

Abstract
Similarly to NHCs, CAACa and BACa react with CO2 to give the corresponding betaines. Based on the carbonyl stretching frequencies of cis-[RhCl(CO)2(L)] complexes, the order of electron donor ability was predicted to be CAACa ≈ BACa > NHCs. When the betaines νasym(CO2) values are used, the apparent ordering is BACa > NHCs ≈ CAACa that indicates a limitation for the use of IR spectroscopy in the ranking of ligand σ-donating ability. Although all carbenes react with carbon disulfide to give the corresponding betaines, a second equivalent of CS2 reacts with the BAC-CS2 leading to a bicyclic thieno[2,3-diamino]-1,3-dithiole-2-thione, which results from a novel ring expansion process. Surprisingly, in contrast to NHCs, CAACa does not react with carbodiimide, whereas BACa exclusively give a ring expanded product, analogous to that obtained with CS2. The intermediate amidinate can be trapped, using the lithium tetrafluoroborate adduct of BACb as a carbene surrogate.
Keywords: Carbenes, Betaines, Carbon dioxide, Carbon disulfide, Carbodiimide, Heterocycles
Introduction
The reactivity of stable cyclic diamino carbenes, the so-called NHCs,[1] towards a variety of organic and organometallic substrates has been widely investigated.[2] In contrast, little is known about the reactivity of the more recently discovered stable cyclic (alkyl)(amino)carbenes (CAACs),[3] and bis(amino)cyclopropenylidenes (BACs)[4] (Figure 1). However, it is already clear that the chemical behavior of CAACs and BACs can be strikingly different from that of NHCs. For examples, CAACs react with CO,[5a] H2,[5b] and NH3,[5b] to give the corresponding adducts, whereas NHCs are inert under the same experimental conditions.[6] CAACs induce the ring opening of white phosphorus giving P4-species,[7a] and BACs promote the degradation of P4 into P1 and P3 fragments,[7b] whereas NHCs induce the aggregation of elemental phosphorus affording novel phosphorus P12 clusters.[7c]
Figure 1.
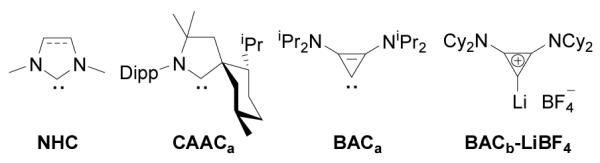
Representative stable carbenes (and lithium adduct) used in this study. Dipp = 2,6-iPr2C6H3.
NHCs are known to react with heteroallenes, such as carbon dioxide,[8] carbon disulfide,[8a,8h,9] carbodiimide,[10] etc.,[9a,11] to afford betaines or spirocyclic bis-adducts. An exhaustive review by DeLaude[12] was recently published highlighting this field, and prompting us to report our findings. We show that the unique steric and electronic parameters of CAACs and BACs induce chemical reactions that can be distinct from those observed with NHCs.
Results and Discussion
The most studied heteroallene, with respect to reactivity towards NHCs, is carbon dioxide.[8] It has been shown that NHC betaines 1 are readily formed and are thermally stable (Scheme 1). Crabtree and others have shown that these betaines can serve as air- and moisture stable free carbene surrogates.[8c,13] Of particular interest, NHC-CO2 adducts have been used to transfer NHCs to a variety of transition metal fragments. More recently, these species have also been involved in catalytic systems, such as in the coupling reaction of carbon dioxide with epoxides[14a] the carboxylative cyclization of propargyl alcohols,[14b] and the conversion of CO2 into methanol.[14c]
Scheme 1.
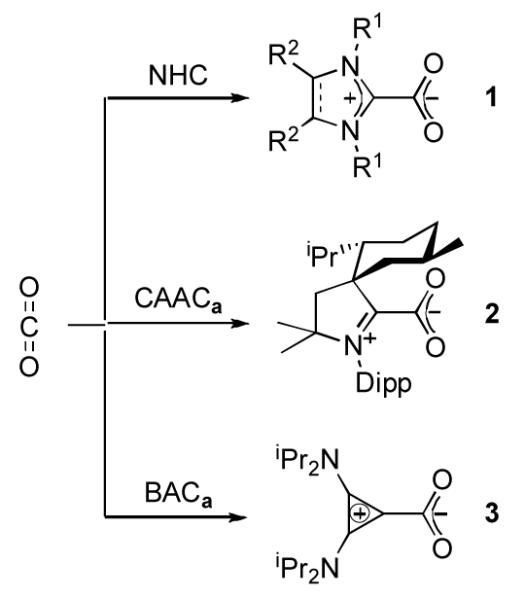
Reaction of NHC, CAACa, and BACa with carbon dioxide.
We began our investigation by bubbling CO2 gas through a room temperature THF solution of either free CAACa or BACa. In both cases, a white precipitate immediately formed. The 13C NMR spectrum of CAAC product 2 displayed resonances at 195 ppm and 160 ppm in the region expected for an iminium salt and a carboxylate carbon, respectively. Similarly, the BAC product 3 give signals at 157 ppm (carboxylate), and at 128 and 112 ppm, typical for a diamino cyclopropenium salt.[15] High resolution mass spectrometry (HRMS) confirmed the monocarboxylated betaine structure of 3; however, the CAAC adduct 2 gave a strong signal at m/z 382.3471 corresponding to decarboxylation under the MS conditions. Both 2 and 3 are not air-sensitive, and are thermally stable in the solid sate [m.p.: 123 °C, dec. (2); 143 °C, dec. (3)].
