

Abstract
Hyperglycemia, a prevalent condition in premature infants, is thought to be a consequence of incomplete suppression of endogenous glucose production and reduced insulin-stimulated glucose disposal in peripheral tissues. However, the molecular basis for these conditions remains unclear. To test the hypothesis that the insulin transduction pathway is underdeveloped with prematurity, fetal baboons were delivered, anesthetized, and euthanized at 125 d gestational age (GA), 140 d GA, or near term at 175 d GA. Vastus lateralis muscle and liver tissues were obtained, and protein content of insulin signaling molecules [insulin receptor (IR)-β, IR substate-1, p85 subunit of phosphatidylinositol 3-kinase, Akt, and AS160] and glucose transporters (GLUT)-1 and GLUT4 was measured by Western blotting. Muscle from 125 d GA baboons had markedly reduced GLUT1 protein content (16% of 140 d GA and 9% of 175 d GA fetuses). GLUT4 and AS160 also were severely reduced in 125 d GA fetal muscle (43% of 175 d GA and 35% of 175 d GA, respectively). In contrast, the protein content of IR-β, IR substate-1, and Akt was elevated by 1.7-, 5.2-, and 1.9-fold, respectively, in muscle from 125 d GA baboons when compared with 175 d GA fetuses. No differences were found in the content of insulin signaling proteins in liver. In conclusion, significant gestational differences exist in the protein content of several insulin signaling proteins in the muscle of fetal baboons. Reduced muscle content of key glucose transport-regulating proteins (GLUT1, GLUT4, AS160) could play a role in the pathogenesis of neonatal hyperglycemia and reduced insulin-stimulated glucose disposal.
Changes in insulin signaling proteins in muscle during normal fetal development are implicated in the development of transient hyperglycemia of premature infants.
The neonate lacks a precise control of glucose homeostasis when compared with the adult (1). In particular, premature infants weighing less than 1000 g have 18 times the risk of developing hyperglycemia when compared with those weighing more than 2000 g (2). Transient neonatal hyperglycemia has been reported in up to 80% of extremely premature infants and has been associated with intraventricular hemorrhage, retinopathy of prematurity, and increased risk of death (2,3,4,5). Our understanding of glucose metabolism in the neonate remains fragmentary. There is evidence that the hyperglycemia of the extremely low birth weight infant (birth weight <1000 g) may be due to inadequate suppression of endogenous glucose production (1,6). In addition, extremely low birth weight infants have elevated plasma insulin levels and an increased proinsulin to insulin ratio (7), indicating that these infants are insulin resistant. Nonetheless, the molecular basis for the reduced insulin-stimulated glucose disposal of prematurity remains unclear. In addition, there is a limited understanding regarding the developmental changes that occur within the insulin signaling pathway.
Skeletal muscle is the main site responsible for insulin-stimulated glucose disposal (8). For insulin to exert its effects on cellular glucose metabolism, this hormone must first bind to its receptor (IR) and initiate a signaling cascade that includes tyrosine phosphorylation of the IR and insulin receptor substrate (IRS)-1, followed by activation of phosphatidylinositol (PI) 3-kinase (9). Insulin-stimulated activation of PI3-kinase leads to translocation of the glucose transporter (GLUT)-4, resulting in an increase in glucose transport rate across the plasma membrane (10). The precise mechanism by which activation of PI 3-kinase causes GLUT4 translocation is still unknown, although phosphorylation/activation of phosphoinositide-dependent kinase 1 (11), Akt (12), protein kinase C (13), and AS160 (14) have been implicated. Several abnormalities distinguish insulin-resistant muscle from normal muscle, including decreased insulin-stimulated GLUT4 translocation, decreased IR and IRS-1 tyrosine phosphorylation, and decreased PI 3-kinase activity (15).
There is evidence suggesting developmental differences in the insulin signaling pathway (16,17). Studies performed in rats have found that insulin-stimulated GLUT4 translocation in muscle is lower in term rats when compared with adult animals (17,18). Others reported that aging is associated with changes in several insulin signaling proteins in the rat, including decreased phosphorylation of IRS-1, Akt, and AS160 (19).
