

Abstract
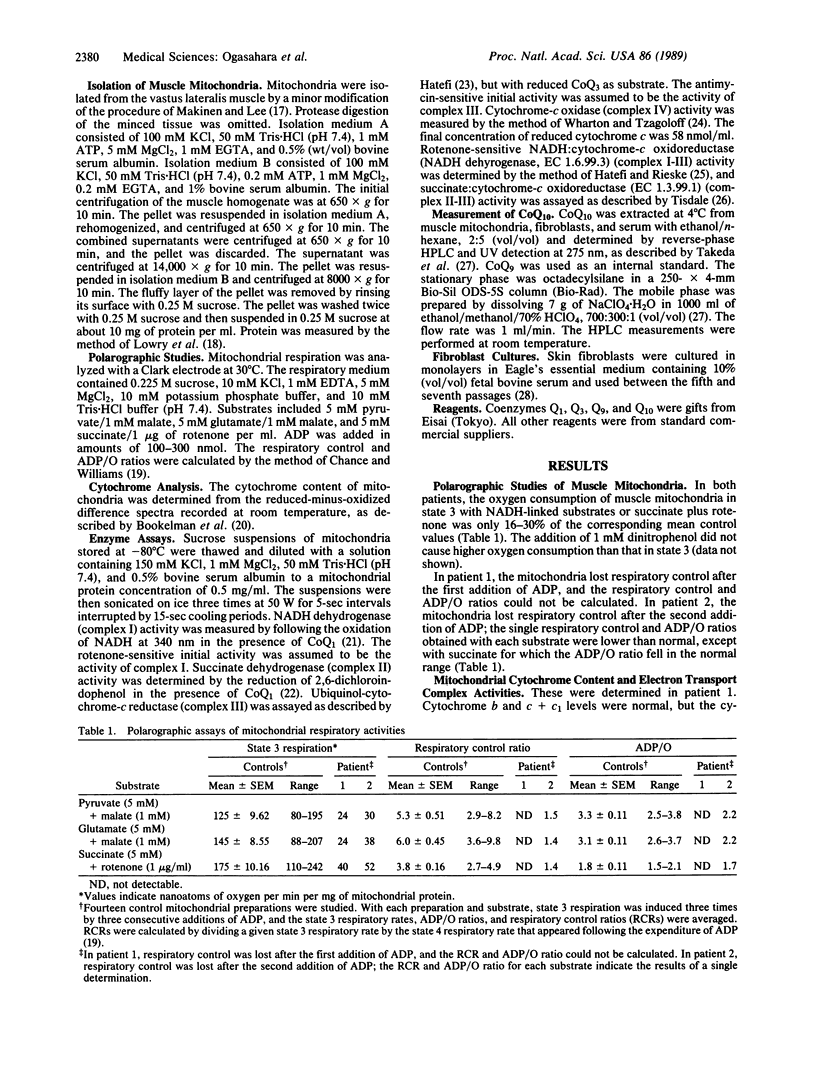
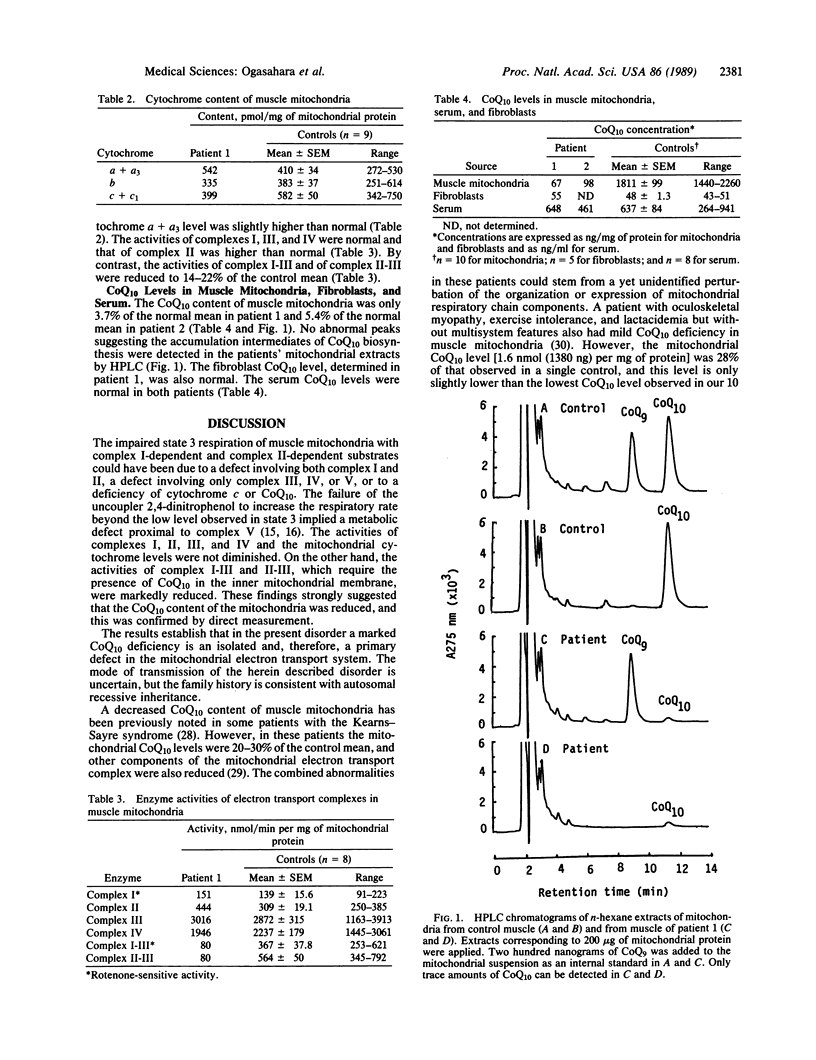
The electron transport system of muscle mitochondria was examined in a familial syndrome of lactacidemia, mitochondrial myopathy, and encephalopathy. The propositus, a 14-year-old female, and her 12-year-old sister had suffered from progressive muscle weakness, abnormal fatigability, and central nervous system dysfunction since early childhood. In the propositus, the state 3 respiratory rate of muscle mitochondria with NADH-linked substrates and with succinate was markedly reduced. The levels of cytochromes a + a3, b, and c + c1 were normal. The activities of complexes I, II, III, and IV of the electron transport chain were normal or increased. By contrast, the activities of complex I-III and of complex II-III, both of which need coenzyme Q10 (CoQ10), were abnormally low. On direct measurement, the mitochondrial CoQ10 content was 3.7% of the mean value observed in 10 controls. Serum and cultured fibroblasts of the propositus had normal CoQ10 contents. In the younger sister, the respiratory activities and CoQ10 level of muscle mitochondria were similar to those observed in the propositus. The findings establish CoQ10 deficiency as a cause of a familial mitochondrial cytopathy and suggest that the disease results from a tissue-specific defect of CoQ10 biosynthesis.
Full text
PDF


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bookelman H., Trijbels J. M., Sengers R. C., Janssen A. J. Measurement of cytochromes in human skeletal muscle mitochondria, isolated from fresh and frozen stored muscle specimens. Biochem Med. 1978 Jun;19(3):366–373. doi: 10.1016/0006-2944(78)90037-6. [DOI] [PubMed] [Google Scholar]
- CHANCE B., WILLIAMS G. R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955 Nov;217(1):383–393. [PubMed] [Google Scholar]
- Capaldi R. A. Arrangement of proteins in the mitochondrial inner membrane. Biochim Biophys Acta. 1982 Nov 30;694(3):291–306. doi: 10.1016/0304-4157(82)90009-0. [DOI] [PubMed] [Google Scholar]
- Clark J. B., Hayes D. J., Morgan-Hughes J. A., Byrne E. Mitochondrial myopathies: disorders of the respiratory chain and oxidative phosphorylation. J Inherit Metab Dis. 1984;7 (Suppl 1):62–68. doi: 10.1007/BF03047377. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Bonilla E., Zeviani M., Nakagawa M., DeVivo D. C. Mitochondrial myopathies. Ann Neurol. 1985 Jun;17(6):521–538. doi: 10.1002/ana.410170602. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Nicholson J. F., Hays A. P., Eastwood A. B., Papadimitriou A., Koenigsberger R., DeVivo D. C. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Ann Neurol. 1983 Aug;14(2):226–234. doi: 10.1002/ana.410140209. [DOI] [PubMed] [Google Scholar]
