

Abstract
We have performed a three-generation, forward genetic screen to identify recessive mutations that affect the patterning of the peripheral nervous system. Using this assay, we identified Sema3AK108N, a novel loss-of-function allele of Sema3A. Class 3 semaphorins, which include Sema3A, are structurally conserved secreted proteins that play critical roles in the development and function of the nervous system. Sema3AK108N mutant mice phenocopy Sema3A-null mice, and Sema3AK108N protein fails to repel or collapse DRG axons in vitro. K108 is conserved among semaphorins, yet the loss-of-function effects associated with K108N are not the result of impaired expression, secretion, or binding of Sema3A to its high-affinity receptor Neuropilin-1 (Npn-1). Using in silico modeling and mutagenesis of other semaphorin family members, we predict that Sema3AK108N interacts poorly with the Npn-1/PlexA holoreceptor and, thus, interferes with its ability to signal at the growth cone. Therefore, through the use of a forward-genetic screen we have identified a novel allele of Sema3A that provides structural insight into the mechanism of Sema3A/Npn-1/PlexinA signaling.
Introduction
During development of the mammalian nervous system billions of neurons elaborate axonal projections that extend through a network of neurons and other cell types to connect with their appropriate synaptic partners. Along their trajectories, axons must survey the external surroundings and appropriately interpret directional cues provided by the environment. The PNS is an excellent model system to study the development of axonal projections and the establishment of neural connectivity. First, many PNS neurons send extremely long axons to distal targets and in the process encounter a myriad of cell and tissue types. Second, the environmental diversity experienced by a single axon during its pathfinding allows one to study a series of choice points for a single axon. Third, neurons within a single ganglion often send their axons to a variety of final targets both in the periphery and CNS. Finally, most PNS neurons develop early and are, thus, amenable to visualization in the context of the whole embryo.
Semaphorins are a large, conserved family of both secreted and integral membrane proteins that canonically act on the growth cone as guidance cues to control the establishment of neural connections. In addition, semaphorins are integral for multiple aspects of neural development and for the development and function of other organ systems (Yazdani and Terman, 2006; Tran et al., 2007; Roth et al., 2009).
Semaphorins share a conserved N-terminal Sema domain but are otherwise divergent and have been classified into eight families on the basis of structure and species (Semaphorin Nomenclature Committee, 1999). Generally, a member of the plexin family of transmembrane proteins serves as the receptor and transducer of the semaphorin signal. Of the eight families, the class 3 semaphorins are the only vertebrate secreted semaphorins. Most members of this secreted subfamily are not capable of direct functional interaction with a plexin receptor; Sema3E is the only member that directly binds to a plexin, PlexinD1 (Gu et al., 2005). Instead, most class 3 semaphorins bind with high affinity to a neuropilin receptor—either neuropilin-1 (Npn-1) or neuropilin-2 (Npn-2)—transmembrane proteins with short cytoplasmic domains lacking obvious signaling motifs. The Npns directly and constitutively associate with Plexin signal transducing subunits (Tran et al., 2007). Thus, a holoreceptor comprised of a Npn and a Plexin is necessary to transduce intracellular signals for most class 3 semaphorins (Takahashi et al., 1999; Rohm et al., 2000). Consistent with this idea, mice with mutations in Npn-1 or Npn-2 receptors (Kitsukawa et al., 1997; Chen et al., 2000; Giger et al., 2000; Gu et al., 2003) and the Sema3-transducing plexin receptors PlexinA3 or PlexinA4 (Cheng et al., 2001; Yaron et al., 2005) display phenotypes that in many ways parallel the major defects observed in mice lacking the ligands.
Here we report an unbiased, genome-wide screen in the mouse for novel loci that encode proteins that control development of connectivity of the PNS. Using forward genetics, we have identified several allelic groups with PNS axonal guidance defects and mapped these mutant lines to discrete chromosomal regions. We describe here one mutant harboring a mutation in Sema3A, Sema3AK108N, in which a single amino acid substitution in the semaphorin domain of Sema3A creates a loss-of-function phenotype. Biochemical analysis of Sema3AK108 indicates that the K108 residue is critical for Sema3A function but is not necessary for Sema3A–Npn-1 interaction demonstrating an in vivo role for the K108 region of Sema3A in transducing downstream signals independent of binding to its high-affinity receptor. Analysis of this novel Sema3A allele serves to define structural parameters that govern Sema3A signaling at the growth cone and validates the power of forward genetic screens to identify factors affecting the growth and development of the PNS.
Materials and Methods
N-Ethyl-N-nitrosourea mutagenesis and screen.
To observe recessive mutations, we designed a three-generation strategy for our mutagenesis screen (Fig. 1). First, 8- to 10-week-old, male, C57BL/6 (BL6) mice (F1) were intraperitoneally injected with 300 mg of N-ethyl-N-nitrosourea (ENU) (1 mg/ml in 95% ethanol; 0.09 m sodium phosphate; 0.045 m sodium citrate; pH 5) per kg of body weight. The ENU was delivered in a fractionated dose of 3 × 100 mg/kg administered weekly. As anticipated, all injected males lost fertility.
Figure 1.

Schematic diagram of the recessive screen for novel alleles affecting neural development. BL6 male mice (8–10 weeks old) were injected with 3 × 100 mg/kg body weight ENU. F1 males that regained fertility were mated to C3H females to produce G1 male mice (lines). Each of these G1 males was in turn mated to C3H females to produce G2 daughters, some of which are presumed heterozygous for a given mutation. After crossing the G2 females to their G1 fathers, recessive mutations were produced and scored for developmental anomalies.
