

Abstract
In primary bovine adrenal glomerulosa cells, the signaling enzyme phospholipase D (PLD) is suggested to mediate priming, the enhancement of aldosterone secretion after pretreatment with and removal of angiotensin II (AngII), via the formation of persistently elevated diacylglycerol (DAG). To further explore PLD’s role in priming, glomerulosa cells were pretreated with an exogenous bacterial PLD. Using this approach, phosphatidic acid (PA) is generated on the outer, rather than the inner, leaflet of the plasma membrane. Although PA is not readily internalized, the PA is nonetheless rapidly hydrolyzed by cell-surface PA phosphatases to DAG, which efficiently flips to the inner leaflet and accesses the cell interior. Pretreatment with bacterial PLD resulted in priming upon subsequent AngII exposure, supporting a role of DAG in this process, because the increase in DAG persisted after exogenous PLD removal. To determine the PLD isoform mediating aldosterone secretion, and presumably priming, primary glomerulosa cells were infected with adenoviruses expressing GFP, PLD1, PLD2, or lipase-inactive mutants. Overexpressed PLD2 increased aldosterone secretion by approximately 3-fold over the GFP-infected control under basal conditions, with a significant enhancement to about 16-fold over the basal value upon AngII stimulation. PLD activity was also increased basally and upon stimulation with AngII. In contrast, PLD1 overexpression had little effect on aldosterone secretion, despite the fact that PLD activity was enhanced. In both cases, the lipase-inactive PLD mutants showed essentially no effect on PLD activity or aldosterone secretion. Our results suggest that PLD2 is the isoform that mediates aldosterone secretion and likely priming.
Lipid signals generated by enzyme phospholipase D (PLD) can mediate priming, strengthening the hypothesis regarding the involvement of endogenous PLD in the priming process.
Aldosterone secretion is regulated primarily by angiotension II (AngII), but also by other agents, such as ACTH and elevated serum potassium levels. Multiple signaling pathways have been suggested to mediate AngII-induced aldosterone secretion. After AngII binds to its receptor, the complex activates a phosphoinositide-specific phospholipase C, which hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate two second messengers, inositol 1,4,5-trisphosphate and diacylglycerol (DAG) (reviewed in Refs. 1 and 2). Inositol 1,4,5-trisphosphate then initiates aldosterone secretion by eliciting a transient increase in cytosolic Ca2+ and Ca2+/calmodulin-dependent protein kinase activation, whereas DAG stimulates protein kinase C (PKC) activity, which has been suggested by some laboratories to underlie sustained aldosterone secretion from bovine adrenal glomerulosa cells (1,3,4). AngII also increases Ca2+ influx, and this enhancement is essential for continued aldosterone secretion as well as PKC activity (5). AngII also activates phospholipase D (PLD), and evidence from our laboratory suggests a role for PLD in AngII-mediated aldosterone secretion (3,6). PLD has the ability to hydrolyze phosphatidylcholine to generate phosphatidic acid (PA), and this PA can be converted to DAG by lipid phosphate phosphatases (LPP or lipin 1–3). Therefore, one possible explanation for the involvement of PLD in aldosterone secretion could be via its ability to form PKC-activating DAG. Mammalian PLD consists of two classic isoforms, namely PLD1 and PLD2, which have approximately 50% overall sequence homology (reviewed in Ref. 7), and bovine adrenal glomerulosa cells express both PLD1 and PLD2 (8). The isoform that mediates AngII-elicited aldosterone secretion in glomerulosa cells, however, is as yet unknown.
Using exogenous bacterial PLD as a tool, our laboratory has demonstrated that PLD-generated PA can be converted to DAG by endogenous LPP to elicit secretion (3). It should be noted that although exogenous PLD alone has only a moderate effect on the aldosterone secretory response, addition of the Ca2+ channel agonist BAY K8644 results in secretion that is essentially comparable to that elicited by AngII (3), consistent with the idea that sustained aldosterone secretion requires both a DAG/PKC and a Ca2+ influx signal, with the two signals interacting (reviewed in Ref. 5). The idea that PLD also plays a role in AngII-induced priming in primary bovine glomerulosa cells formed the basis for the current study. Priming is a type of cellular memory in which pretreatment with AngII, followed by washout of the hormone, sensitizes cells to respond to agents that increase Ca2+ influx [e.g. a second AngII exposure (6), the Ca2+ channel agonist BAY K8644 (6,9), or elevated [K+] (10)] with a greater aldosterone secretory response. Data from our laboratory indicate that the critical window for the observation of priming after AngII removal in primary cultures of bovine adrenal glomerulosa cells is less than 60 min, because extending the washout period from 50 to 60 min results in a second AngII secretory response no different from the first (11). Our previous results also suggest that PLD may underlie priming through triggering the formation of persistent DAG (reviewed in Ref. 11). Thus, both AngII and phorbol 12,13-dibutyrate (PDBu), which each elicit sustained PLD activation, are able to both elicit a persistent increase in DAG after washout and prime glomerulosa cells to respond with enhanced secretion to a subsequent AngII exposure (11). In addition, inhibition of PLD signaling with the primary alcohol 1-butanol, which reduces AngII-stimulated increases in both PA and DAG (12), inhibits the ability of AngII pretreatment to enhance the subsequent aldosterone secretory response to a second AngII exposure or an elevated potassium concentration (11). Nevertheless, PLD activation returns to basal levels by 30 min after washout (11), indicating that the persistent increase in DAG levels with an initial exposure to agonist is not the result of maintained PLD activity after agent removal. Providing a potential link between PLD, priming, and hypertension in vivo is the demonstration that vascular smooth muscle cells from spontaneously hypertensive rats exhibit greater agonist-induced PLD activity than those derived from normotensive control rats (13,14).