- Gibson F., Young I. G. Isolation and characterization of intermediates in ubiquinone biosynthesis. Methods Enzymol. 1978;53:600–609. doi: 10.1016/s0076-6879(78)53061-9. [DOI] [PubMed] [Google Scholar]
- Goewert R. R., Sippel C. J., Grimm M. F., Olson R. E. Identification of 3-methoxy-4-hydroxy-5-hexaprenylbenzoic acid as a new intermediate in ubiquinone biosynthesis by Saccharomyces cerevisiae. Biochemistry. 1981 Sep 15;20(19):5611–5616. doi: 10.1021/bi00522a040. [DOI] [PubMed] [Google Scholar]
- Goewert R. R., Sippel C. J., Olson R. E. Identification of 3,4-dihydroxy-5-hexaprenylbenzoic acid as an intermediate in the biosynthesis of ubiquinone-6 by Saccharomyces cerevisiae. Biochemistry. 1981 Jul 7;20(14):4217–4223. doi: 10.1021/bi00517a041. [DOI] [PubMed] [Google Scholar]
- Hatefi Y. Preparation and properties of NADH: ubiquinone oxidoreductase (complexI), EC 1.6.5.3. Methods Enzymol. 1978;53:11–14. doi: 10.1016/s0076-6879(78)53006-1. [DOI] [PubMed] [Google Scholar]
- Hatefi Y. Preparation and properties of dihydroubiquinone: cytochrome c oxidoreductase (complex III). Methods Enzymol. 1978;53:35–40. doi: 10.1016/s0076-6879(78)53010-3. [DOI] [PubMed] [Google Scholar]
- Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- Kennaway N. G., Buist N. R., Darley-Usmar V. M., Papadimitriou A., Dimauro S., Kelley R. I., Capaldi R. A., Blank N. K., D'Agostino A. Lactic acidosis and mitochondrial myopathy associated with deficiency of several components of complex III of the respiratory chain. Pediatr Res. 1984 Oct;18(10):991–999. doi: 10.1203/00006450-198410000-00017. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- LUFT R., IKKOS D., PALMIERI G., ERNSTER L., AFZELIUS B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest. 1962 Sep;41:1776–1804. doi: 10.1172/JCI104637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen M. W., Lee C. P. Biochemical studies of skeletal muscle mitochondria. I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch Biochem Biophys. 1968 Jul;126(1):75–82. doi: 10.1016/0003-9861(68)90561-4. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Protonmotive redox mechanism of the cytochrome b-c1 complex in the respiratory chain: protonmotive ubiquinone cycle. FEBS Lett. 1975 Aug 1;56(1):1–6. doi: 10.1016/0014-5793(75)80098-6. [DOI] [PubMed] [Google Scholar]
- Mitchell P. The protonmotive Q cycle: a general formulation. FEBS Lett. 1975 Nov 15;59(2):137–139. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]
