

Abstract
Ischemic preconditioning is a phenomenon in which low-level stressful stimuli upregulate endogenous defensive programs, resulting in subsequent resistance to otherwise lethal injuries. We previously observed that signal transduction systems typically associated with neurodegeneration such as caspase activation are requisite events for the expression of tolerance and induction of HSP70. In this work, we sought to determine the extent and duration of oxidative and energetic dysfunction as well as the role of effector kinases on metabolic function in preconditioned cells. Using an in vitro neuronal culture model, we observed a robust increase in Raf and p66Shc activation within 1 h of preconditioning. Total ATP content decreased by 25% 3 h after preconditioning but returned to baseline by 24 h. Use of a free radical spin trap or p66shc inhibitor increased ATP content whereas a Raf inhibitor had no effect. Phosphorylated p66shc rapidly relocalized to the mitochondria and in the absence of activated p66shc, autophagic processing increased. The constitutively expressed chaperone HSC70 relocalized to autophagosomes. Preconditioned cells experience significant total oxidative stress measured by F2-isoprostanes and neuronal stress evaluated by F4-neuroprostane measurement. Neuroprostane levels were enhanced in the presence of Shc inhibitors. Finally, we found that inhibiting either p66shc or Raf blocked neuroprotection afforded by preconditioning as well as upregulation of HSP70, suggesting both kinases are critical for preconditioning but function in fundamentally different ways. This is the first work to demonstrate the essential role of p66shc in mediating requisite mitochondrial and energetic compensation after preconditioning and suggests a mechanism by which protein and organelle damage mediated by ROS can increase HSP70.
Introduction
A short sublethal ischemic event in the brain can lead to subsequent neuronal resistance to a severe stroke, a phenomenon referred to as ischemic preconditioning. Understanding the powerful ability of the CNS to induce the protective pathways observed in ischemic preconditioning could provide critical insight into endogenous pathways to exploit in the design of novel therapeutics for stroke, yet no cohesive understanding of the early events that occur in preconditioning has emerged. We have established, however, that generation of reactive oxygen species (ROS) is one of the earliest requisite signals for the induction of the neuroprotective protein HSP70. Application of antioxidants or free radical spin traps during the preconditioning period blocks both tolerance and chaperone upregulation (McLaughlin et al., 2003).
Once thought as random destructive molecules, ROS have emerged as potent signaling molecules capable of eliciting discrete posttranslational modification of proteins. Indeed, the adaptor protein p66shc is phosphorylated at serine 36 in response to ROS, and loss of p66shc confers resistance to oxidative injury (Migliaccio et al. 1999). p66shc is likely relevant to ischemic preconditioning as it is phosphorylated in muscle after ischemia/reperfusion injury and contributes to cell death (Zaccagnini et al. 2004).
Although other signaling kinases such as ERK (extracellular signal-regulated kinase), Raf, and p38 have been associated with preconditioning (Nishimura et al., 2003; Jones and Bergeron, 2004; Xuan et al., 2005; Scorziello et al., 2007), the mechanism by which these molecules might contribute to the mitochondrial disturbances in redox signaling and energetics after ischemic stress is poorly understood. Moreover, activation of Raf, ERK, and other kinase signaling molecules is far from a universally accepted feature of neuronal models of preconditioning given the failure to link these pathways with downstream mediators of preconditioning such as activation of KATP channel or HSP70 induction.
Lethal energetic failure and redox stress in ischemia has been shown to activate Raf, ERK, p38, as well as p66Shc, yet the relevance to sublethal ischemic signaling and preconditioning is unclear. Indeed, ERK activation has been shown to be a potent inhibitor of heat shock factor 1, the transcription factor for HSP70 (He et al., 1998; Bijur and Jope, 2000; Dai et al., 2000). Similarly, Raf is activated by energetic dysfunction via receptor-dependent mechanisms and by loss of ATP-dependent binding to a sequestering member of the chaperone family (Vossler et al., 1997; Grewal et al., 2000; Song et al., 2001; Peng et al., 2005; Powers and Workman, 2006), and it has been implicated in cardiac preconditioning (Peralta et al., 2003; Rudiger et al., 2003; Xuan et al., 2005).
We believe that understanding the early events in preconditioning that cause kinase activation and how these events link to the defining features of preconditioning is essential to exploiting the neuroprotective potential of this phenomenon. In this work, we sought to understand the extent of the energetic and oxidative dysfunction induced by preconditioning and determine their role in mediating neuroprotection and HSP70 induction. After a nontoxic preconditioning stress, we observed only mild energetic dysfunction. By assessing oxidation of lipids enriched in neurons, we observed a substantial redox stress, yet neither energetic status nor oxidative stress was impacted by blocking Raf. p66Shc was activated early in preconditioned cells and relocalized to mitochondria. Inhibiting this kinase blocked neuroprotection, blocked the upregulation of HSP70, and influenced energetic recovery, ROS production, and diminished formation of autophagosomes.
Preliminary data were presented at the New York Academy of Science meeting in 2007, and an abstract of this work appears in their journal.
Materials and Methods
User-friendly versions of all of the protocols and procedures can be found on our website at http://www.mc.vanderbilt.edu/root/vumc.php?site=mclaughlinlab&doc=17838.
Reagents.
Hyclone Defined FBS was obtained from Fisher Scientific (SH3007003). BSA and 1 m HEPES buffer was procured from Sigma (A1470 and H0887). Poly-l-ornithine hydrobromide from Sigma (P3655) was used to coat 12 mm glass coverslips (63-3029, Carolina Biological Supply) for cultures. All remaining cell culture media and supplements were purchased from Invitrogen unless otherwise noted. Zymed blocking solution, MitoTracker Orange, and ProLong Gold antifade reagents were also procured from Invitrogen (000105, M7511, and P36930). All kinase inhibitors were purchased from Calbiochem. Primary antibodies used in these studies were phospho-ERK (4377, Cell Signaling Technology), HSP70 and HSC70 (SPA-811 and SPA-816, Assay Designs), phospho p66Shc (Calbiochem), phospho Raf S338 (Santa Cruz Biotechnology), LC3 (PD014, MBL International Corp), and MAP2 (M4403, Sigma). All immunoblot supplies unless noted were purchased from Bio-Rad Laboratories. Western Lightning Chemiluminescence Reagent Plus was obtained from PerkinElmer Life Sciences (NEL104001EA), SuperSignal West Dura Extended Duration Substrate Chemiluminescence from Thermo Scientific (34075), and Hybond P polyvinylidene difluoride membranes were purchased from GE Healthcare (RPN303F). Commercial kits used were DC Protein Assay Kit II (500-0112, Bio-Rad), ApoGSH detection kit (K251–100, BioVision Inc), ViaLight Plus Cell Proliferation and Cytotoxicity BioAssay kit (LT07-121, Lonza Rockland Inc), and LDH Toxicology Assay kit from Sigma (TOX7–1KT). All additional chemicals were purchased from Sigma.
Primary cell cultures.
Cortical cultures were prepared from embryonic day 18 Sprague–Dawley rats as previously described (Hartnett et al., 1997). Briefly, cortices were dissociated and the resultant cell suspension was adjusted to 335,000–350,000 cells/ml. Cells were plated 2 ml/well in six-well tissue culture plates containing five 12 mm poly-l-ornithine-coated coverslips per well. Cells were maintained at 37°C, 5% CO2 in growth media composed of a volume to volume mixture of 84% DMEM (11960, Invitrogen), 8% Ham's F12 nutrients (11765, Sigma), 8% fetal bovine serum, 24 U/ml penicillin, 24 mg/ml streptomycin, and 80 μm l-glutamine. Media was partially replaced every 2–3 d, and glial cell proliferation was inhibited after 2 weeks in culture with 2–3 mm cytosine arabinofuranoside, after which the cultures were grown in DMEM-based maintenance medium containing 2% serum, 25 mm HEPES, and 2.5 mm l-glutamine without F12 nutrients. All experiments were conducted 1–2 weeks after mitotic inhibition (27–31 d in vitro) when excitotoxicity is expressed fully in our system (Sinor et al., 1997).