In this report, we sought to determine the role of PLD and PLD-generated DAG in priming, using assays to measure the aldosterone secretory rate, PLD activity, and radiolabeled DAG levels. Pretreatment with exogenous PLD and AngII triggered similar enhanced aldosterone secretion in response to a subsequent AngII treatment. Both AngII and exogenous bacterial PLD elicited an increase in DAG level, with a significant elevation that persisted for 30 min after each was removed. We also investigated which PLD isoform mediated the acute aldosterone secretory response. We showed that overexpressed PLD2, but not PLD1, mediated acute AngII-induced aldosterone secretion, suggesting the possibility that PLD2 promotes priming of bovine adrenal glomerulosa cells.
Materials and Methods
Bovine adrenal glomerulosa cell preparation and culture
Bovine adrenal glomerulosa cells were isolated as reported elsewhere (15). Briefly, glomerulosa cell slices prepared from near-term fetal adrenal glands obtained from a local meat-packing plant were incubated with collagenase, followed by dispersal of adrenal glomerulosa cells using mechanical agitation. Freshly isolated adrenal glomerulosa cells were then cultured overnight in Falcon Primaria dishes (Becton Dickinson Labware, Lincoln Park, NJ) in a DMEM/Ham’s F12 medium (1:1) containing: 10% horse serum (vol/vol), 2% fetal bovine serum (vol/vol), ascorbate (100 μm), α-tocopherol (1.2 μm), Na2SeO3 (0.05 μm), butylated hydroxyanisole (50 μm), metyrapone (5 μm), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (0.25 μg/ml). After replacement of the serum-containing medium with serum-free medium [containing 0.2% BSA with or without adenoviruses and/or 5 μCi/ml [3H]oleic acid (see below)], the cells were incubated for an additional 20–24 h before use.
Aldosterone secretion in a semiperfusion system
Cultured glomerulosa cells incubated for 20–24 h in serum-free medium were rinsed two to three times with bicarbonate-buffered Krebs-Ringer buffer (KRB) containing 2.5 mm sodium acetate (KRB+) and were allowed to equilibrate in this medium for approximately 20 min. KRB+ was added for an additional 10 min, and this medium was removed (represents the 30-min control). Cells were incubated with the appropriate agents for the indicated times, with medium collected and replaced every 10–20 min. At the end of the experiment, cells were solubilized in 0.3 m NaOH to determine protein content using the Bio-Rad assay (Bio-Rad Laboratories, Hercules, CA) with BSA as standard. The supernatants were stored frozen until aldosterone was assayed using a solid-phase RIA kit (Diagnostic Products, Los Angeles, CA).
Measurement of radiolabeled DAG levels
Cells were prelabeled for 2 h with 5 μCi/ml [3H]myristic acid (dried under nitrogen and resuspended by sonication) and pretreated for 30 min in KRB+, followed by incubation in the presence or absence of 10 nm AngII or 0.1 IU/ml PLD from Streptomyces chromofuscus (Sigma, St. Louis, MO) for 30 min. After washing three times with KRB+, cells were incubated with KRB+ for another 30 min with or without AngII or PLD. Reactions were terminated by the addition of 0.2% sodium dodecyl sulfate (SDS) and lipids collected from the upper phase of a chloroform/methanol/aqueous extraction (1:1:0.8 vol/vol), as described elsewhere (2). Samples were resuspended in chloroform/methanol and spotted onto heat-activated silica gel 60 thin-layer chromatography plates. After development in a mobile phase of benzene/ethyl acetate (7:3 vol/vol), radiolabeled lipids were visualized with autoradiography using En3Hance. The spots corresponding to 1,2-DAG, identified by comigration with an authentic standard and visualized with iodine vapor, were cut, placed in liquid scintillation fluid, and counted in a liquid scintillation spectrometer.
Production of virus
Adenovirus particles containing wild-type human PLD1 (hPLD1), mouse PLD2 (mPLD2), and lipase-inactive mutants were produced based on the protocol from B. Vogelstein’s laboratory (http://www.coloncancer.org/adeasy.htm). The lipase-inactive version was made by mutation of a lysine (K898 in PLD1 and K758 in PLD2) in the catalytic site to arginine (16). Virus particles were purified by cesium chloride ultracentrifugation and were dialyzed with storage buffer [10% glycerol, 10 mm Tris base, 0.9% NaCl (pH 8.1)] every hour for three times. Purified viral titer was determined using OD260.
Overexpression of PLD isoforms
Primary bovine adrenal glomerulosa cells were infected with adenovirus possessing hPLD1, lipase-inactive hPLD1 (hPLD1-LI), mPLD2, lipase-inactive mPLD2 (mPLD2-LI), and pShuttle-CMV green fluorescent protein (GFP) for 4 h in serum-free medium at a multiplicity of infection (MOI) of 25. After aspirating the medium, cells were re-fed with serum-free medium for another 16–20 h before processing for Western analysis or PLD activity assay.