Recent literature has suggested the use of IR spectra of carbene-CO2 betaines as a means to assess the relative σ-donor strength of carbenes.[8h] Indeed, the values of the carboxylate asymmetric stretching frequency typically range from 1540 to 1640 cm−1, a region not usually complicated by other signals. Moreover, the absence of possible π back-donation from the CO2 fragment to the carbene moiety should reveal purely the σ-donation effects of the carbene (which is not the case in the method using transition metal carbonyl fragments).[16] The FT-IR spectrum (KBr pellets) of the BAC adduct 3 showed a strong infrared absorption band at 1660 cm−1, shifted to lower frequency relative to that of NHC analogs (1663-1684 cm−1),[8c] confirming the stronger σ-donating capability of BAC, predicted using the corresponding LRh(CO)2Cl complex.[17] Since CAAC ligands have also been predicted, using LRh(CO)2Cl complexes, to be stronger donors than NHC’s,[18] it was surprising to observe the νasym(CO2) for CAAC adduct 2 at 1676 cm−1, a value nearly identical to that reported for the analogous IMes-CO2 adduct (1675 cm−1).[8c] Clearly, this discrepancy indicates a limitation towards the use of IR spectroscopy in the ranking of ligand σ-donating ability.
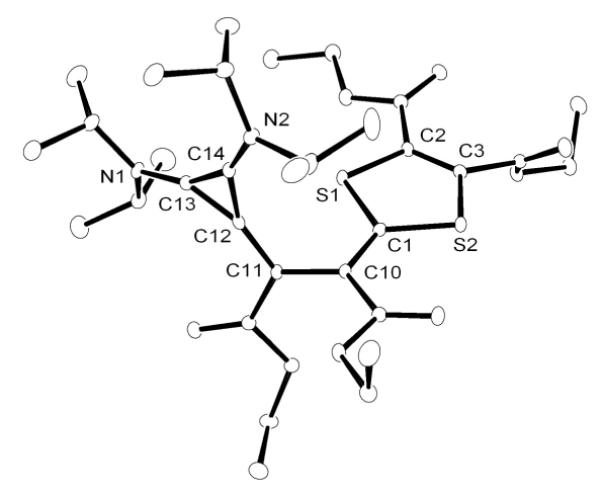
We next turned our attention to carbon disulfide. Addition at room temperature of excess CS2 to a toluene solution of CAACa resulted in the slow formation of an orange solution. Upon workup, the product was identified by 1H and 13C NMR, as well as HRMS, as the betaine 5, which is analogous to the adduct 4 observed with NHCs[8a,8h,9] (Scheme 2). However, in the reaction of excess CS2 with BAC, a different type of product was formed. Indeed, the 13C NMR spectrum showed five low-field signals for quaternary carbons, instead of three as expected for the symmetrical betaine 6. The HRMS data indicated that the observed species 7 was composed of two equivalents of CS2 for one equivalent of BAC. The exact structure of 7 was unambiguously established by a single crystal X-ray diffraction study.[19] It is a ring expanded, bicyclic thieno[2,3-diamino]-1,3-dithiole-2-thione (Figure 2). In the solid state, the two rings are coplanar, while the exocyclic amino groups are twisted out of plane, precluding any significant interaction of the N lone pairs with the ring π-system.
Scheme 2.
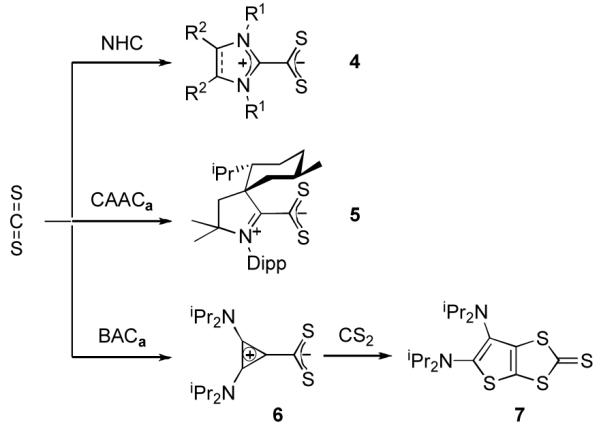
Reaction of NHC, CAACa, and BACa with carbon disulfide.
Figure 2.
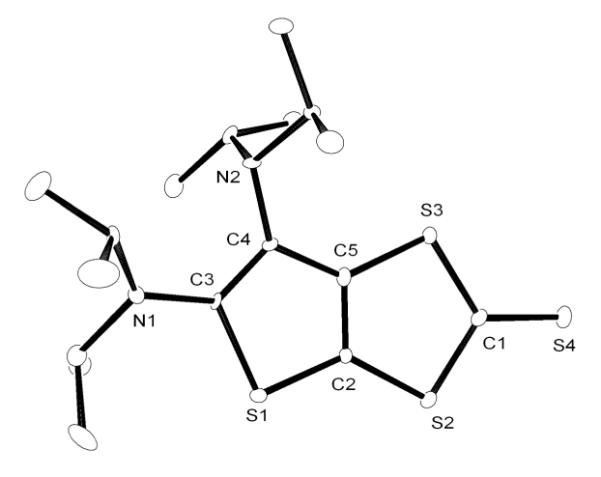
Molecular view of the solid state structure of 7 (hydrogen atoms omitted for clarity).
In order to obtain further insight into the mechanism of the reaction leading to 7, one equivalent of CS2 was added dropwise to a vigorously stirred, room temperature solution of BAC in THF. Under these experimental conditions, the betaine 6 was obtained and isolated in high yield. This species proved to be robust, with a decomposition point of 209-210 °C, and minimal decomposition even in refluxing toluene for 24 hours. Not surprisingly addition of one equivalent of CS2 afforded heterocycle 7 in quantitative yield. Mechanistically, one can envision that betaine 6 can undergo either stepwise addition/cyclization or concerted [3+2]-cycloaddition with CS2 to form the spirocyclic intermediate I (Scheme 3). It should be noted that related spirocyclic species have been isolated in the reaction of NHCs with iso(thio)cyanates.[9a,b,f,j,11a,b,d] Due to ring strain, ring expansion of the cyclopropene occurs yielding the final product 7.
Scheme 3.
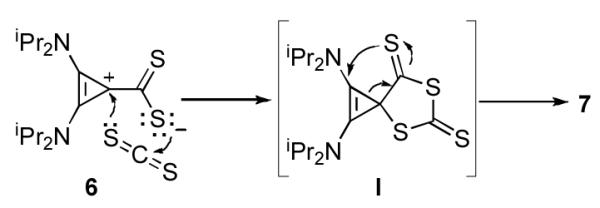
Possible mechanism for the formation of 7.