F1 males that recovered fertility were mated to C3H/He (C3H) females. All mice in this and in subsequent crosses were of the C3H strain to generate the genetic diversity necessary for anticipated mapping of interesting loci. Given that the injected mice were BL6, all mutations would be carried on this background; thus, to establish linkage between a phenotype of interest and the BL6 strain, all BL6 contributions to the cross must be restricted to the injected males. From the series of injections that we performed, we were able to obtain 164 G1 male mice.
Resulting G1 males (BL6 × C3H hybrids harboring multiple single copy mutations) were then mated to C3H females. Of the G2 females produced from this cross, 50% were expected to carry a single copy of any specific mutation. These G2 females were mated to their G1 fathers to produce homozygous mutants at a frequency of 1 in 8 embryos. The crosses were timed, as measured by morning plug dates, and the G3 embryos were killed, stained, and scored at embryonic day 12.5 (E12.5). For each G1 male, 6–8 litters or >40 embryos were screened before a decision was made whether to euthanize or to propagate the line for further analysis.
Wholemount neurofilament assay.
Given the large number of embryos to be processed for the screen, we needed to develop a method to stain not just single embryos but entire litters. Moreover, we needed to develop the ability to process many litters simultaneously. To circumvent these potential problems, we developed a staining assay that allowed us to process each litter in a single 15 ml conical tube throughout the entire staining process. Briefly, embryos were fixed overnight in 10 ml 4% paraformaldehyde at 4°C. All subsequent washes and incubations were in 10 ml, shaking, and at room temperature unless otherwise indicated. The next morning the embryos were washed with PBS for 30 min and then taken through a series of 2–4 h methanol/PBS dehydrations (50%, 75%, 100%). Endogenous peroxidase activity was quenched with 3% H2O2 in 80% methanol/DMSO overnight. The next morning, embryos were washed in five 1 h incubations of TNT (0.1 mm Tris-base, 0.2 mm NaCl, 0.1% Triton X-100) and then incubated in primary 2H3 supernatant antibody 1:150 in TNT with 5% DMSO, 2% Sheep Serum (Millipore Bioscience Research Reagents), 5% milk extract, and 0.025% sodium azide. After 48 h in primary antibody, the embryos were washed using five 1 h incubations of TNT and then incubated in secondary sheep anti-mouse HRP-conjugated antibody (Jackson ImmunoResearch Laboratories) 1:300 in TNT with 2% sheep serum. After 36 h of secondary antibody incubation, the embryos were washed with TBS (0.24 mm Tris-base, 1.37 mm NaCl, 0.002 mm KCl) for two 30 min incubations and then overnight in TBS. The following morning, the horseradish peroxidase reactions were developed with diaminobenzidine and the embryos fixed overnight in 4% paraformaldehyde at 4°C. The next morning, the embryos were washed with PBS for 30 min and then taken through a series of 2–4 h methanol/PBS dehydrations (50%, 75%, 100%). To better visualize the staining, the embryos were cleared in 2:1 benzyl alcohol/benzyl benzoate.
Genotyping.
The Sema3AK108N allele does not encode a restriction-sensitive polymorphism; therefore, standard genotyping methods cannot be used. To assay the mutant versus wild-type single nucleotide polymorphism (SNP), we used a standard allelic discrimination on ABI 7300 using 2× Jump Start PCR mix (Sigma) to assay the ratio of the contribution of each allele. The Sema3AK108N allele is genotyped using the following primers: CTTACACAAGGAGAGATGAATGC, ACGAGAGAAGAGATCACATAGTAG and probes: TGGGCTGGAAAAGATATCCTGGTAAGC (wt), TGGGCTGGAAATGATATCCTGGTAAGC (mut).
DRG repulsion assay.
DRG explants were cocultured in collagen with 293T cells as previously described (Gu et al., 2002, 2003, 2005) with ligand concentration titrated as described previously (Cheng et al., 2001; Yaron et al., 2005).
COS cell binding and morphology assays.
Sema3A and Sema3AK108N binding assays were performed as previously described (Kolodkin et al., 1997; Gu et al., 2005) with minor variations to a 48- and 96-well format. Ligands were produced from AP-fusion proteins to both mouse and human Sema3A. AP-Sema3E and AP-Sema3EK110N collapse assays were performed as previously described (Gu et al., 2005).
Growth cone collapse assay.
Growth cone collapse assays were performed using mouse E12.5 DRG explants grown on laminin/poly-d-lysine-coated coverslips as described previously (Kapfhammer et al., 2007). Growth cones were visualized by staining with rhodamine-phalloidin (Invitrogen) for 1 h then washed and mounted with Vectashield mounting medium (Vector Laboratories).
Results
Recessive screen for axon guidance molecules
To identify novel factors essential for the development of the PNS, we have developed a three-generation, forward genetic screen for recessive mutations affecting the patterning and guidance of PNS axonal projections (Mar et al., 2005). First, 8- to 10-week-old, male, C57BL/6 (BL6) mice (F1) were intraperitoneally injected with 300 mg of ENU to randomly induce point mutations throughout the mouse genome. The ENU was delivered in a fractionated dose of 3 × 100 mg/kg administered weekly. As anticipated, all males lost fertility. Those males that regained fertility were mated to C3H/He (C3H) females to produce BL6:C3H hybrid G1 males (or lines) that are heterozygous for ∼32 gene-inactivating mutations (Justice et al., 1999, 2000; Weber et al., 2000). To isolate the mutations in the BL6 genome, these G1 males were outcrossed to C3H females to produce G2 daughters, 50% of which should also be heterozygous for any given mutation. By mating the G1 males to their G2 daughters, recessive mutations can be produced at a frequency of 12.5% (Kasarskis et al., 1998). To examine the integrity of PNS axonal projections, we performed a wholemount immunological assay using a monoclonal antibody directed against the 165 kDa isoform of neurofilament to stain the ganglia and axons of the developing sensory and motor nervous systems at E11.5–E12 (Fig. 1). By waiting 1.5–2 d later than previous studies (Mar et al., 2005), we are able to identify mutant mice exhibiting aberrations not only in the development and position of the ganglia of the PNS, but also in the growth and guidance of both the central and peripheral axonal projections originating from these ganglia.