Baboons have close (97%) phylogenetic proximity with humans (20). In addition, baboons can develop insulin resistance and diabetes spontaneously, a feature that distinguishes this animals from rodents, a frequently used model in diabetes research that very rarely develops insulin resistance and diabetes spontaneously (20). Previously we have shown that the adult baboon is a pertinent nonhuman primate model to examine the underlying cellular/molecular mechanisms responsible for insulin resistance (20). In this study we describe the incidence of hyperglycemia in a preterm baboon model. We also examined developmental differences of key insulin signaling proteins in skeletal muscle and liver in fetal baboons delivered prematurely at 67, 75, and 94% gestation. We hypothesize that the insulin signal transduction pathway is not fully developed in the liver and muscle of preterm baboons.
Materials and Methods
Incidence of hyperglycemia in preterm baboons
Thirty-two baboons were delivered prematurely at 125 d gestational age (GA) via cesarean section under general anesthesia from healthy, nondiabetic mothers at the Southwest National Primate Research Center at the Southwest Foundation for Biomedical Research (SFBR; San Antonio, TX). All studies were approved by the Institutional Animal Care Committee at the SFBR. Animal experiments were conducted in accord with accepted standards of humane animal care. Animals were intubated immediately after birth and chronically ventilated to maintain alive for up to 14 d. Central lines were placed for fluid management and parenteral nutrition. Minimal enteral feeds were initiated after 5 d of age if bowel gas pattern was considered normal. Sepsis was defined by at least one positive blood, urine, or cerebrospinal fluid culture. Antibiotics were provided accordingly. Plasma glucose concentrations were measured from central arterial lines soon after birth and then at a minimum of every 6 h. Hyperglycemia was defined as a plasma glucose concentration of 150 mg/dl or greater (8.3 mmol/liter) on at least two different occasions during the first 10 d of life; this level has been previously identified as being clinically relevant (5). Intravenous insulin was given at a dose of 0.5 IU/kg to treat plasma glucose greater than 200 mg/dl.
Measurement of insulin signaling and glucose transporter proteins
We studied 18 fetal baboons (10 males, eight females) that were delivered at SFBR. Animals were delivered prematurely via cesarean section under general anesthesia from healthy, nondiabetic mothers at 125 d GA, 140 d GA, or near term at 175 d GA (full term = 185 d GA) and euthanized immediately after birth. Immediately after euthanasia, vastus lateralis muscle and liver tissue samples were obtained, snap frozen in liquid nitrogen, and stored at −80 C.
Materials
Primary antibodies against IR-β, phosphotyrosine, and Akt-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), IRS-1, p85 subunit of PI 3-kinase, Akt-2, GLUT1, and GLUT4 were from Millipore (Chicago, IL), and glyceraldehyde-3-phosphate dehydrogenase, AS160, phospho-IR-β Tyr1361, phospho-Akt Ser473, and total Akt were from Cell Signaling Technology (Danvers, MA). Electrophoresis reagents were purchased from Bio-Rad (Hercules, CA), and enhanced chemiluminescence Western blot reagents and film were from Amersham (Arlington Heights, IL). Serum insulin levels were measured with 1-2-3 UltraSensitive human insulin ELISA (ALPCO Diagnostics, Windham, NH); plasma glucose was measured using the Analox GM9 analyzer (Analox Instruments, London, UK) and the AU 640 immunoanalyzer (Olympus Inc., Center Valley, PA).