Depending on the nature of NHCs, electron deficient alkynes react with NHC-betaines of type 4 to afford either electron-rich alkenes 8 or butadienic species 9[9b] (Scheme 4). The reaction of CAAC betaine 5 with excess acetylene dicarboxylate yielded only the mixed-carbene dimer 10. All attempts to liberate the corresponding carbenes from 10, under thermolytic and photolytic conditions, failed. In the case of BAC-betaine 6, reaction with acetylene dicarboxylate leads directly to the butadienic product 11 by incorporation of two acetylenes. X-Ray analysis of single crystals obtained from a benzene solution of 11 ascertained the structural assignment (Figure 3). It is interesting to note that, at least in the solid state, the double bond between the three-membered ring and the adjacent carbon is twisted by 31°, which indicate a strong polarization of the bond.
Scheme 4.
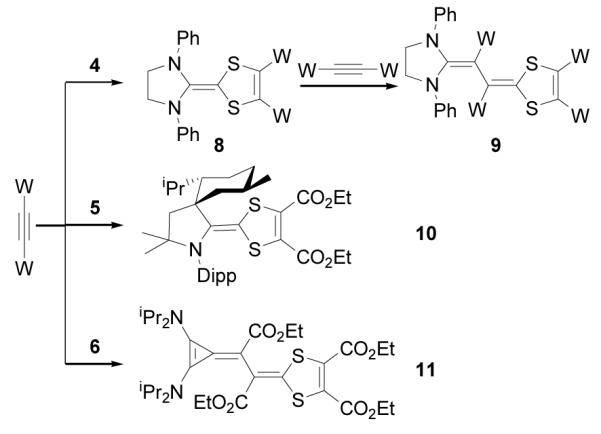
Reactions of carbene-CS2 betaines with electron deficient alkynes.
Figure 3.
Molecular view of the solid state structure of 11 (hydrogen atoms omitted for clarity).
Although NHC reacts with N,N’-diisopropylcarbodiimide to form a stable amidinate 12,[10] CAACa does not react under similar experimental conditions (Scheme 5). In contrast, the addition of one equivalent N,N’-dicyclohexylcarbodiimide to a THF solution of BACa resulted in the complete consumption of the carbodiimide, and the formation of a new compound 13, along with 50% of BACa remaining. Addition to this solution of a second equivalent of carbodiimide, and stirring at RT overnight, resulted in the complete consumption of BACa and quantitative formation of 13. HRMS confirmed the presence of two carbodiimide units with one BACa, and an X-ray diffraction study revealed the bicyclic structure of 13 (Figure 4), which is analogous to that observed with carbon disulfide. Interestingly, in contrast to 7, the fused-ring system has a butterfly structure (folding angle: 17.6°). All attempts to characterize the mono-carbodiimide adduct of BACa, by monitoring the reaction by NMR spectroscopy at low temperatures failed. Thus, BACa is uniquely able to activate carbodimide and subsequently be incorporated into a heterocyclic framework.
Scheme 5.
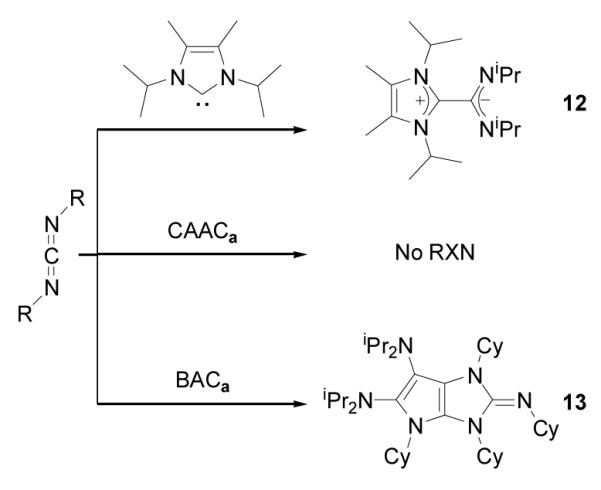
Reaction of NHC, CAACa, and BACa with carbodiimides.
Figure 4.
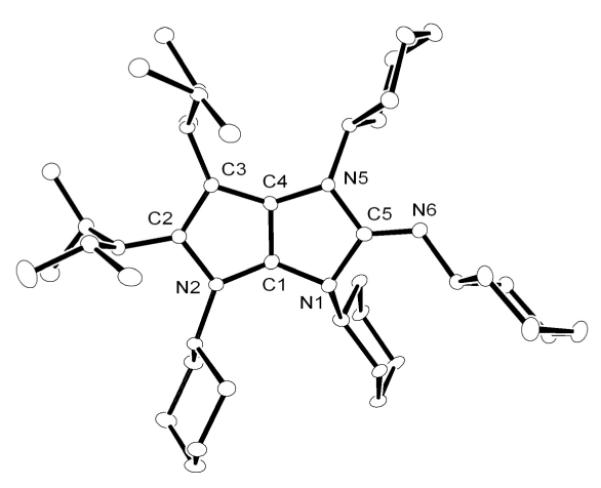
Molecular view of the solid state structure of 13 (hydrogen atoms omitted for clarity).
In order to tame the nucleophilicity of the postulated amidinate inner salt intermediate, we next tested the reaction of a carbodiimide with the BACb-Li salt, reasoning that the presence of the Li cation should limit this reaction to mono-addition. Addition of N,N’-dicyclohexylcarbodiimide to a room temperature suspension of bis(dicyclohexylamino)(lithio)cyclopropenium tetrafluoroborate in THF, resulted in the clean formation of a new product 14 as revealed by 13C NMR, with chemical shifts for the ring carbons at 134 and 103 ppm, indicative of a betaine product (Scheme 6). Single crystals were obtained by slow evaporation of a THF solution, and analyzed by X-ray diffraction (Figure 5). Product 14 appeared to be the BF3 adduct of the expected betaine, the process being accompanied by extrusion of LiF. The overall structural features are consistent with other known cyclopropenium salts with the exception of a possible fluorophilic interaction between C2 of the cyclopropenium ring and a fluorine from BF3 measured at 2.612Å.
