

Introduction
Pulmonary arterial hypertension (PAH) is a syndrome in which pulmonary arterial obstruction increases pulmonary vascular resistance leading to right ventricular (RV) failure and a 15%/year mortality rate. This review highlights recent advances in the Basic Science of pulmonary arterial hypertension (PAH). New concepts clarify the nature of PAH and provide molecular blueprints that explain how PAH is initiated and maintained. Five basic science concepts provide a framework to understand and treat PAH. 1: Endothelial dysfunction creates an imbalance favoring vasoconstriction, thrombosis, and mitogenesis. Restoring this balance by inhibiting endothelin and thromboxane or augmenting nitric oxide and prostacyclin is the paradigm upon which most current therapy is based. 2: PAH has a genetic component. Mutations (Bone morphogenetic protein receptor-2, BMPR-2) and single nucleotide polymorphisms (ion channels and transporter genes) predispose to PAH. 3: Excess proliferation, impaired apoptosis and glycolytic metabolism in pulmonary artery smooth muscle, fibroblasts and endothelial cells suggest analogies to cancer. Many experimental therapies reduce PAH by decreasing the proliferation/apoptosis ratio, including inhibitors of: pyruvate dehydrogenase kinase, serotonin transporters, survivin, 3-hydroxy-3-methylglutaryl-coenzyme A reductase, transcription factors (hypoxia-inducible factor-1α and nuclear factor activating T-lymphocytes) and tyrosine kinases. Augmentation of voltage-gated K+ channels, Kv1.5 and BMPR-2 signaling also address this imbalance. Tyrosine kinase inhibitors used to treat cancer are in Phase-1 PAH trials. 4: Refractory vasoconstriction may occur due to rho kinase activation. Only <20% of PAH patients respond to conventional vasodilators; however refractory vasoconstriction may respond to rho kinase inhibitors. 5: The RV can be therapeutically targeted. Although increased afterload initiates RV failure, the major cause of death/dysfunction in PAH, the RV may be amenable to cardiac-targeted therapies. The RV in PAH has features of ischemic, hibernating myocardium.
Guided by these new concepts and armed with better understanding of disease mechanisms we are poised to identify new therapeutic targets. To achieve balance in a rapidly evolving field, we invited colleagues to contribute figures illustrating pathways in their area of expertise that are important to the pathogenesis and treatment of PAH. These contributors are acknowledged in the Figure legends.
Epidemiology
There are 5 categories of pulmonary hypertension in the latest World Health Organization's classification of pulmonary hypertension (PH): 1: PAH, 2: PH associated with left heart disease, 3: PH associated with lung disease/hypoxia, 4: thromboembolic PH, 5: miscellaneous1. This review focuses on Category 1 (PAH), which includes: idiopathic and familial PAH, as well as PAH associated with a variety of conditions (including connective tissue diseases and congenital heart disease), pulmonary venoocclusive disease, pulmonary capillary hemangiomatosis, and persistent pulmonary hypertension of the newborn. The incidence and prevalence of PAH is estimated at 2.4 cases/million/year and 15 cases/million in France2 and 7.6 cases/million/year and 26 cases/million in Scotland3. The global prevalence of PAH is hard to estimate because accurate diagnosis of PAH is difficult and access to care is limited in many countries. Because diseases which are risk factors for PAH, such as HIV, schistosomiasis and sickle cell disease, are more prevalent in the developing world, the global burden of PAH is likely greater than is currently recognized4. In developed countries prevalence will also likely increase as newer associations with PAH emerge, including dialysis5 and the metabolic syndrome6, and as widespread access to echocardiography identifies PAH earlier and in more individuals.
Definition
PAH is a small subset of pulmonary hypertensive syndromes (WHO categories 2-5). PAH is defined by a resting mean pulmonary artery pressure (PAP) >25mmHg, pulmonary vascular resistance (PVR) >3 Wood units, and pulmonary capillary wedge pressure <15mmHg (in the absence of other causes of PH). Unlike PAH, PH is ubiquitous, the sole diagnostic criterion being a resting mean PAP >25mmHg. This larger PH group often does not have intrinsic pulmonary vascular disease. Their PH is due to high flow, elevated left ventricular end diastolic pressure, lung disease/hypoxia or valve disease. There is no randomized clinical trial evidence that WHO category 2-5 patients benefit from PAH-specific therapies.
Prognosis
The 1-year, incident mortality rate of PAH remains high (15%), despite treatment with prostacyclin, endothelin antagonists and phosphodiesterase-5 inhibitors7. Because PAH is a syndrome, it is not surprising that the prognosis varies depending on the associated comorbid conditions. Prognosis in PAH associated with congenital heart disease tends to be better than in idiopathic PAH (iPAH) (3-year survival rates 77% versus 35%)8, although other cohorts found similar survival in iPAH to be 65% at 3 years9. PAH associated with scleroderma has 3-year survival rates of only 34-47%10, 11.
Current Therapies
Treatment of PAH involves the use of prostanoids (given intravenously, by inhalation, subcutaneously or orally), endothelin receptor blockers, and/or phosphodiesterase 5 inhibitors. L-type calcium channel blockers (e.g. nifedipine) can be effective but are only safe for use in patients who respond to an acute vasodilator challenge with a >20% fall in mean PAP and no decline in cardiac output (a subset representing 12%-20% of PAH patients)12. Most patients empirically receive anticoagulation to prevent thrombosis in situ and diuretics to limit edema. PAH treatments remain expensive and/or difficult to deliver and are more palliative than curative. A year of sildenafil is estimated to cost $13,000 versus ∼$56,000 for bosentan while costs for inhaled iloprost and intravenous prostacyclin exceed $90,000/year. The only randomized PAH study that has shown survival benefit used intravenous Flolan, which, compared to conventional therapy, decreased mortality in 81 WHO Class IV patients13. Thus there is a pressing need for less expensive and more effective therapies.
Most current treatments (prostacylin, endothelin antagonists and warfarin) address endothelial dysfunction by augmenting vasodilator and anti-proliferative mediators and inhibiting vasoconstrictor, prothrombotic and mitogenic pathways. Our increasing knowledge of the cellular and molecular basis of PAH suggests many potential new therapeutic agents.
Histology
The histological findings in PAH include intimal hyperplasia, medial hypertrophy, adventitial proliferation/fibrosis, occlusion of small arteries, thrombosis in situ, and infiltration of inflammatory/progenitor cells. Angioproliferative ‘plexiform’ lesions are found in PAH, but not in other PH categories (Figure 1). Plexiform lesions (and other complex lesions) are often located downstream from occluded arteries and express the transcription and growth factors typically seen in angiogenesis, including vascular endothelial growth factor (VEGF) and hypoxia inducible factor (HIF-1α)14 (Figure 2). PAH typically spares the airway, veins, bronchial circulation, capillaries and systemic vasculature (Figure 1). The various histological abnormalities of PAH are heterogeneous in their distribution and prevalence within the lungs. The natural progression of lesion severity (presumably from medial hypertrophy to plexiform arteriopathy) and the functional relevance of plexiform lesions remain uncertain, although regression of histologically proven PAH has been documented following single lung transplantation15. Human lung tissue is invaluable, offering cells for culture, histological sections for immunohistological assessment of pathogenetic pathways, and tissue to be mined by laser capture microdissection for biomarkers. It remains the gold standard against which to judge animal models.
Figure 1. Histology of PAH.
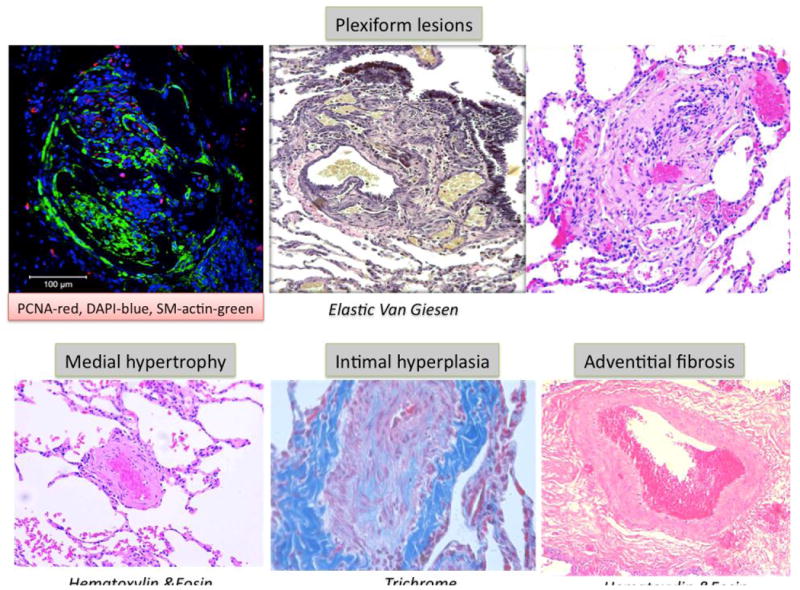
Upper panels show plexiform lesions. Left panel- note the evidence of cell proliferation (red is proliferating cell nuclear antigen, green is smooth muscle actin, blue is DAPI). Lower panels show medial hypertrophy, intimal fibrosis and adventitial proliferation.
Figure and legend contributed by Dr. S.L. Archer
Figure 2. Formation of complex and plexiform lesions in PAH.
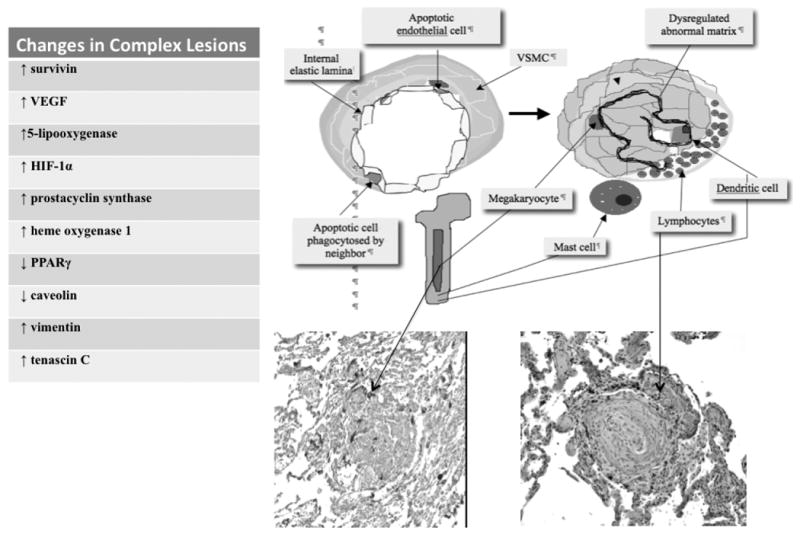
Transformation of an arteriole, into a complex vascular lesion with near total or total lumen obliteration, usually occurs at a vessel bifurcation. The concept depicted is one of initial apoptosis of cells forming the endothelial monolayer (left). Disorganized endovascular angiogenesis results from proliferation of phenotypically abnormal cells due to 1) phagocytosis of apoptotic monolayer endothelial cells by neighboring endothelial cells 2) activation of stem cell-like endothelial cells or 3) attachment of bone-marrow derived “repair cells” to the injured endothelium. Bone marrow participation in the formation of these lesions is postulated because megakaryocytes, mast cells and dendritic cells can be released and attach to the injured vessel. Perivascular lymphocytes may cluster in the lymphatics adjacent to the adventitia. The lesion also shows a dysregulated matrix. Growth factors released by megakaryocytes and mast cells may contribute to the angiogenic growth and T and B-lymphocytes may reflect a local immune response. The table insert lists the phenotypic changes seen in plexiform lesions. Figure and legend contributed by Dr. Norbert Voelkel, Virginia Commonwealth University, Richmond, VA.
Animal Models
The evaluation of these novel targets occasionally involves the off-label use of drugs approved for another indication (e.g. Gleevec), but is largely based on studies in cellular and animal models. Cautious interpretation of preclinical studies is mandatory and one must recognize the strengths/weakness of various animal models and the risks of extrapolation to humans with PAH. Notably, no animal model completely recapitulates human PAH. Promising rodent models include: monocrotaline-treated rats ± pneumonectomy16 or abdominal aortocaval shunt17, fawn-hooded rats (which spontaneously develop PAH and are also hypoxia-sensitive)18, 19 and rats treated with a single dose of VEGF-receptor antagonist (SU5416) plus hypoxia20. Models combining multiple insults yield more severe PAH with better hemodynamic and histological fidelity to human PAH. This may be relevant to the pathogenesis of human PAH, which also appears to require multiple “hits”. Murine models of PAH offer mechanistic insight on the relevance of single genes. Mice that transgenically overexpress serotonin transporters (SERT)21, bone morphogenetic protein receptor (BMPR-2) dominant-negative mutations22 or S100A4/Mts123 develop PH.
New Paradigms
PAH was once regarded largely as a disease of excess vasoconstriction. This view was incomplete and new concepts help us understand the fundamental causes of this syndrome.
PAH is a panvasculopathy (Figure 3)
Figure 3. PAH is a panvasculopathy.
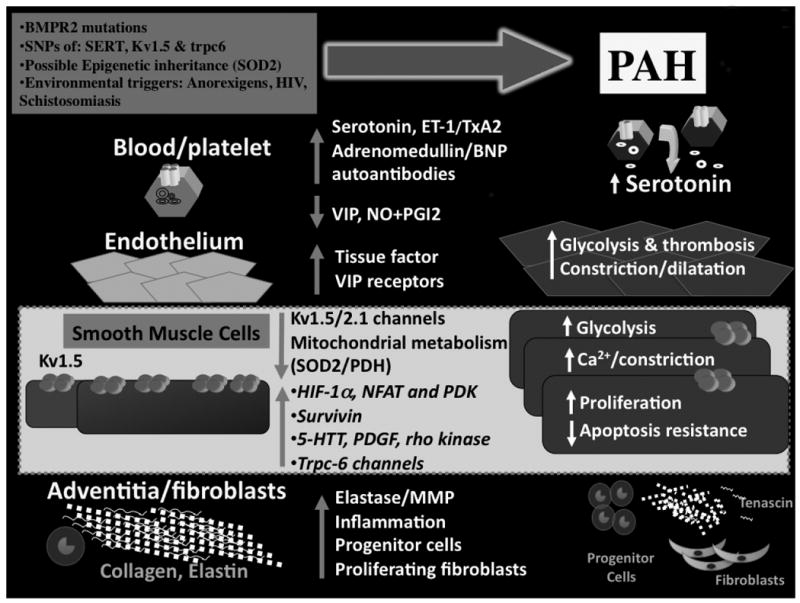
Abnormalities can be seen at each level of the small pulmonary arteries, beginning in the blood and travelling outward to the adventitia. While most of these abnormalities are likely secondary (rather than being the initiating cause of PAH), they nonetheless offer interesting therapeutic targets
Contributed by Dr. S.L. Archer
Let's take a tour of the molecular pathology of PAH, beginning at the lumen of a small pulmonary artery (PA). In the blood, levels of serotonin, a proliferative, fibrogenic, vasoconstrictor, are elevated24 (Figure 4). In the endothelium the vasodilator/vasoconstrictor ratio is decreased 25-27 (Figure 5) whilst prothrombotic factors, including tissue factor28, are increased. It is hypothesized that widespread endothelial apoptosis, early in PAH, culminates in selection of apoptosis-resistant endothelial precursor cells that proliferate and eventually form plexiform lesions20 (Figure 2). In the media, pulmonary artery smooth muscle cell (PASMC) apoptosis is suppressed and proliferation is enhanced38,41,43-45. Many factors drive PASMC proliferation, including mutation29 or downregulation30 of BMPR2 (Figure 6), mitochondrial metabolic abnormalities (Figure 7), de novo expression of the anti-apoptotic protein survivin20, 31, increased expression/activity of SERT21, 32, increased expression/activity of platelet-derived growth factor (PDGF) receptor49, tyrosine kinase activation33 (Figure 8) and decreased expression of Kv1.5, a voltage-gated, O2-sensitive potassium channel. Kv1.5 downregulation occurs in human PAH34, rat PAH models (whether induced by chronic hypoxia35, 36monocrotaline31 or in FHR19) and transgenic mice with PAH due to SERT overexpression21 or BMPR2 mutation37. Loss of Kv1.5, the same channel that is inhibited by hypoxia to initiate hypoxic pulmonary vasoconstriction55, depolarizes the membrane and elevates cytosolic K+ and Ca2+(Figure 9). The resulting calcium overload, later reinforced by activation of transient receptor potential (trp) channels38, leads to Ca2+-calcineurin-dependent activation of the proliferative transcription factor, nuclear factor acting T lymphocytes (NFAT)39. Normoxic activation of HIF-1∝ occurs in FHR19 and human PAH14, 19. In the adventitia, metalloprotease activation causes architectural disruption, permitting cell migration and generating mitogenic peptides (tenascin)40(Figure 10). Adventitial fibroblasts are also hyperproliferative in PH, displaying increased sensitivity to serotonin41. Circulating autoantibodies4 and lung infiltration by inflammatory cells is common, particularly in PAH associated with connective tissue disease and schistosomiasis42 (Figure 11). Finally, there are increased endothelial-precursor cells, and mesenchymal and bone marrow-derived stem cells43, although it is uncertain whether this is harmful or beneficial (Figure 12).
