

SUMMARY
Long bone development depends on endochondral bone formation, a complex process requiring exquisite balance between hypertrophic cartilage (HC) formation and its ossification. Dysregulation of this process may result in skeletal dysplasias and heterotopic ossification. Endochondral ossification requires the precise orchestration of HC vascularization, extracellular matrix remodeling, and the recruitment of osteoclasts and osteoblasts. Matrix metalloproteinase-9 (MMP-9), vascular endothelial growth factor (VEGF) and osteoclasts have all been shown to regulate endochondral ossification, but how their function interrelates is not known. We have investigated the functional relationship among these regulators of endochondral ossification, demonstrating that they have complementary but non-overlapping functions. MMP-9, VEGF and osteoclast deficiency all cause impaired growth plate ossification resulting in the accumulation of HC. VEGF mRNA and protein expression are increased at the MMP-9−/− growth plate, and VEGF activity contributes to endochondral ossification since sequestration of VEGF by soluble receptors results in further inhibition of growth plate vascularization and ossification. However, VEGF bioavailability is still limited in MMP-9 deficiency, as exogenous VEGF is able to rescue the MMP-9−/− phenotype, demonstrating that MMP-9 may partially, but not fully, regulate VEGF bioavailability. The organization of the HC extracellular matrix at the MMP-9−/− growth plate is altered, supporting a role for MMP-9 in HC remodeling. Inhibition of VEGF impairs osteoclast recruitment, whereas MMP-9 deficiency leads to an accumulation of osteoclasts at the chondro-osseous junction. Growth plate ossification in osteoclast-deficient mice is impaired in the presence of normal MMP-9 expression, indicating that other osteoclastic functions are also necessary. Our data delineate the complementary interplay between MMP-9, VEGF and osteoclast function that is necessary for normal endochondral bone formation and provide a molecular framework for investigating the molecular defects contributing to disorders of endochondral bone formation.
INTRODUCTION
The long bones form by the process of endochondral ossification, during which a cartilage template is first laid down, then subsequently removed and replaced by bone (Colnot, 2005; Karsenty and Wagner, 2002; Olsen et al., 2000; Ortega et al., 2004; Provot and Schipani, 2005). During embryogenesis, at the sites of the future skeletal elements, mesenchymal cell condensations form, within which precursor cells differentiate into chondrocytes that proceed through a defined series of proliferative and morphogenetic steps to form a cartilage template. This cartilage template or anlagen contains less mature cells at the two ends and progressively more mature cells toward the middle. As chondrocytes mature, they hypertrophy and secrete a specialized extracellular matrix (ECM) containing collagen X, a collagen that is specific to hypertrophic cartilage (HC). The HC ECM is subsequently mineralized and invaded by blood vessels from the perichondrium. Vascular invasion is immediately preceded by invasion by preosteoclasts that remove the HC ECM. Concurrent with vascular invasion, osteoblasts are recruited and produce bone ECM. The chondrocytes at the ends of the cartilage template continue to proliferate and mature to form more HC, which in turn is continually removed and replaced with bone. This process of endochondral ossification results in longitudinal bone growth and in a thin layer of HC at the two ends of the bone whose size is relatively constant. Dysregulation of this complex and highly coordinated process result in abnormal bone formation seen in many of the skeletal dysplasias, and its abnormal activation may lead to heterotopic ossification (Cohen, 2000a; Cohen, 2000b; Cohen, 2006).
Numerous studies have contributed to characterization of the precise interplay of diverse factors that regulate HC differentiation, vascular invasion, ECM degradation, and bone formation and remodeling (Colnot, 2005). Among them, hypoxia induced factor 1-α (HIF1-α) and one of its targets, vascular endothelial growth factor (VEGF), are crucial in controlling chondrocyte survival, proliferation, angiogenesis and bone formation (Gerber et al., 1999; Schipani et al., 2001; Zelzer et al., 2004). Some ECM molecules are also essential: Col2a1 null mice lack endochondral ossification (Li et al., 1995) and perlecan null mice display chondrodysplasia (Arikawa-Hirasawa et al., 1999; Gustafsson et al., 2003). Several matrix metalloproteinases (MMPs) are also crucial for endochondral ossification and bone formation, notably MMP-9 (Vu et al., 1998), MMP-14 (Holmbeck et al., 1999; Holmbeck et al., 2003; Zhou et al., 2000) and MMP-13 (Inada et al., 2004; Stickens et al., 2004). In humans and mice, MMP-2 contributes to bone homeostasis but not to endochondral ossification (Egeblad et al., 2007; Inoue et al., 2006; Martignetti et al., 2001; Mosig et al., 2007; Tuysuz et al., 2009).
