (pgi), fructose-bisphosphate aldolase (fbaA), triosephosphate isomerase (tpiA), isocitrate dehydrogenase (icdA), malate dehydrogenase (mdh), and isopentenyl diphosphate isomerase (idi). (B) Lycopene production by batch-fed cultivation of WLGB-RPP(pTrcDx-idi-mdh, pLyc184). Closed circles, optical density at 600 nm (OD600); open circles, glucose concentration (conc.); open triangles, acetic acid concentration; closed squares, lycopene concentration. Error bars represent standard deviations.
Among the targets in the MEP and lycopene pathways selected by FSEOF, the idi gene was chosen for amplification because it is a key enzyme that has been shown to exert a positive effect on lycopene production (17); when it was amplified together with the dxs gene, a lycopene concentration and content of 12.85 mg liter−1 and 8.57 mg g DCW−1, respectively, could be achieved. The effects on lycopene production of coamplifying the idi gene with one of the central metabolic genes fbaA, tpiA, or mdh were also examined. Among these, the overexpression of the idi and mdh genes with the dxs gene resulted in the production of lycopene to a concentration of 13.20 mg liter−1 and a content of 9.98 mg g DCW−1, 2.7- and 3.2-fold higher, respectively, than obtained with the control strain DH5α harboring pLyc184 and pTrcDx (see Table S2 in the supplemental material). In addition to these amplification targets, the fluxes that showed a decreasing pattern in FSEOF simulation can be considered as targets for gene knockdown. This can be achieved by amplification of the reverse reaction. One of the knockdown targets identified by FSEOF was the reaction converting phosphoenolpyruvate (PEP) to pyruvate. Thus, the gene pps (encoding PEP synthase) was considered for overexpression (see below and reference 6).
Gene knockout simulation and validation.
The genome-scale MOMA simulation (31) was performed to identify the knockout target genes for further improvement of lycopene production; the knockout of gdhA, cyoA, gpmA, gpmB, icdA, or eno was predicted to enhance lycopene production. This result is slightly different from that previous reported (1), which predicted the gdhA, cyoA, ppc, gpmA, gpmB, eno, glyA, and aceE genes as knockout targets. This might be due to the different genome-scale metabolic network models used in the two studies. In addition, the cofactor required for sequential reactions catalyzed by the Erwinia crtI gene product was set to be flavin adenine dinucleotide (FAD) in this study, rather than NADP as employed in the previous report (1); it was confirmed that the Erwinia crtI gene product uses FAD as a cofactor (Gerhard Sandmann, Goethe Universitat, Frankfurt, Germany, personal communication). Among these targets, knocking out the eno gene or gpmAB genes resulted in the highest increases of flux toward lycopene production. Dual-gene knockout simulation suggested that the most significant enhancement of flux toward lycopene production can be achieved by knocking out the eno-gdhA or gpmAB-gdhA genes (Fig. 2F). However, the eno gene is an essential gene in E. coli (11) and so was removed from the candidate list. The gpmAB and gdhA genes were combinatorially knocked out by using the RED system (5). The gdhA and gpmB knockout strain (WLGB) showed the highest content of lycopene, while the gdhA and gpmAB knockout strains (WLG and WLAB, respectively) produced less lycopene (Fig. 3A). GpmA and GpmB are the isozymes of phosphoglycerate mutase, which have not yet been fully characterized. Simulation of the knockout of gpmAB showed that most of the flux from glucose went through the Entner-Doudoroff (ED) pathway. However, knocking out both genes is not realistic for E. coli as doing so causes no growth under aerobic conditions on glucose (8). Thus, partial deletion of phosphoglycerate mutase is ideal for the increased production of lycopene. The extent of the enhancement of lycopene content and concentration by gene knockout was generally lower than that achieved with gene amplification (Fig. 3A), which suggests that the amplification of genes identified by FSEOF exerted more significant effects on the enhancement of lycopene production than gene knockouts did.
Metabolic engineering based on combined gene amplification and knockout simulations.