Figure 4. Serotonin (5-HT) abnormalities in PAH.
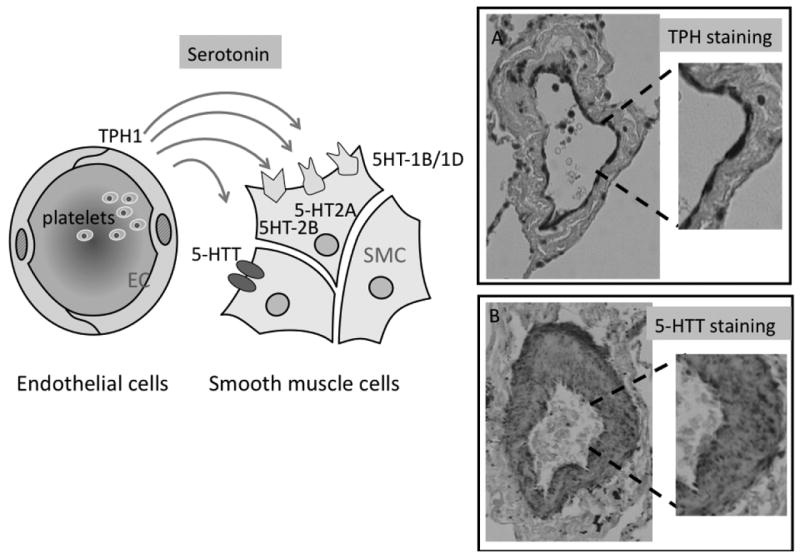
Increased bioavailability of serotonin during progression of PAH results from an increased release of serotonin from platelets and from an increased synthesis of serotonin by endothelial cells which produce serotonin and express tryptophan hydroxylase-1 (TPH1), the key enzyme controling 5-HT synthesis. Overexpression of 5-HTT (SERT) by PASMC is responsible for the increased mitogenic effect of serotonin on these cells. 5-HT receptors, including 5-HT1B/1D and 5-HT2A receptors mediate 5-HT-induced PA contraction of pulmonary vessels. 5-HT2A receptors located on platelets potentiate the aggregation response to various platelet activators. 5-HT2B receptors expressed by PASMC are also involved in the pulmonary vascular remodeling process.
Figure and legend contributed by Dr. Serge Adnot, Département de Physiologie, Hôpital Henri Mondor, CRETEIL, FRANCE
Figure 5. The endothelium and vasodilator/antiproliferative pathways.
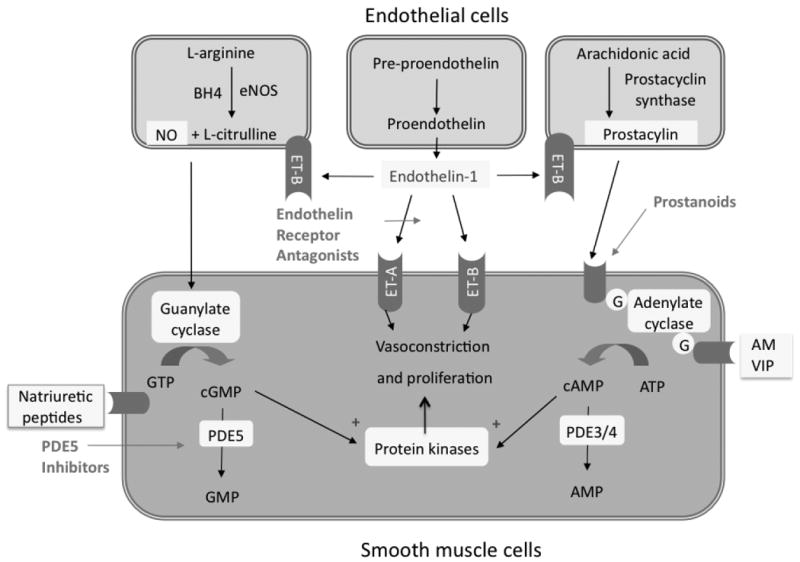
Nitric oxide (NO), generated from L-arginine, and natriuretic peptides stimulate production of cyclic guanosine monophosphate (GMP). cGMP causes vasorelaxation and inhibits proliferation of vascular SMC. Phosphodiesterase type 5 (PDE5) inhibitors (e.g. sildenafil) enhance this vasodilatory mechanism by preventing cGMP degradation. Prostacylin from endothelial cells also promotes relaxation and inhibits cell proliferation, via a cyclic AMP-dependent mechanism. Endothelin is a potent vasoconstrictor and stimulates proliferation via ETA receptors on SMC; while stimulating NO and prostacylin release via endothelial ETB receptors. Adrenomedullin (AM) and vasoactive intestinal polypeptide (VIP) are additional endothelial-derived, cAMP-dependent vasodilators that are dysregulated in PAH.
Contributed by Dr. Martin Wilkins
Figure 6. BMPR2 mutations-a genetic basis for familial PAH.
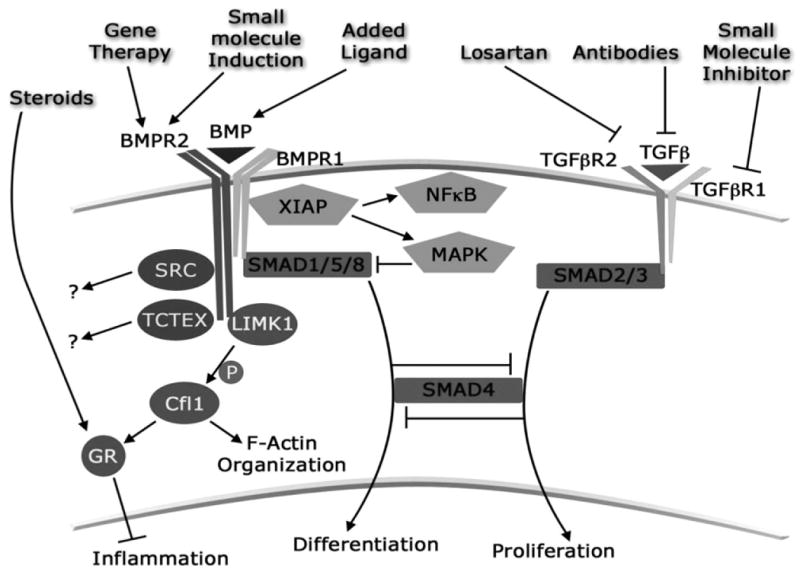
BMPR2 mutations are found throughout the gene, and a universal functional consequence of these mutations has not been identified. Best studied is BMPR1 signaling through SMAD transcription factors. Mutations leading to loss of SMAD signaling decreases cell differentiation, enhance vascular tone, increase TGF-β signaling and likely increase proliferation. Signaling through XIAP (X-linked inhibitor of apoptosis), also requiring BMPR1, can impact both NFkB and MAPK pathways, leading to increased MAPK phosphorylation and presumably pro-inflammatory signaling. BMPR2 has a long, evolutionarily conserved cytoplasmic tail domain unique in the TGF-beta superfamily, which binds SRC, RACK1, and LIMK1. BMPR2 mutation in vivo leads to decreased Cofilin (Cfl1) phosphorylation by LIMK1, with the effect both of alterations in F-actin organization and defects in glucocorticoid receptor (GR) nuclear translocation.
Figure and legend contributed by Drs. James West and John H. Newman, Pulmonary Medicine, Vanderbilt University School of Medicine, Nashville, Tennessee.
Figure 7. Mitochondrial metabolism in PAH.
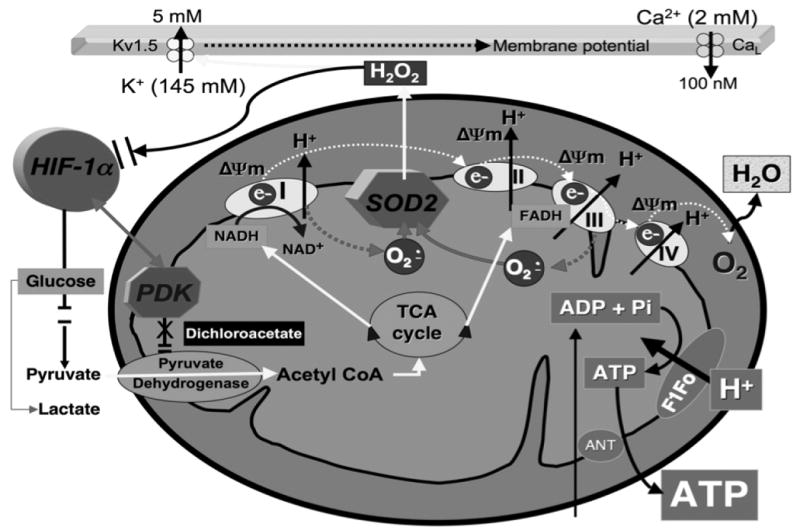
In aerobic metabolism, PDK is inactive, PDH is active and electron donors (mitochondrial NADH and FADH) produced by the TCA (Krebs') cycle pass electrons down a redox-potential gradient in the electron transport chain (ETC) to molecular O2. This electron flux powers H+ ion extrusion, creating the proton-motive force responsible for creating the mitochondria's negative membrane potential (ΔΨm) and powering F1Fo-ATP-synthase. Side reactions between semiquinones and molecular O2, accounting for ∼3% of net electron flux, create superoxide anion in proportion to PO2. Superoxide dismutase (SOD2) rapidly converts superoxide anion (produced at complexes I and III) to H2O2, which serves as a redox messenger signaling “normoxia”. In hypoxia (and PAH and cancer) there is activation of HIF-1α and PDK, which inhibits PDH shifting metabolism toward glycolysis. Therapeutic implications: Dichloroacetate which inactivates PDK by causing conformational changes in its nucleotide- and lipoyl-binding pockets60) regresses experimental PAH7,33.
Contributed by Dr. Stephen L. Archer
Figure 8. Receptor tyrosine kinases and their inhibitors.
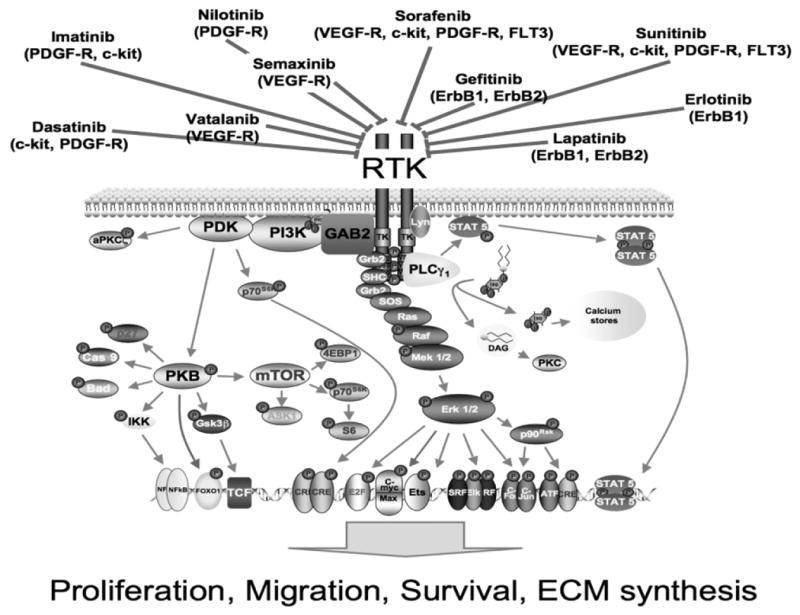
This complex kinase cascade offers many therapeutic targets to treat PAH.
Abbreviations: ATF, activating transcription factor; BAD, BCL-XL/BCL-2 associated death promoter; c-kit, CD117; DAG, diacyl glycerol; EGF-R, epidermal growth factor receptor; ErbB1,2 epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; flt3, fms-like tyrosine kinase receptor-3; GSK, glycogen synthase kinase; JAK, Janus kinase; JNK, Jun N-terminal kinase; MEF, myocyte-specific enhancer-binding nuclear factor; MEK, mitogen-activated protein kinase/ERK kinase; MERM, ezrin/radixin/moezin family of cytoskeletal linkers; mTOR, mammalian target of rapamycin; NHERF, sodium-hydrogen exchange regulatory factor; P, phosphotyrosine; p70S6K, p70 ribosomal S6 kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKB, protein kinase B; PKC, protein kinase C; PLC, phospholipase C; SOS, Son of Sevenless; STAT, signal transducer and activator of transcription; PDGF-R, platelet-derived growth factor receptor: PDK, phosphoinositide-dependent kinase; PI3K, phosphoinositide-3 kinase; PKB, protein kinase B; PKC, protein kinase C; PLC, phospholipase C; SHP, Src homology 2-containing protein tyrosine phosphatase; VEGF-R, vascular endothelial growth factor receptor.
Figure and legend contributed by Dr. Ralph Schermuly, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
Figure 9. Ion Channels in PAH.