Western blot analysis
After infection with the various adenoviruses and incubation with serum-free medium for 16–20 h, primary adrenal glomerulosa cells were processed with hot lysis buffer [0.1875 m Tris-HCl (pH 8.5), 3% SDS, 1.5 mm EGTA], an aliquot was removed for protein determination, and glycerol (10% final concentration), β-mercaptoethanol (5% final concentration), and bromphenol blue were added to constitute Laemmli buffer (17). Protein (15 μg) was loaded onto a 10% SDS gel and separated by electrophoresis. Resolved proteins were transferred to a polyvinylidene difluoride membrane, and the blots were incubated with anti-hemagglutinin (anti-HA) (catalog item 2362 from Cell Signaling Technology, Danvers, MA), anti-PLD2 (catalog item 44-324 from Invitrogen, Camarillo, CA), or anti-actin (sc-1616 from Santa Cruz Biotechnology, Santa Cruz, CA) antibody, washed extensively, and incubated with alkaline phosphatase-conjugated goat antimouse, goat antirat, or goat antirabbit IgG. The bands were visualized using enhanced chemifluorescence (GE Healthcare, Little Chalfont, UK) and a Storm PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
PLD activity assay
PLD activation was measured as an increase in the levels of radiolabeled phosphatidylethanol (PEt) as described elsewhere (2). Briefly, primary adrenal glomerulosa cells infected with hPLD1, hPLD1-LI, mPLD2, mPLD2-LI, or GFP adenoviruses were labeled in serum-free medium containing 5 μCi/ml [3H]oleic acid for 16–20 h. The prelabeled cells were then equilibrated for 30 min in KRB+ before stimulation with the agents of interest (control medium or 10 nm AngII) for 30 min in the presence of 0.5% ethanol. Reactions were terminated by the addition of 0.2% SDS containing 5 mm EDTA, and phospholipids were extracted into chloroform/methanol containing acetic acid (1:2:0.04 vol/vol). After drying under N2, the samples were resuspended in chloroform/methanol containing also 25–50 μg PEt and PA per sample and spotted onto heat-activated silica gel 60 thin-layer chromatography plates (0.25 mm thickness aluminum backed with concentrating zone). Phospholipids were separated using a mobile phase consisting of the upper phase of a solvent system of ethyl acetate/isooctane/acetic acid/water (13:2:3:10 vol/vol) and visualized with autoradiography using En3Hance (PerkinElmer, Waltham, MA). Spots corresponding to PA and PEt, identified by comigration with authentic standards visualized with iodine vapor, were cut, placed in liquid scintillation fluid, and counted.
Immunocytochemistry
Primary bovine glomerulosa cells were plated on 18 × 18 mm coverslips. After infection with mPLD2 adenovirus as above (or no infection for examination of endogenous PLD2 localization), cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100, and blocked with 5% BSA and normal goat serum. The cells were then immunostained using primary antibodies against PLD2 or HA, followed by fluorescent dye-conjugated secondary antibodies. Fixed cells were then mounted with ProLong Gold Anti-fade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) and visualized with a confocal microscope (Zeiss, Jena, Germany). All experiments were performed at least three times with similar results.
Statistical analysis
Experimental values were statistically analyzed by ANOVA using the program Instat (GraphPad Software, San Diego, CA) with a Student-Newman-Keuls post hoc test.
Results
Exogenous bacterial PLD-mediated priming in bovine adrenal glomerulosa cells
Inhibition of PLD-mediated signal generation using 1-butanol results in decreased AngII-induced priming of glomerulosa cells to a second AngII exposure or to an elevated extracellular potassium concentration, suggesting that PLD plays a role in priming (11). However, it has recently been shown that 1-butanol-mediated inhibition of a cellular response does not always translate into an involvement of PLD in that response (18). In this study, the role of PLD-generated signals in aldosterone secretion and priming was investigated using an exogenous bacterial PLD (Fig. 1). Primary bovine glomerulosa cells were pretreated with 0.1 IU/ml exogenous bacterial PLD or 10 nm AngII for 30 min and supernatants collected for assay of aldosterone secretion. As shown in Fig. 1, aldosterone secretion was enhanced to 18.7 ± 2.9% of the maximal aldosterone secretory response by PLD pretreatment and to 56.4 ± 5.1% of maximal aldosterone secretion by AngII pretreatment. After removal of these agents and a 40-min washout period, a subsequent exposure to AngII for another 30 min resulted in enhanced aldosterone secretion with both pretreatments to a similar degree, that is, 96.6 ± 5.9% of maximal aldosterone secretion in the PLD pretreatment group and 93.0 ± 7.6% of the maximal aldosterone secretory response in the AngII-pretreated group. These results suggest that, like AngII, exogenous bacterial PLD can also mediate priming in primary bovine adrenal glomerulosa cells.
Figure 1.

Exogenous bacterial PLD primes glomerulosa cells to respond to a subsequent exposure to AngII with enhanced secretion. Primary cultures of bovine adrenal glomerulosa cells in serum-free medium for 20–24 h were incubated for a 30-min control period. Cells were then treated with 10 nm AngII (□) or 0.1 IU/ml PLD (○) as indicated, followed by a 40-min washout and exposure to 10 nm AngII as illustrated. Aldosterone secretion was assayed using a solid-phase RIA kit. Values represent the means ± sem of three independent experiments, expressed as the percent maximal response. ***, P < 0.001; **, P < 0.01; *, P < 0.05 vs. control (shown at time = 30 min); ††, P < 0.01; †††, P < 0.001 vs. the corresponding time point during the first AngII treatment. The maximal responses, determined in each experiment as the maximum aldosterone secretory rate and occurring in one experiment during the second AngII exposure and in two during the AngII exposure after PLD pretreatment, were 57.5, 115.9, and 27.3 pg/ml · min.