Western blot analysis and immunoprecipitation
Muscle samples were homogenized in ice-cold lysis buffer [containing 20 mmol/liter Tris (pH 7.5), 10 mmol/liter sodium pyrophosphate, 100 mmol/liter sodium fluoride, 2 mmol/liter sodium orthovanadate, 5 mmol/liter EDTA (pH 8.0), 1% Nonidet P-40, 1 mmol/liter phenylmethylsulfonyl fluoride, 3 mmol/liter benzamidine, 10 μg/ml leupeptin, and 10 μg/ml aprotinin]. Homogenates were rotated for 1 h at 4 C and then centrifuged at 14,000 × g for 10 min at 4 C. The supernatants were collected and protein concentrations measured by the Bradford assay. Lysate proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking in Tris-buffered saline with 5% nonfat dry milk, the membranes were incubated overnight at 4 C with the primary antibody indicated above. Bound antibodies were detected with antirabbit immunoglobulin-horseradish peroxidase-linked whole antibody and by using ECL reagents. The intensities of the bands were quantified by densitometry using the NIH imaging program (National Institutes of Health, Bethesda, MD) with the results reported in arbitrary OD units. For measurement of IRS-1, tyrosine phosphorylation proteins were immunoprecipitated with anti-IRS-1 antibody followed by incubation with protein A agarose beads. The beads were washed extensively, and proteins eluted with Laemmli buffer were separated by SDS-PAGE and transferred to nitrocellulose membrane, followed by incubation with an antiphosphotyrosine antibody.
Statistical analysis
Statistical calculations and demographic distributions were performed with SPSS for Windows (version 11.5, SPSS, Inc., Chicago, IL). Differences between groups were determined using one-way ANOVA, followed by the Tukey test. P < 0.05 was considered to be statistically significant.
Results
Hyperglycemia in preterm baboons
Baboons studied had a mean birth weight of 370 ± 43 g and the mean survival was 12.3 ± 3.8 d. Fifty percent of the baboons were males. Twenty-eight of the 32 animals included in this study (87%) developed hyperglycemia. The mean plasma glucose levels per day are depicted in Fig. 1; a total of 1644 plasma glucose measurements were included in the final analysis. Insulin was used for the treatment of hyperglycemia in 60% of all animals (mean 1.7 ± 1.8 d of life). The mean glucose infusion rate during the peak glucose level found on d 1 was 5.0 ± 0.4 mg/kg · min. Three baboons developed sepsis during the study period.
Figure 1.
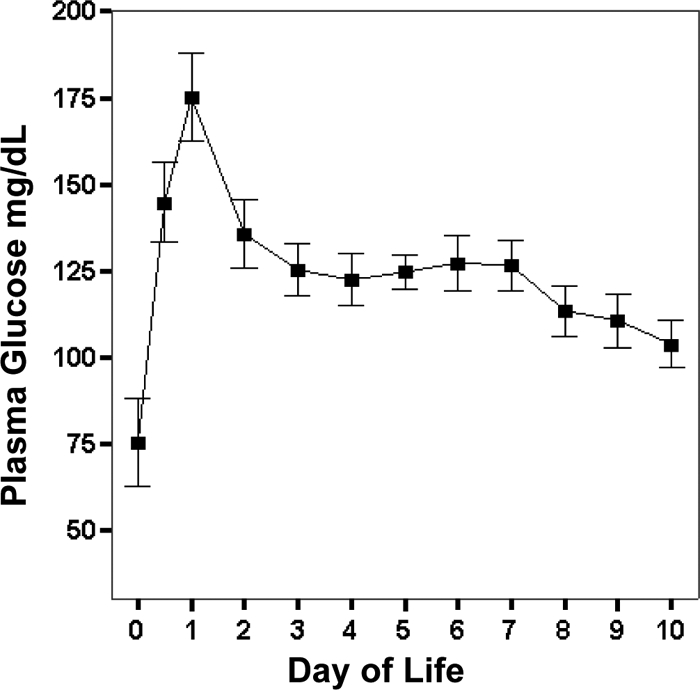
Daily plasma glucose from 125 d GA baboons who survived longer than 7 d. Graphical data are means ± se.
Developmental differences of key insulin signaling proteins in skeletal muscle and liver
Animal characteristics
Eighteen animals were delivered at three different gestations (125 d GA, 140 d GA, and 175 d GA). Animal characteristics are shown in Table 1. Plasma insulin and glucose concentrations at birth were similar between groups (data not shown).
Table 1.