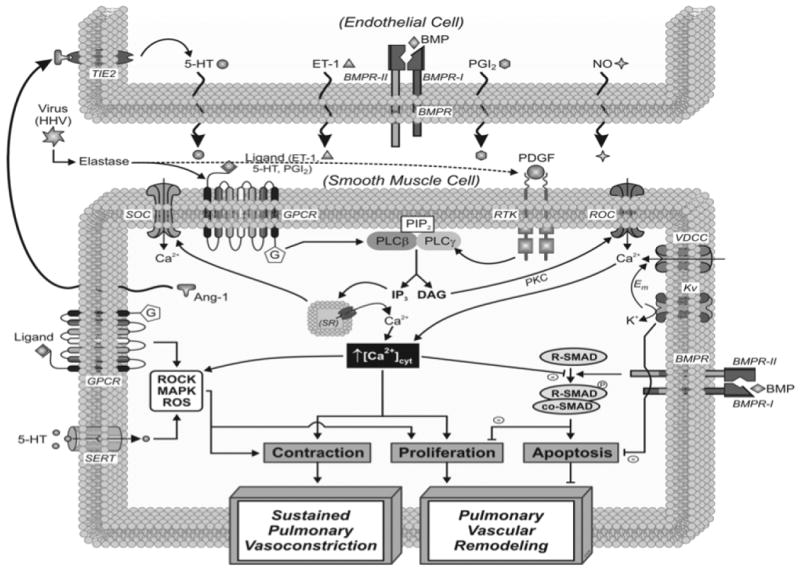
Schematic depiction of the cellular mechanisms linked with the vasoconstriction and remodeling in pulmonary endothelial (PAEC) and PASMC in PAH. Central themes of interest for the development of PAH include: 1) impaired ion channel expression and function in PASMC (Kv, VDCC, SOC, ROC), 2) increased cytosolic calcium ([Ca2+]cyt) in PASMC (mediated by ion channel function and receptor stimulation), 3) altered signaling via membrane receptors (GPCR, TIE-2, BMPR, RTK) and transporters (i.e., SERT) in endothelial cells and PASMC, 4) changes in redox status, 5) enhanced production of vasoconstrictor or mitogenic factors, and 6) viral signaling via GPCR and RTK. Paracrine interactions between PAEC and PASMC are noteworthy.
Abbreviations: Ang-1, angiopoietin-1; ET-1, endothelin-1; GPCR, G protein-coupled receptor; 5-HT, serotonin; MAPK, mitogen-activated protein kinase; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostaglandin I2; ROC, receptor-operated Ca2+ channels; ROCK, Rho-associated kinase; ROS, reactive oxygen species; RTK, receptor tyrosine kinase; SERT, 5-HT transporter; SOC, store-operated Ca2+ channels; SR, sarcoplasmic reticulum; VDCC, voltage-dependent Ca2+ channels.
Figure and Legend Contributed by Drs. Carmelle Remillard and Jason Yuan, University of California, San Diego
Figure 10. Disordered Elastin metabolism and deposition in PAH.
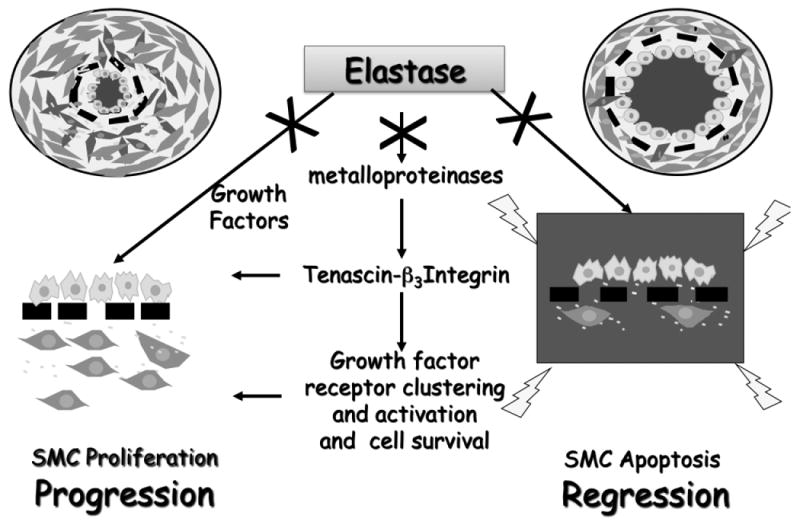
Elastase degrades elastin and other components of the extracellular matrix thereby releasing bound growth factors, that are both mito- and motogenic for PASMC. Heightened elastase activity also activates matrix metalloproteinases (MMPs), which upregulate the glycoprotein tenascin-C. When tenascin-C binds cell surface integrins, such as alpha-v β3 on PASMC, these integrins cluster and cell shape changes in a way that clusters and activates growth factor receptors and increases cell survival signals. Thus, pathway activation causes both release of growth factors and activation of their receptors. Transmission of cell survival signals occurs even in the absence of ligand (growth factor) binding. Blocking elastase activity or growth factor receptors can therefore arrest progression of PASMC by blocking proliferation and induce regression by enhancing apoptosis.
Figure and legend contributed by Marlene Rabinovitch, Stanford University, Palo Alto, CA.
Figure 11. The role of inflammation in the pathogenesis of PAH.

Initial inflammatory stimuli can occur in the form of infectious or foreign antigens or autoimmune disease, leading to an appropriate, but potentially excessive, immune response. The host immune response to these varied stimuli results in the release of pro-inflammatory cytokines, which can recruit bone marrow-derived cells, stimulation of resident inflammatory cells, and endothelial cell dysfunction. Endothelial cell injury and the cellular response can increase endovascular thrombosis. A network of cytokines released by the inflammatory and endothelial cells can also cause aberrant PASMC proliferation. The triad of endothelial cell proliferation, PASMC proliferation, and thrombus formation contributes to PAH. Pro-inflammatory cytokines and cell-cell interactions can potentially be therapeutically targeted.
Abbreviations: EGF: epidermal growth factor; HIMF: hypoxia induced mitogenic factor, also called RELMa and FIZZ1; NO: nitric oxide; PDGF: platelet derived growth factor; RANTES; TNFα: tumor necrosis factor-alpha.
Figure and legend contributed by Drs. Brian Graham and Rubin Tuder, University of Colorado at Denver.
Figure 12. Cellular Basis for Pulmonary Vascular Remodeling-lessons from hypoxia.
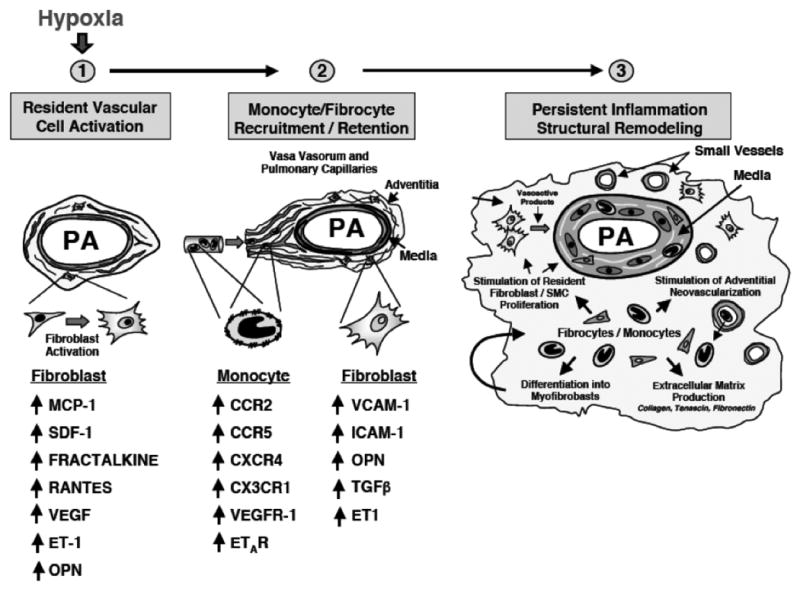
Fibroblasts, monocytes and fibrocytes play critical roles in orchestrating hypoxia-induced pulmonary vascular remodeling. Hypoxia or hypoxia-associated stimuli increase production by resident fibroblasts (and probably PASMC) of chemokines/cytokines including: monocyte chemoattractant protein (MCP)-1, stromal cell-derived factor (SDF)-1, fractalkine (CX3CL1), RANTES, VEGF, osteopontin (OPN) and endothelin. These and other factors stimulate recruitment of monocytes and monocyte-derived mesenchymal precursors (fibrocytes) to the vessel wall. Upregulation of monocyte receptors for these ligands (CCR2, CXCR4, CX3CR1, VEGFR-1 and ETR-A) occurs. Monocytes are retained in the vessel wall by the upregulation of adhesion molecules on fibroblasts, including vascular cell adhesion molecule (VCAM), intracellular adhesion molecule (ICAM) and OPN. As monocytes and fibrocytes accumulate in the vessel wall, they exert potent effects on the proliferative, migratory, matrix-producing and contractile capabilities of resident fibroblasts and PASMC through the secretion of TGF-β, PDGF-A and B, EGF, IL-6, IGF-1, MMP-9 and others. In addition, these cells produce potent proangiogenic molecules such as VEGF, S100A4, βFGF that likely play roles in stimulating further angiogenesis in the vessel wall.
Figure and legend contributed by Kurt Stenmark, University of Colorado at Denver.
PAH has a genetic component (Figure 6)
The bone morphogenetic proteins (BMP) are part of the transforming growth factor-β superfamily (TGF-β. Most (>80%) patients with familial PAH have loss of function mutations in BMPR244-46 that promote cell proliferation. BMPR2 is a constitutively active serine-threonine kinase receptor, which, in response to ligand (BMPs 2, 4, 6, 7, 9 and 10), forms heterodimers with any of 4 type 1 receptors (BMPR1A, BMPR1B, Alk1, Alk2), resulting in phosphorylation of the intracellular portion of the type 1 receptor by BMPR2. Receptor activation initiates a cytosolic Smad protein-signaling cascade. Receptor activated Smads, complex with common partner SMAD (Smad4) and the complex translocates to the nucleus where it regulates gene transcription (Figure 6). The inhibitors of DNA binding (Id) genes are major targets of BMP/Smad signaling47. The Smad DNA interaction is weak and requires co-repressors or activators. BMPs can also act via an alternative, BMPR2-independent pathway involving mitogen activated protein kinases (e.g. p38MAPK, ERK1/2).
Most heterozygous BMPR2 mutations in PAH result in defective Smad signaling, while retaining p38MAPK signaling66,48. The loss of normal BMPR2-Smad activity may exaggerate the susceptibility of vascular cells to proliferate and suppress apoptosis. BMP 2, 4 and 7 suppress PASMC proliferation in normal individuals and patients with secondary PH but are ineffective in PAH29. The BMPR2-Smad pathway may display tissue heterogeneity because it can be regulated by endogenous Smad inhibitors (e.g. chordin and noggin), by inhibitory Smads (6 and 7) and also because of variable heterodimer receptor composition49. This also may explain the restriction of the vascular disease to the pulmonary circulation.
Mice with conditional, endothelial BMPR2 deletions are predisposed to PAH, although PH occurs in only a subset, reminiscent of the incomplete penetrance seen in familial PAH50. Mice with a SMC-specific, overexpression of a BMPR2 dominant-negative mutant develop a vasospastic form of PH that lacks vascular remodeling but is associated with downregulation of Kv1.5 expression. PH in these mice is reversed by nifedipine37. Perhaps disordered BMP signaling, by reducing Kv1.5 transcription, creates an early vasospastic form of PAH that in time becomes fixed by vascular remodeling.
Initial enthusiasm that BMPR2 mutations might represent a “universal” cause of PAH has been tempered. BMPR2 mutation is uncommon (prevalence 10-20%) in the nonfamilial, Category 1 PAH population. Moreover, in familial PAH, penetrance is low (i.e. only ∼25% of carriers in affected families develop PAH)51. While modifier genes, such as SERT and TGFβ, may explain variable penetrance, aberrant BMPR2 function alone is neither a necessary nor a sufficient precondition for most cases of PAH52. Moreover, BMPR2 heterozygous mice do not develop PAH and are not predisposed to hypoxic PH; however they do have an exaggerated hypertensive response to serotonin53.
In genetically normal animal, BMPR2 expression decreases as PH develops54. One posttranscriptional mechanism accounting for downregulation of BMPR2 protein is activation of microRNAs (miRNAs). miRNAs regulate gene expression by inhibiting translation. A computational algorithm on the BMPR2 gene predicted that miRNAs encoded by miRNA cluster 17/92 (miR-17/92) might regulate BMPR2. Overexpression of miR-17/92 did reduce BMPR2 protein, and it appears that BMPR2 is directly targeted by miR-17-5p and miR-20a55.
however, results of BMPR2 rescue therapy are mixed. Intravascular BMPR2 gene therapy, using an endothelial-targeted vector, reduced chronic hypoxic pulmonary hypertension in rats56. However nebulized BMPR2 adenovirus (with a promiscuous promoter) did not regress monocrotaline-induced PAH54. Further study is required and may be productive in identifying BMPR2-related targets for pharmacological manipulation in PAH. For example, inhibition of TGF-β signaling prevents PAH in the monocrotaline model, via inhibition of activin receptor-like kinase-557.
Alternative genetic mechanisms
Work is currently underway searching for modifier genes and for possible epigenetic mechanisms (gene methylation) of inheriting PAH or enhancing disease susceptibility. Single nucleotide polymorphisms (SNP) are genes that differ from normal by a single alternative nucleotide. SNPs can change the function/location of the encoded protein. SNPs occur in a significant proportion of the population and may explain susceptibility to PAH. SNP variants for PAH-relevant genes (including SERT58, Kv1.559 and TRPC6) may predispose to PAH. The consequences of SNPs can be complex, for example the TRPC6 SNP not only increases TRPC6 expression but also creates a binding sequence and activates the inflammatory transcription factor, nuclear factor-kappaB60.
Excess proliferation and impaired apoptosis suggest similarities to cancer in PAH (Figure 7)
Otto Warburg, 1931 Nobel laureate, proposed that a shift in glucose metabolism from oxidative phosphorylation to glycolysis (despite adequate oxygen supply) was central to the cause/maintenance of cancers. Several observations indicate that PAH shares this “Warburg phenotype”19, 61. As highlighted by Tuder and Voelkel62, both cancer and PAH manifest excessive cell proliferation and impaired apoptosis. While PAH does not metastasize or disrupt tissue boundaries, emerging data show it shares a mitochondrial-metabolic abnormality with cancer (Figure 7). Pyruvate dehydrogenase kinase (PDK) is pathologically activated in both conditions19, 63. This enzyme phosphorylates and inhibits pyruvate dehydrogenase (PDH)64. PDH catalyses the irreversible oxidation of pyruvate, yielding acetylCoA and CO2 and is a key enzyme controlling the rate of oxidative metabolism. PDK activation thus impairs the Krebs' cycle and creates a glycolytic shift in glucose metabolism. Subversion of the mitochondrial O2-sensing mechanism, normally used to sense and respond to decreases in PO265, appears to cause the sensor to signal hypoxia despite adequate PO2. These acquired (and reversible) mitochondrial abnormalities of fusion/fission and metabolism61 are postulated to cause the observed normoxic activation of HIF-1∝ in PAH14, 66 and cancer 63. Once active, HIF-1α turns on glycolytic genes and suppresses oxidative metabolism by increasing PDK transcription. The downstream consequences of this mitochondrial-metabolic abnormality include mitochondrial hyperpolarization, reduced production of reactive oxygen species, decreased Kv1.5 expression.
These metabolic abnormalities, which enhance cell proliferation and impair apoptosis, can be partially corrected by a simple, mitochondrial-targeted strategy. Dichloroacetate, a PDK inhibitor, restores PDH activity, increases glucose oxidation, restores mitochondrial membrane potential and reverses normoxic HIF-1α activation67. Dichloroacetate inhibits proliferation, enhances apoptosis and can regress both human cancer, in a xenotransplantation model63, and experimental PAH (chronic hypoxic PH, monocrotaline PAH and FHR PAH)19, 36, 67. Dichloroacetate also regresses spontaneous PH in transgenic mice that over express SERT in PASMC68. Dichloroacetate has been safely used in long-term treatment of inherited lactic acidosis due to mitochondrial diseases, suggesting the potential for translation to the clinic.