Using this approach, we generated and screened 164 G1 lines representing ∼5200 gene-inactivating mutations. Five of these lines (Lines 808, 3329, 687, 3140, and 574) had interesting aberrations in the development and patterning of the PNS. Subsequently, the genetic lesions associated with each of these phenotypes were found to segregate in Mendelian ratios and were localized to discrete chromosomal regions. Each of the five lines exhibited distinct developmental disorders affecting unique subsets of PNS neuronal populations (Fig. 2). Mutants in Line 808 exhibited extreme defasciculation and exuberant overgrowth of axons within the ophthalmic branch of the trigeminal ganglia. In contrast, mutants in Line 3329 displayed dramatic defasciculation of all major peripheral branches of the DRG. This defasciculation observed in Line 3329 was so extreme that, while the major dorsal and lateral projections form and are distinct, all of the secondary and tertiary branches are absent. Instead, upon reaching their superficial targets, the axonal projections are splayed out almost randomly within the dermatome. As with Line 808, the most severe phenotype in 687 mutants was observed for projections of the trigeminal ganglia. These mutants derived from Line 687 were identified based on a defasciculation of the ophthalmic branch of the trigeminal ganglia. Both major branches of the ophthalmic nerve appeared to form and segregate properly and also originated as tightly bound fascicles; however, distal to the initiation point, the axons within the bundle defasciculated, occupying a more loosely defined tract and eventually spreading prematurely within the face. In addition, spinal nerves of 687 mutants, although properly formed with respect to orientation, were underdeveloped. Embryos of Line 3140 had significantly underbranched and underdeveloped spinal nerve axons. Last, the cell bodies of the dorsal root ganglia in Line 574 mutants were improperly positioned along the anterior–posterior (A–P) axis of the embryo. In general, the cell bodies of these neurons are normally located adjacent to the spinal column and coalesce to form ganglia in a regular, metameric pattern with each ganglion occupying a specific dermatome. In 574 mutants, however, the ganglia were not regularly spaced and often fused. The most anterior ganglia, specifically the cervical ganglia, were largely spared; however, all of the ganglia posterior to these seven ganglia were found to be abnormal in all 574 mutants examined. Thus, our wholemount assay allowed us to simultaneously assess the proper development of a wide array of ganglia and their projections, resulting in phenotypes limited to single ganglia as well as those with deficits in a large collection of ganglia. Moreover, our screen allows us to identify interesting phenotypes affecting the development of the PNS at a frequency of ∼1/32 G1 lines.
Figure 2.
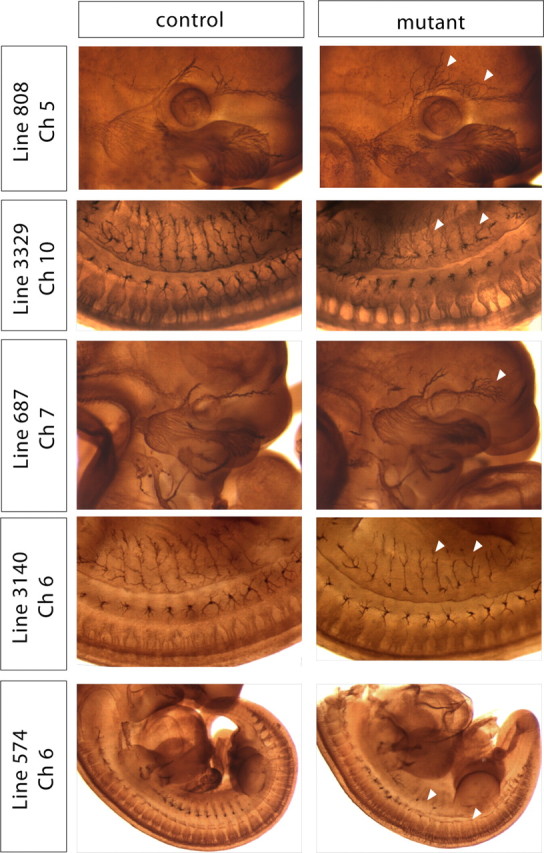
Wholemount neurofilament assay reveals several interesting allelic groups. Mutant and control embryos stained with wholemount neurofilament and examined at E11.5. Arrowheads indicate aberrant axonal projections in the mutants.
Line 808 mutants exhibit severe defasciculation of trigeminal ganglia and DRG axons
Mutants in Line 808 exhibited extreme defasciculation and exuberant overgrowth of axons within the ophthalmic branch of the trigeminal nerve. Compared with their littermate controls, ophthalmic branch axons in mutant embryos grew past their appropriate targets and extended farther anterior into the head of the embryo (Fig. 3A). In addition, the spinal nerves in these mutants were defasciculated and overgrown within their target fields. Last, sensory and motor projections of the limb within the ulnar and radial nerves were found to extend beyond their appropriate targets, traveling in bundles that were disorganized and defasciculated. These phenotypes were robust, completely penetrant and expressive and were easily scored for the purpose of genetic mapping.
Figure 3.

Line 808 harbors a novel allele of Sema3A. A, Wholemount neurofilament staining of 808 mutant and control embryos showing the region of the face containing the trigeminal ganglia and the projections of the ophthalmic branch. B, Schematic diagram of the chromosomal region found to contain the mutation in Line 808, the markers used to diagnose linkage, and the frequency of recombination observed at those markers. C, Sequence data highlighting the mutation Sema3A 324A>T observed in 808 mutants compared with BL6 wild-type DNA. D, Schematic diagram of Sema3A. The 808 point mutation induces a single amino acid substitution K108N in the Sema domain of Sema3A. E, Sema3AK108N fails to complement the null allele. Neurofilament staining of E11.5 embryos from a Sema3A transheterozygous cross. Sema3A−/K108N embryos also display a defasciculation phenotype.