Animal characteristics of fetal baboons used to investigate developmental differences of key insulin signaling proteins in skeletal muscle and liver
| Group | Gestation (% of term) | n | Male/female | Birth weight (g) |
|---|---|---|---|---|
| 125 d GA | 67 | 6 | 4/2 | 412 ± 33 |
| 140 d GA | 75 | 6 | 1/5 | 552 ± 60 |
| 175 d GA | 94 | 6 | 5/1 | 1046 ± 104 |
sd is shown.
Content of IR-β, IRS-1, and p85 subunit of PI3-kinase
Figure 2A shows that the protein content of IR-β in muscle was elevated by 1.9-fold in the most immature animals (P < 0.01). Consistent with this finding, IR-β Tyr1361 phosphorylation also was increased in the muscle of immature animals (P < 0.05) (Fig. 2B). The protein content of IRS-1 was also higher in the muscle of the preterm animals compared with the term animals (P < 0.01) (Fig. 2C). Similarly, IRS-1 tyrosine phosphorylation was elevated in the most immature animals (P < 0.05) (Fig. 2D). The muscle protein content of the p85 subunit of PI3-kinase was similar across all gestational ages (Fig. 2E).
Figure 2.

Insulin signaling protein content and phosphorylation in muscle from fetal baboons born at different gestational ages. IR-β (A), phospho-IR-β Tyr 1361 (B), IRS-1 (C), IRS-1 tyrosine phosphorylation (D), and p85 subunit of PI3-kinase (E) were measured by Western blotting and immunoprecipitation (n = 6/group). Representative blots from three animals per group also are shown. Graphical data are means ± se. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. *, P < 0.05; **, P < 0.01.
Muscle Akt content
Total Akt content was elevated by 1.7-fold in the muscle from 125 d GA baboons (P < 0.001) and 1.4-fold higher in baboons of 140 d GA (P < 0.05) when compared with 175 d GA fetuses (Fig. 3A). In line with Akt protein content, Ser 473 phosphorylation tended to be higher in the muscle of the most immature animals, although this difference did not reach statistical significance (Fig. 3B). Figure 3, C and D, shows that increases in the content of both Akt-1 and Akt-2 account for the elevation in total Akt protein observed in immature baboons.
Figure 3.

Akt protein content and phosphorylation in muscle from fetal baboons. Total Akt (A), phospho-Akt-Ser 473 (B), Akt-1 (C), and Akt-2 (D) were measured by Western blotting (n = 6/group). Representative blots from three animals per group also are shown. Graphical data are means ± se. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase. *, P < 0.05; **, P < 0.01.
Glucose transporter and AS160 content in muscle
In contrast to IR-β, IRS-1, and Akt, the muscle of 125 d GA fetal baboons had markedly lower GLUT1 protein content in the basal state (16% of 140 d GA and 9% of 175 d GA fetuses, P < 0.05) (Fig. 4A). GLUT4 protein also was significantly reduced in the fetal muscle at 125 d GA (35% of 140 d GA and 43% of 175 d GA, P < 0.05) (Fig. 4B). These reductions in GLUT1 and GLUT4 in 125 d GA fetuses were accompanied by lower content of the Rab GTPase activating protein AS160 (47% of 140 d GA and 35% of 175 d GA, P < 0.01) (Fig. 4C).
Figure 4.

Glucose transporter and AS160 protein content in muscle from fetal baboons: GLUT1 (A), GLUT4 (B), and AS160 (C) protein content (n = 6/group). Representative blots from three animals per group also are shown. Graphical data are means ± se. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase. *, P < 0.05; **, P < 0.01.
Insulin signaling molecules in liver
Opposite to our findings in muscle, the liver from fetal baboons had similar content of insulin signaling and glucose transporter proteins across gestational ages (Fig. 5, A–F).
Figure 5.

Protein content of insulin signaling proteins and glucose transporters in liver from fetal baboons born at different gestational ages. GLUT1 (A), GLUT4 (B), IR-β (C), IRS-1 (D), p85 subunit of PI 3-kinase (E), and total Akt protein content (F) was measured by Western analysis (n = 6/group). Representative blots from three animals per group also are shown. Graphical data are means ± se. *, P < 0.05; **, P < 0.01.