The right ventricle (RV) in PAH
The fetal/neonatal RV ejects blood at relatively high pressure into the pulmonary circulation. With maturation the pulmonary circulation develops into a low-pressure circuit and the RV involutes, becoming thin walled. Chronic pressure overload, as occurs in PAH, stimulates RV hypertrophy. Surprisingly little is known about the specific mechanisms underlying RVH and RV dysfunction in the setting of PAH. While the obvious approach to reducing RVH and RV failure is to treat the underlying pulmonary arterial disease, recent experimental evidence suggests that the RV can be therapeutically targeted in PAH36. In RVH phosphodiesterase-5, which was expressed in the fetal RV, is selectively re-expressed. Inhibiting this enzyme (i.e. by sildenafil) enhances RV contractility without affecting the left ventricle69 which lacks phosphodiesterase-5.
In contrast to the normal RV, which can vary its substrate utilization from fatty acids to glucose as needed, metabolism in RVH is reliant on glucose metabolism70. In hypoxia-induced PH, expression of the glucose transporter GLUT4 is significantly increased in the RV, suggesting a metabolic switch to glycolysis. AMP-activated protein kinase (AMPK), which has a key role in the control and regulation of energy metabolism, stimulates fatty acid metabolism and glycolysis, preserving ATP production71, 72. AMPK activation in ventricular hypertrophy73-75 preserves ATP levels by increasing glucose transport and accelerating glycolysis and by inhibiting acetyl CoA carboxylase72. In RVH, there is systolic flow impediment in the right coronary artery that is proportional to RV pressure and mass76. New evidence shows the RV in PAH is glycolytic, due in part to activation of PDK, and behaves as hibernating myocardium, demonstrating enhanced glucose oxidation and improved contractility in response to dichloroacetate77. Future PAH therapies should consider the effects of agents on both the RV and the pulmonary vasculature.
Therapeutic Pathways in PAH (Table 1)
Table 1. Molecular Targets in the Treatment of Pulmonary Hypertension Current Therapies.
| Target | Goal | Drug | Used in Humans | Ref |
|---|---|---|---|---|
| L-type Ca++ channels | Decrease SMC Ca++ | L-type Ca++ channel blockers | Yes | 194 |
| Coagulation cascade | Decrease thrombosis | Warfarin | Yes | 195-197 |
| Prostacyclin receptors | Increase cAMP | epoprostenol | Yes | 198 |
| Endothelin receptors A& B | Inhibit constriction & proliferation | Bosentan | Yes | 199 |
| Endothelin receptor A | Inhibit constriction & proliferation | Sitaxsentan | Yes | 200 |
| Phosphodiesterase 5 inhibitors | Increase cGMP | Sildenafil | Yes | 201 |
| Guanylate cyclase activators | Increase cGMP | iNO | Yes | 202 |
| Novel Targets | ||||
| Rho Kinase | Decrease Ca++ sensitivity in SMCs | Rho kinase inhibitors-fasudil | Yes Acute trial | 140, 141 |
| Rho A prenylation | Decrease Ca++ sensitivity in SMCs | Statins | Yes | 16 |
| Serine elastases | Decrease MMP activation | Elastase inhibitors | No | 40 |
| Kinase associated receptors | Inhibit PDGF or EGF activity | Tyrosine kinase inhibitors Publication pending | imatinib sorafenib | 165, 168 |
| PDH kinase | Normalize mitochondrial function | Dichloroacetate | Yes* | 36 |
| NFAT | Decrease antiapoptotic bcl-2; slow proliferation | Cyclosporine A | Yes* | 39 |
| Immune system | Immunosuppression | Mycophenolate mofetil | Yes* | 203 |
| Survivin | Inhibits antiapoptotic effect of survivin | Transfection of dominant-negative | Yes* | 31 |
| Guanylate cyclase | Increase cGMP |
|
Phase III trial | 107, 204 |
| Cyclooxygenase | Inhibit TxA2 | aspirin | Yes* | 28 |
| Ornithine decarboxylase | Inhibit polyamine synthesis | α-difluoromethylornithine | No | |
| Cyclin-dependent-kinase inhibitor γ27 | Inhibit SMC proliferation | Heparin | Yes* | 205 |
| Peroxisome proliferator-activated receptor –Y | Increase PPARγ activity | Rosiglitazone | Yes* | 206 |
| Angiopoietin & TIE2 | Inhibit SMC proliferation | Adenoviral gene transfection | No-and conflicting results | 173 |
| Serotonin transporter | Inhibit SMC proliferation | SSRI | Yes† | 134 |
| VPAC 1 & 2 receptors | Inhibit SMC proliferation | Vasoactive intestinal peptide, inhaled | Yes | 125 |
| Adrenomedullin receptors | Inhibit SMC proliferation, vasodilatation | Adrenomedullin | Yes++ | 207, 208 |
| BMPR2 | Enhance BMPR2 signaling | Adenoviral transfection and/or enhancing receptor trafficking to membrane | No conflicting results | 30, 126, 209 |
| eNOS | Increase cGMP and NO signaling | eNOS transfected EPCs | Phase I trial in progress | 188 |
| eNOS | Increase cGMP and NO signaling | VEGF transfected Fibroblasts | No | 191 |
Abbreviations: DHEA: dihydroepiandrosterone. MMPs: matrix metalloproteinases. PDH: pyruvate dehydrogenase kinase. SSRI: Selective serotonin reuptake inhibitors
Human Use qualifiers:
Yes*=used in human but in a disease other than PAH
Yes†=used in human with PAH but retrospective data
Yes++†=used in human with PAH but only in an acute hemodynamic study
Prostanoids and prostanoid receptors (Figure 5, Table 1)
One of the most successful therapeutic strategies for PAH has been to augment endogenous prostacyclin production with exogenous prostanoids. Fatty acid cyclooxygenase (COX) converts arachidonic acid to prostaglandin H2 (PGH2), a substrate for both PGI2 (prostacyclin) synthase and thromboxane synthase. PGI2 synthase is expressed in pulmonary vascular endothelium and generates prostacyclin, which relaxes pulmonary artery smooth muscle cells (PASMC) and inhibits platelet aggregation through interaction with IP receptors and stimulation of cyclic adenosine monophosphate (cAMP). Thromboxane synthase, in platelets and endothelium, produces thromboxane A2 (TXA2). TXA2 stimulates vasoconstriction and platelet aggregation through TP receptors. Endothelial dysfunction and platelet activation in PAH reduce prostacyclin levels and increases TXA2 production.
Continuous intravascular infusion of epoprostenol (Flolan®) decreases pulmonary vascular resistance (PVR), increases cardiac output, and improves life expectancy13. Epoprostenol's poor stability, expense and requirement for continuous intravenous infusion have fostered development of more stable analogs and alternative routes of administration: iloprost (inhalation), treprostinil (subcutaneous) and beraprost (oral). New prostacylin agonists and thromboxane antagonists are in clinical trials. The combination of a prostanoid (such as iloprost) and a phosphodiesterase-5 (PDE5) inhibitor enhances pulmonary hemodynamic effects and improves exercise capacity in PAH78, 79.
Nitric oxide (NO) and cyclic guanosine monophosphate (cGMP)-(Figure 5, Table 1)
NO is a radical, synthesized from L-arginine by three nitric oxide synthases (NOS). Endothelial NOS (eNOS) is the principle mediator of endothelium-dependent vasodilatation in the pulmonary circulation. Endothelium-derived NO diffuses into PASMC where it stimulates soluble guanylate cyclase (sGC) to produce cGMP. cGMP's cardiovascular effects are mediated by interaction with at least three groups of proteins: cGMP-dependent protein kinases (PKG), cGMP-regulated PDE, and cyclic nucleotide-gated ion channels. PDE5, the molecular target of sildenafil (Viagra®), decreases intracellular cGMP levels and opposes PKG-dependent signaling elicited by NO and natriuretic peptides.
The NO pathway is impaired in several ways in PAH. NOS expression80 and NO biovailability81 are depressed. Moreover, PDE5 is induced both in PASMC82 and the right ventricle (RV)69, which hastens inactivation of cGMP. Finally, production of endogenous NOS inhibitors, asymmetric and symmetric dimethylarginines (ADMA and SDMA), is enhanced in pulmonary hypertension (PH)83, 84.
Pharmacological or genetic perturbations of the NO pathway demonstrate the pivotal role of the cGMP pathway in regulating PVR. Mice develop PH if they are rendered deficient in eNOS, GTP cyclohydrase-1 (the rate-limiting enzyme in synthesis of the NOS co-factor, tetrahydrobiopterin,) or dimethylarginine dimethylaminohydrolase (DDAH, the enzyme responsible for eliminating endogenous NOS inhibitors) 85-87. Inhibition of NO production in humans, using a competitive NOS antagonist (L-NMMA), increases pulmonary vascular resistance (PVR) 88, 89. Chronic pharmacological NOS inhibition causes PH in rats, although there is disproportionate systemic hypertension90.
NONOates
Inhalation of exogenous NO gas (iNO, 0.1-100 parts per million) decrease pulmonary arterial pressure (PAP) and improves oxygenation and hemodynamics in children and adults with diverse forms of PH91. Although chronic iNO therapy for PAH is feasible92, delivery is complicated by the instability of NO, which mandates continuous inhalation. In addition, higher concentrations of NO and especially its oxidation products are toxic. Consequently iNO dosing must be carefully monitored to prevent exposure to toxic nitrogen oxides and methemoglobin. Alternative strategies that exploit the specificity of iNO, but utilize more stable NO sources are appealing. One such strategy uses NO/nucleophile adducts, such as diethylenetriamine/NO (DETA/NO). NONOates spontaneously release predictable amounts of NO when exposed to physiological pH. Daily nebulization of DETA/NO (half-time of NO release >20 hours) for ∼1 week reduces PH in monocrotaline-induced PAH without causing systemic hypotension93. DETA/NO has been used effectively to improve pulmonary hemodynamics in intubated patients with adult respiratory distress syndrome94. It may be valuable to investigate the many NONOates for long-term, ambulatory use in PAH.
Phosphodiesterase Inhibitors (Figure 5)
Eleven PDE families are known; however, they vary in substrate affinity, selectivity and regulatory mechanisms95. In the pulmonary circulation, PDE5 and PDE1 are highly relevant. PDE1 has 3 isoforms that are regulated by calcium-calmodulin and can hydrolyse both cAMP and cGMP. Both PDE1A and PDE1C are upregulated in pulmonary arteries (PAs) from idiopathic (IPAH) patients96. Infusion of the PDE1 inhibitor 8-methoxymethyl-isobutyl-1-methylxanthine reduces PVR and RV hypertrophy (RVH) in rodent PH models96.
PDE5 expression, normally absent in cardiac myocytes, is upregulated in the right ventricle (RV) in PAH69. PDE5 inhibition in PAH models increases RV contractility through a cGMP-mediated inhibition of PDE369. Thus in PAH sildenafil has an effect on the RV similar to the PDE3 inhibitor, milrinone. A single dose of sildenafil (75mg) reduces PVR without lower systemic vascular resistance in PAH patients and simultaneously lowers wedge pressure and increases cardiac output97. Sildenafil causes sustained improvement in hemodynamics and functional capacity in PAH and it has been approved as a first-line, oral treatment for PAH (reviewed in 98). Combining a PDE inhibitor (PDE3/4 or PDE5), even at subtherapeutic doses, with a prostanoid augments the hemodynamic/functional benefit of the prostanoid79, 99, 100.
Soluble Guanylate Cyclase activators (Figure 5)
sGC is a heterodimer, consisting of α-and β-subunits. sGC expression is upregulated as a compensatory mechanism in human PAH101. Experimental hypoxic-PH is exacerbated in mice lacking the sGCα1102. Thus, augmentation of sGC activation is an attractive therapeutic strategy. There are NO independent, haem-dependent sGC stimulators (e.g. BAY 41-2272) and NO and haem-independent sGC activators (e.g. BAY 58-2667 and HMR-1766)103. BAY 41-2272 stimulates sGC directly but also sensitizes the enzyme to NO, resulting in synergism. It improves pulmonary hemodynamics in models of persistent pulmonary hypertension of the newborn (PPHN)104, 105. NO-independent sGC activators, such as BAY 58-2667, HMR-1766 and S-3448 provide an additive, rather than synergistic, effect when combined with NO donors. Both BAY 41-2272 and BAY 58-2667 reverse established PH in rodent models, although this benefit is partially dependent on endogenous NOS activity106. BAY 63-2521 (Riociguat), a sGC stimulator, is available orally, has a favorable safety profile and has entered phase III trials in PAH107.
Enhancing NOS activity – tetrahydrobiopterin and transcription enhancers (Figure 5)
Tetrahydrobiopterin (BH4) is an important NOS cofactor, essential for dimerization and for the oxygenation of L-arginine to create NO and L-citrulline. Without BH4, NOS becomes uncoupled and produces superoxide anion which rapidly reacts with NO, producing peroxynitrite, further attenuating NO bioavailability108.
The rate-determining step for the de novo production of BH4 is catalyzed by GTP-cyclohydrolase 1 (GTP-CH1). Mice with impaired GTP-CH1 activity exhibit reduced lung BH4 and spontaneously develop PH with vascular remodelling86. Conversely, congenital over-expression of GTP-CH1 in vascular endothelium protects mice from hypoxic-PH86. In a porcine PPHN model, combined therapy with BH4 and a superoxide dismutase mimetic (which enhances survival of endogenous NO) restores endothelial function109.
While there is no evidence for GTP-CH1 deficiency in PAH, GTP-CH1 polymorphisms are associated with variations in NO bioavailability and systemic hypertension. Some PAH patients show increased markers of oxidative stress110 and this may result in conversion of BH4 to dihydrobiopterin. BH4 is a co-factor for several enzymes and is well tolerated when administered in its synthetic form, sapropterin, to patients with phenylketonuria111. This observation paves the way for studies in PAH patients.
eNOS transcription enhancers, such as AVE9488 and AVE3085, similarly aim to increase NO signaling. Theoretically, increasing eNOS without corresponding increases in cofactors such as BH4 could lead to uncoupling and the formation of superoxide ions. However AVE9488-treatment also increases BH4 levels and improves eNOS coupling in apolipoprotein E-knockout mice112. To our knowledge this agent has not been used in vivo in humans.
Vasoactive peptides and endopepitase inhibitors (Figure 5)
Endothelin receptor antagonists (ERA) (Figure 5)
Endothelin-1 is a vasoconstrictor that acts via two receptors, ETA and ETB, to regulate vascular tone and cell proliferation. Both receptor subtypes are found on PASMC and mediate vasoconstriction, while the ETB receptor on endothelial cells mediates nitric oxide and prostacyclin release, causing vasodilatation. Lung and circulating endothelin-1 levels are increased in PAH patients113. ERAs, such as bosentan, ambrisentan and sitaxsentan cause a significant, but modest, improvement in pulmonary hemodynamics, exercise capacity (6-minute walk distance) and symptoms and are approved for management of PAH. There are no trial data to indicate whether selective ETA antagonism offers advantages over combined ETA and ETB antagonism (Bosentan) nor is the relative efficacy compared to PDE5 inhibitors known (although a small trial suggests some benefits of sildenafil)114. Liver toxicity and teratogenicity are class effects. While comparisons with historical control data suggest that bosentan monotherapy increases survival115 there are no robust survival data from appropriately designed clinical trials.