MMP-9−/− mice show impeded vascularization and ossification of HC at the growth plate, resulting in a lengthened HC zone with accumulation of late HC (Ortega et al., 2005; Vu et al., 1998). The delayed ossification may be the result of an imbalance between angiogenic and anti-angiogenic activities at the MMP-9−/− growth plate, since MMP-9−/− HC explants show delayed release of angiogenic activities (Vu et al., 1998). One of these angiogenic activities may be VEGF function. Sequestration of VEGF activity by soluble VEGF receptor results in inhibition of endochondral ossification and accumulation of HC similar to, albeit more severe than, that seen in MMP-9 deficiency (Gerber et al., 1999). Decreasing VEGF expression in HC by gene targeting using Cre-lox technology also gives similar results (Haigh et al., 2000). Mice that express only the VEGF120 soluble isoform also show defects in cartilage vascularization and endochondral ossification (Maes et al., 2002; Zelzer et al., 2002). However, mice expressing only VEGF164, or only VEGF188, have normal endochondral ossification of metaphyseal growth plates, although the VEGF188 mice have abnormal epiphyses with increased cell apoptosis and delayed secondary ossification (Maes et al., 2004). These studies suggest a model whereby normal endochondral ossification requires restricted VEGF bioavailability that might be regulated by MMPs through ECM remodeling.
Based on this model, we proposed that VEGF synthesized by hypertrophic chondrocytes would be sequestered in the HC ECM, and that MMP-9 degrades ECM molecules leading to the release of bound VEGF. Indeed, this seems to be the case in models of pancreatic islet carcinogenesis and glioblastoma (Bergers et al., 2000; Du et al., 2008). However, despite the expected matrix remodeling function of MMP-9, there are no direct data defining the relationship between VEGF and MMP-9 in the context of matrix remodeling in postnatal endochondral ossification. In this study we explore the mechanisms whereby VEGF and MMPs synergize to drive endochondral bone formation.
RESULTS
VEGF accumulates in late hypertrophic chondrocytes in MMP-9-deficient mice during postnatal endochondral ossification
Since there is defective growth plate ossification in both MMP-9 and VEGF deficiency, and an apparent imbalance in angiogenic activities in the MMP-9−/− HC (Gerber et al., 1999; Vu et al., 1998), we first asked whether VEGF expression or function is limiting at the MMP-9−/− growth plates. We previously showed that Vegf (also known as Vegfa) mRNA is expressed in the most mature HC in normal mice (Gerber et al., 1999; Vu et al., 1998). Here, we compared the VEGF mRNA and protein patterns in wild-type (WT) mice with MMP-9−/− mice. In situ hybridization showed strong expression of Vegf mRNA by HC in both WT and MMP-9 null growth plates (Fig. 1A–D). However, the area of Vegf expression in MMP-9−/− growth plates is expanded compared with that in WT growth plates, consistent with the expanded zone of HC. VEGF immunostaining showed that the zone of VEGF protein expression is also expanded in the MMP-9−/− growth plate (Fig. 1E,F). Notably, the zone of the VEGF protein expression was larger compared with that of Vegf mRNA expression, and included the upper HC in both WT and MMP-9−/− growth plates. This is probably the result of persistence of the VEGF protein in the surrounding ECM, even when the cells have decreased their expression of mRNA. The increase in VEGF protein expression in the MMP-9 growth plates was confirmed by western blotting of protein extracts from WT and MMP-9−/− growth plate HC, which, in addition, showed that the main isoform of VEGF that was expressed was VEGF164 (Fig. 1G). Interestingly, we observed smaller forms of VEGF migrating around 28 kDa in HC from both WT and MMP-9−/− HC (Fig. 1G). Considering its molecular weight, this form may be the plasmin-processed form of VEGF, since the plasminogen system is expressed in fetal bones and plays a role in endochondral ossification (Daci et al., 2003; Hackel et al., 1995). These smaller forms of VEGF were noted only in the samples with intact perichondrium, suggesting that this is where this processing takes place (Fig. 1G).
Fig. 1.