Line 808 is a novel allele of Sema3A
The defasciculated ophthalmic branch phenotype segregated in Mendelian ratios with 55% (31/56) of G2/N3 females carrying the allele and the expected 12% (56/459) of embryos screened displaying the defasciculation phenotype (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To generate recombinant meioses, Line 808 was mapped via backcrossing to C3H mice. Given the similarity of phenotypes observed among Line 808 and Sema3A mutants (Behar et al., 1996; Taniguchi et al., 1997) and in a mutant allele of the Sema3A receptor, Npn-1, that is incapable of binding Sema3A (Gu et al., 2003), we first checked for linkage within regions of the mouse genome that contain these genes: proximal chromosome 5 and distal chromosome 8, respectively. Using PCR polymorphisms, Npn-1 was eliminated and linkage was instead established to a large (∼25 Mb) chromosomal region containing several class 3 semaphorins (between D5MIT346 and D5MIT148; Fig. 3B).
Additional polymorphisms contained in this region of chromosome 5 allowed us to identify a smaller region between rs3714258 and rs6404215 (<2 Mb) that contains Sema3A and in which no recombinants were observed (Fig. 3B). This clearly established Sema3A as an excellent candidate for the Line 808 gene; thus, we sequenced all of the coding exons of Sema3A. DNA was amplified from two mutants and also from BL6 DNA as a control. The mutant locus was found to contain a single base pair substitution in the end of exon 3 coded by 324A>T (Fig. 3C). This nucleotide change results in a single amino acid substitution, K108N, located in the Sema domain of Sema3A (Fig. 3D). To verify that this amino acid substitution was indeed responsible for the phenotypes we observed in this mutant line, we crossed Sema3A heterozygotes (Sema3A+/−) (Behar et al., 1996) with the Sema3A+/K108N mouse line to obtain embryos that were Sema3A−/K108N. Indeed, in these trans-heterozygous embryos we observed the same ophthalmic nerve phenotype found in Sema3A−/− and, most importantly, in Sema3AK108N/K108N mutant embryos (Fig. 3E). Interestingly, homozygous Sema3AK108N/K108N mice are viable, survive into adulthood, and are fertile when maintained in a C3H genetic background. Two null alleles of Sema3A have been made−one (Behar et al., 1996) was found to be embryonic lethal and the other (Taniguchi et al., 1997) to be adult viable. These lines have been propagated in different backgrounds suggesting that this variation is due to the genetic background of the inbred mouse strains.
In vitro analysis of Sema3A K108N function
Based on the similarity between Sema3AK108N mutant embryos and age-matched Sema3A-null mutant embryos, we assayed the ability of Sema3AK108N to function as a repulsive guidance cue in vitro. Alkaline phosphatase fusion proteins with mouse Sema3A and mouse Sema3AK108N (AP-Sema3A and AP-Sema3AK108N, respectively) were overexpressed in 293T cells and cocultured in a collagen gel matrix with wild-type DRG explants (Messersmith et al., 1995; Yaron et al., 2005). Both fusion proteins were highly expressed in 293T cells and were secreted, suggesting that K108N does not grossly affect the expression, stability or secretion of the ligand. Using this assay, all dilutions of AP-Sema3A-expressing cells tested repelled DRG axons on the proximal side of the ganglia (Fig. 4A–C,G,H). In contrast, DRG explants show very little responsiveness to Sema3AK108N [proximal to distal (P/D) ratio, Sema3A 0.1 ± 0.04, Sema3AK108N 0.59 ± 0.11, p < 0.001, Student's t test] (Fig. 4D–F,H). At a 1:8 dilution of ligand-expressing cells, Sema3A (Fig. 4C,H) still repels DRG axons (P/D ratio 0.68 ± 0.1, p < 0.001, Student's t test), however Sema3AK108N does not (Fig. 4F,H) (P/D ratio 0.96 ± 0.08, p > 0.05, Student's t test). Furthermore, similar results were observed with respect to DRG growth cone collapse. DRG explants from E12.5 mouse embryos were incubated with medium containing AP (Fig. 4I), Sema3A (Fig. 4J) or Sema3AK108N (Fig. 4K) at concentrations of 0.1, 1.0 or 10.0 nm (Fig. 4L). While all concentrations of Sema3A led to robust collapse of DRG neuron growth cones, Sema3AK108N did not induce growth cone collapse at concentrations of 0.1 or 1.0 nm (collapsed growth cone Sema3AK108N 27 ± 8%, AP control 23 ± 4%, p > 0.05, Student's t test). Even at a very high concentration of 10 nm, Sema3AK108N induced only extremely modest growth cone collapse (Sema3AK108N 38 ± 8%, AP control 25 ± 4%, p < 0.05, Student's t test). Thus, Sema3AK108N is greatly impaired in its ability to repel axons and collapse the actin cytoskeleton of DRG sensory neuron growth cones.
Figure 4.