Preconditioning with chemical ischemia.
Preconditioning reagents were prepared in sterile glucose-free balanced salt solution (150 mm NaCl, 2.8 mm KCl, 1 mm CaCl2, and 10 mm HEPES; pH 7.3) containing 3 mm potassium cyanide (KCN) to block Complex IV of the electron transport chain. Cortical cultures were preconditioned (PC) in this oxygen glucose deprivation (OGD) media for 90 min as previously described (McLaughlin et al., 2003). OGD treatment was terminated by rinsing cells (200:1) and replacing wash solution with maintenance medium. To test the efficacy of various agents at altering the neuroprotective effects of preconditioning, compounds were added 30 min to 1 h before OGD treatment (preincubation), during the 90 min OGD treatment (coincubation), and during recovery (postincubation). Raf1 kinase inhibitor I (Rafi 5 nm), a potent inhibitor of the Src kinases, PP2 (Shci 50 nm), the free radical spin trap N-tert-butyl-α-phenylnitrone (PBN 500 μm), and PD98059 (10 μm), a MAP kinase kinase inhibitor, as well as the p38 inhibitor SB239063 (20 μm), were all added preincubation, coincubation, and postincubation.
Twenty-four hours after the preconditioning stimulus, cells were treated with the glutamate receptor agonist NMDA. Immediately before agonist treatment, cells were rinsed (200:1) in MEM with Earle's salt solution supplemented with 0.01% bovine serum albumin (BSA) and 25 mm HEPES without phenol red. Cells were then exposed for 60 min to 100 μm NMDA in the presence of 10 μm glycine at 37°C, 5% CO2. Treatment was halted by serial dilution (200:1) in MEM. Five hundred microliters of MEM was then added per well, and the cultures were returned to the incubator. Neuronal viability was determined 18 to 24 h later by measuring lactate dehydrogenase (LDH) release with the LDH in vitro toxicology assay kit. Forty microliter samples of medium were assayed spectrophotometrically (490 nm absorbance, 630 nm emission) in duplicate according to the manufacturer's protocol to obtain a measure of cytoplasmic LDH released from dead and dying neurons (Hartnett et al., 1997). LDH results were confirmed qualitatively by visual inspection of the cells and, in several instances, quantitatively by cell counts using our previously described method (McLaughlin et al., 2003).
To compare across treatments with various compounds, data are expressed as “relative toxicity” as previously described (McLaughlin et al., 2003). Given that most of the agents used to block preconditioning have the potential to block glutamate toxicity directly or indirectly, all naive cultures for these experiments received the same agents to ensure they did not block NMDA toxicity. Naive cells were transferred into 24-well plates in the same manner as OGD cells, but remained in maintenance media with or without inhibitors until they were washed and NMDA was applied. For these experiments, LDH values for NMDA exposure in cells that had not been treated with OGD, but were given drugs, were considered 100% cell death. Statistical significance was assessed by parametric comparison between means.
Immunoblotting.
At various times after preconditioning treatment, both naive and PC cultures were harvested for immunoblots by placing plates on ice, then washing them twice with ice-cold PBS (4.3 mm Na2HPO4 · 7 H2O, 1.4 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl; pH 7.4). Cells were scraped from the dish using a rubber policeman in 350–500 μl of TNEB (50 mm Tris-Cl, pH 7.8, 2 mm EDTA, 100 mm NaCl, and 1% NP-40) with added protease inhibitor. Up to 200 μl of this suspension was saved for protein determination and the remaining lysate was resuspended in an equal volume of Laemmli buffer (Bio-Rad) with β-mercaptoethanol (1:20), heated to 95°C for 5 min, and stored at −20°C. Protein concentrations were determined spectrophotometrically by using a Bio-Rad microprotein assay kit.
Equal protein concentrations were separated using Criterion Tris-HCl or Bis-Tris gels followed by transfer to Hybond polyvinylidene difluoride membranes and then blocked in methanol for 5 min. After 10 min of drying, the membranes were incubated at 4°C overnight with their respective antibody in 5% nonfat dry milk dissolved in Tris-buffered saline containing 0.1% Tween 20 (TBS-Tween). Primary antibodies were used at the following concentrations: phospho-ERK (1:1000), phospho-p38 (1:1000), Heat shock protein 70 (HSP70;1:1000), the constitutive form of HSP70 (HSC70; 1:2000), and S36 phospho-p66shc (1:100). Membranes were washed three times with TBS-Tween, and incubated for 1 h at room temperature with 1:5000 horseradish peroxidase–conjugated secondary antibody dissolved in milk block. After three additional washes in TBS-Tween, protein bands were visualized using Western Lightning chemiluminescence reagent plus enhanced luminol reagents and exposed to Kodak BioMax light film.
The Raf1 S338 antibody required a different block and chemiluminescence kit for optimal signal. Membranes were blocked for 1 h in Zymed blocking solution then exposed overnight at 4°C to phospho-Raf S338 diluted 1:1000 in the Zymed blocking solution. Membranes were washed for a total of 35 min in TBS-Tween and incubated in 1:5000 horseradish peroxidase–conjugated anti-rabbit secondary made up in Zymed blocking solution. The bands were exposed to SuperSignal West Dura Extended Duration Substrate Chemiluminescence and visualized as above. Western bands were quantified using the NIH ImageJ analysis program as previously described (McLaughlin et al., 2003).
MitoTracker and immunostaining.
A working concentration of 790 nm MitoTracker Orange was added to maintenance media to stain the mitochondria of the living cells. After 45–60 min of incubation at 37°C, 5% CO2 media was removed, the coverslips washed with PBS, and fixed with 4% formaldehyde. The remainder of the immunocytochemistry was performed essentially as we previously described (McLaughlin et al., 2003). Briefly, the cells were permeabilized with 0.1% Triton X-100, washed with PBS, and blocked with 8% BSA diluted in PBS. After 25 min, the coverslips were coincubated in phospho-p66Shc (Ser 36) primary antibody (1:100) and MAP2 primary antibody (1:2000) overnight at 4°C. Cells were then washed in PBS for a total of 30 min and incubated in Alexa Fluor anti-mouse (1:500) and cy-2 anti-rabbit (1:1000) secondary antibodies for 60 min. After 25 min of additional washing, coverslips were mounted and fluorescence was visualized with a Zeiss Axioplan microscope equipped with an Apotome optical sectioning slider.
ATP assays.
Measurement of ATP content was performed 3 or 24 h after preconditioning by bioluminescent detection of light in the presence of luciferin. Briefly, a coverslip was removed from the toxicity plate and added to a new plate containing 300 μl of Cell Lysis reagent from the ViaLight Plus Kit. After 10 min, 80 μl of cell lysate along with 100 μl of ATP monitoring reagent was added to a 96-well transparent white plate. Addition of this reagent leads to the formation of light from the interaction of the enzyme luciferase with ATP present in the cell and luciferin. The resulting bioluminescent measurements are linearly related to ATP concentration and were taken on a SPECTRAfluor Plus Tecan plate reader after a 2 min incubation using an integration time of 1000 ms and a gain of 150. Measurements were obtained in duplicate for each sample and normalized for protein levels after a protein assay. ATP levels are expressed as the mean from three independent experiments ± SEM.