Scheme 6.
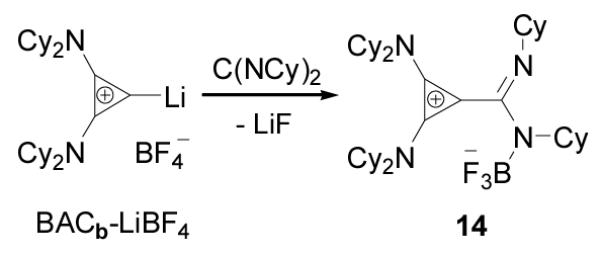
Reactions of BACb-LiBF4 adduct with N,N’dicyclohexylcarbodiimide
Figure 5.
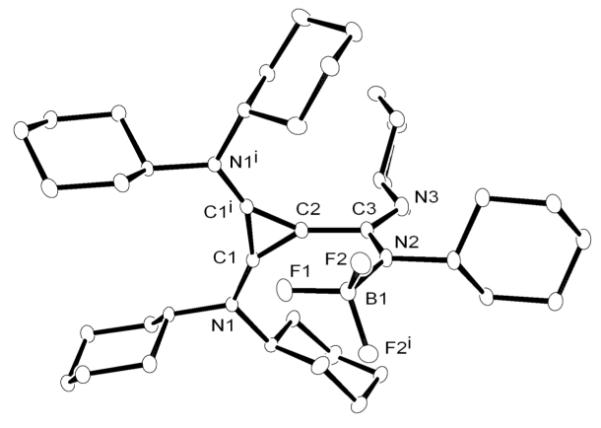
Molecular view of the solid state structure of 14 (hydrogen atoms omitted for clarity).
Conclusion
Based on these results, it is apparent that stable CAACs, BACs, and BAC-Li salts react as strong nucleophiles in the activation of heteroallenes. Based on the asymmetric C-O stretching frequency of the BAC-CO2 adduct 3, BAC appears to provide greater donation to activated substrates relative to their N-heterocyclic carbene counterparts. However, the result observed for CAAC-CO2 adduct 2 calls into question the generality of this type of analysis. Like NHCs, both CAACs and BACs form stable mono-adduct betaines with both carbon dioxide and carbon disulfide. However, due to increased ring strain, and lack of heteroatoms bearing lone pairs adjacent to the carbene center, BAC adducts display an increased electrophilicity. Reaction of these species with an excess of either CS2 or carbodiimide leads to the formation of electron rich, fused ring heterocycles, via a spirocyclic intermediate. The use of a cyclopropenylidene-lithium adduct hinders formation of this spirocyclic species by trapping of the intermediate amidinate by BF3. Efforts to capitalize upon the spiro to ring expansion sequence using various partners are ongoing and should lead to a more generalized synthetic methodology towards a wide range of electron rich, fused ring systems.
Experimental Section
All manipulations were performed in an inert atmosphere of dry argon using standard Schlenk techniques or in an mBraun glovebox. Dry, oxygen-free solvents were employed. NMR spectra were recorded on a Bruker Avance 300, or Varian Inova 400 and 500 spectrometers. 1H and 13C chemical shifts are reported relative to SiMe4 or referenced to residual solvent peaks. 19F NMR chemical shifts are reported relative to CFCl3. FTIR spectra were recorded on a Bruker Equinox 55 spectrometer from KBr pellets.
2: Carbon dioxide was bubbled through a room temperature, stirred solution of CAACa (300 mg, 0.788 mmol) in THF (10 mL) for 30 min. affording an off-white precipitate. Volatiles were removed under vacuum and the residue washed with hexanes to afford a white powder. Yield: 322 mg (96 %). m.p. 123 °C, dec; IR (KBr, ν cm−1): 3453, 2956, 2953, 2947, 1676 (CO2), 1567, 1473, 1343, 1147, 1053, 810, 747; 1H NMR (300 MHz, CD3CN/THF, 25 °C) δ = 7.48-7.33 (m, 3 H), 2.87-2.77 (m, 2 H), 2.61-2.51 (broad, overlapping m, 3 H), 2.14-2.05 (broad, overlapping m, 2 H), 1.51-0.88 (overlapping m, 33 H); 13C{1H} NMR (75 MHz, CD3CN/THF, 25 °C) δ = 194.8, 160.5, 147.6, 147.4, 131.5, 130.9, 127.0, 126.5, 78.0, 58.8, 53.8, 53.7, 50.6, 35.7, 31.1, 30.8, 30.7, 29.6, 29.4, 28.7, 27.3, 27.0, 26.9, 25.4, 24.9, 24.3, 23.0, 19.2; ESI-MS m/z 382.3471 (382.3468 calcd for C27H44N, MH+ - CO2).
3: At room temperature, carbon dioxide was bubbled through a vigorously stirred solution of BACa (501 mg, 2.12 mmol) in THF (20 mL) for 1 hour. Solvent was removed under vacuum, and the resulting white precipitate washed with 10 mL hexanes to afford 6 as a white powder. Yield: 576 mg (97%). mp 143 °C, dec; IR (KBr, ν cm−1): 2982, 2935, 2880, 1898, 1660 (CO2), 1540, 1468, 1387, 1371, 1333, 1292, 1217, 1161, 1053, 1019, 893, 802; 1H NMR (300 MHz, CDCl3) δ = 3.81 (overlapping m, 4 H), 1.40 (d, J = 6.8 Hz, 12 H), 1.26 (d, J = 6.8 Hz, 12 H); 13C{1H} NMR (75 MHz, CDCl3) δ = 157.2, 128.3, 112.2, 55.3, 49.6, 21.3, 21.2; ESI MS m/z 281.2230 (281.2224 calcd for C16H29N2O2, MH+).