Discussion
Hyperglycemia is a common condition that leads to significant morbidity in premature infants (2,3,4). However, the molecular basis for the hyperglycemia of prematurity is poorly understood. The purpose of this study was to examine whether developmental differences exist in the content of key insulin signaling proteins in muscle and liver during the fetal period. We used a baboon model because these animals share 97% homology with humans at the DNA level and spontaneously develop many diseases similar to those in man, including obesity and diabetes (20). In addition, we recently sequenced genes and proteins of several insulin signaling molecules in the baboon, which were found to be about 98% identical with those of man (20).
We found gestational differences in the protein content of several insulin signaling and glucose transporter proteins in the muscle of fetal baboons. The most striking differences observed were reductions in the content of GLUT1 and GLUT4 in immature baboons. GLUT4 is the predominant insulin-sensitive transporter in muscle and its translocation from an intracellular compartment to the plasma membrane is required for the ability of insulin to promote glucose transport (21). The physiological importance of GLUT4 in muscle is highlighted by studies in GLUT4 knockout mice, which showed that these animals are glucose intolerant and have severely depressed insulin-stimulated glucose transport in muscle (22). Because GLUT4 is ultimately responsible for transporting glucose into the cell in response to insulin, the reduction in GLUT4 content observed in immature animals likely plays an important role in the alterations of glucose homeostasis that occur in prematurity.
In line with a reduction in GLUT4 in more preterm baboons, these animals also had severely reduced content in GLUT1. Whereas GLUT4 is the main glucose transporter involved in postprandial glucose transport, when insulin concentrations in the circulation are the highest, GLUT1 is responsible for the constitutive, insulin-independent glucose transport that takes place in all cells, including muscle (23). Studying the role that GLUT1 plays on maintaining glucose homeostasis using knockout models has been limited because deletion of this gene is embryonically lethal (24). However, GLUT1 overexpression results in a 3- to 4-fold increase in basal glucose transport in muscle ex vivo, in fasting hypoglycemia and improved glucose tolerance (25), suggesting that GLUT1 also plays an important role in maintaining whole-body glucose homeostasis.
Along with reductions in GLUT1 and GLUT4, the most immature baboons had reduced content of AS160 in muscle. AS160 is a Rab-GAP (GTPase-activating protein), and phosphorylation of AS160 on phospho-Akt substrate motifs on insulin stimulation is thought to remove its inhibitory GAP activity and increases GLUT4 trafficking to the cell membrane (26,27). Therefore, our finding of reduced AS160 content in muscle from immature baboons is consistent with the observed reductions in the content of distal proteins involved in glucose transport.
In contrast to the reductions observed in distal signaling molecules, we found that some proximal signaling proteins such as IR-β, IRS-1, and Akt are increased in muscle from the more preterm animals. Akt, the IR, and IRS-1 play other roles besides insulin signaling. During embryonic development, IGFs function to promote growth, as evidenced by mutants lacking IGF-I or IGF-II, which develop severe intrauterine growth restriction (28). This growth-promoting function is mediated by two receptors, IR and the IGF-I receptor, with the role of the former being predominantly related to IGF-II signaling and growth control during most of the embryonic period (29). The role of IR in growth is further evidenced in infants with leprechaunism, a disease that results from mutations in the IR gene and leads to growth restriction and death (30). Regarding IRS-1, mice with targeted deletion of this protein are born alive but retarded in embryonal and postnatal growth (31). In human fetuses, variations in insulin concentrations contribute to alterations in fetal growth. For example, elevated insulin concentrations, as seen during uncontrolled maternal diabetes, is known to promote fetal overgrowth, whereas isolated fetal insulin deficiency under a stable maternal environment can cause fetal growth restriction (32,33). In addition, insulin promotes skeletal muscle protein synthesis and growth, which decreases progressively with maturity (34). Therefore, the increase in IR-β and IRS-1 content seen in immature animals could be related to the accelerated growth that occurs during early gestation.