Immunofluorescence and quantification of activated LC3-containing neurons.
Twenty-four hours after mixed neuronal cultures were exposed to OGD or naive conditions in the presence or absence of 50 nm PP2. Immunocytochemistry was performed as described above using rabbit anti-LC3 (microtubule-associated protein 1 light chain 3, 1:250) and mouse anti-MAP2 (microtubule-associated protein 2, 1:1000) at 4°C overnight. Cells were washed with PBS for a total of 30 min and incubated in Cy3 anti-rabbit (1:500) and Cy2 anti-mouse (1:500) secondary antibodies for 1 h. Cells were then washed for a total of 25 min in PBS and incubated in 1.4 μm DAPI for 10 min, followed by further washes. Coverslips were mounted on microscope slides, and fluorescence was visualized with a Zeiss Axioplan microscope equipped with an Apotome optical sectioning slider. Using a modification of the method of Tan et al. (2009), the percentage of neurons containing activated LC3 was determined by counting the number of activated LC3 immunostained neurons and dividing by the total number of MAP2- and DAPI-stained cells. Neurons with activated LC3 were identified by a change from baseline dispersed staining to a more condensed brighter staining indicative of LC3-II interaction with autophagosomes (Kabeya et al., 2000; Tan et al., 2009). All data points represent the average ± SEM from three independent experiments. Data are combined values for all experiments from two independent observers. Statistical significance was determined by two-tailed ANOVA and post hoc analysis performed on a group effect with p < 0.05.
Measurement of F2-IsoPs and F4-neuroprostanes.
Lipid peroxidation was assessed through quantification of both F2-isoprostanes (F2-IsoPs), prostaglandin-like molecules generated from the free radical-mediated peroxidation of arachidonic acid, and F4-neuroprostanes (F4-NeuroPs), F2-IsoP-like compounds generated similarly via the peroxidation of docosahexaenoic acid (DHA), a polyunsaturated fatty acid enriched in neurons (see Fig. 5A). F2-IsoPs and F4-NeuroPs can be measured simultaneously using gas chromatography–mass spectrometry as previously described (Milne et al., 2007). Briefly, cells were harvested and 500 μl of the lysate mixed with methanol containing 0.05% butylated hydroxytoluene (BHT) to prevent auto-oxidation. The remaining lysate was saved for a protein assay to normalize to protein concentrations. F2-IsoPs and F4-NeuroPs esterified to phospholipids were hydrolyzed by chemical saponification, after which total F2-IsoPs and F4-NeuroPs were extracted using C-18 and silica Sep-Pak cartridges, purified by thin-layer chromatography, converted to pentaflurobenzyl ester trimethylsilyl ether derivatives, and quantified by stable isotope dilution techniques using gas chromatography/negative ion chemical ionization mass spectrometry using [2H4]-8-iso-PGF2α (m/z 573) as an internal standard. F2-IsoPs were separated from F4-NeuroPs based upon the respective m/z detected in the mass spectrometer; F2-IsoPs are detected at m/z 569 while F4-NeuroPs are detected at m/z 593.
Figure 5.

Shc activation regulates the extent of oxidative stress in preconditioning. Formation of F2-Isoprostanes (F2-IsoP) and F4-Neuroprostanes (NeuroP) is dependent upon lipid environment and initiated by oxidative stress. A, The pathway by which each class of compounds is formed is outlined. B, Primary cultures were exposed to preconditioning (PC) and harvested 24 h later to evaluate total F2-IsoP content to test the effects of the Shc inhibitor PP2 (50 nm) on preconditioning-induced oxidative stress. Control cultures incubated in 100 μm NMDA for 1 h, which causes 100% neuronal death, were used as an index of oxidative injury. Data represent the mean ± SEM for three to six independent experiments and were analyzed using one-way ANOVA. *p < 0.05 versus naive. C, Neuroprostane levels were measured at 24 h using incubation conditions outlined in the prior panel. Data represent the mean ± SEM of five to eight independent experiments performed in duplicate. Asterisks are used to demonstrate a significant effect compared with naive cells; statistical analysis between groups is denoted by Δ where post hoc analysis of data using Tukey test was performed comparing groups to PC alone. Both asterisks and Δ represent p < 0.05.
Analysis and statistics.
Except where otherwise noted data were summarized and are represented as mean ± SEM. The statistical significance of differences between means was assessed using one-way ANOVA at the 95% level (p < 0.05), followed by the Tukey multiple-comparison tests using GraphPad Prism software.
Results
Mild mitochondrial stress results in neuronal survival in an in vitro model of preconditioning
We have previously established a powerful, reproducible, and accessible in vitro neuronal culture model to address the cellular and molecular pathways which contribute to the expression of preconditioning-induced neuroprotection (McLaughlin et al., 2003; McLaughlin, 2004). Activation of KATP channels, induction of HSP70, and new protein synthesis are all conserved features of preconditioning in this system (McLaughlin et al., 2003). In this model, mature cortical cultures consisting of neurons (20%) and glia (80%) are exposed to a preconditioning stress for 90 min, and 24 h later cells are subjected to an otherwise lethal dose of NMDA (Fig. 1). Representative photomicrographs taken 24 h after exposure illustrate the extent of neuronal protection by this preconditioning paradigm (Fig. 1). Naive cultures not exposed to KCN or NMDA, and preconditioned cultures had many phase-bright neurons that remained morphologically intact (Fig. 1A,B). Using toxicity assays of LDH release, we saw no evidence of death indicating preconditioning is not toxic in and of itself (Fig. 1C). In contrast, cultures, which were not preconditioned but subsequently exposed to the lethal dose of NMDA, demonstrated massive neuronal cell death as evident by the loss of phase bright neurons and high levels of released LDH (Fig. 1D,F). Notably, preconditioned cultures exhibited much less cell death after NMDA exposure with many intact phase-bright neurons and a 50% decrease in LDH release (Fig. 1E,F). Thus, using this preconditioning paradigm we see significant neuronal survival after an otherwise lethal event.
Figure 1.

Mild mitochondrial stress protects primary neuronal cultures against excitotoxicity. Cortical cultures were chemically preconditioned for 90 min before exposure to an otherwise lethal dose of NMDA. A–F, Representative photos and LDH measurement of viability were taken 24 h after preconditioning alone (A–C) or 24 h after NMDA exposure (D–F). A, Naive cultures were washed but not exposed to preconditioning or 100 μm NMDA and exhibit phase bright neuronal somas with little or no cell death. B, Cells that were preconditioned with 3 mm KCN and oxygen glucose deprivation but not exposed to NMDA are also intact and healthy, demonstrating that preconditioning is not toxic to cells. C, Lack of cell death induced by preconditioning is demonstrated by LDH release in naive versus preconditioning alone. D, Cells, which were not preconditioned but were exposed to 100 μm NMDA for 1 h experienced near total neuronal death after 24 h. E, After 24 h, many of the preconditioned cells remained phase bright and morphologically intact after 1 h exposure to an otherwise lethal NMDA dose. F, LDH data comparing the effects of NMDA toxicity in naive and preconditioned cells demonstrate that prior preconditioning significantly decreases cell death. Pooled data are expressed as the ratio of the LDH released in NMDA-treated cultures to that released in glycine-only-treated sister cultures. LDH data are pooled from >12 experiments and were analyzed using a two-way ANOVA. *p < 0.01.