5: Carbon disulfide (48 μL, 0.798 mmol) was added to a room temperature solution of CAACa (300 mg, 0.788 mmol) in THF (10 mL) and stirred overnight affording an orange solution. Volatiles were removed under vacuum, and the residue washed with hexanes to give an orange powder. Yield: 331 mg (92 %). m.p. 219 °C, dec; 1H NMR (300 MHz, C6D6, 25 °C) δ = 7.02-6.94 (m, 3 H), 3.21 (m, 1 H), 2.99 (broad, overlapping m, 2 H), 2.76 (broad s, 1 H), 2.53 (broad d, 2 H), 2.01 (broad m, 2 H), 1.68-0.86 (broad, overlapping m, 32 H); 13C{1H} NMR (75 MHz, C6D6, 25 °C) δ = 229.7, 187.2, 147.6, 147.5, 131.0, 130.7, 126.9, 126.5, 74.9, 59.6, 55.3, 54.8, 51.8, 35.2, 31.1, 30.7, 30.4, 29.6, 29.3, 29.2, 28.8, 28.7, 28.6, 26.6, 26.1, 25.5, 22.9, 22.3; ESI MS m/z 458.2913 found (458.2921 calcd for C28H44NS2, M+).
6. Carbon disulfide (55 μL, 0.910 mmol) was added dropwise to a stirred room temperature solution of BACa (214 mg, 0.905 mmol) in hexanes (10 mL) forming an immediate deep red precipitate. After one hour, volatiles were removed under vacuum, and the product washed with hexanes yielding a pink powder. Yield: 230 mg (81 %). Deep red, blocky crystals of 3 formed readily from a hexanes/THF solution at −20 °C overnight. m.p. 209-210 °C, dec; 1H NMR (300 MHz, CDCl3) δ = 3.87-3.78 (overlapping m, 2 H), 1.44 (d, J = 6 Hz, 6 H), 1.33 (d, J = 6 Hz, 6 H); 13C{1H} NMR (75 MHz, CDCl3) δ = 231.6, 122.9, 120.8, 55.6, 49.1, 22.5, 21.3; ESI MS m/z 313.1781 found (313.1772 calcd for C16H29N2S2, MH+).
7. An excess of carbon disulfide (95 μL, 1.572 mmol) was added to a room temperature solution of BACa (119 mg, 0.503 mmol) in THF (5 mL), rapidly yielding a bright yellow solution. Volatiles were removed under vacuum to afford an orange powder. Yield: 188 mg (96 %); Single crystals of 7 were obtained from a diethyl ether solution at −20 °C. m.p. 97-99 °C, dec; 1H NMR (500 MHz, C6D6) δ = 3.55 (sept, J = 7 Hz, 2 H), 3.16 (sept, J = 6 Hz, 2 H), 1.04 (d, J = 7 Hz, 12 H), 0.92 (d, J = 6 Hz, 12 H); 13C{1H} NMR (125 MHz, C6D6) δ = 214.0, 153.0, 138.2, 133.1, 121.8, 51.2, 51.1, 22.7, 22.1; ESI-MS m/z 389.1219 (389.1208 calcd for C17H29N2S4, MH+).
10: Diethylacetylene dicarboxylate (75 μL, 0.469 mmol) was added dropwise to a stirred solution of 5 (210 mg, 0.459 mmol) in toluene resulting in an immediate darkening of the red solution. Volatiles were removed under vacuum to obtain 10 as a purple powder. 1H NMR (300 MHz, C6D6) δ = 7.15-7.01 (overlapping m, 3 H), 3.90 (q, J = 7 Hz, 2 H), 3.73 (dq, J = 1, 7 Hz, 2 H), 3.63 (m, 1 H), 3.07-2.84 (overlapping m, 3 H), 2.79 (d, J = 13 Hz, 1 H), 2.22 (m, 1 H), 2.00 (m, 1 H), 1.89 (d, J = 13 Hz, 1 H), 1.80-1.68 (overlapping m, 3 H), 1.63 (d, J = 7 Hz, 3 H), 1.56 (d, J = 7 Hz, 3 H), 1.46-1.40 (m, 1 H), 1.36 (s, 3 H), 1.31 (d, J = 7 Hz, 3 H), 1.25 (d, J = 7 Hz, 3 H), 1.18 (d, J = 7 Hz, 6 H), 0.97-0.83 (overlapping m, 12 H), 0.76 (t, J = 7 Hz, 3 H); 13C{1H} NMR (75 MHz, C6D6) δ = 161.2, 161.1, 151.1, 149.5, 149.4, 137.9, 135.5, 130.8, 129.1, 125.3, 125.2, 88.7, 64.3, 62.0, 61.8, 60.0, 53.1, 50.2, 46.2, 31.7, 30.2, 29.6, 29.5, 29.0, 28.5, 26.4, 26.3, 25.9, 25.5, 24.2, 23.0, 22.6, 22.3, 14.2, 14.1; ESI-MS m/z 628.3466 (628.3489 calcd for C36H54NO4S2, MH+).
11: Diethylacetylene dicarboxylate (173 μL, 1.08 mmol) was added dropwise to a stirred solution of 6 (331 mg, 1.06 mmol) in THF (10 mL) resulting in an immediate darkening of the red solution. The solution was stirred at room temperature overnight and volatiles removed under vacuum to afford a deep red powder. After washing with hexanes (20 mL) a purple powder was obtained. Yield: 313 mg (48 %); Single crystals of 11 were obtained from a hexanes solution at −20 °C. m.p. 112-114 °C, dec; 1H NMR (300 MHz, C6D6) δ = 4.36-3.54 (overlapping m, 12 H), 1.44-0.81 (overlapping m, 36 H); 13C{1H} NMR (75 MHz, C6D6) δ = 168.4, 168.2, 160.8, 160.5, 155.2, 136.7, 131.1, 122.0, 121.7, 116.5, 68.2, 62.5, 60.6, 58.6, 51.2, 26.2, 23.1, 22.5, 15.9, 15.1, 14.1, 14.0; ESI-MS m/z 653.2826 (653.2925 calcd for C32H49N2O8S2, MH+).