VEGF is expressed in the MMP-9−/− mice. (A–D) Bright-field (A,B) and dark-field (C,D) photographs of WT (A,C) and MMP-9−/− (B,D) sections of 1-week-old metatarsal growth plates hybridized with a Vegf antisense probe. Vegf is expressed in a subpopulation of hypertrophic chondrocytes (hc). (E,F) Immunostaining of tissue sections of 1-week-old WT (E) and MMP-9−/− (F) growth plates with VEGF antibody. VEGF protein is found in the ECM surrounding the hypertrophic chondrocytes (arrows). (G) Western blot of protein extracts from 2-week-old WT and MMP-9−/− growth plate HC probed with an anti-VEGF antibody showing that the major VEGF isoform expressed is VEGF164. Note the smaller forms of VEGF migrating around 28 kDa in both WT and MMP-9−/− HC with intact perichondrium (arrows). (H) Quantification by ELISA of VEGF protein secreted into the medium by cultured 2-week-old WT and MMP-9−/− growth plate ossification fronts. Bars, 200 μm (A–F).
MMP-9 deficiency does not impair VEGF release and activity
Although there is abundant expression of VEGF, the impaired ossification at the MMP-9−/− growth plates may still be because of limiting VEGF activity if VEGF is sequestered and not released efficiently. To determine whether VEGF release from HC is impaired in MMP-9 deficiency, we compared VEGF released from the ossification fronts (including the bone-cartilage interface and the last few rows of hypertrophic chondrocytes) of WT and MMP-9−/− growth plate HC. We found that there was no difference in the amount of VEGF released into growth medium in the absence of MMP-9 (Fig. 1H). Addition of active, purified MMP-9 did not increase VEGF release from the cartilage explants (data not shown).
We next determined whether VEGF activity contributes to vascularization and ossification of the MMP-9−/− growth plate. We inhibited VEGF activity in vivo using soluble VEGF receptors. Administration of mFlt(1–3)-IgG systemically to MMP-9−/− mice by daily intraperitoneal injection for 10 days resulted in further accumulation of HC in the treated mice. The treated HC zone was approximately 20% longer than control HC (Fig. 2A–C). As has been seen with mFlt(1–3)-IgG treatment of WT mice (Gerber et al., 1999), there was inhibition of vascular growth into HC. Platelet/endothelial cell adhesion molecule-1 (PECAM-1) immunostaining showed a reduced number of metaphyseal capillaries running parallel to hypertrophic chondrocyte columns in the treated mice (Fig. 2D,E). Bone formation was also inhibited, with a shortened trabecular bone region. There were few primary trabeculae, and the secondary trabeculae were shortened and thicker in the treated mice (Fig. 2F,G). These data show that active VEGF is present and contributes to the vascularization and ossification of HC in the absence of MMP-9.
Fig. 2.

Inhibition of VEGF activity causes further accumulation of HC in the MMP-9−/− growth plate. (A,B) Hematoxylin and eosin (H&E)-stained sections of 1-week-old MMP-9−/− mice treated for 10 days with either control IgG (A) or mFlt(1–3)-IgG (B) showing lengthening of the HC zone (hc) in the treated mice. (C) Bar graphs of the mean and standard deviation of the HC zone lengths of six control and mFlt-treated bones; the differences between control and treated samples were significant (P<0.05). (D,E) Immunostaining of tissue sections with PECAM-1 antibody showing metaphyseal vessels running parallel to hypertrophic chondrocyte columns in the control-IgG-treated mice (D, arrows) and a reduced number of these vessels in the mFlt(1–3)-IgG-treated mice (E, arrows). (F,G) H&E-stained sections showing primary (F, arrowheads) and secondary (F, arrows) trabeculae in the control-IgG-treated mice, and thickened and shortened secondary trabeculae (G, arrows) in the mFlt(1–3)-IgG-treated mice. Bars, 400 μm (A,B); 100 μm (D–G).