Activation of p66Shc and Raf rapidly occurs after preconditioning
At the very core of preconditioning is the ability of neurons to integrate and adapt to energetic and oxidative stress via multiple stress-sensitive signaling pathways that contribute to this endogenous neuroprotection. Using our in vitro model, we next sought to determine the temporal activation of signal transduction pathway kinases including ERK, p38, Raf, and p66Shc that have been linked to neuroprotection. At various time points after initiation of preconditioning, cells were harvested and the phosphorylated forms of these proteins were analyzed by Western blot (Fig. 2A,B). As HSC70 levels remain constant in preconditioning (Ohtsuka and Suzuki, 2000), we used this protein as our loading control. Both ERK and p38 demonstrated only modest changes in their phosphorylation state (Fig. 2A). S338 phosphorylation, which is required for activation of Raf, occurred within minutes but was no longer evident within an hour (Fig. 2A). At the time when cells would be exposed to the secondary stress, Raf phosphorylation rebounded (Fig. 2A; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). p66shc phosphorylation occurred within an hour of preconditioning initiation while cells were still experiencing OGD and was maximal at 2–4 h. However, at 24 h, when maximal expression of HSP70 occurs, p66shc phosphorylation was significantly decreased in contrast to Raf (Fig. 2B).
Figure 2.

p66Shc and Raf are rapidly and independently activated in preconditioned cells. Whole-cell extracts of neuronal cultures were harvested at various time points after initiation of preconditioning. Proteins were separated on SDS-PAGE gels and probed with antibodies specific to the phosphorylated forms of p42/44 ERK, p38, S338 Raf, and p66Shc or HSC70 as a loading control. A, Lysates from 5 min to 24 h after preconditioning reveal a rapid as well as a delayed increase in Raf activation and modest increases in ERK and p38. B, Phosphorylation of p66Shc S36 was increased within 1 h of initiation of preconditioning and remained high for several hours. C, Cells harvested 3 h after the initiation of preconditioning revealed no effect of Raf inhibition on phospho-p66Shc activation, whereas the free radical spin trap N-tert-butyl-α-phenylnitrone (PBN) decreased p66Shc phosphorylation. D, Raf phosphorylation was evaluated at 30 min (first 4 lanes) or 24 h (last lane). Quantification of these data is presented in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). Similar results were obtained in three to five additional independent experiments.
To determine whether Raf and p66shc activation are dependent upon one another in this model, we used kinase inhibitors specific to each protein and evaluated their effect on the phosphorylation state of the alternate protein at the time when each protein was maximally activated. p66shc phosphorylation was assessed at 3 h and was still evident in the presence of the Raf kinase inhibitor (Fig. 2C). Similarly, blocking p66shc phosphorylation had no effect on Raf phosphorylation at 30 min or 24 h (Fig. 2D), indicating that these two proteins are acting independently of one another. In support of a model in which free radicals activate p66shc, use of the free radical spin trap PBN led to a decrease in p66shc phosphorylation at 3 h (Fig. 2C).
Energetic status is impaired after preconditioning and is dependent on reactive oxygen species and phosphorylation of p66shc
Previous studies have demonstrated that Raf is directly activated by energetic dysfunction, indirectly activated by ATP loss via failed association and sequestering of chaperones, and possibly activated by redox stress (Vossler et al., 1997; Grewal et al., 2000; Peng et al., 2005). Strong evidence also exists linking p66shc to redox stress and determining metabolic energy demands (Nemoto et al., 2006). We sought to determine whether the early activation of these kinases was associated with energetic and redox status in preconditioning. We first evaluated energetic status of our cortical cultures after preconditioning and observed a small but significant loss of energetic stores within 3 h, which despite the substantial respiratory challenge, were recovered 24 h later (Fig. 3A). Neither Raf inhibition nor the KATP channel blocker had any significant effect on ATP levels. However, exposure to a free-radical spin trap PBN or p66shc inhibitor blocked the reduction in ATP content at 3 h, suggesting a link between ROS generation and p66shc activation for energetic status after preconditioning (Fig. 3B).
Figure 3.

Acute impairment of energetic status of preconditioned cultures is dependent on redox stress and Shc activation. A, Primary cultures were exposed to preconditioning and harvested 3 or 24 h later, and total ATP content was measured. Data represent the raw luminescent units ± SEM. B, To test the efficacy of various agents on ATP content at the 3 h time point, the KATP channel blocker glibenclamide (1 μm), the free radical spin trap (PBN; 500 μm), the Raf kinase inhibitor 1 (Rafl 5 nm), or the Shc inhibitor PP2 (Shci 50 nm) were added as described in Materials and Methods. Preconditioning alone decreased total ATP by 20%, an effect that was blocked only by the free radical spin trap or Shc inhibitor. Data represent the mean ± SEM of five to eight independent experiments performed in duplicate. Asterisks are used to demonstrate a significant effect compared with naive cells. Statistical significance between groups compared with preconditioning is denoted with a bracket and Δ. Data were analyzed using Tukey test after two-way ANOVA by comparing groups to PC alone. Both asterisks and Δ represent p < 0.05.
p66shc localizes to mitochondria and the nucleus after preconditioning
The impact of p66shc inhibitor on ATP content suggests that p66shc is altering metabolic tone in preconditioned cells. Using immunostaining, we next evaluated the subcellular sites of phosphorylated p66shc 3–4 h after the initiation of preconditioning when the highest levels are evident. As our cultures consist of a mix of neurons and glia, we used the neuron specific marker microtubule associated protein 2 (MAP2) to identify neurons, whereas the cell permeant mitochondria-specific Mitotracker probe was used to visualize the mitochondria, a possible site of p66shc activation (Orsini et al., 2004; Nemoto et al., 2006; Pellegrini et al., 2007). The Mitotracker staining was more diffuse after preconditioning exposure, potentially because of alterations in mitochondrial membrane potential and oxidant stress (Fig. 4, top) (Buckman et al., 2001). Increased p66shc phosphorylation was evident after preconditioning compared with naive cells, in keeping with our Western blot data (Fig. 4, middle). Phosphorylated p66shc was adjacent to the mitochondria. Several neurons also had perinuclear activated p66shc staining as well (Fig. 4, bottom and area detail).
Figure 4.

Phosphorylated Shc is rapidly activated and relocalizes after preconditioning. Primary cultures were incubated with the potentiometric dye Mitotracker (red) and then fixed 3–4 h after the initiation of preconditioning for immunofluorescent detection of phosphorylated p66Shc. Mitochondria (red) are well dispersed in both naive and preconditioned neurons (green, MAP2 staining). Processes are intact in both conditions and mitochondria are present in both soma and processes. Phosphorylated p66Shc (blue) is markedly increased after preconditioning with staining evident in both the nucleus and adjacent to mitochondria.
Oxidative stress and autophagy are regulated by p66shc phosphorylation after preconditioning
We have previously demonstrated that if free radical stress is halted during the preconditioning period, neurons will no longer be protected from the subsequent stress (McLaughlin et al., 2003). Studies have also shown that p66shc moves to the mitochondria where it interacts with cytochrome c, a component of the electron transport chain, and increases ROS generation (Pinton et al., 2007). To determine the extent of oxidative stress after preconditioning and p66shc involvement, we measured F2-isoprostane (IsoP) and F4-neuroprostane (NeuroP) formation via mass spectrometry in the presence or absence of the p66shc inhibitor. F2-IsoPs are a family of prostaglandin-like molecules formed nonenzymatically as a result of free radical–mediated peroxidation of arachidonic acid (Morrow et al., 1990; Roberts and Morrow, 2002) and have been shown to be accurate and reliable indices of oxidative stress (Kadiiska et al., 2005a, 2005b). NeuroPs are IsoP like compounds formed by the oxidation of docosahexaenoic acid, which is highly enriched in neurons. This allows us to assess neuronal lipid injury more specifically (Fig. 5A). Both F2-IsoP and F4-NeuroP levels are significantly increased after preconditioning with an even further enhancement of NeuroPs after p66shc inhibition (Fig. 5B,C). After NMDA-only exposure, F2-IsoP levels were similar to naive cells whereas NeuroP levels increased. As F2-IsoPs represent a combination of glia and neuronal oxidative stress, these data suggest that neurons experience a greater oxidative stress burden than glia.