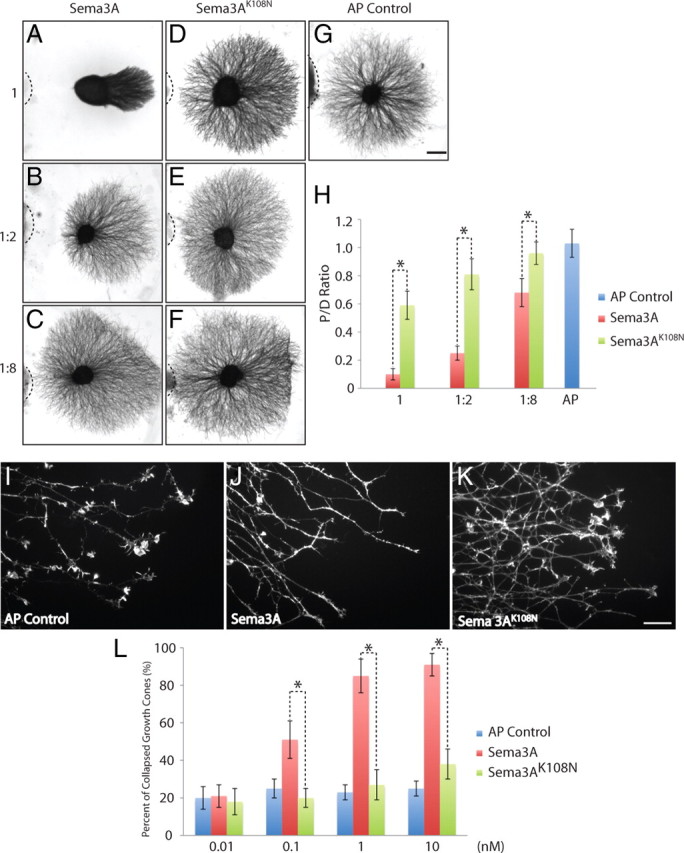
Sema3AK108N is deficient in DRG axonal repulsion and growth cone collapse in vitro. A–C, Coculture of DRG explants with different dilutions of Sema3A transfected cell aggregates. D–F, Coculture of DRG explants with different dilutions of Sema3AK108N transfected cell aggregates. G, DRG axon outgrowth in the presence of AP control construct transfected cell aggregates. H, Quantitation of P/D outgrowth ratios for explants cocultured with different dilutions of Sema3A and Sema3AK108N. At all three tested dilutions, Sema3AK108N showed a statistically significant difference from Sema3A in its effects on DRG axon repulsion (p < 0.001, Student's t test). I–K, Morphology of DRG growth cones after exposure to 1 nm AP control, Sema3A or Sema3AK108N medium for 30 min. L, Quantitation of the dose-dependent collapse of DRG growth cones in response to AP control, Sema3A and Sema3AK108N. Percentage of collapsed growth cones are shown. Sema3AK108N does not collapse DRG growth cones at concentrations of 0.01, 0.1 and 1 nm (p > 0.05, Student's t test), and shows significant differences compared with Sema3A at all concentrations tested (p < 0.01, Student's t test). All experiments were repeated three times, each time using at least four explants; for growth cone collapse, ∼100 growth cones were counted from each of at least four separate explants. Representative images are shown. Scale bars: A–G (in G), 200 μm; I–K (in K), 20 μm.
Receptor binding capacity of Sema3A K108N
Given the in vivo loss-of-function phenotype and the failure of Sema3AK108N to induce axon repulsion and growth cone collapse in vitro, we sought to test the role of the K108N substitution on the ability of Sema3A to function as a ligand. To determine whether Sema3AK108N could bind Npn-1, its high-affinity binding partner in the holoreceptor complex, AP-Sema3A or AP-Sema3AK108N-containing media was applied to COS-1 cells overexpressing Npn-1. Strikingly, we observed no apparent difference in the level of AP-Sema3A-Npn-1 and AP-Sema3AK108N-Npn-1 binding using this assay (Fig. 5A). To determine whether there were more subtle differences between AP-Sema3A and AP-Sema3AK108N binding to Npn-1, we quantified the amount of bound ligand as a function of concentration of ligand applied to uniformly transfected COS-1 cells (Gu et al., 2005). Again, we were unable to observe a difference between AP-Sema3A and AP-Sema3AK108N binding to Npn-1 (Fig. 5B). The sensitivity of this assay does not allow us to rule out very subtle differences in binding between AP-Sema3A-Npn-1 and AP-Sema3AK108N-Npn-1, but it does indicate that these molecules are capable of binding Npn-1 with comparable affinities. Additionally, we compared the ability of AP-Sema3A and AP-Sema3AK108N to bind to endogenous receptor complexes expressed on the surface of cultured DRG neurons. In contrast to findings in the COS cell binding experiments, we observed a modest, 35% reduction in surface labeling of AP-Sema3AK108N compared with AP-Sema3A (Fig. 5C). Thus, AP-Sema3AK108N binds normally to Npn-1 but is impaired in its ability to engage receptor complexes expressed on the cell surface of DRG sensory neurons.
Figure 5.

Sema3A and Sema3AK108N exhibit similar binding affinities to Npn-1. A, COS-1 cells transfected with Npn-1, or mock transfected, and incubated with AP-Sema3A or AP-Sema3AK108N. Enzymatic detection of binding indicates that at 1 nm concentrations both ligands bind to the transfected cells, but not to the untransfected cells, with comparable affinity. B, Graphical representation of the amount of AP-Sema3A or AP-Sema3AK108N bound to COS-1 cells transfected with Npn-1 as a function of the amount of ligand applied. C, Differences between levels of AP-Sema3A and AP-Sema3AK108N binding to dissociated DRG neurons grown in culture. Dissociated DRG neurons were incubated with AP control ligand, AP-Sema3A or AP-Sema3AK108N (1 nm). Results shown are averages and SEM of three independent experiments. The background AP control ligand binding levels were <14% that of Sema3A ligands, and were subtracted from the values for AP-Sema3A and AP-Sema3AK108N; 1 U = change OD405/h.