Exogenous VEGF partially normalizes the MMP-9 deficiency phenotype
We next asked whether bioavailable VEGF could still be limiting in MMP-9 deficiency. We determined whether exogenous VEGF could rescue the MMP-9−/− phenotype. We administered recombinant human VEGF165 systemically to 1-week-old MMP-9 null mice by daily intraperitoneal injection. After 1 week of treatment, there was a 35% reduction in the length of the HC zone in the treated mice (Fig. 3A,B,F). Continuing treatment with VEGF165 for 2 weeks did not further reduce the length of MMP-9−/− HC, which was still significantly larger than WT HC (Fig. 3C–F). An increase in the size of the trabecular bone region in the treated mice accompanied the reduction in the size of the HC zone (Fig. 3G,H). The decrease in HC zone size was not because of a decrease in chondrocyte proliferation. The populations of proliferative chondrocytes that incorporated bromodeoxyuridine (BrdU) were similar in both untreated and treated mice (Fig. 3I,J). Thus, exogenous VEGF was able to partially overcome the effects of MMP-9 deficiency, and enhance ossification of MMP-9−/− HC, thus stimulating HC exit from the growth plate, ECM remodeling and trabecular bone formation.
Fig. 3.

Treatment with VEGF rescues endochondral ossification defects in MMP-9 −/− mice. (A–D) H&E-stained sections of metatarsals of 1-week-old MMP-9−/− mice treated for 1 week (A,B) or 2 weeks (C,D) with either vehicle or recombinant human VEGF165, showing shortening of the HC zone (hc) in VEGF-treated mice (B,D) compared with vehicle-treated mice (A,C). (E) H&E-stained section of 3-week-old WT metatarsals showing the size of the normal HC zone (hc). (F) Bar graphs of the mean and standard deviation of the HC zone lengths of six vehicle- and VEGF-treated bones after 1 and 2 weeks of treatment; the differences between control and treated samples at each time point were significant (P<0.01). (G,H) H&E-stained sections of the metatarsals of MMP-9−/− mice treated for 2 weeks with either vehicle (G) or VEGF (H) showing lengthening of the trabecular bone region (tb) in VEGF-treated mice. (I,J) BrdU labeling of proliferating cells in the metatarsal growth plates of 1-week-old MMP-9−/− mice treated for 1 week with VEGF. BrdU-positive cells stain brown. Bars, 400 μm (A–D); 200 μm (G,H); 100 μm (I,J).
MMP-9 deficiency affects the organization of the hypertrophic chondrocyte matrix
The partial normalization obtained with VEGF treatment, and the fact that we could not detect a defect in VEGF release from the MMP-9−/− growth plate suggests that MMP-9 contributes an additional function besides regulating VEGF bioavailability. Angiogenesis at the growth plate requires both endothelial cell growth stimulated by angiogenic factors and ECM degradation to allow for vascular invasion. The activity of MMP-9 may be necessary to degrade the ECM for path clearing, as well as for regulation of sequestered VEGF. We therefore determined whether there are alterations in ECM remodeling at the MMP-9−/− growth plates. A detailed analysis of the hypertrophic area of postnatal metatarsals from WT or MMP-9−/− mice stained with Safranin-O showed that accumulation of HC correlates with a decrease of Safranin-O staining in MMP-9−/− mice (Fig. 4A,B), indicating a loss of matrix proteoglycans. Semi-thin sections stained with Toluidine Blue showed disorganized columns and thickening of the ECM pericellular to the hypertrophic chondrocytes, and delimiting the lacuna, in MMP-9−/− growth plates (Fig. 4C,D). Electron microscopy studies confirmed these differences: granular staining characteristic of proteoglycans was less dense in MMP-9−/− samples, concomitant with a clear increase in collagen fibril density (Fig. 4E,F). In the most distal rows of hypertrophic chondrocytes, the transverse septa showed striking differences between WT and MMP-9−/− samples. In WT samples, a progressive lysis of the last transverse septa eventually allows vascular invasion and elimination of condensed or apoptotic chondrocytes (Fig. 4G,I). In MMP-9−/− samples, transverse septa were thicker and denser with accumulation of large collagen type I fibers right under the septa (Fig. 4H,J). Higher magnification of the last transverse septa showed that, in WT samples, collagen fibrils are completely degraded on the bone marrow side, whereas in MMP-9−/− samples, collagen fibrils were still visible and dense (Fig. 4I,J). These results suggest that ECM remodeling is impaired in MMP-9 deficiency.
Fig. 4.