Given the extent of oxidative dysfunction and p66shc involvement in preconditioning-mediated protection, we sought to determine whether oxidized organelles and proteins were subject to autophagy after preconditioning. We have previously shown that caspase 3 activation is required for this protection suggesting mitochondrial impairment (McLaughlin et al., 2003). Moreover, chaperones are essential mediators of preconditioning and contribute to autophagy (Chen et al., 1996; Currie et al., 2000; Kiffin et al., 2004; Majeski and Dice, 2004). We therefore monitored autophagy in preconditioned cells with and without the p66Shc inhibitor by staining for the microtubule-associated protein1 light chain 3 (LC3) which is the mammalian homolog of Atg8. This ubiquitin-like protein undergoes a conjugation process during autophagy and is localized, and its concentration is used as a hallmark feature of autophagosomal membrane formation (Klionsky et al., 2008). Preconditioned neurons experienced an increase in autophagy compared with naive cells (Fig. 6A,B,D). LC3 staining was further enhanced on blocking p66shc activation after preconditioning (Fig. 6C,D). These data are consistent with the decreased levels of F2-IsoP and NeuroP levels upon p66shc inhibition.
Figure 6.

Preconditioning increases neuronal autophagic signaling. Cultures were fixed 24 h after the initiation of preconditioning and stained with the nuclear dye DAPI (blue), the neuronal cytoskeletal marker MAP2 (green), and LC3 (red), a hallmark of autophagic activation. A, Control cultures had well defined processes, and light dispersed LC3 activation. B, Autophagosome punctae containing LC3 increased in response to preconditioning (bottom left, in red). C, Exposure to the Shci PP2 (50 nm) exacerbated this effect. D, Cell counts by two independent investigators of >400 neurons from three independent experiments revealed a 20% increase in intensely labeled LC3II-positive cells in preconditioned cells, and a further increase of 15% when Shci was present.
In previous work. we demonstrated that HSC70 plays an essential role in blocking proapoptotic signaling from overwhelming preconditioned cells. We stained preconditioned cultures before the onset of HSP70 induction and observed a substantial colocalization of HSC70 with LC3 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) suggesting that the constitutive chaperone is also contributing to autophagic processing.
Raf and p66shc activation are both required for preconditioning protection and upregulation of HSP70
To determine whether the increased phosphorylation of both Raf and p66shc are required for preconditioning, we next assessed the contribution of these kinase-signaling pathways to neuronal protection. Cells were preconditioned in the presence of inhibitors of MEK, p38, Raf, or p66Shc and 24 h later received the normally lethal exposure to NMDA. LDH-based toxicity assays indicated that the MAP kinase inhibitors had no effect on the protection seen after preconditioning. In contrast, inhibition of Raf and p66shc activation blocked cellular tolerance to the NMDA exposure and resulted in massive neuronal death (Figs. 7A, 8B).
Figure 7.

Raf activation is required for preconditioning protection and upregulation of HSP70. Inhibitors of Raf, p42/44 ERK, or p38 were added during preconditioning as outlined in Materials and Methods. Twenty-four hours after preconditioning, cultures were washed and exposed to 100 μm NMDA with 10 μm glycine for 1 h. A, Cell death was assessed 24 h later by LDH release from dead and dying neurons. Data represent the mean ± SEM for five independent experiments and were analyzed using one-way ANOVA. Post hoc analysis was done using Tukey test comparing groups to PC alone. Δ is used to denote statistical significance compared with preconditioning alone. Both asterisks and Δ represent p < 0.05. B, The upper blot demonstrates that HSP70 expression is increased at 24 h and that Raf inhibition blocks HSP70 upregulation. HSC70 is constitutively expressed and is used as a loading control.
Figure 8.
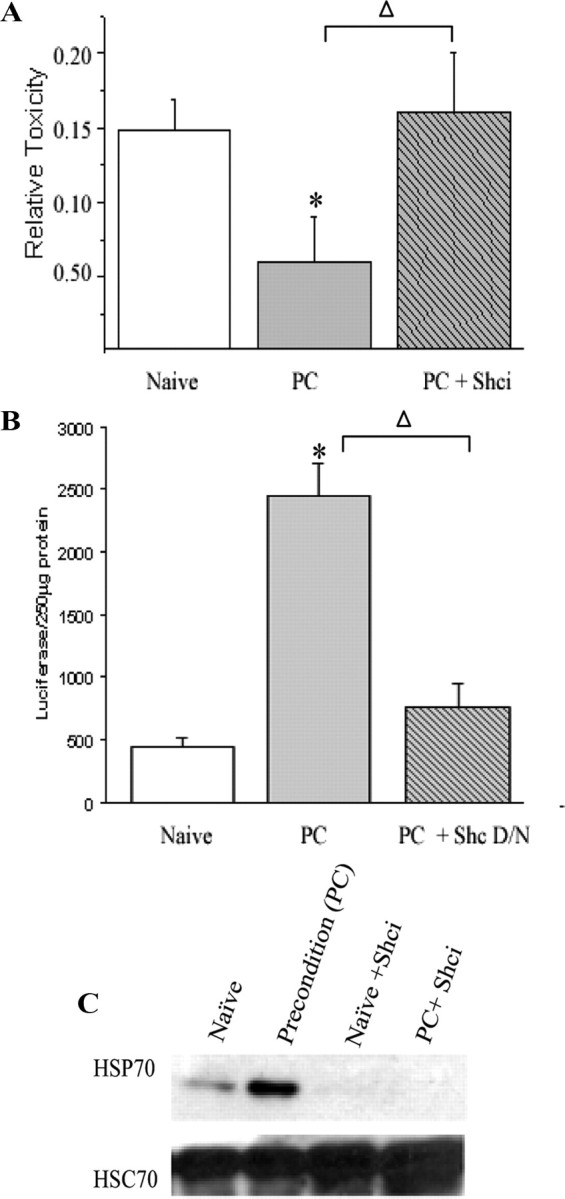
Shc activation is required for preconditioning protection and upregulation of HSP70. The Shc inhibitor PP2 was added during preconditioning as outlined in Materials and Methods. Twenty-four hours after preconditioning, cultures were washed and exposed to 100 μm NMDA with 10 μm glycine for 1 h. A, Cell death was assessed 24 h later by LDH release from dead and dying neurons. B, A redox-insensitive dominant-negative form of p66shc (S36A) was also used to verify that loss of function of this kinase blocked neuroprotection. Cells were transfected with vectors, which included a firefly luciferase reported 24 h prior to preconditioning and then were treated with preconditioning stress or washed. The following day, NMDA exposure was performed, and 24 h later cell survival was assessed by measuring luciferase signal from surviving cells. Data from A represent the mean ± SEM for five independent experiments and from B represent three experiments performed in duplicate. All averages were analyzed using one-way ANOVA. Post hoc analysis was done using Tukey test comparing groups to PC alone. Δ is used to denote statistical significance compared with preconditioning alone. Both asterisks and Δ represent p < 0.05. C, The upper blot demonstrates that HSP70 expression is increased at 24 h and that Shc inhibition blocks HSP70 upregulation. HSC70 is constitutively expressed and is used as a loading control.