In silico modeling of K108 in semaphorin signaling
K108 is conserved among all members of the class 3 secreted semaphorins (Fig. 6A) and among all known plexin-binding semaphorins but is not found in Sema7a, which interacts with integrins and may not signal through a plexin (Fig. 6B). Thus, we used the crystal structure of Sema3A (Antipenko et al., 2003) to evaluate the function of K108 in silico. K108 (blue) is found in an interblade helical insertion between blades 1 and 2 and is present on the surface of Sema3A (Fig. 7A). The side chain nitrogen of K108 (blue) is not within salt-bridge or hydrogen bond donor distance of any typical acceptor. Instead, K108 packs the aliphatic portion of its side-chain against the aromatic side-chains of F137 and F194 (yellow), from blades 2 and 3 of the propeller, respectively (Fig. 7B). The interblade insertion is internally stabilized by a disulfide bond (red C103-C114; Fig. 7B); therefore, any perturbation caused by mutation of K108 is likely to be local rather than global. Indeed, computational analysis of the K108N mutation predicts that the point mutation would be energetically favorable (ΔΔG = 1.49 kcal/mol) (Parthiban et al., 2006). Mutation to asparagine could function by altering the precise orientation of the basic nitrogen at the end of K108. Alternatively, K108 could contribute to the interaction surface between the interblade insertion and the core Sema domain, and K108 may function in a global sense as a “snap” to orient the interblade insertion in a fixed conformation with respect to the rest of the protein.
Figure 6.

K108N is conserved between all secreted and plexin-binding semaphorins. A, ClustalW alignment of class 3 semaphorins showing K108 (red highlight) conserved among all family members. B, ClustalW alignment of vertebrate semaphorins showing K108 (red highlight) conservation among all family members excluding Sema7a. This conservation extends to invertebrate semaphorins (data not shown).
Figure 7.

K108N is located in an interblade insertion and directly contacts the core Sema domain. A, K108 (blue) is within an insertion located between blades 1 and 2 of Sema3A and oriented parallel to one face of the Sema domain (PDB = 1Q47). B, K108 interacts with F137 and F194 (yellow) from blades 2 and 3, respectively. Additionally, the interblade insertion is stabilized by a disulfide bond (red).
Sema3AK108N maintains high affinity binding to Npn-1 yet is greatly impaired in its ability to signal for growth cone collapse, suggesting that K108 may be an essential residue for the secondary semaphorin–plexin interaction within the holoreceptor complex. Antipenko et al. (2003) used sequence conservation patterns and mutagenesis to identify a region of Sema3A necessary for its interaction with Npn-1. The identified site overlaps with the semaphorin dimerization region but is distant from K108, making direct interaction between K108 and the major binding partner of the holoreceptor complex unlikely. However, regions of blade 3 may function as specificity determinants for direct plexin–semaphorin interactions in other secreted semaphorins. Interestingly, with a segment of the sema domain containing K108 was previously shown to impart specificity to secreted semaphorins (Koppel et al., 1997). K108 is adjacent to blade 3—K108 directly contacts F194 from this region—suggesting that the K108N substitution disrupts a plexin–semaphorin interaction in the context of the functional neuropilin–plexin–semaphorin complex (Fig. 8).
Figure 8.

K108N is remote from the Npn-1 binding site of Sema3A but near to the predicted plexin-binding region of the protein. Surface features of Sema3A reveal the location of K108 (blue) relative to the known semaphorin dimerization and Npn binding loops (yellow and red) and predicted blade 3 plexin interaction surface (green).
K108 signaling in other class 3 semaphorins
Given that the K108 residue is essential for Npn-dependent semaphorin signaling, we experimentally tested whether K108 functions as an essential part of the ligand–receptor interaction surface of other semaphorins. To investigate this possibility, we induced homologous substitutions in a Npn-dependent class 3 semaphorin (Sema3FK109N) and in a Npn-independent class 3 semaphorin (Sema3EK110N). Using AP fusion proteins, we assessed the effect of these mutations on the ability of these ligands (AP-Sema3FK109N and AP-Sema3EK110N) to bind their high-affinity receptors, Npn-2 and PlexinD1, respectively. We observed no difference in the ability of AP-Sema3F or AP-Sema3FK109N to bind COS-1 cells transfected with Npn-2. Furthermore, we observed no difference in the ability of AP-Sema3E and AP-Sem3EK110N to bind to COS-1 cells transfected with PlexinD1.
We then tested the ability of the mutant secreted semaphorin proteins to activate downstream signaling. To this end, SCG explants from postnatal day 1 (PN1) mice were dissected and cocultured with cell aggregates transfected with either wild-type Sema3F or mutant Sema3F for 72 h (Fig. 9). The response of SCG axons to Sema3FK109N (Fig. 9C) is significantly abrogated compared with Sema3F (Fig. 9B) (P/D ratio, Sema3F 0.18 ± 0.05, Sema3FK109N 0.96 ± 0.07, p < 0.001, Student's t test). Similar to AP–control transfected cell aggregates, Sema3FK109N cell aggregates do not repel axons of SCG explants (Fig. 9A,C) (P/D ratio, AP control 0.98 ± 0.11, p > 0.05, Student's t test). Next, we tested the possibility that mutant AP-Sema3E is able to bind PlexinD1 but not activate the PlexinD1 receptor, by assaying the ability of AP-Sema3EK110N to induce collapse in PlexinD1-expressing COS-7 cells (Gu et al., 2005). Strikingly, mutant Sema3E potently induced COS-7 cell collapse. We observed no difference in the ability of either AP-Sema3E or AP-Sema3EK110N to induce collapse in COS cells when 0.1, 0.5 or 1 nm concentrations were incubated with the cells transfected with PlexinD1 (Fig. 9E). Thus, the residue corresponding to Sema3A K108 is essential for Npn-dependent signaling but is dispensable for Npn-independent signaling. These data support the idea that the conserved region containing K108 is a secondary semaphorin-plexin binding site in the holoreceptor complex.
Figure 9.