Exogenous PLD increases radiolabeled DAG levels in bovine adrenal glomerulosa cells
The mechanism underlying the development of this priming response is unknown. It has been proposed that the AngII priming effect is related to the induction of a persistent association of PKC with the plasma membrane (19). Physiologically, PKC association with the plasma membrane is promoted by DAG (reviewed in Ref. 20), suggesting the possibility that a persistent increase in DAG levels could retain PKC at the membrane to underlie priming. We postulated that this persistent DAG is derived from PLD-mediated phosphatidylcholine hydrolysis upon AngII stimulation, based upon our finding that myristate-containing DAG persists after removal of AngII and myristate is preferentially incorporated into phosphatidylcholine (11), the substrate of PLD (20), in primary bovine glomerulosa cells. To determine whether persistent myristate-containing DAG is a mechanism by which exogenous PLD induces priming, [3H]myristate-labeled DAG levels were measured upon treatment with or without exogenous PLD or AngII for 30 min, followed by washout of the agent and an additional 30-min incubation. As shown in Fig. 2A, AngII treatment increased radiolabeled DAG levels, and this increase persisted after AngII removal, consistent with our previous findings (11). Similarly, exogenous PLD significantly increased DAG levels, and after washout and a subsequent 30-min incubation, radiolabeled DAG levels remained significantly elevated relative to the control value (Fig. 2B).
Figure 2.

AngII and exogenous bacterial PLD increase [3H]myristate-labeled DAG that persists after agent removal. Primary cultures of bovine adrenal glomerulosa cells in serum-free medium for 20–24 h were labeled for 2 h in KRB+ containing 5 μCi/ml [3H]myristic acid. The KRB+ was removed and replaced with KRB+ containing no additions (control, Con) or with 10 nm AngII (A) or 0.1 IU/ml PLD (B) for 30 min as shown. Adrenal glomerulosa cells were then washed three times with KRB+ before incubation for an additional 30 min with KRB+ alone or KRB+ containing 10 nm AngII (A) or 0.1 IU/ml PLD (B) as indicated. Radiolabeled DAG was then extracted into chloroform/methanol, separated by thin-layer chromatography, and quantified as described in Materials and Methods. Values represent the means ± sem of 10 samples from five separate experiments. ***, P < 0.001; *, P < 0.05 vs. control (without agent in both the first and second 30-min time periods).
Overexpressed PLD1 and PLD2 increase PLD activity in bovine adrenal glomerulosa cells
Based on studies in the literature suggesting a role for PLD2 in AngII-induced PLD activation in SHR spontaneously hypertensive rats (14), we hypothesized that PLD2 was the isoform that mediates acute aldosterone secretion. To identify the isoform underlying acute AngII-elicited aldosterone secretion in bovine adrenal glomerulosa cells, primary cultures were infected with hPLD1, mPLD2, or lipase-inactive mutants, all with an HA tag at their N terminus. Infection efficiency was monitored by GFP expression, which was observed in about 80–90% of the cells. As shown in Fig. 3A, all of the constructs were overexpressed successfully as measured by Western analysis using an anti-HA antibody. Western analysis using an anti-PLD2 antibody demonstrated an approximate 50-fold overexpression of wild-type PLD2 and about a 25-fold increase in the lipase-inactive mutant relative to endogenous PLD2 (Fig. 3B). Overexpressed PLD2 (Fig. 3, C and D) and PLD1 (Fig. 3, E and F) both increased the levels of radiolabeled PEt, a marker of PLD activity (21), when compared with control, whereas neither lipase-inactive mutant affected PEt levels.
Figure 3.

Overexpressed PLDs increase PLD activity under both basal conditions and upon AngII stimulation. Primary bovine adrenal glomerulosa cells were infected with adenoviruses expressing GFP, hPLD1, PLD1-LI, mPLD2, or PLD2-LI for 4 h in serum-free medium at an MOI of 25. After aspiration of the medium containing virus, cells were re-fed with serum-free medium (containing [3H]oleate) for another 16–20 h before processing. A, Immunoblots (IB) for GFP and HA-tagged PLD1, PLD1-LI, PLD2, and PLD2-LI verify infection and overexpression of the appropriate proteins; B, immunoblots of cells infected with GFP, PLD2, or PLD2-LI were visualized with an anti-PLD2 antibody and quantified to determine the degree of PLD2 overexpression; C, PLD activity was assayed after PLD1 and PLD1-LI overexpression; D, cumulative data from three experiments performed in duplicate; E, PLD activity was assayed after PLD2 and PLD2-LI overexpression; F, cumulative data from three experiments performed in duplicate. Values represent the means ± sem of three separate experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 vs. GFP.
Overexpressed PLD2, but not PLD1, mediates acute aldosterone secretion upon AngII stimulation of bovine adrenal glomerulosa cells
Because both overexpressed PLD1 and PLD2 increased PLD activity as measured by elevated [3H]PEt formation in [3H]oleate-prelabeled primary bovine glomerulosa cells, aldosterone secretion was monitored upon PLD1 or PLD2 overexpression. As illustrated in Fig. 4A, PLD2 overexpression induced a slight but statistically insignificant increase in basal aldosterone secretion (of approximately 3-fold above the GFP control). AngII itself increased aldosterone secretion about 7-fold compared with the control level. In combination with AngII stimulation for 30 min, overexpression of PLD2 resulted in enhanced aldosterone secretion to a value about 16-fold greater than the GFP control (P < 0.05). On the other hand, overexpression of the PLD2-LI mutant had no effect on aldosterone secretion by itself or upon AngII stimulation (Fig. 4A).
Figure 4.