As the upregulation of HSP70 is a hallmark feature of ischemic preconditioning, we next determined whether the kinase inhibitors that blocked preconditioning would also block HSP70 induction. The MEK inhibitor slightly decreased HSP70, whereas the p38 inhibitor had no effect (Fig. 7B). In contrast, both Raf and p66shc inhibition robustly prevented an increase in HSP70 expression (Figs. 7B, 8B). Together, this work highlights the importance of both Raf and p66shc signaling for endogenous neuroprotective pathways necessary for ischemic tolerance.
Discussion
The ability of tissue to withstand a normally lethal ischemic event by being previously “primed” with a subtoxic ischemic challenge was first described >20 years ago in the heart (Murry et al., 1986) and later observed in the brain (Kitagawa et al., 1990). Neuronal preconditioning can be achieved in vivo and in vitro by highly divergent stimuli such as ROS, β amyloid, limited exposure to mitochondrial toxins, and hypoxia (Pérez-Pinzón et al., 1997; Wiegand et al., 1999; Bergeron et al., 2000; Sharp et al., 2004). Because of the sheer number of stressors and models of preconditioning, defining the conserved early events of this phenomena and linking them to known mediators of protection has proven difficult. In this work, we describe the first evidence that preconditioning activates a discreet pathway mediated by p66shc. Shc activation is an essential mediator of metabolic tone in preconditioning, and blocking this kinase alters ATP content, increases neuron specific oxidative stress, as well as autophagosome formation, and blocks the upregulation of the neuroprotective protein HSP70.
Short-term interruption of aerobic respiration or other means of depleting ATP is a clear requirement for induction of tolerance in cardiomyocytes and neuronal systems (Liu et al., 1998; Obrenovitch, 2008). Indeed, the convergence of energetic dysfunction, oxidative stress, and molecular signaling at the level of the mitochondria is widely believed to mediate preconditioning (Dirnagl and Meisel, 2008). In this work, we observed that our preconditioned cultures were able to recover energetic content after 90 min removal of oxygen and glucose coupled with blockade of the electron transport chain with only modest loss of total ATP at 3 h. Raf inhibition did not alter energetic status whereas blocking p66shc phosphorylation reversed ATP depletion. These data are in keeping with recent work from two studies that suggest adaptations at the level of the respiratory chain are essential determinants of cell fate in ischemic tolerance. The in vivo work of Dave et al. (2008) demonstrated multiple complexes of the electron transport chain are phosphorylated in synaptosomes from preconditioned animals, and that increased oxygen consumption is observed at the time when animals are protected. A second collaborative study between Nichols and Prehn reported that neuronal cultures that survive glutamatergic stress were likely to have hyperpolarized mitochondria, increased glucose uptake, and NADPH availability (Ward et al., 2007). Together, these data suggest that strong mechanisms of neuronal compensation exist to adapt to nonlethal anaerobic stress.
ATP depletion is thought to result in opening KATP channels, which is also facilitated by free radicals (Teshima et al., 2003; O'Rourke, 2004), which we have previously shown to be a required event for the preconditioning protection in this model (McLaughlin et al., 2003). By measuring both total oxidative burden as well as the injury to docosahexaenoic acid enriched membranes which typify neurons (Sastry, 1985), we were able to determine the extent of oxidative stress. F2-IsoPs, stable and biologically inert compounds formed via the ROS-mediated oxidation of arachidonic acid, a membrane fatty acid, were used to measure total oxidative stress. Measurement of F2-IsoPs is considered the “gold standard” index of oxidative stress (Kadiiska et al., 2005a, 2005b).
Increased F2-IsoP levels have been observed in CNS as a result of a variety of insults and are increased in infarcted brain tissue from mice subjected to middle cerebral artery occlusion (Marin et al., 2000; Montine et al., 2004). The increase of total oxidative stress in preconditioned cells assessed by F2-IsoPs supports an essential role of redox dysfunction stress in initiating defensive pathways in preconditioning (Cohen et al., 2000). The observation that F2-IsoPs are higher in preconditioned cells compared with levels obtained in an excitotoxic challenge that induces 100% neuronal death may initially be puzzling. However, given that glia constitutes 80% of our cultures and are impervious to NMDA, the net oxidative burden to all lipids after excitotoxin challenge would likely be less dramatic than that induced by removal of oxygen and glucose in the presence of an inhibitor of the electron transport chain, which presents a significant challenge to both neurons and glia. This hypothesis is supported by measurements of redox damage to docosahexaenoic acid–enriched neuronal membranes, in which we observed the largest increase in F4-NeuroPs in our lethal NMDA stress. We believe that as culture methodology continues to evolve to increase the alanine and DHA precursors to more fully capture in vivo DHA levels, these differences may become even more striking (Kaduce et al., 2008).
Although both F4-NeuroPs and F2-IsoPs are excellent biomarkers of stress, it is worth noting that lipid peroxidation results in both inactive products like F2-IsoPs as well as bioactive molecules. Indeed, in two recent studies, we demonstrated that reactive cyclopentenone IsoPs with PGA2- and PGJ2-like ring structures are formed in ischemic stroke in postmortem human tissue. When purified compounds are applied to neurons, they exert potent bioactivity which increases ischemic injury and oxidative insults via a mitochondrial p66shc-dependent pathways (Musiek et al., 2006; Zeiger et al., 2009).
p66shc S36 phosphorylation is caused by ROS, and severe hypoxia induces both phosphorylation of this residue and Ras/Raf activation within 30 min, which is maintained for hours (Jung et al., 2002), and blocking Raf inhibits oxidative stress–induced death in neural cells (Stanciu et al., 2000). We observed an early rise in both Raf and Shc activation within 3 h of initiation of preconditioning treatment. As Shc and Raf inhibition did not alter the phosphorylation of the alternate kinase, any conserved signaling would be at the level of downstream effector molecules.
Downstream targets of Raf include Akt and MAP kinases, which have both been independently implicated in preconditioning (Wick et al., 2002; Jones and Bergeron, 2004; Lange-Asschenfeldt et al., 2004; Zhang et al., 2007). ERK has, however, been shown to be an inhibitor of the transcription factor for HSP70 (He et al., 1998; Bijur and Jope, 2000; Dai et al., 2000; Seo et al., 2006), and our work demonstrated that ERK inhibition did not block preconditioning protection while only modestly impacting total HSP70 induction. HSPs play crucial roles in protein refolding and degradation, and induction of HSP70 is one of the hallmark features of preconditioning (Currie et al., 2000). We observed a biphasic increase in S338 phosphorylation of Raf with a robust early increase coupled with a high level of phosphorylation at the time of secondary stress. Ser338 of c-Raf corresponds to a similar site in B-Raf (Ser445), which is constitutively phosphorylated (Mason et al., 1999) and has been associated with ERK-dependent and -independent pathways and mitochondrial translocation. The bioenergetic and redox consequences of this subcellular redistribution are not yet clear (Chen et al., 2001), but Raf inhibition in our model had no impact on energetic status of preconditioned cells after 3 h. Dawson and colleagues used a neuron-enriched model of preconditioning and defined nitrosylative stress and Ras/Raf-dependent preconditioning (Gonzalez-Zulueta et al., 2000), and our work supports a conserved role for Raf in mediating preconditioning but provides the first evidence that Raf inhibition also blocks HSP70 induction.
p66shc activation enhances mitochondrial hydrogen peroxide production when the cell is challenged with a proapoptotic stimulus (Giorgio et al., 2005). p66Shc expression is also indispensable for the upregulation of intracellular ROS during ischemia-induced apoptosis (Zaccagnini et al., 2004; Cosentino et al., 2008). Prolonged activation of S36-dependent pathways of p66shc can lead to apoptosis, and ablation of the p66shc increases resistance to ROS and dramatically increases life span (Migliaccio et al., 1999). Unlike the other two ShcA isoforms, p66shc can be activated by receptors without evoking a Ras/Raf/ERK signalosome (Migliaccio et al., 1997). In addition to direct mitochondrial targeting and redox activity, p66shc activation is critical in mediating responsiveness to glucose challenge. Deletion of p66Shc in diabetic mice dampens ROS generation suggesting that p66Shc is responsive to redox and energetic stress and can modulate each of these variables. Given that p66Shc gene expression and F2-IsoP levels were increased in blood monocytes from diabetic patients (Camici et al., 2007), this suggests that these two molecules may also be clinical markers of stress in other disorders involving glucose and redox dysregulation including stroke and transient ischemic attack.