Against our hypothesis, we found that total Akt protein content decreased progressively with age. Insulin-responsive tissues are enriched in Akt (35), and this protein is thought to be a key mediator of both cell growth and metabolism (12,36). Akt has three isoforms; in muscle, the most abundant are Akt-1 and Akt-2 (37,38). It has been proposed that Akt-1 and Akt-2 transmit distinct biological signals (39). In particular, Akt-1 is implicated in cell growth, whereas Akt-2 is thought to play a more important role in regulating glucose metabolism (39). Upon our observation of increased total Akt content in immature baboons, we expected that the increase in total Akt content would be accounted mainly by elevated Akt-1 due to the rapid rate of growth that occurs during early gestation. Nonetheless, we found significant increases in the content of both Akt isoforms in early gestation. Even though Akt-2 is thought to play a role in regulating glucose metabolism, there is scarce evidence of its specific physiological implications in animal models and humans. In addition, because insulin action seems to change with maturity, developmental differences in Akt isoforms may coexist (20). We speculate that the up-regulation in IR-β and Akt protein content in the muscle of the most immature baboons may be related to preferential insulin signaling toward growth and/or compensation to overcome the reductions in the content of distal insulin signaling proteins/glucose transporters.
In this study, we demonstrate that hyperglycemia is a common condition in preterm baboons, affecting up to 87% of the animal population studied. The incidence of hyperglycemia is similar of that reported in extremely premature infants (2,3,4,5,6). Moreover, fetal baboons were delivered at comparable gestational ages as seen in humans. In clinical practice, infants of youngest gestation are commonly delivered between 24–27 wk and have a birth weight less than 1000 g (60–67% gestation, compared with 125 d baboons delivered at 67% gestation); premature infants delivered at 30 wk weigh between 1000 and 1500 g (75% gestation, equivalent to 140 d baboons); and infants delivered at term are at least 37 wk of gestation (92% gestation, compared with 94% gestation of 175 d baboons). Due to the high incidence of hyperglycemia and reduced insulin-stimulated glucose disposal reported in premature infants of lowest gestation (24–27 wk, or <1000 g) (2,3,4,6), finding a significant down-regulation of GLUT1, GLUT4, and AS160 proteins in the immature baboons when compared with older gestations is very relevant to the hyperglycemia that occurs in premature infants.
It is possible that developmental changes in insulin signaling protein content in the muscle of preterm baboons could be due to concurrent variations in fetal insulin and/or glucose concentrations. However, plasma glucose and insulin levels measured at birth in the preterm animals evaluated for insulin signaling protein content were the same as the levels measured at birth in term animals.
In contrast to our findings in muscle, there were no differences in the content of insulin signaling proteins and glucose transporters in the liver across the different gestational ages. The lack of differences is consistent with the findings that hepatic dysfunction is a late event in the development and progression of diabetes (40).
In summary, this study provides evidence that hyperglycemia is a common condition in preterm baboons and that variations in insulin signaling proteins exist in muscle during normal fetal development. No differences were present in the liver from preterm animals. Down-regulation of AS160, GLUT1, and GLUT4 muscle protein content may be implicated in the development of transient hyperglycemia of premature infants. Future studies will be needed to evaluate whether preterm baboons have defects in the insulin transduction pathways under insulin-stimulated conditions.
Acknowledgments
We thank the personnel at the University of Texas Health Science Center at San Antonio, the Texas Diabetes Institute, and the Southwest Foundation for Biomedical Research for their dedication and support for this project.