Altered hypertrophic cartilage ECM in MMP-9 null growth plates. (A,B) Safranin-O staining of 3-week-old metatarsals from WT (A) and MMP-9−/− (B) mice, showing a decrease in the staining of the lower portion of the hypertrophic chondrocyte area in MMP-9−/− mice (outlined by dotted lines). (C,D) Semi-thin sections of Epon-embedded ossification fronts from WT (C) and MMP-9−/− (D) mice, stained with Toluidine Blue, showing the presence of hypertrophic chondrocytes in the last rows at the ossification fronts in MMP-9−/− mice compared with WT mice. The longitudinal septae of the lacunae surrounding the last rows of hypertrophic chondrocytes are thicker in the MMP-9−/− mice (D, arrow) than in the WT mice (C, arrow). (E,F) Transmission electron micrographs of the areas indicated by arrows in C and D, showing the ultrastructure of the ECM in these areas. The ECM in these septae in WT mice is rich in proteoglycans, as indicated by numerous black dots, and collagen fibers are sparse (E). By contrast, in MMP-9−/− mice, the ECM of these septae contains dense collagen fibers and fewer black dots, indicating a decrease in proteoglycan content, which was also observed with Safranin-O staining in A and B. (G,H) Transmission electron micrographs showing the ultrastructure of the ECM of the last transverse septa in WT (G) and MMP-9−/− (H) mice. The boxed areas are shown under higher magnification in I and J. Transverse septae in the MMP-9−/− samples (J) show more collagen fibers and there is an accumulation of collagen type I fibers under the septae compared with the transverse septae in WT samples (I). Bars, 500 μm (A,B); 50 μm (C,D); 20 nm (E,F); 16 μm (G,H); 2 μm (I,J).
Either MMP-9 deficiency or VEGF inhibition results in an accumulation of cleaved collagen II at the cartilage-bone junction
Besides stimulating endothelial cell growth and morphogenesis, VEGF recruits osteoclasts, which in turn secrete ECM degrading enzymes (Gerber et al., 1999). Thus, the activities of VEGF and MMP-9 may complement each other. In previous work we showed that MMP-9 and MMP-13 co-operate in the degradation of cartilage type II collagen (Engsig et al., 2000; Stickens et al., 2004). To further determine the relationship between VEGF and MMP-9 function in ECM degradation during endochondral ossification, we analyzed samples for the presence of a degraded MMP substrate, cleaved type II collagen, at the ossification front. Immunostaining with an antibody that specifically recognizes a neoepitope on the N-terminal 3/4-length collagen fragments resulting from collagenase cleavage (Billinghurst et al., 1997) demonstrated the presence of the specific cleaved type II collagen fragments in a thin line at the cartilage-bone junction in the WT growth plate (Fig. 5A). The zone of staining slightly increased in the MMP-9−/− mice, suggesting that cleaved collagen fragments accumulate as a result of MMP-9 deficiency (Fig. 5B). Both MMP-13 and MMP-14 (MT1-MMP) can cleave type II collagen and both are expressed at the cartilage-bone junction. Thus, the accumulation may be secondary to the lack of MMP-9 activity, which would further degrade and solubilize the collagen fragments cleaved by MMP-13 and/or MMP-14. Alternatively, it may be secondary to the induction of MMP-13 or other enzyme activities that can generate these cleaved type II collagen fragments. Indeed, as shown below (Fig. 8), we observed the increased expression of MMP-13 in the expanded HC zone in the MMP-9−/− growth plates, which may account for this increase in collagen II cleavage.
Fig. 5.

MMP-9 deficiency and inhibition of VEGF alter the accumulation of cleaved collagen at the growth plate. (A–D) Immunostaining with an antibody against cleaved fragments of type II collagen on sections of metatarsals of 2-week-old WT (A) and MMP-9−/− (B) mice, and of sections of 1-week-old MMP-9−/− mice treated with cont-IgG (C) or mFlt(1–3)-IgG (D) for 1 week, showing the presence of cleaved collagen at a thin line along the cartilage-bone junction in WT mice (A, arrows), a larger zone of cleaved collagen in MMP-9−/− mice (B, arrows), and a larger zone with an irregular border in MMP-9−/− mice treated with mFlt(1–3)-IgG (D, arrows). (E,F) Immunostaining for MMP-9 and substrate staining for tartrate-resistant acidic phosphatase (TRAP) activity on the same tissue sections from WT growth plates showing MMP-9-positive cells that are both TRAP positive (F, arrows) and TRAP negative (F, arrowheads). Bars, 100 μm (A–E); 32 μm (F).
Fig. 8.