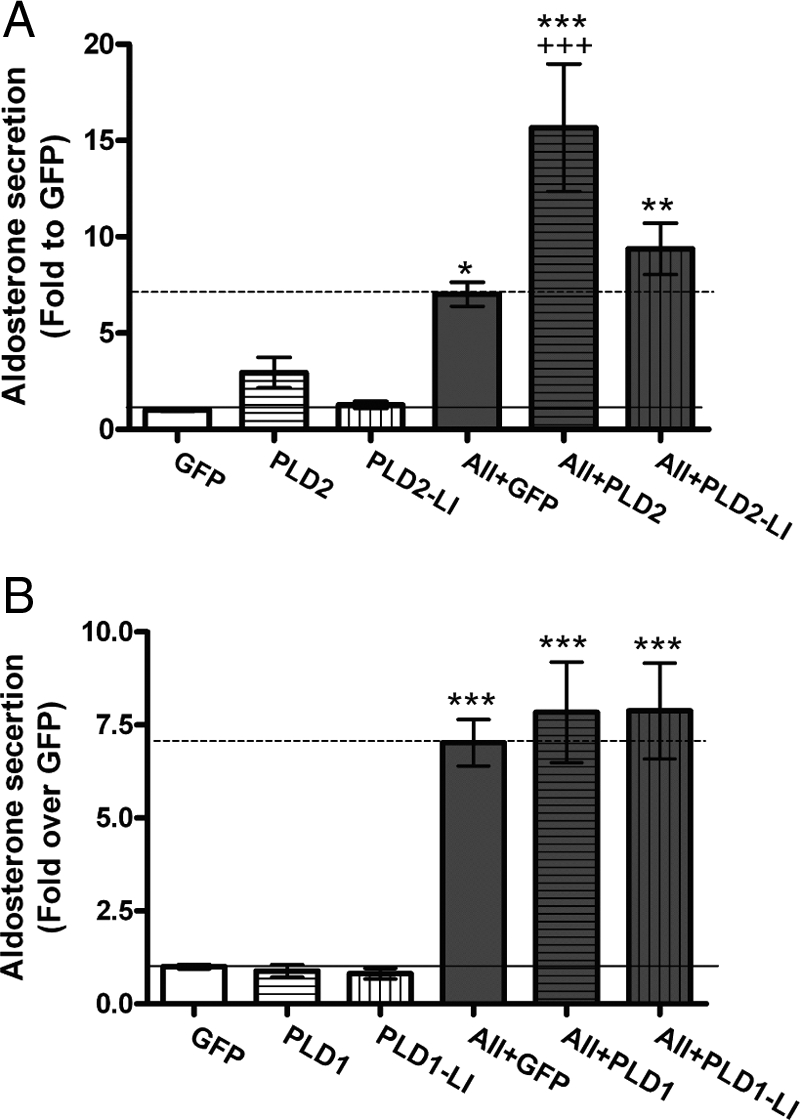
Overexpressed PLD2, but not PLD1, induces aldosterone secretion from primary bovine glomerulosa cells upon AngII exposure. Primary bovine adrenal glomerulosa cells were infected with adenoviruses expressing GFP, mPLD2 or PLD2-LI (A) or GFP, hPLD1, or PLD1-LI (B) for 4 h in serum-free medium at an MOI of 25, followed by incubation with serum-free medium for 16–20 h. Cells were then treated with or without 10 nm AngII (AII) for 30 min as indicated. Aldosterone secretion was assayed using a solid-phase RIA kit. Values are expressed as the fold change relative to the GFP control and represent the means ± sem of six samples from three separate experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 vs. GFP and +++, P < 0.001 vs. AII + GFP. Please note the difference in scale between A and B.
In contrast, overexpression of PLD1 did not increase aldosterone secretion in either the basal or AngII treatment group (Fig. 4B). Thus, although both overexpressed PLD1 and PLD2 increase PLD activity in bovine glomerulosa cells, only PLD2 promotes increased acute aldosterone secretion.
Overexpressed PLD2 and endogenous PLD2 show similar localization
One potential caveat of overexpression studies is that overexpressed proteins can localize to areas to which they have limited or no access under normal conditions. To address the possibility that overexpressed PLD2 mediated aldosterone secretion by generating PA/DAG at a nonphysiological subcellular localization, immunocytochemistry was performed to investigate the distribution of endogenous and overexpressed PLD2. Staining with a PLD2 antibody showed that endogenous PLD2 localized largely in a cytoplasmic and perinuclear vesicular pattern (Fig. 5A); upon treatment with AngII, endogenous PLD2 exhibited a similar distribution with some additional localization to plasma membrane processes (Fig. 5B). Staining with an anti-HA antibody was used to distinguish the recombinant protein from endogenous PLD2 because an HA tag was included in the N terminus of the PLD2 gene in the adenoviral construct. As with endogenous PLD2, overexpressed PLD2 also localized basally in a cytoplasmic and perinuclear vesicular pattern (Fig. 5C), and treatment with AngII for 30 min induced the movement of a small portion of the exogenous PLD2 to plasma membrane processes (Fig. 5D). Staining of AngII-stimulated PLD2-overexpressing cells with an anti-PLD2 antibody to detect both endogenous and overexpressed PLD2 showed similar cytoplasmic, perinuclear vesicular, and plasma membrane process localization (Fig. 5E).
Figure 5.

Overexpressed PLD2 localizes similarly to endogenous PLD2. Primary bovine adrenal glomerulosa cells were uninfected (A and B) or infected with adenoviruses expressing mPLD2 (C–E) for 4 h in serum-free medium at an MOI of 25, followed by incubation in serum-free medium for 16–20 h. After treatment with or without 10 nm AngII for 30 min as indicated, cells were fixed and incubated with primary antibodies as indicated. Cells were then incubated with a C3-labeled secondary antibody and imaged by confocal immunofluorescent microcopy. The upper part of E represents a magnification of the region shown in D as indicated. Figures are representative of results from three separate experiments.