13: A room temperature solution of N,N’-dicyclohexylcarbodiimide (200 mg, 0.969 mmol) in THF (1 ml) was slowly added to a stirred solution of BACa (115 mg, 0.486 mmol) in THF (1 ml) after which the solution became bright yellow. After stirring at room temperature overnight, solvent was removed under vacuum to afford a sticky orange powder. Slow evaporation of a benzene solution of the product affords yellow crystals of 13.Yield: 293 mg (93 %). m.p. 143-147 °C; 1H NMR (300 MHz, C6D6) δ = 3.74-3.13 (broad overlapping m, 8 H), 2.19-1.11 (broad overlapping m, 64 H); 13C{1H} NMR (75 MHz, C6D6) δ = 155.5, 136.4, 122.6, 119.8, 115.6, 58.4, 57.3, 55.5, 54.4, 51.7, 36.5, 32.6, 31.2, 29.4, 27.1, 27.0, 26.7, 26.6, 26.0, 25.7, 25.2, 24.4, 23.3; ESI-MS m/z 649.5895 (649.5891 calcd for C41H73N6, MH+).
Synthesis of BACb-LiBF4. Procedure adapted from Lavallo, et al.[20] BACbClBF4: Dicyclohexylamine (22 mL, 0.133 mol) was added dropwise at 0 °C to a stirred solution of tetrachlorocyclopropene (2.7 mL, 0.0215 mol) in CH2Cl2 (300 mL). After warm up to room temperature and stirring for six hours. a pale yellow suspension was formed. NaBF4 (2.36 g, 0.0215 mmol) was added and the suspension stirred vigorously for 16 hours. 1H NMR (300 MHz, 25 °C, CDCl3) δ = 3.55 (pseudo t, J = 11.8 Hz, 2 H) 3.33 (pseudo t, J = 11.7 Hz, 2 H), 2.00-1.11 (broad overlapping m, 40 H); 13C{1H} NMR (75 MHz, 25 °C, CDCl3) δ = 132.4, 93.6, 65.8, 56.9, 32.7, 30.8, 25.7, 25.5, 24.9, 24.7. BACb-HBF4: Triphenylphosphine (5.64 g, 0.021 mol) was added, followed immediately by deionized water (250 mL), and the suspension was stirred at room temperature for 10 hours with a vent to open air. The aqueous layer was decanted and the resulting suspension washed with deionized water (4 × 250 mL) to afford a yellow solution which was dried over MgSO4. Volatiles were removed under vacuum at 50 °C for 6 hours to afford a yellow, sticky solid. BACb-HBF4 was purified by two recrystallizations from refluxing THF. 1H NMR (300 MHz, 25 °C, CDCl3) δ = 7.49 (s, 1 H), 3.53-3.45 (m, 2 H), 3.35-3.31 (m, 2 H), 1.92-1.21 (overlapping m, 40 H); 13C{1H} NMR (75 MHz, 25 °C, CDCl3) δ = 134.3, 100.3, 64.9, 58.1, 31.2, 30.8, 25.6, 25.5, 24.7, 24.6. BACb-LiBF4: To suspension of CyBAC-HBF4 (2.0 g, 4.128 mmol) in Et2O (30 mL) at −78 °C, a 2.5M solution of nBuLi in hexanes (1.65 mL, 4.128 mmol) was added. The suspension was stirred for one hour, and allowed to warm to room temperature for an additional one hour. Volatiles were removed under vacuum and the resulting sticky, yellow solid washed with hexanes to afford an off-white powder. Yield 1.34 g (66%), 1H NMR (300 MHz, 25 °C, C6D6) δ = 3.43 (broad s, 2 H), 2.54 (broad s, 4 H), 2.15-0.88 (broad, overlapping m, 40 H); 13C{1H} NMR (75 MHz, 25 °C, THF) δ = 167.3, 155.5, 61.1, 58.1, 32.3, 26.6, 25.7.
14: A room temperature solution of N,N’-dicyclohexylcarbodiimide (52 mg, 0.252 mmol) in THF (1 mL) was slowly added to a stirred suspension of BACb-LiBF4 (124 mg, 0.253 mmol) in THF (1 mL). After stirring for 10 hrs, a pale yellow solution with a white precipitate was formed. After filtration, volatiles were removed under vacuum to afford an off-white powder. Yield 137 mg (81 %). Single crystals of 14 formed readily by slow evaporation of a THF solution. mp 185-187 °C, dec; 1H NMR (300 MHz, CDCl3, 25 °C) δ = 3.67 (broad s, 4 H), 3.55 (broad m, 1 H), 3.22 (broad m, 1 H), 1.86-1.19 (broad, overlapping m, 60 H); 13C{1H} NMR (75 MHz, CDCl3, 25 °C) δ = 141.5, 134.3, 103.4, 64.8, 61.3, 57.9, 49.1, 35.8, 34.9, 32.0, 31.7, 31.5, 31.3, 30.2, 25.7, 25.6, 24.9, 24.7; 19F{1H} NMR (282 MHz, CDCl3, 25 °C) δ = 152.9. ESI MS m/z 603.5363 (603.5371 calcd for C40H67N4, [M – BF3]+).
Crystal structure determination of complexes 7, 11, 13, and 14
The Bruker X8-APEX X-ray diffraction instrument with Mo-radiation was used for data collection.[21a] All data frames were collected at low temperatures (T = 100 K) using an ω, φ-scan mode (0.5° ω-scan width, hemisphere of reflections) and integrated using a Bruker SAINTPLUS software package.[21b] The intensity data were corrected for Lorentzian polarization. Absorption corrections were performed using the SADABS program.[21c] The SIR97 was used for direct methods of phase determination, and Bruker SHELXTL software package for structure refinement and difference Fourier maps.[21d] Atomic coordinates, isotropic and anisotropic displacement parameters of all the non-hydrogen atoms of compounds were refined by means of a full matrix least-squares procedure on F2. All H-atoms were included in the refinement in calculated positions riding on the C atoms.
Acknowledgements
Financial support from the NIH (R01 GM 68825) and NSF (CHE-0924410) is gratefully acknowledged.