MMP expression at the growth plate. Dark-field photographs of tissue sections of metatarsals from 1-week-old WT (A,C) and MMP-9−/− mice (B,D) hybridized with antisense probes against MMP-13 (A,B) and MMP-14 (C,D). MMP-13 is expressed in cells at the cartilage-bone junction, consistent with the previously reported expression in terminal hypertrophic chondrocytes (A,B, arrowheads) and in the metaphysis (A,B, asterisks), in both WT and MMP-9−/− mice. In addition, MMP-13 is also expressed in some lower hypertrophic chondrocytes in MMP-9−/− mice (B, arrows). MMP-14 is expressed in cells at the cartilage-bone junction (C,D, arrowheads) and the metaphysis (C,D, asterisks). It is also expressed in the perichondrium (C,D, arrows). Bars, 200 μm.
Inhibition of VEGF activity in MMP-9−/− growth plates by treatment with mFlt(1–3)-IgG resulted in a slight further enlargement of the zone of cleaved type II collagen staining (Fig. 5C,D). These data suggest that VEGF activity also modulates the local balance of ECM degrading activities at the ossification front.
MMP-9 is not sufficient for normal endochondral ossification without osteoclasts
MMP-9 is expressed not only by osteoclasts and their progenitors, but also by TRAP-negative cells at the chondro-osseous junction (Fig. 5E,F). We previously showed that reconstitution of MMP-9 expression in the hematopoietic, and thus the osteoclast, lineage by bone marrow transplantation is sufficient to restore normal endochondral ossification of the MMP-9−/− growth plates (Vu et al., 1998). We therefore postulated that MMP-9 expression by non-osteoclasts is not crucial in this process. To test this hypothesis, we investigated mice with null mutations in c-Fos, Csf1, PU.1 (also known as Sfpi1) and Rank (also known as Tnfrsf11a), which are required for osteoclast development. Mice with null mutations in any of these genes are osteopetrotic owing to the absence of osteoclasts (Dougall et al., 1999; Grigoriadis et al., 1994; Li et al., 2000; Marks and Lane, 1976; McKercher et al., 1996; Tondravi et al., 1997; Yoshida et al., 1990). We found that mice that were deficient in any of these factors displayed increased HC zones at the growth plates, consistent with defective endochondral ossification (Fig. 6A,C,E,G,I,K,M,O).
Fig. 6.

Osteopetrotic mice have growth plate abnormalities with normal expression of MMP-9. Histological sections of metatarsal growth plates from c-Fos+/+ (A), c-Fos−/− (C), Csf1+/+ (E), Csf1op/op (G), PU.1+/+ (I), PU.1−/− (K), Rank+/+ (M) and Rank−/− (O) mice showing enlarged HC zones in the mutant growth plates. Immunostaining for MMP-9 showed similar expression of MMP-9 in the Csf1+/+ (F), Csf1op/op (H), PU.1+/+ (J), PU.1−/− (L), Rank+/+ (N) and Rank −/− (P) growth plates. The c-Fos−/− growth plate showed decreased immunostaining for MMP-9 (D) compared with the control c-Fos+/+ growth plate (B). Bars, 100 μm.
We next asked whether this phenotype was associated with reduced MMP-9 expression at the growth plates. Except for the c-Fos null growth plates, which showed decreased MMP-9 expression (Fig. 6B,D), all the other osteoclast-deficient growth plates showed normal levels of MMP-9 protein expression by immunohistochemistry (Fig. 6F,H,J,L,N,P), suggesting that there may be a compensatory increase in expression of MMP-9 by the TRAP-negative non-osteoclastic cells. The decreased expression of MMP-9 in c-Fos−/− growth plates may reflect the regulation of MMP-9 expression by c-Fos (Bischof et al., 2003). The normal expression of MMP-9, coupled with impaired endochondral ossification in the osteopetrotic mice, indicates that MMP-9 expression by non-osteoclasts is not sufficient to drive the ossification process normally and that another osteoclastic function is required for normal endochondral ossification.