The endothelins are produced from big endothelin by endothelin converting enzyme (ECE). ECE inhibitors are an alternative approach to reducing endothelin levels. Although studies with this drug class (e.g. daglutril) have been conducted in patients with systemic hypertension and heart failure, data for pulmonary hypertension are limited.
Natriuretic peptides (Figure 5)
The natriuretic peptides (ANP and BNP) are synthesized in and released from myocardial tissue in response to stretch and their elevation in the blood in PAH indicates the extent of RV dysfunction116. CNP is produced in vascular tissue. These peptides interact with the extracellular domain of the natriuretic peptide receptors, NPR-A and NPR-B, which are transmembrane guanylate cyclases. Upon binding, the intracellular domain hydrolyses GTP to cGMP117. Genetic inactivation of NPR-A is associated with PH while chronic administration of ANP attenuates PAH in animal models118. Short-lived natriuretic peptides are not feasible agents for chronic therapy. An alternative approach is to inhibit metabolism of endogenous natriuretic peptides using neutral endopeptidase (NEP) inhibitors. NEP inhibitors have demonstrated efficacy in animal models and in combination with PDE5 inhibition119, but this combination is untested in patients.
Adrenomedullin (Figure 5)
This vasodilator peptide activates several signaling pathways, such as cAMP, NO-cGMP and PI3K/Akt. It decreases mean PAP and RVH in hypoxic rats, and exhibits anti-proliferative properties120. Adrenomedullin-2, a novel peptide, acts by the same receptors as adrenomedullin and its levels are also elevated in the RV of rats with hypoxic-PH. When aerosolized, adrenomedullin-2 reduces monocrotaline-induced PAH in rats and improves survival121. In humans with PAH, inhaled adrenomedullin causes a modest reduction in PVR and increases peak O2-consumption during exercise without significant effects on the systemic vasculature122.
Vasoactive Intestinal Polypeptide (VIP)
VIP is a 28 amino-acid peptide that increases cardiac output, scavenges oxygen free radical species, inhibits platelet activation and is a potent vasodilator. VIP's effects are mediated by G protein-coupled receptors, VPAC1 and VPAC2. Receptor activation stimulates both adenylate and guanylate cyclase signaling pathways. VIP-knockout mice spontaneously develop PH123. They over-express pro-inflammatory genes and genes involved in pulmonary vascular remodeling, and under-express anti-proliferative genes124, including eNOS/NOS3, prostacyclin synthase, GTP cyclohydrolase-1 and BMP-2. Thus VIP is also a key regulator of multiple genes that control the process of vascular remodeling.
VIP receptor expression (particularly the VPAC-2 subtype) and receptor-binding affinity are increased in PASMC from PAH patients; conversely, serum and lung VIP levels are low in PAH. VIP inhibits the proliferation of PASMC from PAH patients125. Nebulized VIP (200μg daily) improves pulmonary hemodynamics in PAH patients and, when continued for 3 months, reduces PVR and improves 6-minute walk, with little effect on the systemic circulation. The medical use of peptides in general, and VIP specifically, is complicated by their rapid degradation by endogenous proteases. A sustained release liposomal VIP preparation has extended pharmacological effects and may facilitate the development of VIP as a PAH treatment.
BMPR2-targeted treatment strategies
Loss of BMPR2 function after germ-line mutation has been linked strongly to the development and progression of familial and sporadic forms of iPAH. This has directed attention to strategies targeted at repairing BMPR2 signaling in patients with proven mutations. Gene mutations can directly inactivate BMPR2 (rescue strategies using viral vectors discussed in part 1) or can suppress function by impairing its trafficking to the cell surface. Substitution of cysteine residues in the ligand-binding domain prevents BMPR2 trafficking to the cell membrane and this can be rescued (in a cell model)126. In cystic fibrosis, where impaired protein trafficking also occurs, sodium 4-phenylbutyrate can improve membrane trafficking of the chloride channel127. Mutant BMPR2 protein that is trapped intracellularly can be ‘rescued’ using chemical chaperones (thapsigargin, glycerol or sodium 4-phenylbutyrate), increasing membrane expression126. It remains uncertain how much mutant BMPR2 must reach the cell membrane to induce a clinically relevant effect.
An alternative to restoring BMPR2 function is to inhibit pro-proliferative pathways that are unchecked by BMPR2 dysfunction. PASMC in familial PAH demonstrate increased sensitivity to TGF-β/activin receptor-like kinase 5 (ALK5) signaling, suggesting TGF-β blockade as a therapeutic strategy. The ALK5 inhibitor, SB525334, reverses PAH and RVH in a rodent model, indicating that strategies that inhibit ALK5-signaling may have therapeutic benefit128
Inhibitors of Serotonin and the Serotonin Transporter (SERT)-Figure 4
Plasma serotonin is increased in iPAH patients, even after lung transplantation129, suggesting that serotonin is either an etiologic factor in iPAH or is associated with such a factor. In addition, PAH endothelial cells do generate more serotonin than controls. SERT expression is increased in PASMC from iPAH patients and these cells proliferate more rapidly in response to serotonin than control cells130. In some patients with severe iPAH, the LL SERT polymorphism is associated with greater SERT expression and higher mean PAP than LS or SS genotypes130; however, in two separate iPAH cohorts this relationship was not detectedl131, 132. The proliferation of bovine and iPAH PASMC in response to serotonin depends on serotonin internalization via SERT and is blocked by selective serotonin reuptake inhibitors (SSRIs), such as fluoxetine (Prozac®)32. Fluoxetine reduces hypoxic-PH in rats133. In a retrospective cohort study of PAH patients, the use of SSRIs was associated with a trend towards reduced risk of death134. The time is right for a randomized clinical trial of fluoxetine versus placebo, on a background of conventional PAH therapy.
Another target is tryptophan hydroxylase, the enzyme that synthesizes serotonin. Deletion of tryptophan hydroxylase 1 reduces pulmonary vascular remodeling and hypoxic-PH135. The 5-HT2A receptor mediates serotonin-induced proliferation in rat PA fibroblasts.41 Genetic deficiency of the 5-HT2B serotonin receptor reduces hypoxic-PH in mice136. Terguride, a potent antagonist of 5-HT2B and 5-HT2A receptors, and partial dopamine agonist, is currently in a Phase II study in PAH patients and has received orphan drug status from the European Medicines Agency. PRX-08066, a selective 5-HT2B antagonist, is in Phase II (NCT00345774), having demonstrated evidence of efficacy in inhibiting hypoxia-induced rises in PAP in humans. SERT and the 5-HT receptors may act in concert to mediate the proliferative effects of serotonin on PASMC, suggesting simultaneously inhibiting the receptor and transporter as a strategy.
Much has been written about the potential role of serotonin in the etiology of PAH associated with anorexigens, such as dexfenfluramine. Transgenic mice lacking tryptophan hydroxylase are protected from dexfenfluramine-induced PAH137. Given that PAH was still uncommon even amongst those who consumed anorexigens138, it seems likely that a combination of factors is required to cause disease. For example, serotonin's effects on the pulmonary vasculature are modified by interaction between the serotonin pathway and BMPR2 signaling. Chronic serotonin infusion causes exaggerated PAH and pulmonary vascular remodeling in BMPR2 haploinsufficient mice compared with wild-type mice139. There is also a link to mitochondrial metabolism and Kv1.5 channel downregulation as SERT overexpressing mice respond favorably to therapy with the pyruvate dehydrogenase kinase inhibitor, dichloroacetate68.
Rho Kinase Inhibitors
In response to calcium/calmodulin, MLC kinase phosphorylates myosin light chain (MLC) causing PASMC contraction; conversely, MLC phosphatase dephosphorylates MLC, causing relaxation. Rho kinase inhibits MLC phosphatase, leading to prolonged, refractory vasoconstriction. Rho kinase participates in the vasoconstriction elicited by many vasoactive agents involved in PAH, such as serotonin, endothelin-1 and thromboxane A2. Rho-kinase inhibitors (Y-27632, fasudil) also markedly reduces PH in PAH models: the fawn-hooded rat (FHR), the chronic hypoxia/SUGEN 5416 model, and the monocrotaline model, illustrating the critical role of refractory vasoconstriction models140. In humans with PAH, fasudil, a rho kinase inhibitor, causes modest, acute reductions in PVR141. The challenge with the use of rho kinase inhibitors is avoidance of systemic vasodilatation. Airway nebulization offers a potential means of selectively inhibiting rho kinase in the lung. Rho kinase also participates in the vascular SMC proliferation. There is a rho kinase-dependent mechanism by which serotonin transactivates the PASMC BMPR1A receptor and downstream signaling Smads 1/5/8142. In SERT over-expressing mice, Rho kinase inhibition reduces PAH and vascular remodeling and this is associated with suppression of ERK phosphorylation in PA fibroblasts143.
Restoration of Potassium Channels (Figures 7 and 9)
Downregulation of the expression and activity of voltage-gated K+ channels, notably Kv1.5, is a finding common to human PAH and all rodent PAH models. Kv channels not only regulate the EM but are also involved in survival signaling, suggesting that K+ channel activation or augmentation therapy could be beneficial in PAH144.
Potassium channels are tetrameric, membrane-spanning proteins that selectively conduct K+. K+ leaks from PASMC down its intracellular/extracellular concentration gradient (145/5 mM) helping to establish resting membrane potential (EM) at approximately -60mV. EM controls vascular tone by regulating the gating of large conductance, voltage-gated calcium channels (the target of nifedipine, a clinically important PAH treatment177). Depolarization, in response to K+ channel inhibition/downregulation, activates these channels, elevating cytosolic calcium and causing constriction. By regulating intracellular K+ and calcium, K+ channels also regulate cell proliferation and apoptosis and thus vascular remodeling.
PASMC express a diverse array of K+ channels (including voltage-gated (Kv) channels). Several channels are germane to PAH, most notably Kv1.5. Acute inhibition of Kv1.5 by hypoxia initiates hypoxic pulmonary vasoconstriction65. Interestingly anorexigens such as dexfenfluramine, which promote PAH, also acutely inhibit PASMC K+ current and block Kv1.5. Expression of Kv1.5 increases longitudinally in the pulmonary circulation and is maximal in resistance arteries, the major site of pathology in PAH. Selective loss of Kv channel expression (and membrane depolarization) is a hallmark of human145 and experimental35, 144, 146, 147 PAH. Restoring Kv1.5 expression reduces hypoxic-PH144.
K+ channel downregulation increases PASMC proliferation and reduces apoptosis, contributing to obstructive vascular remodeling31, 36, 148, 149. Increased cell proliferation reflects, in part, activation of the Ca2+-calcineurin-dependent proliferative transcription factor, Nuclear Factor for Activated T-lymphocytes (NFAT)39. There are several theories for how Kv downregulation impairs apoptosis (essentially by preventing cell shrinkage and by elevating cytosolic K+, which inhibits capases). Kv channel downregulation also occurs in cancer, the prototypic proliferative, anti-apoptotic disease63.
Inhibition of transcription factors
A variety of transcription factors (HIF-1α, NFAT and c-Jun150) govern the expression of Kv1.5 in PASMC and regulate other factors important to the pathogenesis of PAH. HIF-1α is activated even during normoxia in the PASMC of patients and FHR with PAH19. HIF-1α activation promotes cell survival and inhibition of HIF-1α may be beneficial. Inhibition of HIF-1α restores Kv1.5 expression and Kv current in experimental PAH19. The high cytosolic calcium in PAH PASMC results in nuclear translocation (activation) of NFAT. NFAT promotes PASMC proliferation and decreases Kv1.5 expression39. NFAT inhibition, using either cyclosporine or the more specific peptide inhibitor VIVIT, regresses experimental PAH39. NFAT activation also likely contributes to the hyperpolarized mitochondria seen in PAH PASMC. The antiapoptotic protein, bcl-2, which promotes mitochondrial hyperpolarization, is upregulated in iPAH151. Inhibitors of NFAT increase Kv1.5 expression152 and inhibit bcl-2 expression in monocrotaline-induced PAH39. NFAT inhibition also decreases hypoxic-PH153. Moreover, NFATc3 knock-out mice do not show PA remodeling after chronic hypoxia154. HIF-1α and NFAT inhibition are promising therapeutic strategies.
Inhibition of transient receptor potential (trp) channels (Figure 9)
Upregulation of TRPC6, a nonselective cation channel, occurs in PAH and is another mechanism by which excess amounts of extracellular calcium enter the cells in PAH, independent of L-type calcium channel38, 155-158. Chronic increases in calcium, in part via trp channels, and in part via calcineurin-dependent pathways involving NFAT activation39, drive PASMC proliferation, making trp channel inhibition an interesting therapeutic strategy.
Mitochondria-Metabolic dysfunction in PAH (Figure 7)
PASMC from FHR, and PASMC19 and endothelial cells159 from human PAH exhibit dysmorphic and hyperpolarized mitochondria and a glycolytic shift in metabolism. Such a shift to glycolysis, occurring independent of PO2, was first described in cancer cells (the Warburg phenotype) and is thought to confer resistance to apoptosis. Key molecular contributors to this metabolic phenotype are activation of HIF-1α, which in turn activates transcription of pyruvate dehydrogenase kinase (PDK).
Increased expression of HIF-1α activates a panel of glycolytic genes (such as the glucose transporter, glut 1). HIF-1α simultaneously suppresses the activity of the mitochondrial electron transport chain (ETC) by transactivating the PDK gene, which phosphorylates and inhibits the PDH complex64. PDH catalyses the irreversible oxidation of pyruvate, yielding acetylCoA and CO2. Phosphorylation of any of PDH's 3 regulatory serines by PDK completely inhibits PDH160.
Dichloroacetate inhibits all four PDK isoforms, thereby activating PDH and promoting glucose oxidation. In PAH PASMC (but not normal PASMC), dichloroacetate depolarizes the mitochondria, increasing hydrogen peroxide production and restoring Kv1.5 expression. The net effect of inhibiting PDK is an induction of apoptosis and decrease in proliferation. Interestingly, there is little effect of dichloroacetate on normal cells because PDK is normally relatively inactive. Dichloroacetate regresses many forms of experimental PAH (chronic hypoxic PHT, monocrotaline PAH and FHR PAH)90, 19, 63. An advantage in translating the use of dichloroacetate from rats to humans is that it has be used safely as a treatment of lactic acidosis in children and has been tested acutely in adults with heart failure. New, isoform selective PDK inhibitors are in development for diseases such as diabetes.
Tyrosine kinase inhibition (Figure 8, Table 1)
Excessive expression/activity of a variety of growth factors contribute to obstructive pulmonary vascular remodeling in PAH, including PDGF, basic fibroblast growth factor, epidermal growth factor, and vascular endothelial growth factor (VEGF). Most growth factor receptors are transmembrane receptor tyrosine kinases and they activate diverse signaling pathways (Figure 8)161, 162. Inhibition of EGF and PDGF receptors have beneficial effects on hemodynamics, remodeling, and survival in experimental PAH163, 164. In humans there are case reports of a beneficial effect of adding imatinib to baseline therapy165. The mechanism(s) for the potential beneficial effect of imatinib are unclear because it inhibits the tyrosine kinases, PDGF receptors, BCR-ABL, and c-kit.