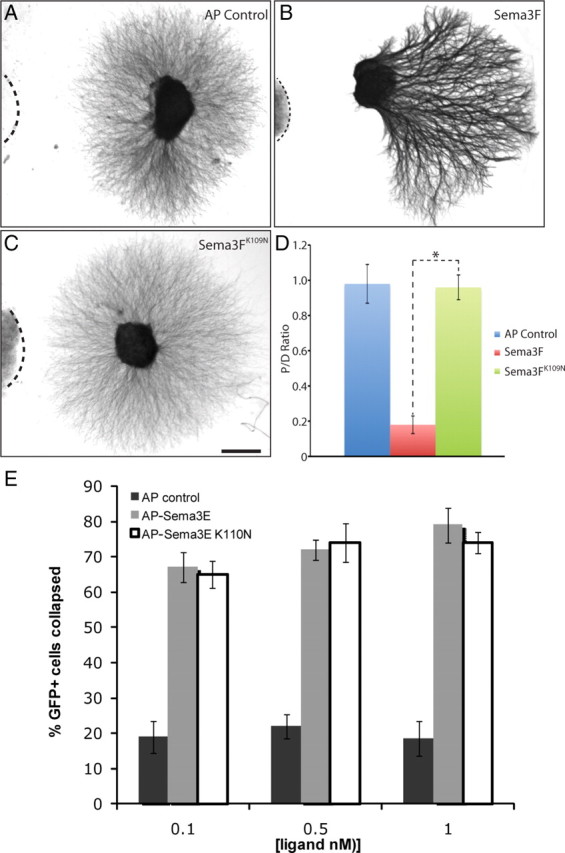
Sema3FK109N is unable to repel SCG axons whereas Sema3EK110N is able to collapse PlexinD1-expressing COS-7 cells. A–C, Coculture of SCG explants with COS cell aggregates transfected with indicated constructs. D, Quantitation of P/D axon outgrowth ratio in SCG explants. Sema3FK109N showed a significantly reduced ability to repel SCG axons in vitro compared with Sema3F (p < 0.001, Student's t test). All experiments were repeated twice, each time with at least 3 explants. E, COS-7 cells transfected with expression vectors encoding EGFP and PlexinD1 were incubated with AP control ligand (AP alone), AP-Sema3E or AP-Sema3EK110N. Treatment of cells with either AP-Sema3E or AP-Sema3EK110N led to collapse of COS-7 cells. Representative images of SCG explants are shown. Scale bar: A–C (in C), 200 μm.
Discussion
The connectivity of the adult nervous system underscores the complexity of processes necessary to establish this pattern. The number of cell-intrinsic and extrinsic factors required for neural development is large. Current tools of molecular biology and genomics allow us to survey the entire transcriptional profile of single cells allowing for a catalog of all the genes expressed in a particular cell type throughout the course of development to be determined. This list can be culled into interesting candidates, and animals genetically mutant at each of these independent loci can be created. Yet, the task of surveying each cell type and creating thousands of animals is daunting, particularly given that only a small subset of the genome is likely to be involved in any one specific process. As an alternative to candidate-based approaches for surveying developmental effectors, a phenotype-based, forward genetic approach can be used to survey the contribution of both spontaneous and induced mutations toward a developmental event.
We have designed and implemented a forward genetic screen to identify novel alleles critical for the developing PNS. Using a wholemount assay to label PNS neuron axons, we have identified several allelic groups with a wide array of phenotypes each affecting subsets of neurons. With each G1 mouse line, we anticipated the capacity to screen ∼32 novel inactivating mutations. Thus, for 164 lines generated and screened using our assay we sampled the roles of ∼5248 gene-inactivating mutations within the developing mouse embryo. These mutations are random and can include multiple sampling events within the same locus. Nonetheless, we have recovered interesting phenotypes in 5 of 164 of our G1 lines. We mapped each of these phenotypes to small chromosomal regions and observed clear segregation according to Mendelian ratios. Our analysis of mice exhibiting these aberrant phenotypes uncovered mutations in previously uncharacterized genes as well as interesting mutations in genes encoding known axon guidance molecules. Using this screen, we identified Sema3AK108N, a novel loss-of-function allele of Sema3A. Mice harboring the Sema3AK108N allele are homozygous viable but share guidance deficits within the PNS that are comparable to those observed in homozygous embryos in Sema3A-null alleles (Behar et al., 1996; Taniguchi et al., 1997). Previous characterized alleles of Sema3A vary with respect to viability, and these effects are likely the result of variation in mouse genetic background (Taniguchi et al., 1997). Sema3AK108N mutants, like both Sema3A-null mutants, have exuberant overgrowth of the sensory neurons within the developing DRG and the trigeminal ganglia.
Sema3AK108N encodes a mutant form of Sema3A that is stable and secreted. Given the robust loss-of-function phenotypes seen of Sema3AK108N mutants, it was surprising to find that Sema3AK108N retains its ability to bind with high affinity to its receptor Npn-1 in vitro. Indeed, in silico modeling suggests that K108 is distal to the site of Sema3A–Npn-1 interaction. Instead, we suggest that K108 is an essential component of a Sema3A–Plexin interaction site. Further experiments are needed to compare and contrast the semaphorin–plexin interaction surfaces in both Npn-independent and -dependent systems. It is possible that two distinct surfaces on the semaphorin exist, representing high- and low-affinity sites, that mediate Npn-independent and -dependent binding to plexins, respectively. Alternatively, it is possible that the high-affinity semaphorin–plexin interaction site in Npn-independent systems is simply an extended version of a secondary low-affinity site found in Npn-dependent semaphorins. This second possibility seems likely given that K108 is conserved not only among all members of the class 3 secreted semaphorins but among all known plexin-binding semaphorins. Interestingly, K108 is not found in Sema7a, which signals guidance responses via interactions with integrins.