In order to construct the final production strain by combining the beneficial effects of gene amplification and knockout, strain WLGB(pTrcDx-idi-mdh, pLyc184) was developed. Flask culture of WLGB (pTrcDx-idi-mdh, pLyc184) resulted in a lycopene concentration and content of 26.77 mg liter−1 and 8.50 mg g DCW−1, respectively (Fig. 3A), showing a significant increase in the concentration but not in the content. To further increase the lycopene content, the ispA gene, which is an amplification target in the lycopene biosynthetic pathway, was chosen for overexpression. Due to the already large size of the plasmid, however, the ispA gene was overexpressed from the chromosome by promoter replacement. Since the dxs and ispA genes form a single operon, replacing its promoter with the strong trc promoter results in constitutive overexpression of both the dxs and the ispA gene in the lacI mutant strain. To balance the flux to lycopene (Fig. 2A), the promoter of the idi gene was also replaced with the trc promoter to construct strain WLGB-RP.
As described earlier, FSEOF also identified the PEP-to-pyruvate conversion step as a knockdown target, which can be achieved by overexpressing the pps gene which encodes the enzyme responsible for the reverse reaction. This was achieved by replacing the promoter of the pps gene with the trc promoter to construct strain WLGB-RPP. The lycopene contents obtained by flask cultivation of WLGB-RP and WLGB-RPP were 8.80 and 9.51 mg g DCW−1, respectively, higher than those of WLGB (3.65 mg g DCW−1) and WLGB (pTrcDx-idi-mdh) (8.50 mg g DCW−1) (Fig. 3A). Flask cultures of strains WLGB-RP(pTrcDx-idi-mdh, pLyc184) and WLGB-RPP(pTrcDx-idi-mdh, pLyc184) resulted in lycopene concentrations of 23.97 and 19.35 mg liter−1 and contents of 12.32 and 10.63 mg g DCW−1, respectively. These results are slightly lower than that previously obtained with a strain that was developed by combined rational metabolic engineering and genomic library screening in flask cultures (12). However, batch-fed fermentation of WLGB-RP(pTrcDx-idi-mdh, pLyc184) and WLGB-RPP(pTrcDx-idi-mdh, pLyc184) resulted in the production of 206.7 and 283.0 mg liter−1 of lycopene, respectively (Fig. 3B; also see Fig. S2 in the supplemental material). The latter titer achieved with this 100% rationally designed strain is slightly higher than that obtained by batch-fed culture of an engineered E. coli strain which was optimized by combined rational metabolic engineering and random mutagenesis (2). While the previous report showed a similar performance using different methods, such as random mutagenesis, the method presented here allows us to rationally find nonintuitive targets, such as mdh, instead of having to screen thousands of mutant strains.
In conclusion, FSEOF allows the in silico identification of fluxes (gene amplification targets) to be amplified for the enhanced production of desired bioproducts through the analysis of flux trends in response to varying product formation fluxes. Also, metabolic performance could be further enhanced by combination with gene knockouts. It is true that not all the targets predicted by FSEOF resulted in enhanced product formation upon their amplification, due to the limitations in our ability to predict real flux values using the current genome-scale metabolic models. However, it is important to note that gene amplification targets that are difficult if not impossible to predict could be identified by FSEOF, and most of them were indeed successfully used to develop improved strains for the production of lycopene. Thus, the strategy reported here should be useful in developing industrial strains capable of enhanced production of desired bioproducts, and in combination with other methods, further improvement will be possible.
Supplementary Material
Acknowledgments
We thank H. O. Bang for the construction of pLyc184.
This work was supported by the Korean Systems Biology Research Project (grant no. 20090065571) of the Ministry of Education, Science, and Technology (MEST) through the National Research Foundation of Korea (NRF). Further support, from the World Class University Program (grant no. R32-2008-000-10142-0) of the MEST through the NRF and by the LG Chem Chair Professorship, the IBM SUR program, and Microsoft, is appreciated.