In addition to receptor tyrosine kinases, serine/threonine kinases, such as the Raf family and its downstream pathways, offer targets for intervention in PAH166, 167. Sorafenib is a “multikinase inhibitor”, blocking the serine/threonine kinases Raf-1 and b-Raf, tyrosine kinases, PDGF and VEGF receptors, c-kit, and Flt-3, with IC50 values between 6 and 70 nmol/L. Sorafenib is approved for the treatment of renal and hepatocellular carcinoma. Sorafenib prevents and reverses PAH and cardiac remodeling in monocrotaline-treated rats and may have more pronounced effects on right ventricular function than imatinib168. Phase 1 clinical trials with both imatinib and sorafenib (NCT00452218) have been completed but not yet published.
Elastase and Matrix Metalloproteinases (MMPs)
Increased elastolytic activity may be an early feature of PH and serum elastase levels are elevated in experimental PAH169. Endogenous elastases may contribute to the development of PAH by liberating mitogens (e.g. tenascin c40), and growth factors from the matrix and activating growth factor receptors in a ligand-independent manner (Figure 10). Elastase inhibitors can attenuate or reverse experimental PAH170 but synthetic inhibitors with acceptable toxicity in humans have yet to be developed. Augmenting expression of endogenous elastin inhibitors, such as elafin171, may prove a better strategy.
Peroxisome proliferator-activated receptor activation (PPARs, Table 1)
PPARs are ligand-activated transcription factors, belonging to the nuclear receptor superfamily. Upon ligand activation, PPARs heterodimerize with the retinoid X receptor and bind to PPAR response elements in regulatory promoter regions of their target genes. A series of recent observations suggests that PPARγ could be a drug target in PAH172, 173. PPARγ is a downstream target of BMP2 in human PASMC173. PPARγ is important for BMP2-mediated inhibition of PDGF-induced vascular SMC proliferation172. Mice lacking SMC PPARγ develop PAH172. PPARγ activation stimulates apolipoprotein E expression. Recombinant apolipoprotein E inhibits PDGFR-β–mediated SMC proliferation and migration174 PPARγ targets, independent of apolipoprotein E, may also important in suppressing pulmonary vascular remodeling as male apolipoprotein E (-/-) mice fed a high fat diet develop PAH which is reversed by rosiglitazone (a PPARγ agonist)173. PPARγ agonists have direct anti-inflammatory and pro-apoptotic effects. PPARs can also interact with signaling molecules to regulate gene expression, independent of DNA binding. PPARγ can impair the phosphorylation of extracellular signal-regulated protein kinase (ERK)175 which is implicated in PASMC proliferation and migration. iPAH patients have reduced lung expression of PPARγ and apolipoprotein E mRNA. Because the thiazolidinedione rosiglitazone is widely used in the treatment of type II diabetes, a trial in PAH would be feasible. Despite this promise, rosiglitazone failed to ameliorate pulmonary hypertension in hypoxic-PH rats, although it did reduce RVH and pulmonary vascular remodeling176.
Inflammation (Figure 11)
Aside from the association of PAH with several collagen vascular autoimmune disorders (e.g. scleroderma, systemic lupus erythematosus and mixed connective disease) and schistosomiasis, several observations argue for a role of inflammation in the pathogenesis of PAH. These include: the presence of T cells, B cells and macrophages in plexiform lesions; the detection of autoantibodies to endothelial cells and fibroblasts; raised blood cytokine and chemokine levels; and the association of PAH with certain infections such as human herpes virus 8. Mice that overexpress S100A4/Mts1 develop extensive and severe neointimal lesions following injection of the γ murine herpes virus-68 (the murine homolog of HHV-8)177. PAH also develops in a subset of patients with HIV disease. The HIVnef gene was also recently implicated in plexogenic pulmonary vascular lesions associated with PAH in HIV-infected patients and SIV-infected nonhuman primates178.
Athymic nude rats, lacking T cells, appear more sensitive than normal rats to the development of PAH when challenged with the VEGF-receptor antagonist SU-5416179. A protective role for T cells was established by the administration of splenocytes from euthymic rats. In iPAH, regulatory Treg cells are increased whilst CD8+ cytotoxic T cells are decreased180. Treg cells maintain immunotolerance and they are potent inhibitors of antitumour and possibly antiviral immune response. The increase in Treg cells may be a normal counter-regulation or compensation for an initial inflammatory response.
Can the immune system be therapeutically targeted in PAH? Mycophenolate mofetil, a potent immunosuppressant used in humans, prevents monocrotaline-induced PAH in rats181; however, regression trials (a more clinically relevant standard for experimental PAH therapies) are needed.
Endothelial Progenitor Cells (EPCs)
EPCs arise from mesodermal stem cells or haemangioblasts in the bone marrow. Circulating in plasma, they home to sites of ischemia or endothelial injury and differentiate into mature endothelial cells in situ, contributing to revascularization and vascular homeostasis. EPCs can be considered a potential therapeutic target, a predictive biomarker182 or as a vector for cell-based therapy. Circulating EPC numbers (defined by CD34+/KDR+ and CD34+/CD133+/KDR+ positive cells) are significantly lower in patients with Eisenmenger's syndrome compared with normal controls183. Some, but not all, investigators have reported reduced EPCs in iPAH patients. Differences in the markers used to identify and quantify EPCs complicates interpretation of the data.
The in vitro functions of endothelial-like, mononuclear cells (e.g. colony forming capacity, adherence, migration and sensitivity to apoptosis) isolated from the blood of iPAH patients differ from those of healthy controls183-186. Whether these difference are beneficial, promoting revascularization in the hypertensive lung187, or contribute to the pathology, by augmenting pulmonary vascular remodelling185, is unclear. This distinction is important given that some treatments (e.g. sildenafil) are associated with a dose-dependent increase in the abundance of circulating EPCs183 and potential new therapies for PAH, such as statins and PPAR-γ agonists, also induce the mobilization and differentiation of EPCs.
Administration of EPCs has produced improvements in pulmonary hemodynamics, vascular remodeling and survival in monocrotaline-induced PAH188. Cell therapy has been less effective in hypoxia-induced PAH and may contribute to the pathologic vascular remodeling (Figure 12). The benefits of cell therapy may be enhanced by the expression of genes that inhibit smooth muscle cell proliferation and/or stimulate angiogenesis (e.g. eNOS)187, 189, 190. Even fibroblasts can be made somewhat therapeutic when they are transfected with VEGF. These modified fibroblasts prevent worsening of monocrotaline-induced PAH191.
Two small pilot studies, in which adults and children with IPAH were given a single intravenous infusion of autologous mononuclear cells provide support for the therapeutic potential of cell-based therapy in patients192, 193. A therapeutic trial (PHACeT, NCT00469027) to assesses the safety of administrating autologous, cultured, eNOS-transduced mononuclear cells in iPAH patients has commenced.
There remains much to be done in the field of cell-based therapies for PAH, particularly as it remains uncertain whether influx of progenitor cells into the lung in PAH is beneficial or harmful. In Figure 12 lessons learned from remodeling in hypoxia are reviewed. In hypoxia, inflammatory and progenitor cells appear to contribute to the pathological remodeling; however, it is not certain whether this applies to PAH.
Miscellaneous pathways with therapeutic implications
Statins, heparins, dihydroepiandrosterone (DHEA) and inhibitors of Angiopoietin 1, STAT3, Polyamines, Survivin, and the cell cycle offer potential therapeutics for PAH and are discussed, due to page limits, in the on-line supplement.
Conclusions
In this review of the basic science of PAH we assessed emerging concepts of the molecular mechanisms of PAH and identified the novel therapeutic targets suggested by this science. New therapeutic strategies include: enhancing endothelial function/vasodilatation using guanylate cyclase activators or vasodilator peptides, such as adrenomedullin and vasointestinal peptide, augmenting the BMPR2/SMAD pathway, inhibiting serotonin and the serotonin transporter, modulating expression/activity of ion channels (Kv1.5 and TRPC6), inhibiting transcription factors (NFAT and HIF-1∝), increasing apoptosis (survivin inhibitors), inhibiting tyrosine kinases, inhibiting the contractile apparatus (rho kinase inhibitors), preserving elastin, and modulating influx of inflammatory and progenitor cells. Opportunity also exists to accelerate drug development using molecules approved for the management cancer, vascular dysfunction and metabolic disorders. These conditions share PAH's pathophysiologic abnormalities (endothelial dysfunction, excessive cell proliferation, disordered apoptosis, and inflammation). Repurposed drugs that have potential in PAH include: phosphodiesterase-5 inhibitors (for erectile dysfunction), Imatinib (for chronic myelogenous leukemia), Sorafenib (for renal carcinoma) and dichloroacetate (for mitochondrial diseases). We do not endorse the off-label application of these agents in clinical practice; however there is a compelling need to study these potentially curative agents in preclinical and, when appropriate, clinical trials. This is an exciting time in the search for a cure for PAH and it is time for Physicians, armed with a Basic Science Playbook, to take the field.
Supplementary Material
Acknowledgments
The authors thank Dr. Nick Morrell, Cambridge, U.K. for his comments on the BMPR2 signaling portion of this review. Dr. Archer is supported by NIH-RO1-HL071115 and 1RC1HL099462-01, the American Heart Association and the Roche Foundation for Anemia Research. Dr. Weir is supported by R01 HL 65322. Dr Wilkins is supported by grants from the British Heart Foundation and Medical Research Council.
Footnotes
Conflict of Interest Disclosure: The authors have no conflict of interest to disclose other than the acknowledged grant support. All authors had full access to the manuscript and approved the final version.
References
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–1030. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 3.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30(1):104–109. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- 4.Butrous G, Ghofrani HA, Grimminger F. Pulmonary vascular disease in the developing world. Circulation. 2008;118(17):1758–1766. doi: 10.1161/CIRCULATIONAHA.107.727289. [DOI] [PubMed] [Google Scholar]
- 5.Fruchter O, Yigla M. Underlying aetiology of pulmonary hypertension in 191 patients: a single centre experience. Respirology. 2008;13(6):825–831. doi: 10.1111/j.1440-1843.2008.01364.x. [DOI] [PubMed] [Google Scholar]
- 6.Robbins I, Newman J, Johnson R, Hemnes A, Fremont R, Piana R, Zhao D, Byrne D. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009 doi: 10.1378/chest.08-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30(6):1103–1110. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. Journal of Heart and Lung Transplantation. 1996;15:100–105. [PubMed] [Google Scholar]
- 9.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167(4):580–586. doi: 10.1164/rccm.200204-333OC. [DOI] [PubMed] [Google Scholar]
- 10.Condliffe R, Kiely D, Peacock A, Corris P, Gibbs J, Vrapi F, Das C, Elliot C, Johnson M, DeSoyza J, Torpy C, Goldsmith K, Hodgkins D, Hughes R, Pepke-Zaba J, Coghlan J. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179:91–92. doi: 10.1164/rccm.200806-953OC. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd J, Robbins I. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. AJRCCM. 2003;167(4):580–586. doi: 10.1164/rccm.200204-333OC. [DOI] [PubMed] [Google Scholar]
- 12.Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–3111. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 13.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296–302. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 14.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195(3):367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 15.Levy NT, Liapis H, Eisenberg PR, Botney MD, Trulock EP. Pathologic regression of primary pulmonary hypertension in left native lung following right single-lung transplantation. J Heart Lung Transplant. 2001;20(3):381–384. doi: 10.1016/s1053-2498(00)00153-4. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura T, Faul JL, Berry GJ, Vaszar LT, Qiu D, Pearl RG, Kao PN. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med. 2002;166(10):1403–1408. doi: 10.1164/rccm.200203-268OC. [DOI] [PubMed] [Google Scholar]
- 17.van Albada ME, du Marchie Sarvaas GJ, Koster J, Houwertjes MC, Berger RM, Schoemaker RG. Effects of erythropoietin on advanced pulmonary vascular remodelling. Eur Respir J. 2008;31(1):126–134. doi: 10.1183/09031936.00035607. [DOI] [PubMed] [Google Scholar]
- 18.Sato K, Webb S, Tucker A, Rabinovitch M, O'Brien RF, McMurtry IF, Stelzner TJ. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145(4 Pt 1):793–797. doi: 10.1164/ajrccm/145.4_Pt_1.793. [DOI] [PubMed] [Google Scholar]
- 19.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet SN, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir E, Archer SL. An abnormal mitochondrial-HIF-1-Kv channel pathway disrupts oxygen-sensing and triggers pulmonary arterial hypertension (PAH) in fawn-hooded rats: similarities to human PAH. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 20.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. Faseb J. 2005 doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- 21.Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res. 2006;98(10):1323–1330. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- 22.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–1114. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 23.Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164(1):253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herve P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, Poubeau P, Cerrina J, Duroux P, Drouet L. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99(3):249–254. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 25.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 26.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res. 1997;81(1):34–41. doi: 10.1161/01.res.81.1.34. [DOI] [PubMed] [Google Scholar]
- 27.Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114(6):464–469. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- 28.White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL, Miller CM, Blaxall BC, Hall CM, Pierce RA, Cool CD, Taubman MB. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L583–590. doi: 10.1152/ajplung.00321.2006. [DOI] [PubMed] [Google Scholar]
- 29.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 30.McMurtry MS, Moudgil R, Hashimoto K, Bonnet S, Michelakis ED, Archer SL. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L872–878. doi: 10.1152/ajplung.00309.2006. [DOI] [PubMed] [Google Scholar]
- 31.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115(6):1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eddahibi S, Raffestin B, Hamon M, Adnot S. Is the serotonin transporter involved in the pathogenesis of pulmonary hypertension? J Lab Clin Med. 2002;139(4):194–201. doi: 10.1067/mlc.2002.122181. [DOI] [PubMed] [Google Scholar]
- 33.Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, Desai AA, Singleton PA, Sammani S, Sam L, Liu Y, Husain AN, Lang RM, Ratain MJ, Lussier YA, Garcia JG. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33(2):278–291. doi: 10.1152/physiolgenomics.00169.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351(9104):726–727. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- 35.Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol. 2001;90(6):2249–2256. doi: 10.1152/jappl.2001.90.6.2249. [DOI] [PubMed] [Google Scholar]
- 36.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105(2):244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 37.Young KA, Ivester C, West J, Carr M, Rodman DM. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L841–848. doi: 10.1152/ajplung.00158.2005. [DOI] [PubMed] [Google Scholar]
- 38.Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. 2004;19:44–50. doi: 10.1152/nips.01457.2003. [DOI] [PubMed] [Google Scholar]
- 39.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105(1):21–34. doi: 10.1172/JCI6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welsh DJ, Harnett M, MacLean M, Peacock AJ. Proliferation and signaling in fibroblasts: role of 5-hydroxytryptamine2A receptor and transporter. Am J Respir Crit Care Med. 2004;170(3):252–259. doi: 10.1164/rccm.200302-264OC. [DOI] [PubMed] [Google Scholar]