Together with our time course and inhibitor studies, the observation that blocking p66shc enhanced F4-NeuroPs production suggests that inactivation of Shc-mediated energetic and oxidative stress may involve preexisting defenses. One mechanism that may contribute to this is the mitochondrial HSP70 complex that associates with Tim44, a component of the high-molecular-weight complex that cooperates with mtHSP70 to inactivate p66Shc within the mitochondrion. Interestingly, overexpression of Tim44 normalize ROS generation (Matsuoka et al., 2005; Orsini et al., 2006; Schiller et al., 2008). Oxidative stress has also been shown to increase the permeability of the mitochondrial membrane increasing autophagic activation to prevent cytochrome c release and promote survival (Elmore et al., 2001; Xue et al., 2001) in vitro. Autophagy has been linked to activation of oxidative and ischemic cell death in vivo (Yan et al., 2005; Carloni et al., 2008; Zhang et al., 2008). Given that overexpression of HSP70 suppresses autophagic cell death (Park et al., 2008), the ability of preexisting chaperones including HSC70 and mtHSP70 to dampen p66shc may be essential in mediating protection and suggests that a delicate balance exists in determining the role of Shc in cellular survival versus death (Baehrecke, 2005).
In conclusion, we report the first evidence that p66shc phosphorylation occurs early in neuronal preconditioning and acts in a Raf-independent manner to elicit HSP70 induction and neuroprotection. It is clear that activation of proapoptotic pathways like p66shc is tightly temporally and spatially regulated in preconditioning, akin to our prior observation with activated caspase 3 (McLaughlin et al., 2003) The current data are consistent with a model in which preexisting chaperones in the cell, including HSC70, are recruited to a subpopulation of activated caspases, damaged proteins and organelles (Fig. 9; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We have previously shown that loss of functional pools of HSC70 is an essential signal to drive expression of neuroprotective levels of HSP70 (McLaughlin et al., 2003). Based on our current data and existing literature (McLaughlin et al., 2003; Park et al., 2008), we propose a model (Fig. 9) in which existing chaperones such as mitochondrial HSP70 and HSC70 block neurotoxic p66shc signaling and activated caspases. Our new data suggest that recruitment of HSC70 binding partners to autophagosomes may contribute to loss of this neuroprotective protein and subsequent upregulation of HSP70. Although many of the signals of preconditioning manifest as cell death triggers, including ROS production, caspase cleavage, autophagy, and protein damage, there is clearly a need to couple multiple measures of stress to understand whether cells are fated to die. Finally, we suggest that overaggressive management of mild caspase activation, ROS production, and p66shc after clinical stresses like angina or transient ischemic attacks that induce clinical preconditioning may block cytoprotective pathways.
Figure 9.

Model of Raf and p66Shc contribution to oxidative and energetic dysfunction in preconditioning. Preconditioning oxygen glucose deprivation induces an early but modest decrease in total ATP content with a larger increase in total oxidative stress assessed by formation of F2-IsoPs and F4-NeuroPs. Raf and p66Shc kinases are rapidly activated, and p66Shc relocates to subcellular organelles including the mitochondria. In the absence of p66Shc, cellular ATP, oxidative and protein stress persist, resulting in increased formation of F4-NeuroPs and formation of autophagosomes. We hypothesize that the limited autophagy experienced by preconditioning functions to sequester damaged organelles and proteins via HSC70 and other pathways which ultimately enhance the induction of HSP70 and other protective proteins upregulated at the time of secondary stressors.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS050396 (to B.M.), training grants from Pharmaceutical Research and Manufacturers of America (to E.S.M.), and National Institutes of Health Grant MH065215 (to S.L.H.Z.). Statistical and graphical support was provided by P30HD15052 (Vanderbilt Kennedy Center). We thank Jeannette Stankowski and Drs. Pat Levitt, Laura Lillien, and Gregg Stanwood for helpful comments and suggestions, as well as Karen Hartnett and the members of the Aizenman and McLaughlin laboratories for help in establishing and characterizing the preconditioning model.
References
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3{beta} in the regulation of HSF-1 activity. J Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- Buckman JF, Hernández H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, Luscher TF, Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:566–577. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci U S A. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor to KATP channel. Ann Rev Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Francia P, Camici GG, Pelicci PG, Volpe M, Lüscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66Shc protein. Arterioscler Thromb Vasc Biol. 2008;28:622–628. doi: 10.1161/ATVBAHA.107.156059. [DOI] [PubMed] [Google Scholar]
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res. 2000;863:169–181. doi: 10.1016/s0006-8993(00)02133-8. [DOI] [PubMed] [Google Scholar]
- Dai R, Frejtag W, He B, Zhang Y, Mivechi NF. c-Jun NH2-terminal kinase targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J Biol Chem. 2000;275:18210–18218. doi: 10.1074/jbc.M000958200. [DOI] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase Cε. J Neurosci. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55:334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cyctochrome c and p66 shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, Horgan AM, York RD, Withers GS, Banker GA, Stork PJ. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J Biol Chem. 2000;275:3722–3728. doi: 10.1074/jbc.275.5.3722. [DOI] [PubMed] [Google Scholar]
- Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- He B, Meng YH, Mivechi NF. Glycogen synthase kinase 3 beta and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of transcriptionally active granules after heat shock. Mol Cell Biol. 1998;18:6624–6633. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NM, Bergeron M. Hypoxia-induced ischemic tolerance in neonatal rat brain involves enhanced ERK1/2 signaling. J Neurochem. 2004;89:157–167. doi: 10.1111/j.1471-4159.2004.02324.x. [DOI] [PubMed] [Google Scholar]
- Jung F, Haendeler J, Hoffmann J, Reissner A, Dernbach E, Zeiher AM, Dimmeler S. Hypoxic induction of the hypoxia-inducible factor is mediated via the adaptor protein Shc in endothelial cells. Circ Res. 2002;91:38–45. doi: 10.1161/01.res.0000024412.24491.ca. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Graham LB, Parker CE, Ames BN, Basu S, Fitzgerald GA, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, 2nd, Rokach J, Shigenaga MK, Sun J, Walter PB, Tomer KB, Barrett JC, et al. Biomarkers of oxidative stress study III. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in CCl4 poisoning. Free Radic Biol Med. 2005a;38:711–718. doi: 10.1016/j.freeradbiomed.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, et al. Biomarkers of oxidative stress study II. Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med. 2005b;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kaduce TL, Chen Y, Hell JW, Spector AA. Docosahexaenoic acid synthesis from n-3 fatty acid precursors in rat hippocampal neurons. J Neurochem. 2008;105:1525–1535. doi: 10.1111/j.1471-4159.2008.05274.x. [DOI] [PubMed] [Google Scholar]