- Moreadith R. W., Batshaw M. L., Ohnishi T., Kerr D., Knox B., Jackson D., Hruban R., Olson J., Reynafarje B., Lehninger A. L. Deficiency of the iron-sulfur clusters of mitochondrial reduced nicotinamide-adenine dinucleotide-ubiquinone oxidoreductase (complex I) in an infant with congenital lactic acidosis. J Clin Invest. 1984 Sep;74(3):685–697. doi: 10.1172/JCI111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Hughes J. A., Darveniza P., Kahn S. N., Landon D. N., Sherratt R. M., Land J. M., Clark J. B. A mitochondrial myopathy characterized by a deficiency in reducible cytochrome b. Brain. 1977 Dec;100(4):617–640. doi: 10.1093/brain/100.4.617. [DOI] [PubMed] [Google Scholar]
- Morgan-Hughes J. A., Darveniza P., Landon D. N., Land J. M., Clark J. B. A mitochondrial myopathy with a deficiency of respiratory chain NADH-CoQ reductase activity. J Neurol Sci. 1979 Sep;43(1):27–46. doi: 10.1016/0022-510x(79)90071-6. [DOI] [PubMed] [Google Scholar]
- Ogasahara S., Yorifuji S., Nishikawa Y., Takahashi M., Wada K., Hazama T., Nakamura Y., Hashimoto S., Kono N., Tarui S. Improvement of abnormal pyruvate metabolism and cardiac conduction defect with coenzyme Q10 in Kearns-Sayre syndrome. Neurology. 1985 Mar;35(3):372–377. doi: 10.1212/wnl.35.3.372. [DOI] [PubMed] [Google Scholar]
- Schneider H., Lemasters J. J., Höchli M., Hackenbrock C. R. Liposome-mitochondrial inner membrane fusion. Lateral diffusion of integral electron transfer components. J Biol Chem. 1980 Apr 25;255(8):3748–3756. [PubMed] [Google Scholar]
- Schotland D. L., DiMauro S., Bonilla E., Scarpa A., Lee C. P. Neuromuscular disorder associated with a defect in mitochondrial energy supply. Arch Neurol. 1976 Jul;33(7):475–479. doi: 10.1001/archneur.1976.00500070017003. [DOI] [PubMed] [Google Scholar]
- Takada M., Ikenoya S., Yuzuriha T., Katayama K. Studies on reduced and oxidized coenzyme Q (ubiquinones). II. The determination of oxidation-reduction levels of coenzyme Q in mitochondria, microsomes and plasma by high-performance liquid chromatography. Biochim Biophys Acta. 1982 Feb 17;679(2):308–314. doi: 10.1016/0005-2728(82)90301-2. [DOI] [PubMed] [Google Scholar]
- Trumpower B. L. New concepts on the role of ubiquinone in the mitochondrial respiratory chain. J Bioenerg Biomembr. 1981 Apr;13(1-2):1–24. doi: 10.1007/BF00744743. [DOI] [PubMed] [Google Scholar]
- Van Biervliet J. P., Bruinvis L., Ketting D., De Bree P. K., Van der Heiden C., Wadman S. K. Hereditary mitochondrial myopathy with lactic acidemia, a De Toni-Fanconi-Debré syndrome, and a defective respiratory chain in voluntary striated muscles. Pediatr Res. 1977 Oct;11(10 Pt 2):1088–1093. doi: 10.1203/00006450-197711100-00005. [DOI] [PubMed] [Google Scholar]
- Yorifuji S., Ogasahara S., Takahashi M., Tarui S. Decreased activities in mitochondrial inner membrane electron transport system in muscle from patients with Kearns-Sayre syndrome. J Neurol Sci. 1985 Nov;71(1):65–75. doi: 10.1016/0022-510x(85)90037-1. [DOI] [PubMed] [Google Scholar]