Footnotes
Published ahead of print on 26 March 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Alper, H., Y. S. Jin, J. F. Moxley, and G. Stephanopoulos. 2005. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab. Eng. 7:155-164. [DOI] [PubMed] [Google Scholar]
- 2.Alper, H., K. Miyaoku, and G. Stephanopoulos. 2006. Characterization of lycopene-overproducing E-coli strains in high cell density fermentations. Appl. Microbiol. Biotechnol. 72:968-974. [DOI] [PubMed] [Google Scholar]
- 3.Becker, J., C. Klopprogge, O. Zelder, E. Heinzle, and C. Wittmann. 2005. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 71:8587-8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burgard, A. P., P. Pharkya, and C. D. Maranas. 2003. OptKnock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84:647-657. [DOI] [PubMed] [Google Scholar]
- 5.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farmer, W. R., and J. C. Liao. 2000. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat. Biotechnol. 18:533-537. [DOI] [PubMed] [Google Scholar]
- 7.Feist, A. M., C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis, and B. O. Palsson. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer, E., and U. Sauer. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880-891. [DOI] [PubMed] [Google Scholar]
- 9.Fowler, Z. L., W. W. Gikandi, and M. A. Koffas. 2009. Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl. Environ. Microbiol. 75:5831-5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrgard, M. J., B. S. Lee, V. Portnoy, and B. O. Palsson. 2006. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 16:627-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillman, J. D., and D. G. Fraenkel. 1975. Glyceraldehyde 3-phosphate dehydrogenase mutants of Escherichia coli. J. Bacteriol. 122:1175-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin, Y. S., and G. Stephanopoulos. 2007. Multi-dimensional gene target search for improving lycopene biosynthesis in Escherichia coli. Metab. Eng. 9:337-347. [DOI] [PubMed] [Google Scholar]
- 13.Kim, S. W., and J. D. Keasling. 2001. Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol. Bioeng. 72:408-415. [DOI] [PubMed] [Google Scholar]
- 14.Kirschner, M. W. 2005. The meaning of systems biology. Cell 121:503-504. [DOI] [PubMed] [Google Scholar]
- 15.Lee, D. Y., H. Yun, S. Park, and S. Y. Lee. 2003. MetaFluxNet: the management of metabolic reaction information and quantitative metabolic flux analysis. Bioinformatics 19:2144-2146. [DOI] [PubMed] [Google Scholar]
- 16.Lee, K. H., J. H. Park, T. Y. Kim, H. U. Kim, and S. Y. Lee. 2007. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol. Syst. Biol. 3:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, P. C., and C. Schmidt-Dannert. 2002. Metabolic engineering towards biotechnological production of carotenoids in microorganisms. Appl. Microbiol. Biotechnol. 60:1-11. [DOI] [PubMed] [Google Scholar]
- 18.Lee, S. Y. 1996. High cell-density culture of Escherichia coli. Trends Biotechnol. 14:98-105. [DOI] [PubMed] [Google Scholar]
- 19.Lee, S. Y., D. Y. Lee, and T. Y. Kim. 2005. Systems biotechnology for strain improvement. Trends Biotechnol. 23:349-358. [DOI] [PubMed] [Google Scholar]
- 20.Ma, H., and A. P. Zeng. 2003. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics 19:270-277. [DOI] [PubMed] [Google Scholar]
- 21.Mahadevan, R., and C. H. Schilling. 2003. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5:264-276. [DOI] [PubMed] [Google Scholar]
- 22.Matthews, P. D., and E. T. Wurtzel. 2000. Metabolic engineering of carotenoid accumulation in Escherichia coli by modulation of the isoprenoid precursor pool with expression of deoxyxylulose phosphate synthase. Appl. Microbiol. Biotechnol. 53:396-400. [DOI] [PubMed] [Google Scholar]
- 23.Misawa, N., M. Nakagawa, K. Kobayashi, S. Yamano, Y. Izawa, K. Nakamura, and K. Harashima. 1990. Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J. Bacteriol. 172:6704-6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park, J. H., K. H. Lee, T. Y. Kim, and S. Y. Lee. 2007. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. U. S. A. 104:7797-7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pharkya, P., A. P. Burgard, and C. D. Maranas. 2003. Exploring the overproduction of amino acids using the bilevel optimization framework OptKnock. Biotechnol. Bioeng. 84:887-899. [DOI] [PubMed] [Google Scholar]
- 26.Pharkya, P., and C. D. Maranas. 2006. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 8:1-13. [DOI] [PubMed] [Google Scholar]
- 27.Price, N. D., J. L. Reed, and B. O. Palsson. 2004. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2:886-897. [DOI] [PubMed] [Google Scholar]
- 28.Reed, J. L., T. D. Vo, C. H. Schilling, and B. O. Palsson. 2003. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 4:R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 30.Sanchez, A. M., G. N. Bennett, and K. Y. San. 2006. Batch culture characterization and metabolic flux analysis of succinate-producing Escherichia coli strains. Metab. Eng. 8:209-226. [DOI] [PubMed] [Google Scholar]
- 31.Segre, D., D. Vitkup, and G. M. Church. 2002. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. U. S. A. 99:15112-15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.