References
- [1] a).Hahn FE, Jahnke MC. Angew. Chem. 2008;120:3166–3216. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]; Hahn FE, Jahnke MC. Angew. Chem. Int. Ed. 2008;47:3122–3172. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]; b) Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem. Rev. 2000;100:39–91. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]; c) Arduengo AJ., III Acc. Chem. Res. 1999;32:913–921. [Google Scholar]
- [2] a).Díez-González S, Marion N, Nolan SP. Chem. Rev. 2009;109 doi: 10.1021/cr900074m. DOI: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]; b) Lin JCY, Huang RTW, Lee CS, Bhattacharyya A, Hwang WS, Lin IJB. Chem. Rev. 2009;109 doi: 10.1021/cr8005153. DOI: 10.1021/cr8005153. [DOI] [PubMed] [Google Scholar]; c) Samojłowicz C, Bieniek M, Grela K. Chem. Rev. 2009;109 doi: 10.1021/cr800524f. DOI: 10.1021/cr800524f. [DOI] [PubMed] [Google Scholar]; d) Grubbs RH. Angew. Chem. 2006;118:3845–3850. [Google Scholar]; Grubbs RH. Angew. Chem. Int. Ed. 2006;45:3760–3765. doi: 10.1002/anie.200600680. [DOI] [PubMed] [Google Scholar]; e) Marion N, Diez-Gonzalez S, Nolan SP. Angew. Chem. 2007;119:3046–3058. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; Marion N, Diez-Gonzalez S, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; f) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; g) Kamber NE, Jeong W, Waymouth RM, Pratt RC, Lohmeijer BGG, Hedrick JL. Chem. Rev. 2007;107:5813. doi: 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- [3] a).Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew. Chem. 2005;117:5851–5855. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:5705–5709. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jazzar R, Dewhurst RD, Bourg JB, Donnadieu B, Canac Y, Bertrand G. Angew. Chem. 2007;119:2957–2960. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:2899–2902. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zeng X, Frey GD, Kinjo R, Donnadieu B, Bertrand G. J. Am. Chem. Soc. 2009;131:8690–8696. doi: 10.1021/ja902051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4] a).Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Science. 2006;312:722–724. doi: 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Holschumacher D, Hrib CG, Jones PG, Tamm M. Chem. Commun. 2007:3661–3663. doi: 10.1039/b706708a. [DOI] [PubMed] [Google Scholar]
- [5] a).Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Angew. Chem. 2006;118:3568–3571. doi: 10.1002/anie.200600987. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:3448–3491. [Google Scholar]; b) Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439–441. doi: 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]
- [6] a).Dixon DA, Arduengo AJ, III, Dobbs KD, Khasnis DV. Tetrahedron Lett. 1995;36:645. [Google Scholar]; b) Denk MK, Rodezno JM, Gupta S, Lough AJ. J. Organomet. Chem. 2001;617:242. [Google Scholar]; c) Herrmann WA, Elison M, Fischer J, Köcher C, Artus GRJ. Chem. Eur. J. 1996;2:772. [Google Scholar]
- [7] a).Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. Angew. Chem. 2007;119:7182–7185. doi: 10.1002/anie.200703055. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:7052–7055. doi: 10.1002/anie.200703055. [DOI] [PubMed] [Google Scholar]; b) Back O, Kuchenbeiser G, Donnadieu B, Bertrand G. Angew. Chem. 2009;121:5638–5641. doi: 10.1002/anie.200902344. [DOI] [PubMed] [Google Scholar]; Back O, Kuchenbeiser G, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2009;48:5530–5533. doi: 10.1002/anie.200902344. [DOI] [PubMed] [Google Scholar]; c) Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. J. Am. Chem. Soc. 2007;129:14180–14181. doi: 10.1021/ja077296u. [DOI] [PubMed] [Google Scholar]
- [8] a).Kuhn N, Steimann M, Weyers G. Z Naturforsch B. 1999;54:427–433. [Google Scholar]; b) Duong HA, Tekavec TN, Arif AM, Louie J. Chem. Commun. 2004:112–113. doi: 10.1039/b311350g. [DOI] [PubMed] [Google Scholar]; c) Tudose A, Demonceau A, Delaude L. J. Organomet. Chem. 2006;691:5356–5365. [Google Scholar]; d) Ishiguro K, Hirabayashi K, Nojima T, Sawaki Y. Chem Lett. 2002:796–797. [Google Scholar]; e) Schmidt A, Merkel L, Eisfeld W. Eur. J. Org. Chem. 2005:2124–2130. [Google Scholar]; f) Holbrey JD, Reichert WM, Tkatchenko I, Bouajila E, Walter O, Tommasi I, Rogers RD. Chem. Commun. 2003:28–29. doi: 10.1039/b211519k. [DOI] [PubMed] [Google Scholar]; g) Tommasi I, Sorrentino F. Tetrahedron Lett. 2006;47:6453–6456. [Google Scholar]; h) Delaude L, Demonceau A, Wouters J. Eur. J. Inorg. Chem. 2009:1882–1891. [Google Scholar]