In addition to providing insights into the possible site of sema–plexin interaction within the holoreceptor complex, Sema3AK108N may provide an interesting mouse model in which to study Sema3A plexin-independent developmental events. Previous studies have proposed a Sema3A–Npn-1–L1 receptor complex critical for the development of DRG projection and neurons of the developing cortex. It is possible that comparisons between Sema3AK108N and Sema3A-null alleles might further elucidate the role of L1 in semaphorin signaling.
In conclusion, we have identified a novel allele of Sema3A, Sema3AK108N, which abolishes Sema3A signaling in vivo. Our in vitro analysis suggests that this amino acid substitution does not globally affect the expression or stability of the protein. Moreover, Sema3AK108N appears to be able to bind its receptor, Npn-1, with high affinity. We hypothesize that K108N alters the interblade region between blades 1 and 2 of the Sema domain and affects the ability of the mutant Sema3A to interact with PlexinA in the Npn-1–PlexA holoreceptor complex. Thus, this novel Sema3A allele establishes a new mouse model system for understanding the roles of Sema3-Npn-1-Plexin signaling during mouse development and in the adult.
Footnotes
This work was supported by National Institute of Mental Health Grant R01 MH59199 (D.D.G., A.L.K.), the Johns Hopkins Brain Sciences Institute (D.D.G., A.L.K.), National Cancer Institute Grant R01 CA090466 (D.J.L.), and National Center for Research Resources Grant P20 RR020171 from the National Institutes of Health (C.W.V.K.). D.D.G. and A.L.K. are investigators of the Howard Hughes Medical Institute. We thank members of the Ginty and Kolodkin laboratories for assistance and discussions throughout the course of this project. The 2H3 monoclonal antibody developed by T. M. Jessell and J. Dodd was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
References
- Antipenko A, Himanen JP, van Leyen K, Nardi-Dei V, Lesniak J, Barton WA, Rajashankar KR, Lu M, Hoemme C, Püschel AW, Nikolov DB. Structure of the semaphorin-3A receptor binding module. Neuron. 2003;39:589–598. doi: 10.1016/s0896-6273(03)00502-6. [DOI] [PubMed] [Google Scholar]
- Behar O, Golden JA, Mashimo H, Schoen FJ, Fishman MC. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature. 1996;383:525–528. doi: 10.1038/383525a0. [DOI] [PubMed] [Google Scholar]
- Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, Lowenstein DH, Skarnes WC, Chédotal A, Tessier-Lavigne M. Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron. 2000;25:43–56. doi: 10.1016/s0896-6273(00)80870-3. [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, Tessier-Lavigne M. Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron. 2001;32:249–263. doi: 10.1016/s0896-6273(01)00478-0. [DOI] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- Gu C, Limberg BJ, Whitaker GB, Perman B, Leahy DJ, Rosenbaum JS, Ginty DD, Kolodkin AL. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002;277:18069–18076. doi: 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin A, Ginty DD. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307:265–268. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8:1955–1963. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Justice M, Carpenter D, Favor J, Neuhauser-Klaus A, Hrabé de Angelis M, Soewarto D, Moser A, Cordes S, Miller D, Chapman V, Weber JS, Rinchik EM, Hunsicker PR, Russell WL, Bode VC. Effects of ENU dosage on mouse strains. Mamm Genome. 2000;11:484–488. doi: 10.1007/s003350010094. [DOI] [PubMed] [Google Scholar]
- Kapfhammer JP, Xu H, Raper JA. The detection and quantification of growth cone collapsing activities. Nat Protoc. 2007;2:2005–2011. doi: 10.1038/nprot.2007.295. [DOI] [PubMed] [Google Scholar]
- Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci U S A. 1998;95:7485–7490. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, Yagi T, Fujisawa H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997;19:995–1005. doi: 10.1016/s0896-6273(00)80392-x. [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- Koppel AM, Feiner L, Kobayashi H, Raper JA. A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron. 1997;19:531–537. doi: 10.1016/s0896-6273(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Mar L, Rivkin E, Kim DY, Yu JY, Cordes SP. A genetic screen for mutations that affect cranial nerve development in the mouse. J Neurosci. 2005;25:11787–11795. doi: 10.1523/JNEUROSCI.3813-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith EK, Leonardo ED, Shatz CJ, Tessier-Lavigne M, Goodman CS, Kolodkin AL. Semaphorin III can function as a selective chemorepellent to pattern sensory projections in the spinal cord. Neuron. 1995;14:949–959. doi: 10.1016/0896-6273(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239–W242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohm B, Ottemeyer A, Lohrum M, Püschel AW. Plexin/neuropilin complexes mediate repulsion by the axonal guidance signal semaphorin 3A. Mech Dev. 2000;93:95–104. doi: 10.1016/s0925-4773(00)00269-0. [DOI] [PubMed] [Google Scholar]
- Roth L, Koncina E, Satkauskas S, Crémel G, Aunis D, Bagnard D. The many faces of semaphorins: from development to pathology. Cell Mol Life Sci. 2009;66:649–666. doi: 10.1007/s00018-008-8518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semaphorin Nomenclature Committee. Unified nomenclature for the semaphorins/collapsins. Cell. 1999;97:551–552. doi: 10.1016/s0092-8674(00)80766-7. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/s0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, Yagi T. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron. 1997;19:519–530. doi: 10.1016/s0896-6273(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Tran TS, Kolodkin AL, Bharadwaj R. Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol. 2007;23:263–292. doi: 10.1146/annurev.cellbio.22.010605.093554. [DOI] [PubMed] [Google Scholar]
- Weber JS, Salinger A, Justice MJ. Optimal N-ethyl-N-nitrosourea (ENU) doses for inbred mouse strains. Genesis. 2000;26:230–233. [PubMed] [Google Scholar]
- Yaron A, Huang PH, Cheng HJ, Tessier-Lavigne M. Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron. 2005;45:513–523. doi: 10.1016/j.neuron.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]