Discussion
The experiments presented here were designed to determine whether exogenous bacterial PLD could induce priming in bovine glomerulosa cells, whether the mechanism of such priming involved a persistent increase in DAG, and which isoform of PLD mediated acute aldosterone secretion. Using exogenous bacterial PLD as a tool, we observed an induction of priming in response to a subsequent AngII exposure (Fig. 1). Indeed, after the washout of exogenous bacterial PLD or AngII, the second AngII exposure elicited similar priming in both groups (93.0 ± 7.6 vs. 96.6 ± 5.9% of maximal aldosterone secretion). Although with this approach, PA is generated within the outer leaflet of the plasma membrane rather than the inner leaflet, and PA does not readily flip-flop between leaflets, PA phosphatases present on the cell surface can hydrolyze PA to DAG, which can easily traverse the plasma membrane (22). Therefore, this result provides evidence that lipid signals generated by PLD can mediate priming and further strengthens our hypothesis about the involvement of endogenous PLD in this process. It should be noted that the initial aldosterone secretion induced by treatment with exogenous bacterial PLD is less than that elicited by AngII, because PLD provides only the DAG/PKC signal required for sustained aldosterone secretion, whereas AngII triggers both the DAG/PKC and enhanced Ca2+ influx signals (reviewed in Ref. 5).
Our previous study demonstrated that both PDBu- and AngII-induced PLD activity returned to basal levels within 30 min of removal of either agent, thus negating the likelihood of a persistent PLD activity mediating priming, because the second AngII treatment occurred after a 40-min washout of PDBu, AngII, or exogenous PLD (Fig. 1). The question remains as to how PLD mediates priming. PLD hydrolyzes phosphatidylcholine to generate PA, and this PA can be converted to DAG by LPP or lipin. Our previous studies have shown that there is a persistent increase in DAG levels after AngII (or PDBu) removal but not after the removal of carbachol, which does not induce priming (23). We hypothesized that PLD mediates priming through a persistent elevation in DAG. If so, the DAG derived from PLD activity is likely metabolized differently from that derived from phospholipase C activity, to allow it to persist after agonist removal (1,3,4). Indeed, we have previously shown that DAG labeled with radioactive arachidonate decreases rapidly upon inhibition of AngII receptor signaling with an antagonist, whereas myristate-labeled DAG levels decline slowly (9). A possible explanation involves a known DAG kinase that preferentially phosphorylates arachidonate-containing DAG (24). Likewise, adrenal glomerulosa cells express a DAG lipase that preferentially hydrolyzes arachidonate-DAG (25,26), suggesting the likelihood that DAG derived from hydrolysis of phosphoinositides, which are enriched in arachidonic acid (9,27), may be more rapidly metabolized than DAG derived from phosphatidylcholine via PLD activity. Although this idea awaits further investigation, our results in this study demonstrate that both AngII and exogenous bacterial PLD induced a persistent increase in [3H]myristate-labeled DAG (Fig. 2). These findings support our idea that a persistent increase in DAG levels underlies AngII-induced priming.
A novel feature of the results presented here is that overexpressed PLD2 but not PLD1 mediated aldosterone secretion from primary bovine adrenal glomerulosa cells. Although both overexpressed PLD1 and PLD2 increased the production of [3H]PEt, a specific marker of PLD activity, PLD1 did not alter aldosterone secretion either under basal conditions or upon AngII exposure (Fig. 4B). In contrast, overexpressed PLD2 increased AngII-stimulated aldosterone secretion in PLD2-overexpressing cells and slightly, but not significantly, enhanced basal secretion. Importantly, overexpression of mouse PLD2 did not change the localization of endogenous (bovine) PLD2. This report is the first to distinguish the PLD isoform that mediates acute aldosterone secretion from primary bovine adrenal glomerulosa cells.
There is evidence to suggest the involvement of changes in PLD2 activity in the hypertensive response to AngII. For example, in rabbit vascular smooth muscle cells (VSMC), overexpression of the PLD2-LI mutant, but not the PLD1-LI mutant, markedly reduces AngII-stimulated PLD activity (28). However, in our current study, the PLD2-LI mutant did not function in a dominant-negative manner. The reason for this is unclear but could be related to disparities in PLD2 regulation in different cell types. For example, if PLD2, and not a regulator of its activity, is rate limiting in a particular cell type, then overexpression of the PLD2-LI mutant may not be able to prevent endogenous PLD2 activity/activation because of the surfeit of regulators available. Second, in VSMC expressing wild-type PLD2 but not wild-type PLD1, AngII-stimulated PLD activity was enhanced, suggesting that PLD2 is the major isoform activated by AngII in VSMC (29). Together, these data suggest that PLD2 is the main signaling isoform involved in AngII-mediated PLD activity in these cells (14). Moreover, AngII stimulates PLD2 through PKCζ activation in VSMC (30). A link between PKCζ and PLD2 activation could also provide a mechanism by which exogenous bacterial PLD could elicit endogenous PLD2 activity: S. chromofuscus PLD elevates ceramide levels in some cell types (31,32). Because ceramide activates PKCζ (33), the increased ceramide levels could result in enhanced activity of endogenous PLD2 via PKCζ. PLD, in turn, plays a role in VSMC functions, such as adhesion, spreading, and hypertrophy, processes associated with the pathogenesis of atherosclerosis and malignant hypertension (30). Similarly, our study in bovine glomerulosa cells suggests an important role for PLD2 in regulating acute AngII-induced secretion of aldosterone, the hormone that controls sodium, and thus water, balance in the body.