- [9] a).Winberg HE, Coffman DD. J. Am. Chem. Soc. 1965;87:2776–2777. [Google Scholar]; b) Schössler W, Regitz M. Chem. Ber. 1974;107:1931–1948. [Google Scholar]; c) Sheldrick WS, Schönberg A, Singer E, Eckert P. Chem. Ber. 1980;113:3605–3609. [Google Scholar]; d) Krasuski W, Nikolaus D, Regitz M. Liebigs Ann. Chem. 1982:1451–1465. [Google Scholar]; e) Kuhn N, Bohnen H, Henkel G. Z. Naturforsch., Teil B. 1994;49:1473–1480. [Google Scholar]; f) Küçükbay H, Cetinkaya E, Durmaz R. Arzneim. Forsch. 1995;45-2:1331–1334. [PubMed] [Google Scholar]; g) Enders D, Breuer K, Runsink J, Teles JH. Liebigs Ann. 1996:2019–2028. [Google Scholar]; h) Kuhn N, Niquet E, Steimann M, Walker I. Z Naturforsch B. 1999;54:1181–1187. [Google Scholar]; i) Faust R, Göbelt B. Chem. Commun. 2000:919–920. [Google Scholar]; j) Küçükbay H, Durmaz R, Orhan E, Günal S. Farmaco. 2003;58:431–437. doi: 10.1016/S0014-827X(03)00068-5. [DOI] [PubMed] [Google Scholar]; k) Nyce GW, Csihony S, Waymouth RM, Hedrick JL. Chem. Eur. J. 2004;10:4073–4079. doi: 10.1002/chem.200400196. [DOI] [PubMed] [Google Scholar]
- [10].Kuhn N, Steimann M, Weyers G, Henkel G. Z Naturforsch B. 1999;54:434–440. [Google Scholar]
- [11] a).Regitz M, Hocker J. Synthesis. 1970:301–302. [Google Scholar]; b) Regitz M, Hocker J, Schössler W, Weber B, Liedhegener A. Liebigs Ann. Chem. 1971;748:1–19. [Google Scholar]; c) Hoffmann RW, Hagenbruch B, Smith DM. Chem. Ber. 1977;110:23–36. [Google Scholar]; d) Enders D, Breuer K, Runsink J, Teles JH. Liebigs Ann. Chem. 1996:2019–2028. [Google Scholar]; e) Enders D, Breuer K, Kallfass U, Balensiefer T. Synthesis. 2003:1292–1295. [Google Scholar]; f) Liu MF, Wang B, Cheng Y. Chem. Commun. 2006:1215–1217. doi: 10.1039/b517700f. [DOI] [PubMed] [Google Scholar]; g) Cheng Y, Liu MF, Fang DC, Lei XM. Chem-Eur J. 2007;13:4282–4292. doi: 10.1002/chem.200601482. [DOI] [PubMed] [Google Scholar]; h) Zhu Q, Liu MF, Wang B, Cheng Y. Org. Bioml. Chem. 2007;5:1282–1286. doi: 10.1039/b701326d. [DOI] [PubMed] [Google Scholar]; i) Ma YG, Cheng Y. Chem. Commun. 2007:5087–5089. doi: 10.1039/b712098b. [DOI] [PubMed] [Google Scholar]; j) Cheng Y, Ma Y-G, Wang X-R, Mo J-M. J. Org. Chem. 2009;74:850–855. doi: 10.1021/jo802289s. [DOI] [PubMed] [Google Scholar]; k) Zhang J-H, Cheng Y. Org. Biomol. Chem. 2009;7:3264–3270. doi: 10.1039/b904575a. [DOI] [PubMed] [Google Scholar]
- [12].Delaude L. Eur. J. Inorg. Chem. 2009:1681–1699. [Google Scholar]
- [13] a).Voutchkova AM, Appelhans LN, Chianese AR, Crabtree RH. J. Am. Chem. Soc. 2005;127:17624–17625. doi: 10.1021/ja056625k. [DOI] [PubMed] [Google Scholar]; b) Voutchkova AM, Feliz M, Clot E, Eisenstein O, Crabtree RH. J. Am. Chem. Soc. 2007;129:12834–12846. doi: 10.1021/ja0742885. [DOI] [PubMed] [Google Scholar]; c) Tudose A, Delaude L, Andre B, Demonceau A. Tetrahedron Lett. 2006;47:8529–8533. [Google Scholar]
- [14] a).Zhou H, Zhang WZ, Liu CH, Qu JP, Lu XB. J. Org. Chem. 2008;73:8039–8044. doi: 10.1021/jo801457r. [DOI] [PubMed] [Google Scholar]; b) Kayaki Y, Yamamoto M, Ikariya T. Angew. Chem. 2009;121:4258–4261. doi: 10.1002/anie.200901399. [DOI] [PubMed] [Google Scholar]; Kayaki Y, Yamamoto M, Ikariya T. Angew. Chem. Int. Ed. 2009;48:4194–4197. doi: 10.1002/anie.200901399. [DOI] [PubMed] [Google Scholar]; c) Riduan SN, Zhang Y, Ying JY. Angew. Chem. 2009;121:3372–3375. doi: 10.1002/anie.200806058. [DOI] [PubMed] [Google Scholar]; Riduan SN, Zhang Y, Ying JY. Angew. Chem. Int. Ed. 2009;48:3322–3325. doi: 10.1002/anie.200806058. [DOI] [PubMed] [Google Scholar]
- [15].Komatsu K, Kitagawa T. Chem. Rev. 2003;103:1371–1427. doi: 10.1021/cr010011q. [DOI] [PubMed] [Google Scholar]
- [16] a).For recent examples see: Kelly RA, Clavier H, Giudice S, Scott NM, Stevens ED, Bordner J, Samardjiev I, Hoff CD, Cavallo L, Nolan SP. Organometallics. 2008;27:202–210.; Furstner A, Alcarazo M, Krause H, Lehmann CW. J. Am. Chem. Soc. 2007;129:12676–12677. doi: 10.1021/ja076028t.; Frey GD, Rentzsch CF, von Preysing D, Scherg T, Muhlhofer M, Herdtweck E, Herrmann WA. J. Organomet. Chem. 2006;691:5725–5738..
- [17].Kuchenbeiser G, Donnadieu B, Bertrand G. J. Organomet. Chem. 2008;693:899–904. doi: 10.1016/j.jorganchem.2007.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lavallo V, Canac Y, DeHope A, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2005;44:7236–7239. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].CCDC 742786 (7), 742787 (11), 742788 (13), and 742789 (14) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre doi: 10.6028/jres.101.038. via www.ccdc.cam.ac.uk/data_request/cif..
- [20].Lavallo V, Ishida Y, Donnadieu B, Bertrand G. Angew. Chem. 2006;118:6804–6807. doi: 10.1002/anie.200602701. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lavallo V, Ishida Y, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2006;45:6652–6655. doi: 10.1002/anie.200602701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21] a).APEX2. version 1.0-22 ed Bruker AXS Inc.; Madison, Wisconsin, USA: 2004. [Google Scholar]; b) SAINT. version V7.06A ed Bruker AXS Inc.; Madison, Wisconsin, USA: 2003. [Google Scholar]; c) SADABS. version 2004/1 ed Bruker AXS Inc.; Madison, Wisconsin, USA: 2004. [Google Scholar]; d) SHELXTL. version 6.14 ed Bruker AXS Inc.; Madison, Wisconsin, USA: 2003. [Google Scholar]