- 42.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22(2):358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 43.Davie NJ, Crossno JT, Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L668–678. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 44.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37(10):741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26(1):81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Davies RJ, Southwood M, Long L, Yang X, Sobolewski A, Upton PD, Trembath RC, Morrell NW. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102(10):1212–1221. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L740–754. doi: 10.1152/ajplung.00284.2002. [DOI] [PubMed] [Google Scholar]
- 49.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296(5573):1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 50.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118(7):722–730. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura C, Phillips JA, 3rd, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P, Nichols WC, Elliott G. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43(12 Suppl S):33S–39S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 52.Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, Le Gall C, Parent F, Garcia G, Herve P, Barst RJ, Simonneau G. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2003;167(10):1433–1439. doi: 10.1164/rccm.200204-330OC. [DOI] [PubMed] [Google Scholar]
- 53.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98(6):818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 54.McMurtry M, Moudgil R, Hashimoto K, Bonnet S, Michelakis E, Archer S. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L872–878. doi: 10.1152/ajplung.00309.2006. [DOI] [PubMed] [Google Scholar]
- 55.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104(10):1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 56.Reynolds AM, Xia W, Holmes MD, Hodge SJ, Danilov S, Curiel DT, Morrell NW, Reynolds PN. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1182–1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
- 57.Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK, Morrell NW. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119(4):566–576. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- 58.Eddahibi S, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet A, Dartevelle P, Housset B, Hamon M, Weitzenblum E, Adnot S. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation. 2003;108:1839–1844. doi: 10.1161/01.CIR.0000091409.53101.E8. [DOI] [PubMed] [Google Scholar]
- 59.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O'Connor DT, Yuan JX. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292(5):C1837–1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 60.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A Functional Single-Nucleotide Polymorphism in the TRPC6 Gene Promoter Associated With Idiopathic Pulmonary Arterial Hypertension. Circulation. 2009;119(17):2313–2322. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1{alpha}-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294(2):H570–578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 62.Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1998;114(3 Suppl):225S–230S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 63.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 64.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 65.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353(19):2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial Metabolism, Redox Signaling and Fusion…a mitochondria-ROS-HIF-1{alpha}-Kv1.5 oxygen-sensing pathway intersecting pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2007 doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 67.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95(8):830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 68.Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22{alpha}-targeted overexpression of the serotonin transporter. FASEB J. 2009 doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
- 69.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116(3):238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 70.Sharma S, Taegtmeyer H, Adrogue J, Razeghi P, Sen S, Ngumbela K, Essop MF. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2004;286(3):H1185–1192. doi: 10.1152/ajpheart.00916.2003. [DOI] [PubMed] [Google Scholar]
- 71.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15(3):110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 72.Evans AM. AMP-activated protein kinase and the regulation of Ca2+ signalling in O2-sensing cells. J Physiol. 2006;574(Pt 1):113–123. doi: 10.1113/jphysiol.2006.108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allard MF, Parsons HL, Saeedi R, Wambolt RB, Brownsey R. AMPK and metabolic adaptation by the heart to pressure overload. Am J Physiol Heart Circ Physiol. 2007;292(1):H140–148. doi: 10.1152/ajpheart.00424.2006. [DOI] [PubMed] [Google Scholar]
- 74.Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004;44(5):662–667. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- 75.Bayrak F, Komurcu-Bayrak E, Mutlu B, Kahveci G, Basaran Y, Erginel-Unaltuna N. Ventricular pre-excitation and cardiac hypertrophy mimicking hypertrophic cardiomyopathy in a Turkish family with a novel PRKAG2 mutation. Eur J Heart Fail. 2006;8(7):712–715. doi: 10.1016/j.ejheart.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 76.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29(1):120–127. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 77.Piao L, Fang YH, Cadete VJJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. Journal of Molecular Medicine. 2009 doi: 10.1007/s00109-009-0524-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghofrani HA, Wiedemann R, Rose F, Olschewski H, Schermuly RT, Weissmann N, Seeger W, Grimminger F. Combination therapy with oral sildenafil and inhaled iloprost for severe pulmonary hypertension. Ann Intern Med. 2002;136(7):515–522. doi: 10.7326/0003-4819-136-7-200204020-00008. [DOI] [PubMed] [Google Scholar]
- 79.Simonneau G, Rubin LJ, Galie N, Barst RJ, Fleming TR, Frost AE, Engel PJ, Kramer MR, Burgess G, Collings L, Cossons N, Sitbon O, Badesch DB. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149(8):521–530. doi: 10.7326/0003-4819-149-8-200810210-00004. [DOI] [PubMed] [Google Scholar]
- 80.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333(4):214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 81.Archer SL, Djaballah K, Humbert M, Weir KE, Fartoukh M, Dall'ava-Santucci J, Mercier JC, Simonneau G, Dinh-Xuan AT. Nitric oxide deficiency in fenfluramine- and dexfenfluramine-induced pulmonary hypertension. Am J Respir Crit Care Med. 1998;158(4):1061–1067. doi: 10.1164/ajrccm.158.4.9802113. [DOI] [PubMed] [Google Scholar]
- 82.Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol. 2002;137(8):1187–1194. doi: 10.1038/sj.bjp.0704984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bulau P, Zakrzewicz D, Kitowska K, Leiper J, Gunther A, Grimminger F, Eickelberg O. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L18–24. doi: 10.1152/ajplung.00076.2006. [DOI] [PubMed] [Google Scholar]
- 84.Pullamsetti S, Kiss L, Ghofrani HA, Voswinckel R, Haredza P, Klepetko W, Aigner C, Fink L, Muyal JP, Weissmann N, Grimminger F, Seeger W, Schermuly RT. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. 2005;19(9):1175–1177. doi: 10.1096/fj.04-3223fje. [DOI] [PubMed] [Google Scholar]
- 85.Fagan KA, McMurtry I, Rodman DM. Nitric oxide synthase in pulmonary hypertension: lessons from knockout mice. Physiol Res. 2000;49(5):539–548. [PubMed] [Google Scholar]
- 86.Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, Wilkins MR, Channon KM. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation. 2005;111(16):2126–2133. doi: 10.1161/01.CIR.0000162470.26840.89. [DOI] [PubMed] [Google Scholar]
- 87.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13(2):198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 88.Celermajer DS, Dollery C, Burch M, Deanfield JE. Role of endothelium in the maintenance of low pulmonary vascular tone in normal children. Circulation. 1994;89(5):2041–2044. doi: 10.1161/01.cir.89.5.2041. [DOI] [PubMed] [Google Scholar]
- 89.Stamler JS, Loh E, Roddy MA, Currie KE, Creager MA. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation. 1994;89(5):2035–2040. doi: 10.1161/01.cir.89.5.2035. [DOI] [PubMed] [Google Scholar]
- 90.Hampl V, Archer SL, Nelson DP, Weir EK. Chronic EDRF inhibition and hypoxia: effects on pulmonary circulation and systemic blood pressure. J Appl Physiol. 1993;75(4):1748–1757. doi: 10.1152/jappl.1993.75.4.1748. [DOI] [PubMed] [Google Scholar]
- 91.Bloch KD, Ichinose F, Roberts JD, Jr, Zapol WM. Inhaled NO as a therapeutic agent. Cardiovasc Res. 2007;75(2):339–348. doi: 10.1016/j.cardiores.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez-Penate GM, Julia-Serda G, Ojeda-Betancort N, Garcia-Quintana A, Pulido-Duque J, Rodriguez-Perez A, Cabrera-Navarro P, Gomez-Sanchez MA. Long-term inhaled nitric oxide plus phosphodiesterase 5 inhibitors for severe pulmonary hypertension. J Heart Lung Transplant. 2008;27(12):1326–1332. doi: 10.1016/j.healun.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 93.Hampl V, Tristani-Firouzi M, Hutsell TC, Archer SL. Nebulized nitric oxide/nucleophile adduct reduces chronic pulmonary hypertension. Cardiovasc Res. 1996;31(1):55–62. [PubMed] [Google Scholar]
- 94.Lam CF, Van Heerden PV, Blott J, Roberts B, Ilett KF. The selective pulmonary vasodilatory effect of inhaled DETA/NO, a novel nitric oxide donor, in ARDS-a pilot human trial. J Crit Care. 2004;19(1):48–53. doi: 10.1016/j.jcrc.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 95.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 96.Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N, Ghofrani HA, Kaulen C, Dunkern T, Schudt C, Voswinckel R, Zhou J, Samidurai A, Klepetko W, Paddenberg R, Kummer W, Seeger W, Grimminger F. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: target for reverse-remodeling therapy. Circulation. 2007;115(17):2331–2339. doi: 10.1161/CIRCULATIONAHA.106.676809. [DOI] [PubMed] [Google Scholar]
- 97.Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105(20):2398–2403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- 98.Archer SL, Michelakis ED. Phosphodiesterase Type 5 Inhibitors for Pulmonary Arterial Hypertension. N Engl J Med. 2009;361:1862–1869. doi: 10.1056/NEJMct0904473. [DOI] [PubMed] [Google Scholar]
- 99.Schermuly RT, Ghofrani HA, Enke B, Weissmann N, Grimminger F, Seeger W, Schudt C, Walmrath D. Low-dose systemic phosphodiesterase inhibitors amplify the pulmonary vasodilatory response to inhaled prostacyclin in experimental pulmonary hypertension. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1500–1506. doi: 10.1164/ajrccm.160.5.9901102. [DOI] [PubMed] [Google Scholar]
- 100.Schermuly RT, Roehl A, Weissmann N, Ghofrani HA, Schudt C, Tenor H, Grimminger F, Seeger W, Walmrath D. Subthreshold doses of specific phosphodiesterase type 3 and 4 inhibitors enhance the pulmonary vasodilatory response to nebulized prostacyclin with improvement in gas exchange. J Pharmacol Exp Ther. 2000;292(2):512–520. [PubMed] [Google Scholar]
- 101.Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, Schluter KD, Dingendorf A, Hackemack S, Kolosionek E, Kaulen C, Dumitrascu R, Weissmann N, Mittendorf J, Klepetko W, Seeger W, Ghofrani HA, Grimminger F. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J. 2008;32(4):881–891. doi: 10.1183/09031936.00114407. [DOI] [PubMed] [Google Scholar]
- 102.Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, Pellens M, Gillijns H, Swinnen M, Graveline A, Collen D, Dewerchin M, Brouckaert P, Bloch KD, Janssens S. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation. 2007;116(8):936–943. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- 103.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5(9):755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deruelle P, Grover TR, Abman SH. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L798–806. doi: 10.1152/ajplung.00119.2005. [DOI] [PubMed] [Google Scholar]
- 105.Deruelle P, Grover TR, Storme L, Abman SH. Effects of BAY 41-2272, a soluble guanylate cyclase activator, on pulmonary vascular reactivity in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L727–733. doi: 10.1152/ajplung.00409.2004. [DOI] [PubMed] [Google Scholar]
- 106.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113(2):286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 107.Mittendorf J, Weigand S, Alonso-Alija C, Bischoff E, Feurer A, Gerisch M, Kern A, Knorr A, Lang D, Muenter K, Radtke M, Schirok H, Schlemmer KH, Stahl E, Straub A, Wunder F, Stasch JP. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension. ChemMedChem. 2009 doi: 10.1002/cmdc.200900014. [DOI] [PubMed] [Google Scholar]
- 108.Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24(3):413–420. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 109.Nandi M, Leiper J, Arrigoni F, Hislop A, Vallance P, Haworth S. Developmental regulation of GTP-CH1 in the porcine lung and its relationship to pulmonary vascular relaxation. Pediatr Res. 2006;59(6):767–772. doi: 10.1203/01.pdr.0000219301.19958.a0. [DOI] [PubMed] [Google Scholar]
- 110.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169(6):764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 111.Sanford M, Keating GM. Spotlight on sapropterin in primary hyperphenylalaninemia. BioDrugs. 2009;23(3):201–202. doi: 10.2165/00063030-200923030-00007. [DOI] [PubMed] [Google Scholar]
- 112.Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hubschle T, Mang C, Strobel H, Suzuki T, Kleinert H, Forstermann U, Ruetten H, Li H. Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J Pharmacol Exp Ther. 2008;325(2):370–379. doi: 10.1124/jpet.107.128009. [DOI] [PubMed] [Google Scholar]
- 113.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 114.Wilkins MR, Paul GA, Strange JW, Tunariu N, Gin-Sing W, Banya WA, Westwood MA, Stefanidis A, Ng LL, Pennell DJ, Mohiaddin RH, Nihoyannopoulos P, Gibbs JS. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171(11):1292–1297. doi: 10.1164/rccm.200410-1411OC. [DOI] [PubMed] [Google Scholar]
- 115.McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galie N, Rainisio M, Simonneau G, Rubin LJ. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25(2):244–249. doi: 10.1183/09031936.05.00054804. [DOI] [PubMed] [Google Scholar]
- 116.Nagaya N, Nishikimi T, Okano Y, Uematsu M, Satoh T, Kyotani S, Kuribayashi S, Hamada S, Kakishita M, Nakanishi N, Takamiya M, Kunieda T, Matsuo H, Kangawa K. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31(1):202–208. doi: 10.1016/s0735-1097(97)00452-x. [DOI] [PubMed] [Google Scholar]
- 117.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27(1):47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 118.Zhao L, Winter RJ, Krausz T, Hughes JM. Effects of continuous infusion of atrial natriuretic peptide on the pulmonary hypertension induced by chronic hypoxia in rats. Clin Sci (Lond) 1991;81(3):379–385. doi: 10.1042/cs0810379. [DOI] [PubMed] [Google Scholar]
- 119.Baliga RS, Zhao L, Madhani M, Lopez-Torondel B, Visintin C, Selwood D, Wilkins MR, MacAllister RJ, Hobbs AJ. Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(8):861–869. doi: 10.1164/rccm.200801-121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi JG, Ding YG, Tang CS, Du JB. Chronic administration of adrenomedullin attenuates hypoxic pulmonary vascular structural remodeling and inhibits proadrenomedullin N-terminal 20-peptide production in rats. Peptides. 2007;28(4):910–919. doi: 10.1016/j.peptides.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 121.Shirai M, Pearson JT, Shimouchi A, Nagaya N, Tsuchimochi H, Ninomiya I, Mori H. Changes in functional and histological distributions of nitric oxide synthase caused by chronic hypoxia in rat small pulmonary arteries. Br J Pharmacol. 2003;139(5):899–910. doi: 10.1038/sj.bjp.0705312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nagaya N, Kyotani S, Uematsu M, Ueno K, Oya H, Nakanishi N, Shirai M, Mori H, Miyatake K, Kangawa K. Effects of adrenomedullin inhalation on hemodynamics and exercise capacity in patients with idiopathic pulmonary arterial hypertension. Circulation. 2004;109(3):351–356. doi: 10.1161/01.CIR.0000109493.05849.14. [DOI] [PubMed] [Google Scholar]
- 123.Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation. 2007;115(10):1260–1268. doi: 10.1161/CIRCULATIONAHA.106.681718. [DOI] [PubMed] [Google Scholar]
- 124.Hamidi SA, Prabhakar S, Said SI. Enhancement of pulmonary vascular remodelling and inflammatory genes with VIP gene deletion. Eur Respir J. 2008;31(1):135–139. doi: 10.1183/09031936.00105807. [DOI] [PubMed] [Google Scholar]