Footnotes
This work was supported by grants from the American Diabetes Association (to N.M. and R.A.D.), Grant HL52636 from the National Institutes of Health (to Bronchopulmonary Dysplasia Resource Center), Grants AG030979 and DK080157 (to N.M.), Grant DK24092 (to R.A.D), Grant P51RR13986 for facility support at the Southwest Foundation for Biomedical Research, the San Antonio Nathan Shock Center (to N.M), the University of Texas Health Science Center at San Antonio Executive Research Committee (to N.M.), the South Texas Health Research Center (to N.M.), and the U.S. Department of Veterans Affairs (to R.A.D.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 16, 2010
Abbreviations: GA, Gestational age; GLUT, glucose transporter; IR, insulin receptor; IRS, IR substrate; PI, phosphatidylinositol; SFBR, Southwest Foundation for Biomedical Research.
References
- Farrag HM, Nawrath LM, Healey JE, Dorcus EJ, Rapoza RE, Oh W, Cowett RM 1997 Persistent glucose production and greater peripheral sensitivity to insulin in the neonate vs. the adult. Am J Physiol Endocrinol Metab 272:E86–E93 [DOI] [PubMed] [Google Scholar]
- Garg R, Agthe AG, Donohue PK, Lehmann CU 2003 Hyperglycemia and retinopathy of prematurity in very low birth weight infants. J Perinatol 23:186–194 [DOI] [PubMed] [Google Scholar]
- Blanco CL, Baillargeon JG, Morrison RL, Gong AK 2006 Hyperglycemia in extremely low birth weight infants in a predominantly Hispanic population and related morbidities. J Perinatol 26:737–741 [DOI] [PubMed] [Google Scholar]
- Hall NJ, Peters M, Eaton S, Pierro A 2004 Hyperglycemia is associated with increased morbidity and mortality rates in neonates with necrotizing enterocolitis. J Pediatr Surg 39:898–901 [DOI] [PubMed] [Google Scholar]
- Pildes RS 1986 Neonatal hyperglycemia. J Pediatr 109:905–907 (Review) [DOI] [PubMed] [Google Scholar]
- Farrag HM, Cowett RM 2000 Glucose homeostasis in the micropremie. Clin Perinatol 27:1–22, v (Review) [DOI] [PubMed] [Google Scholar]
- Mitanchez-Mokhtari D, Lahlou N, Kieffer F, Magny JF, Roger M, Voyer M 2004 Both relative insulin resistance and defective islet β-cell processing of proinsulin are responsible for transient hyperglycemia in extremely preterm infants. Pediatrics 113:537–541 [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP1981 The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30:1000–1007 [DOI] [PubMed] [Google Scholar]
- White MF, Yenush L 1998 The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Curr Top Microbiol Immunol 228:179–208 (Review) [DOI] [PubMed] [Google Scholar]
- Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M 1996 Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J Cell Biol 135:415–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick KL, Liu F 2001 A new molecular target of insulin action: regulating the pivotal PDK1. Curr Drug Targets Immune Endocr Metabol Disord 1:209–221 (Review) [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ 2001 Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352 [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML 2005 Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans 33:350–353 [DOI] [PubMed] [Google Scholar]
- Zeigerer A, McBrayer MK, McGraw TE 2004 Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 15:4406–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson AC 2001 Regulation of muscle glucose uptake in vivo. In: Mandarino LR, Bonadonna O, McGuinness A, Halseth AE, Wasserman D, eds. Handbook of physiology. New York: The Oxford University Press; 803–848 [Google Scholar]
- Hughes SJ 1994 The role of reduced glucose transporter content and glucose metabolism in the immature secretory responses of fetal rat pancreatic islets. Diabetologia 37:134–140 [DOI] [PubMed] [Google Scholar]
- Santalucía T, Camps M, Castelló A, Muñoz P, Nuel A, Testar X, Palacin M, Zorzano A 1992 Developmental regulation of GLUT-1 (erythroid/Hep G2) and GLUT-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 130:837–846 [DOI] [PubMed] [Google Scholar]
- He J, Thamotharan M, Devaskar SU 2003 Insulin-induced translocation of facilitative glucose transporters in fetal/neonatal rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol 284:R1138–R1146 [DOI] [PubMed] [Google Scholar]