Although the importance of priming in vivo has not yet been demonstrated, there are suggestions in the literature that such a mechanism might regulate aldosterone secretion and contribute to cardiovascular pathologies, such as hypertension. For example, Romero et al. (14) have shown that in the VSMC of spontaneously hypertensive rats, AngII induces a greater increase in PLD activity than in VSMCs from normotensive control rats (13,14). If a similarly enhanced PLD response to AngII were to occur in adrenal glomerulosa cells, the result would be enhanced aldosterone secretion, which could potentially contribute to the hypertension in this model. In addition, alterations in tissue production of AngII and local AngII concentrations have been hypothesized to be involved in the altered adrenal responsiveness to this hormone in some forms of hypertension (34). In vivo glomerulosa cells are likely exposed to AngII and other aldosterone secretagogues, both simultaneously and sequentially. Thus, priming in response to AngII may allow enhanced responsiveness to elevated serum potassium levels even once AngII levels decrease. This effect may represent a beneficial response, for instance, under conditions of salt restriction. On the other hand, if priming to AngII were excessive (as occurs in spontaneously hypertensive rats) or occurred under inappropriate conditions, e.g. during high salt intake (34), priming could potentially contribute to the development of hypertension. In addition, because priming is a type of cellular memory, understanding the mechanism of its generation may provide insight into other forms of memory events, for example, in the central nervous or immune systems (35).
Acknowledgments
We gratefully acknowledge the expert technical assistance of Mr. Peter M. Parker and Ms. Mariya V. George as well as the Medical College of Georgia Imaging Core Facility and the Veterans Affairs Confocal Microscopy Core Facility for assistance with two-photon immunofluorescent microscopy.
Footnotes
This work was supported by National Institutes of Health (NIH) Award HL70046 and American Heart Association Grant-in-Aid Award 0350166N (to W.B.B.) and by NIH Award GM71520 (to M.A.F.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 10, 2010
Abbreviations: AngII, Angiotensin II; DAG, diacylglycerol; GFP, green fluorescent protein; HA, hemagglutinin; hPLD1, human PLD1; hPLD1-LI, lipase-inactive hPLD1; KRB, bicarbonate-buffered Krebs-Ringer buffer; LPP, lipid phosphate phosphatase; MOI, multiplicity of infection; mPLD2, mouse PLD2; PA, phosphatidic acid; PEt, phosphatidylethanol; PDBu, phorbol 12,13-dibutyrate; PKC, protein kinase C; PLD, phospholipase D; SDS, sodium dodecyl sulfate; VSMC, vascular smooth muscle cells.
References
- Barrett PQ, Bollag WB, Isales CM, McCarthy RT, Rasmussen H 1989 Role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev 10:496–518 [DOI] [PubMed] [Google Scholar]
- Ganguly A, Davis JS 1994 Role of calcium and other mediators in aldosterone secretion from the adrenal glomerulosa cells. Pharmacol Rev 46:417–447 [PubMed] [Google Scholar]
- Bollag WB, Barrett PQ, Isales CM, Liscovitch M, Rasmussen H 1990 A potential role for phospholipase-D in the angiotensin-II-induced stimulation of aldosterone secretion from bovine adrenal glomerulosa cells. Endocrinology 127:1436–1443 [DOI] [PubMed] [Google Scholar]
- Kapas S, Purbrick A, Hinson JP 1995 Role of tyrosine kinase and protein kinase C in the steroidogenic actions of angiotensin II, α-melanocyte-stimulating hormone and corticotropin in the rat adrenal cortex. Biochem J 305(Pt 2):433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen H, Isales CM, Calle R, Throckmorton D, Anderson M, Gasalla-Herraiz J, McCarthy R 1995 Diacylglycerol production, Ca2+ influx, and protein kinase C activation in sustained cellular responses. Endocr Rev 16:649–681 [DOI] [PubMed] [Google Scholar]
- Jung E, Betancourt-Calle S, Mann-Blakeney R, Foushee T, Isales CM, Bollag WB 1998 Sustained phospholipase D activation in response to angiotensin II but not carbachol in bovine adrenal glomerulosa cells. Biochem J 330(Pt 1):445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA, Morris AJ 1999 Phospholipase D structure and regulation. Chem Phys Lipids 98:127–140 [DOI] [PubMed] [Google Scholar]
- Zheng X, Bollag WB 2003 AngII induces transient phospholipase D activity in the H295R glomerulosa cell model. Mol Cell Endocrinol 206:113–122 [DOI] [PubMed] [Google Scholar]
- Bollag WB, Barrett PQ, Isales CM, Rasmussen H 1991 Angiotensin-II-induced changes in diacylglycerol levels and their potential role in modulating the steroidogenic response. Endocrinology 128:231–241 [DOI] [PubMed] [Google Scholar]
- Betancourt-Calle S, Mann-Blakeney RS, Isales CM, Calle RA, Bollinger Bollag W 2001 Angiotensin II priming of aldosterone secretion with agents that enhance Ca2+ influx. Mol Cell Endocrinol 177:61–70 [DOI] [PubMed] [Google Scholar]
- Bollag WB, Kent P, White S, Malinova M, Isales CM, Calle RA 2007 Characterization and phospholipase D mediation of the angiotensin II priming response in adrenal glomerulosa cells. Endocrinology 148:585–593 [DOI] [PubMed] [Google Scholar]