- 125.Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, Funk G, Hamilton G, Novotny C, Burian B, Block L. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111:1339–1346. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, Morrell NW. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet. 2008;17(20):3180–3190. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- 127.Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee CK, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther. 2002;6(1):119–126. doi: 10.1006/mthe.2002.0639. [DOI] [PubMed] [Google Scholar]
- 128.Thomas M, Docx C, Holmes AM, Beach S, Duggan N, England K, Leblanc C, Lebret C, Schindler F, Raza F, Walker C, Crosby A, Davies RJ, Morrell NW, Budd DC. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am J Pathol. 2009;174(2):380–389. doi: 10.2353/ajpath.2009.080565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hervé P, Lanuay J, Scrobahaci M, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–254. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 130.Eddahibi S, Humbert M, Fadel E, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Machado RD, Koehler R, Glissmeyer E, Veal C, Suntharalingam J, Kim M, Carlquist J, Town M, Elliott CG, Hoeper M, Fijalkowska A, Kurzyna M, Thomson JR, Gibbs SR, Wilkins MR, Seeger W, Morrell NW, Gruenig E, Trembath RC, Janssen B. Genetic association of the serotonin transporter in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173(7):793–797. doi: 10.1164/rccm.200509-1365OC. [DOI] [PubMed] [Google Scholar]
- 132.Willers ED, Newman JH, Loyd JE, Robbins IM, Wheeler LA, Prince MA, Stanton KC, Cogan JA, Runo JR, Byrne D, Humbert M, Simonneau G, Sztrymf B, Morse JA, Knowles JA, Roberts KE, McElroy JJ, Barst RJ, Phillips JA., 3rd Serotonin transporter polymorphisms in familial and idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173(7):798–802. doi: 10.1164/rccm.200509-1361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mitani Y, Mutlu A, Russell J, Brindley D, DeAlmeida J, Rabinovitch M. Dexfenfluramine protects against pulmonary hypertension in rats. J Appl Physiol. 2002;93:1770–1778. doi: 10.1152/japplphysiol.00500.2002. [DOI] [PubMed] [Google Scholar]
- 134.Kawut S, Horn E, Berekashvili K, Lederer D, Widlitz A, Rosenzweig E, Barst R. Selective serotonin reuptake inhibitor use and outcomes in pulmonary arterial hypertension. Pulm Pharmacol Ther. 2006;19:370–374. doi: 10.1016/j.pupt.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 135.Morecroft I, Dempsie Y, Bader M, et al. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension. 2007;49:232–236. doi: 10.1161/01.HYP.0000252210.58849.78. [DOI] [PubMed] [Google Scholar]
- 136.Launay JM, Herve P, Peoc'h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, Maroteaux L. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med. 2002;8(10):1129–1135. doi: 10.1038/nm764. [DOI] [PubMed] [Google Scholar]
- 137.Dempsie Y, Morecroft I, Welsh D, MacRitchie N, Herold N, Loughlin L, Nilsen M, Peacock A, Harmar A, Bader M, MacLean M. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation. 2008;117:2928–2937. doi: 10.1161/CIRCULATIONAHA.108.767558. [DOI] [PubMed] [Google Scholar]
- 138.Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Begaud B. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609–616. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- 139.Long L, MacLean M, Jeffery T, Morecroft I, Yang X, Rudarkanchana N, Southwood M, James V, Trembath R, Morrell N. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-Deficint mice. Circ Res. 2006;98:818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 140.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res. 2007;100(6):923–929. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 141.Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J. 2006;70(2):174–178. doi: 10.1253/circj.70.174. [DOI] [PubMed] [Google Scholar]
- 142.Liu Y, Ren W, Warburton R, Toksoz D, Fanburg B. Serotonin induces Rho/ROCK-dependent activation of Smads 1/5/8 in pulmonary artery smooth muscle cells. FASEB J. 2009 doi: 10.1096/fj.08-127910. [DOI] [PubMed] [Google Scholar]
- 143.Mair K, MacLean M, Morecroft I, Dempsie Y, Palmer T. Novel interactions between the 5-HT transporter, 5-HT1B receptors and Rho kinase in vivo and in pulmonary fibroblasts. Br J Pharmacol. 2008;155:606–616. doi: 10.1038/bjp.2008.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pozeg ZI, Michelakis ED, McMurtry MS, Thebaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A, Archer SL. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation. 2003;107(15):2037–2044. doi: 10.1161/01.CIR.0000062688.76508.B3. [DOI] [PubMed] [Google Scholar]
- 145.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98(14):1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 146.Smirnov SV, Robertson TP, Ward JPT, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol. 1994;266(1 Part 2):H365–H370. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- 147.Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases K(V) channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol. 2001;280(4):L801–812. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- 148.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68(2):75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 149.Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol. 2004;286(1):L49–67. doi: 10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- 150.Yu Y, Platoshyn O, Zhang J, Krick S, Zhao Y, Rubin LJ, Rothman A, Yuan JX. c-Jun decreases voltage-gated K(+) channel activity in pulmonary artery smooth muscle cells. Circulation. 2001;104(13):1557–1563. doi: 10.1161/hc3801.095662. [DOI] [PubMed] [Google Scholar]
- 151.Geraci M, Moore M, Gesell T, Yeager M, Alger L, Golpon H, Gao B, Loyd J, Tuder R, Voelkel N. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- 152.Bonnet S, Rochefort G, Sutendra G, Archer S, Haromy A, Webster L, Hashimoto K, Bonnet S, Michelakis E. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. PNAS. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Koulmann N, Novel-Chate V, Peinnequin A, Chapot R, Serrurier B, Simler N, Richard H, Ventura-Clapier R, Bigard X. Cyclosporin A inhibits hypoxia-induced pulmonary hypertension and right ventricle hypertrophy. Am J Respir Crit Care Med. 2006;174:699–705. doi: 10.1164/rccm.200512-1976OC. [DOI] [PubMed] [Google Scholar]
- 154.deFrutos S, Spangler R, Alo D, Bose L. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha-actin up-regulation. J Biol Chem. 2007;282:15081–15089. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(6):L1233–1245. doi: 10.1152/ajplung.00445.2002. [DOI] [PubMed] [Google Scholar]
- 156.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280(2):H746–755. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 157.Zhang S, Patel HH, Murray F, Remillard CV, Schach C, Thistlethwaite PA, Insel PA, Yuan JX. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1202–1210. doi: 10.1152/ajplung.00214.2006. [DOI] [PubMed] [Google Scholar]
- 158.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2004;101(38):13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104(4):1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001;70:33–75. doi: 10.1016/s0079-6603(01)70013-x. [DOI] [PubMed] [Google Scholar]
- 161.Roberts KE, McElroy JJ, Wong WP, Yen E, Widlitz A, Barst RJ, Knowles JA, Morse JH. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24(3):371–374. doi: 10.1183/09031936.04.00018604. [DOI] [PubMed] [Google Scholar]
- 162.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11(2):211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 163.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 165.Ghofrani H, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 166.Yogi A, Callera GE, Montezano AC, Aranha AB, Tostes RC, Schiffrin EL, Touyz RM. Endothelin-1, but not Ang II, activates MAP kinases through c-Src independent Ras-Raf dependent pathways in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27(9):1960–1967. doi: 10.1161/ATVBAHA.107.146746. [DOI] [PubMed] [Google Scholar]
- 167.Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004;110(6):718–723. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- 168.Moreno-Vinasco L, Gomber-Maitland M, Maitland M, Desai A, Singleton P, Sammani S, Sam L, Liu Y, Husain A, Lang R, Ratain M, Lussier Y, Garcia J. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33:278–291. doi: 10.1152/physiolgenomics.00169.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rabinovitch M. Elastase and the pathobiology of unexplained pulmonary hypertension. Chest. 1998;114(3 Suppl):213S–224S. doi: 10.1378/chest.114.3_supplement.213s. [DOI] [PubMed] [Google Scholar]
- 170.Ye CL, Rabinovitch M. Inhibition of elastolysis by SC-37698 reduces development and progression of monocrotaline pulmonary hypertension. Am J Physiol. 1991;261(4 Pt 2):H1255–1267. doi: 10.1152/ajpheart.1991.261.4.H1255. [DOI] [PubMed] [Google Scholar]
- 171.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105(4):516–521. doi: 10.1161/hc0402.102866. [DOI] [PubMed] [Google Scholar]
- 172.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(5):1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115(10):1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 174.Ishigami M, Swertfeger DK, Granholm NA, Hui DY. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J Biol Chem. 1998;273(32):20156–20161. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- 175.Wakino S, Kintscher U, Liu Z, Kim S, Yin F, Ohba M, Kuroki T, Schonthal AH, Hsueh WA, Law RE. Peroxisome proliferator-activated receptor gamma ligands inhibit mitogenic induction of p21(Cip1) by modulating the protein kinase Cdelta pathway in vascular smooth muscle cells. J Biol Chem. 2001;276(50):47650–47657. doi: 10.1074/jbc.M108719200. [DOI] [PubMed] [Google Scholar]
- 176.Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L885–897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 177.Spiekerkoetter E, Alvira CM, Kim YM, Bruneau A, Pricola KL, Wang L, Ambartsumian N, Rabinovitch M. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L276–289. doi: 10.1152/ajplung.00414.2007. [DOI] [PubMed] [Google Scholar]
- 178.Marecki JC, Cool CD, Parr JE, Beckey VE, Luciw PA, Tarantal AF, Carville A, Shannon RP, Cota-Gomez A, Tuder RM, Voelkel NF, Flores SC. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am J Respir Crit Care Med. 2006;174(4):437–445. doi: 10.1164/rccm.200601-005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175(12):1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75(3):272–280. doi: 10.1159/000111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114(17):1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 182.Smadja DM, Gaussem P, Mauge L, Israel-Biet D, Dignat-George F, Peyrard S, Agnoletti G, Vouhe PR, Bonnet D, Levy M. Circulating endothelial cells: a new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation. 2009;119(3):374–381. doi: 10.1161/CIRCULATIONAHA.108.808246. [DOI] [PubMed] [Google Scholar]
- 183.Diller GP, van ES, Okonko DO, Howard LS, Ali O, Thum T, Wort SJ, Bedard E, Gibbs JS, Bauersachs J, Hobbs AJ, Wilkins MR, Gatzoulis MA, Wharton J. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117(23):3020–3030. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]
- 184.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–217. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 185.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, Comhair SA, Xu W, Licina L, Huang L, nand-Apte B, Yoder MC, Tuder RM, Erzurum SC. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172(3):615–627. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Junhui Z, Xingxiang W, Guosheng F, Yunpeng S, Furong Z, Junzhu C. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med. 2008;102(7):1073–1079. doi: 10.1016/j.rmed.2007.12.030. [DOI] [PubMed] [Google Scholar]
- 187.Zhao YD, Courtman DW, Ng DS, Robb MJ, Deng YP, Trogadis J, Han RN, Stewart DJ. Microvascular regeneration in established pulmonary hypertension by angiogenic gene transfer. Am J Respir Cell Mol Biol. 2006;35(2):182–189. doi: 10.1165/rcmb.2005-0115OC. [DOI] [PubMed] [Google Scholar]
- 188.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96(4):442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 189.Nagaya N, Kangawa K, Kanda M, Uematsu M, Horio T, Fukuyama N, Hino J, Harada-Shiba M, Okumura H, Tabata Y, Mochizuki N, Chiba Y, Nishioka K, Miyatake K, Asahara T, Hara H, Mori H. Hybrid cell-gene therapy for pulmonary hypertension based on phagocytosing action of endothelial progenitor cells. Circulation. 2003;108(7):889–895. doi: 10.1161/01.CIR.0000079161.56080.22. [DOI] [PubMed] [Google Scholar]
- 190.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96(4):442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 191.Zhao YD, Courtman DW, Ng DS, Robb MJ, Deng YP, Trogadis J, Han RN, Stewart DJ. Microvascular regeneration in established pulmonary hypertension by angiogenic gene transfer. Am J Respir Cell Mol Biol. 2006;35(2):182–189. doi: 10.1165/rcmb.2005-0115OC. [DOI] [PubMed] [Google Scholar]
- 192.Wang XX, Zhang FR, Shang YP, Zhu JH, Xie XD, Tao QM, Zhu JH, Chen JZ. Transplantation of autologous endothelial progenitor cells may be beneficial in patients with idiopathic pulmonary arterial hypertension: a pilot randomized controlled trial. J Am Coll Cardiol. 2007;49(14):1566–1571. doi: 10.1016/j.jacc.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 193.Zhu JH, Wang XX, Zhang FR, Shang YP, Tao QM, Zhu JH, Chen JZ. Safety and efficacy of autologous endothelial progenitor cells transplantation in children with idiopathic pulmonary arterial hypertension: Open-label pilot study. Pediatr Transplant. 2008 doi: 10.1111/j.1399-3046.2007.00863.x. [DOI] [PubMed] [Google Scholar]
- 194.Rich S, Brundage B. High dose calcium blocking therapy for primary pulmonary hypertension: Evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76:135–141. doi: 10.1161/01.cir.76.1.135. [DOI] [PubMed] [Google Scholar]
- 195.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580–587. doi: 10.1161/01.cir.70.4.580. [DOI] [PubMed] [Google Scholar]
- 196.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 197.Rich S, Kaufmann E, Levy P. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 198.Rubin L, Mendoza J, Hood M, McGoon M, Barst R, Williams W, Diehl J, Crow J, Long W. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112:485–491. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- 199.Channick R, Simonneau G, Sitbon O, Robbins I, Frost A, Tapson V, Badesch D, Roux S, Rainisio M, Bodin F, Rubin L. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–1123. doi: 10.1016/S0140-6736(01)06250-X. [DOI] [PubMed] [Google Scholar]
- 200.Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, Naeije R, Galie N. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47(10):2049–2056. doi: 10.1016/j.jacc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 201.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 202.Perez-Penate G, Julia-Serda G, Ojeda-Betancort N, Garcia-Quintanna A, Pulido-Duque J, Rodriguez-Perez A, Cabrera-Navarro P, Gomez-Sanchez M. Long-term inhaled nitric oxide plus phosphodiesterase 5 inhibitors for severe pulmonary hypertension. J Heart Lung Transplant. 2008;27:1326–1332. doi: 10.1016/j.healun.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 203.Suzuki C, Takahashi M, Morimoto H, Izawa A, Ise H, Hongo M, Hoshikawa Y, Ito T, Miyashita H, Kobayashi E, Shimada K, Ikeda U. Mycophenolate mofetil attenuates pulmonary arterial hypertension in rats. Biochem Biophys Res Commun. 2006;349(2):781–788. doi: 10.1016/j.bbrc.2006.08.109. [DOI] [PubMed] [Google Scholar]
- 204.Bonnet S, Dumas-de-La-Roque E, Begueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci U S A. 2003;100(16):9488–9493. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Olson J, Hacker A, Altiere R, Gillelspie M. Polyamines and the development of monocrotaline-induced pulmonary hypertension. Am J Physiol. 1984;247:H682–685. doi: 10.1152/ajpheart.1984.247.4.H682. [DOI] [PubMed] [Google Scholar]
- 206.Thompson BT, Spence CR, Janssens SP, Joseph PM, Hales CA. Inhibition of hypoxic pulmonary hypertension by heparins of differing in vitro antiproliferative potency. Am J Respir Crit Care Med. 1994;149(6):1512–1517. doi: 10.1164/ajrccm.149.6.8004307. [DOI] [PubMed] [Google Scholar]
- 207.Nagaya N, Kyotani S, Uematsu M, Ueno K, Oya H, akanishi N, Shirai M, Mori H, Miyatake K, Kangawa K. Effects of adrenomedullin inhalation on hemodynamics and exercise capacity in patients with idiopathic pulmonary arterial hypertension. Circulation. 2004;109:351–356. doi: 10.1161/01.CIR.0000109493.05849.14. [DOI] [PubMed] [Google Scholar]
- 208.Nagaya N, Okumura H, Uematsu M, Shimzu W, Ono F, Shirai M, Mori H, Miyatake K, Kangawa K. Repeated inhalation of adrenomedullin ameliorates pulmonary hypertension and survival in monocrotaline rats. Am J Physiol Heart Circ Physiol. 2003;285:H2125–2131. doi: 10.1152/ajpheart.00548.2002. [DOI] [PubMed] [Google Scholar]
- 209.Reynolds A. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol. 2007;292:L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.