- Gupte AA, Bomhoff GL, Geiger PC 2008 Age-related differences in skeletal muscle insulin signaling: the role of stress kinases and heat shock proteins. J Appl Physiol 105:839–848 [DOI] [PubMed] [Google Scholar]
- Chavez AO, Lopez-Alvarenga JC, Tejero ME, Triplitt C, Bastarrachea RA, Sriwijitkamol A, Tantiwong P, Voruganti VS, Musi N, Comuzzie AG, DeFronzo RA, Folli F 2008 Physiological and molecular determinants of insulin action in the baboon. Diabetes 57:899–908 [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Kahn BB 1999 Glucose transporters and insulin action—implications for insulin resistance and diabetes mellitus. N Engl J Med 341:248–257 [DOI] [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB 2000 Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 6:924–928 [DOI] [PubMed] [Google Scholar]
- Pessin JE, Bell GI 1992 Mammalian facilitative glucose transporter family: structure and molecular regulation. Annu Rev Physiol 54:911–930 [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Kikkawa T, Hori S, Terasaki T 2006 Modulation and compensation of the mRNA expression of energy related transporters in the brain of glucose transporter 1-deficient mice. Biol Pharmaceut Bull 29:1587–1591 [DOI] [PubMed] [Google Scholar]
- Marshall BA, Ren JM, Johnson DW, Gibbs EM, Lillquist JS, Soeller WC, Holloszy JO, Mueckler M 1993 Germline manipulation of glucose homeostasis via alteration of glucose transporter levels in skeletal muscle. J Biol Chem 268:18442–18445 [PubMed] [Google Scholar]
- Thong FS, Bilan PJ, Klip A 2007 The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 56:414–423 [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE 2003 Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278:14599–14602 [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A 1993 Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75:73–82 [PubMed] [Google Scholar]
- Louvi A, Accili D, Efstratiadis A 1997 Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol 189:33–48 [DOI] [PubMed] [Google Scholar]
- Krook A, Brueton L, O'Rahilly S 1993 Homozygous nonsense mutation in the insulin receptor gene in infant with leprechaunism. Lancet 342:277–278 [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Tamemoto H, Tobe K, Terauchi Y, Ueki K, Kaburagi Y, Yamauchi T, Satoh S, Sekihara H, Aizawa S, Yazaki Y 1996 Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1 and identification of insulin receptor substrate-2. Diabet Med 13(9 Suppl 6):S103–S108 [PubMed] [Google Scholar]
- Fowden AL, Hughes P, Comline RS 1989 The effects of insulin on the growth rate of the sheep fetus during late gestation. Q J Exp Physiol 74:703–714 [DOI] [PubMed] [Google Scholar]
- Philipps AF, Rosenkrantz TS, Clark RM, Knox I, Chaffin DG, Raye JR 1991 Effects of fetal insulin deficiency on growth in fetal lambs. Diabetes 40:20–27 [DOI] [PubMed] [Google Scholar]
- Kimball SR, Farrell PA, Nguyen HV, Jefferson LS, Davis TA 2002 Developmental decline in components of signal transduction pathways regulating protein synthesis in pig muscle. Am J Physiol Endocrinol Metab 282:E585–E592 [DOI] [PubMed] [Google Scholar]
- Calera MR, Martinez C, Liu H, Jack AK, Birnbaum MJ, Pilch PF 1998 Insulin increases the association of Akt-2 with Glut4-containing vesicles. J Biol Chem 273:7201–7204 [DOI] [PubMed] [Google Scholar]
- Brozinick Jr JT, Roberts BR, Dohm GL 2003 Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes 52:935–941 [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME 1999 Cellular survival: a play in three Akts. Genes Dev 13:2905–2927 [DOI] [PubMed] [Google Scholar]
- Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR 1998 Activation of protein kinase Bβ and γ isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase Bα. Biochem J 331:299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SS, Cho H, Mu J, Birnbaum MJ 2003 Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem 278:49530–49536 [DOI] [PubMed] [Google Scholar]
- Home PD, Pacini G 2008 Hepatic dysfunction and insulin insensitivity in type 2 diabetes mellitus. Diabetes Obes Metab 10:699–718 [DOI] [PubMed] [Google Scholar]