- Bollag WB, Jung E, Calle RA 2002 Mechanism of angiotensin II-induced phospholipase D activation in bovine adrenal glomerulosa cells. Mol Cell Endocrinol 192:7–16 [DOI] [PubMed] [Google Scholar]
- Kondo T, Inui H, Konishi F, Inagami T 1994 Enhanced phospholipase D activity in vascular smooth muscle cells derived from spontaneously hypertensive rats. Clin Exp Hypertens 16:17–28 [DOI] [PubMed] [Google Scholar]
- Andresen BT, Jackson EK, Romero GG 2001 Angiotensin II signaling to phospholipase D in renal microvascular smooth muscle cells in SHR. Hypertension 37:635–639 [DOI] [PubMed] [Google Scholar]
- Betancourt-Calle S, Bollag WB, Jung EM, Calle RA, Rasmussen H 1999 Effects of angiotensin II and adrenocorticotropic hormone on myristoylated alanine-rich C-kinase substrate phosphorylation in glomerulosa cells. Mol Cell Endocrinol 154:1–9 [DOI] [PubMed] [Google Scholar]
- Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA 1997 Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J 16: 4519–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- Su W, Yeku O, Olepu S, Genna A, Park JS, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA 2009 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol 75:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PQ, Kojima I, Kojima K, Zawalich K, Isales CM, Rasmussen H 1986 Short term memory in the calcium messenger system. Evidence for a sustained activation of protein kinase C in adrenal glomerulosa cells. Biochem J 238:905–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y 1995 Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 9:484–496 [PubMed] [Google Scholar]
- Thompson NT, Bonser RW, Garland LG 1991 Receptor-coupled phospholipase D and its inhibition. Trends Pharmacol Sci 12:404–408 [DOI] [PubMed] [Google Scholar]
- Pagano RE, Longmuir KJ 1985 Phosphorylation, transbilayer movement, and facilitated intracellular transport of diacylglycerol are involved in the uptake of a fluorescent analog of phosphatidic acid by cultured fibroblasts. J Biol Chem 260:1909–1916 [PubMed] [Google Scholar]
- Bollag WB, Barrett PQ, Isales CM, Liscovitch M, Rasmussen H 1992 Signal transduction mechanisms involved in carbachol-induced aldosterone secretion from bovine adrenal glomerulosa cells. Mol Cell Endocrinol 86:93–101 [DOI] [PubMed] [Google Scholar]
- Tang W, Bunting M, Zimmerman GA, McIntyre TM, Prescott SM 1996 Molecular cloning of a novel human diacylglycerol kinase highly selective for arachidonate-containing substrates. J Biol Chem 271:10237–10241 [PubMed] [Google Scholar]
- Natarajan R, Stern N, Nadler J 1988 Diacylglycerol provides arachidonic acid for lipoxygenase products that mediate angiotensin II-induced aldosterone synthesis. Biochem Biophys Res Commun 156:717–724 [DOI] [PubMed] [Google Scholar]
- Natarajan R, Dunn WD, Stern N, Nadler J 1990 Key role of diacylglycerol-mediated 12-lipoxygenase product formation in angiotensin II-induced aldosterone synthesis. Mol Cell Endocrinol 72:73–80 [DOI] [PubMed] [Google Scholar]
- Martinson EA, Goldstein D, Brown JH 1989 Muscarinic receptor activation of phosphatidylcholine hydrolysis. Relationship to phosphoinositide hydrolysis and diacylglycerol metabolism. J Biol Chem 264:14748–14754 [PubMed] [Google Scholar]
- Shome K, Rizzo MA, Vasudevan C, Andresen B, Romero G 2000 The activation of phospholipase D by endothelin-1, angiotensin II, and platelet-derived growth factor in vascular smooth muscle A10 cells is mediated by small G proteins of the ADP-ribosylation factor family. Endocrinology 141:2200–2208 [DOI] [PubMed] [Google Scholar]
- Parmentier JH, Muthalif MM, Nishimoto AT, Malik KU 2001 20-Hydroxyeicosatetraenoic acid mediates angiotensin II-induced phospholipase D activation in vascular smooth muscle cells. Hypertension 37:623–629 [DOI] [PubMed] [Google Scholar]
- Parmentier JH, Pavicevic Z, Malik KU 2006 ANG II stimulates phospholipase D through PKCζ activation in VSMC: implications in adhesion, spreading, and hypertrophy. Am J Physiol Heart Circ Physiol 290:H46–H54 [DOI] [PubMed] [Google Scholar]
- Jung EM, Griner RD, Mann-Blakeney R, Bollag WB 1998 A potential role for ceramide in the regulation of mouse epidermal keratinocyte proliferation and differentiation. J Invest Dermatol 110:318–323 [DOI] [PubMed] [Google Scholar]
- Imamura S, Horiuti Y 1979 Purification of Streptomyces chromofuscus phospholipase D by hydrophobic affinity chromatography on palmitoyl cellulose. J Biochem 85:79–95 [DOI] [PubMed] [Google Scholar]
- Fox TE, Houck KL, O'Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M 2007 Ceramide recruits and activates protein kinase Cζ (PKCζ) within structured membrane microdomains. J Biol Chem 282:12450–12457 [DOI] [PubMed] [Google Scholar]
- Williams GH, Hollenberg NK 1991 Functional derangements in the regulation of aldosterone secretion in hypertension. Hypertension 18:III143–III149 [DOI] [PubMed] [Google Scholar]
- Bollag WB, Xie D 2008 Phospholipase D, aldosterone secretion and priming: mechanism of a type of cellular memory. Curr Trends Endocrinol 3:77–87 [Google Scholar]