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Breclesen DE, Brodsky JL, Brumell JH, Brunk UT, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Pérez-Pinzón MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Marin JG, Cornet S, Spinnewyn B, Demerlé-Pallardy C, Auguet M, Chabrier PE. BN 80933 inhibits F2-isoprostane elevation in focal cerebral ischaemia and hypoxic neuronal cultures. Neuroreport. 2000;11:1357–1360. doi: 10.1097/00001756-200004270-00041. [DOI] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T, Wada J, Hashimoto I, Zhang Y, Eguchi J, Ogawa N, Shikata K, Kanwar YS, Makino H. Gene delivery of Tim44 reduces mitochondrial superoxide production and ameliorates neointimal proliferation of injured carotid artery in diabetic rats. Diabetes. 2005;54:2882–2890. doi: 10.2337/diabetes.54.10.2882. [DOI] [PubMed] [Google Scholar]
- McLaughlin B. The kinder side of killer proteases: caspase activation contributes to neuroprotection and CNS remodeling. Apoptosis. 2004;9:111–121. doi: 10.1023/B:APPT.0000018793.10779.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin BA, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in ischemic preconditioning. Proc Natl Acad Sci U S A. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. Opposite effects of the p52(shc)/p46(shc) and p66(shc) splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F-2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-Like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Combs CA, French S, Ahn BH, Fergusson MM, Balaban RS, Finkel T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J Biol Chem. 2006;281:10555–10560. doi: 10.1074/jbc.M511626200. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Sugino T, Nozaki K, Takagi Y, Hattori I, Hayashi J, Hashimoto N, Moriguchi T, Nishida E. Activation of p38 kinase in the gerbil hippocampus showing ischemic tolerance. J Cereb Blood Flow Metab. 2003;23:1052–1059. doi: 10.1097/01.WCB.0000084251.20114.65. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- Ohtsuka K, Suzuki T. Roles of molecular chaperones in the nervous system. Brain Res Bull. 2000;53:141–146. doi: 10.1016/s0361-9230(00)00325-7. [DOI] [PubMed] [Google Scholar]
- O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–432. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- Orsini F, Moroni M, Contursi C, Yano M, Pelicci P, Giorgio M, Migliaccio E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66(Shc) Biol Chem. 2006;387:1405–1410. doi: 10.1515/BC.2006.176. [DOI] [PubMed] [Google Scholar]
- Park MA, Curiel DT, Koumenis C, Graf M, Chen CS, Fisher PB, Grant S, Dent P. PERK-dependent regulation of HSP70 expression and the regulation of autophagy. Autophagy. 2008;4:364–367. doi: 10.4161/auto.5593. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Finetti F, Petronilli V, Ulivieri C, Giusti F, Lupetti P, Giorgio M, Pelicci PG, Bernardi P, Baldari CT. p66SHC promotes T cell apoptosis by inducing mitochondrial dysfunction and impaired Ca2+ homeostasis. Cell Death Differ. 2007;14:338–347. doi: 10.1038/sj.cdd.4401997. [DOI] [PubMed] [Google Scholar]
- Peng X, Guo X, Borkan SC, Bharti A, Kuramochi Y, Calderwood S, Sawyer DB. Heat shock protein 90 stabilization of ErbB2 expression is disrupted by ATP depletion in myocytes. J Biol Chem. 2005;280:13148–13152. doi: 10.1074/jbc.M410838200. [DOI] [PubMed] [Google Scholar]
- Peralta C, Serafin A, Fernández-Zabalegui L, Wu ZY, Roselló-Catafau J. Liver ischemic preconditioning: a new strategy for the prevention of ischemia-reperfusion injury. Transplant Proc. 2003;35:1800–1802. doi: 10.1016/s0041-1345(03)00571-2. [DOI] [PubMed] [Google Scholar]
- Pérez-Pinzón MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R. Protein kinase C {beta} and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer. 2006;13:S125–S135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger HA, Graf R, Clavien PA. Sub-lethal oxidative stress triggers the protective effects of ischemic preconditioning in the mouse liver. J Hepatol. 2003;39:972–977. doi: 10.1016/s0168-8278(03)00415-x. [DOI] [PubMed] [Google Scholar]
- Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24:69–176. doi: 10.1016/0163-7827(85)90011-6. [DOI] [PubMed] [Google Scholar]
- Schiller D, Cheng YC, Liu Q, Walter W, Craig EA. Residues of Tim44 involved in both association with the translocon of the inner mitochondrial membrane and regulation of mitochondrial Hsp70 tethering. Mol Cell Biol. 2008;28:4424–4433. doi: 10.1128/MCB.00007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorziello A, Santillo M, Adornetto A, Dell'aversano C, Sirabella R, Damiano S, Canzoniero LM, Di Renzo GF, Annunziato L. NO-induced neuroprotection in ischemic preconditioning stimulates mitochondrial Mn-SOD activity and expression via RAS/ERK1/2 pathway. J Neurochem. 2007;103:1472–1480. doi: 10.1111/j.1471-4159.2007.04845.x. [DOI] [PubMed] [Google Scholar]
- Seo HR, Chung DY, Lee YJ, Lee DH, Kim JI, Bae S, Chung HY, Lee SJ, Jeoung D, Lee YS. Heat shock protein 25 or inducible heat shock protein 70 activates heat shock factor 1: dephosphorylation on serine 307 through inhibition of ERK1/2 phosphorylation. J Biol Chem. 2006;281:17220–17227. doi: 10.1074/jbc.M600062200. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1:26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor JD, Boeckman FA, Aizenman E. Intrinsic redox properties of N-methyl-d-aspartate receptor can determine the developmental expression of excitotoxicity in rat cortical neurons in vitro. Brain Res. 1997;747:297–303. doi: 10.1016/s0006-8993(96)01237-1. [DOI] [PubMed] [Google Scholar]
- Song J, Takeda M, Morimoto RI. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat Cell Biol. 2001;3:276–282. doi: 10.1038/35060068. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Tan YK, Kusuma CM, St John LJ, Vu HA, Alibek K, Wu A. Induction of autophagy by anthrax lethal toxin. Biochem Biophys Res Comm. 2009;379:293–297. doi: 10.1016/j.bbrc.2008.12.048. [DOI] [PubMed] [Google Scholar]
- Teshima Y, Akao M, Li RA, Chong TH, Baumgartner WA, Johnston MV, Marbán E. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34:1796–1802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisová P, Düssmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand F, Liao W, Busch C, Castell S, Knapp F, Lindauer U, Megow D, Meisel A, Redetzky A, Ruscher K, Trendelenburg G, Victorov I, Riepe M, Diener HC, Dirnagl U. Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1229–1237. doi: 10.1097/00004647-199911000-00007. [DOI] [PubMed] [Google Scholar]
- Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, Bolli R. Role of the protein kinase C-{epsilon}-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation. 2005;112:1971–1978. doi: 10.1161/CIRCULATIONAHA.105.561522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, Capogrossi MC. p66ShcA modulates tissue response to hindlimb ischemia. Circulation. 2004;109:2917–2923. doi: 10.1161/01.CIR.0000129309.58874.0F. [DOI] [PubMed] [Google Scholar]
- Zeiger SL, Musiek ES, Zanoni G, Vidari G, Morrow JD, Milne GJ, McLaughlin B. Neurotoxic lipid peroxidation species formed by ischemic stroke increase injury. Free Radic Biol Med. 2009;47:1422–1431. doi: 10.1016/j.freeradbiomed.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol. 2007;292:H2573–H2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]