MMP-9 deficiency leads to accumulation of TRAP-positive cells at the chondro-osseous junction, which is reversed by VEGF treatment
The requirement of osteoclast function suggests their recruitment might be an essential regulatory mechanism of endochondral bone formation. During primary ossification, osteoclast recruitment into the cartilage anlagen requires MMP-9 activity (Engsig et al., 2000). We therefore determined whether impaired osteoclast recruitment might contribute to the defect in HC ossification at the MMP-9−/−growth plates. We compared the number of multinucleated (fully differentiated osteoclasts) and mononucleated (osteoclast precursors) TRAP-positive cells along the chondro-osseous junction between WT and MMP-9−/− mice. Surprisingly, compared with WT, MMP-9−/− mice showed significantly increased numbers of both multinucleated osteoclasts (WT, 3.0±0.4, versus MMP-9−/−, 6.5±0.8; P<0.01) and mononucleated cells (WT, 4.8±0.4, versus MMP-9−/−, 8.5±0.9; P<0.01) along the total length of the chondro-osseous junction (Fig. 7A–C). These results show that MMP-9 is not necessary for osteoclast recruitment during growth plate ossification, and that impaired osteoclast recruitment is not a cause of the defective ossification in the MMP-9−/− growth plates. This is in contrast to the function of VEGF, which is necessary for osteoclast recruitment in both primary ossification of the cartilage anlagen (Maes et al., 2002; Zelzer et al., 2002) and during growth plate ossification (Gerber et al., 1999).
Fig. 7.

Osteoclast recruitment to the chondro-osseous junction. (A,B) TRAP staining of tissue sections showing more TRAP+ cells at the chondro-osseous junction in MMP-9−/− growth plates (B) compared with WT growth plates (A). (C) Quantification of mononuclear and multinuclear TRAP+ cells at the WT and MMP-9−/− growth plates. (D,E) TRAP staining of tissue sections showing a reduced number of TRAP+ cells at the chondro-osseous junction in MMP-9−/− growth plates treated with VEGF (E) compared with untreated MMP-9−/− growth plates (D). (F) Quantification of mononuclear and multinuclear TRAP+ cells at the untreated and VEGF-treated MMP-9−/− growth plates. For C and F, mononucleated and multinucleated TRAP+ cells were counted under the microscope at a 40× magnification and expressed as the total number of TRAP+ cells along the total length of the chondro-osseous junction. The counting was performed on a minimum of three mice per group, using both hind limbs, and on three to six different sections, with three metatarsals per section, for each hind limb. The results are expressed as mean ± standard error of the mean (S.E.M.). The results were statistically significant (P<0.01) between WT and MMP-9−/− samples for both cell populations in C, and between VEGF treated and untreated samples for the mononuclear population in F. The results were not statistically significant between VEGF treated and untreated samples for the multinuclear population in F.
We next determined the effect of exogenous VEGF on osteoclast recruitment at the MMP-9−/− growth plate. We assessed the number of TRAP-positive cells at the chondro-osseous junction of 1-week-old MMP-9−/− mice treated for 1 week with either vehicle alone or VEGF. Surprisingly, MMP-9−/− mice treated with VEGF had a decreased number of mononuclear TRAP-positive cells compared with mice treated with vehicle alone (control, 7.7±0.7, versus VEGF treated, 3.8±0.4; P<0.01) (Fig. 7D–F). Thus, the improvement in vascularization and ossification of MMP-9−/− HC by exogenous VEGF is associated with normalization of osteoclast number at the chondro-osseous junction.
Other MMPs contribute to endochondral ossification in the absence of MMP-9
Since MMP-9 alone is not sufficient for normal endochondral ossification without osteoclasts, which also express other MMPs, we asked whether the activity of other MMPs might be necessary. We found high expression of MMP-13 and MMP-14 at the cartilage-bone junction by in situ hybridization (Fig. 8A–D). MMP-13 expression was observed in terminal hypertrophic chondrocytes and in cells on the trabecular bone surfaces (Fig. 8A), consistent with previously reported results (Inada et al., 2004; Stickens et al., 2004). MMP-14 expression was seen in cells at the cartilage-bone junction, which may be (pre)osteoclasts, as well as in cells along the trabecular bone, and in the perichondrium (Fig. 8C). The pattern of expression of MMP-13 and MMP-14 in the MMP-9 null growth plate resembled that in the WT mice, except that their expression levels were increased slightly (Fig. 8B,D). In addition, there was a larger zone of expression of MMP-13 in the late hypertrophic chondrocytes in the expanded HC zone in the MMP-9−/− mice (Fig. 8D).
To determine whether inhibition of all MMP activity would further inhibit endochondral ossification, we treated MMP-9−/−mice systemically by daily intraperitoneal injection with GM6001, a broad-spectrum inhibitor of MMPs. After 1 week of treatment, the HC zone in the treated mice further lengthened, increasing by approximately 25% in length (Fig. 9A,B,E). This became more pronounced after 2 weeks of treatment, with a 65% length increase (Fig. 9C–E). Vascularization was completely inhibited in the treated mice. PECAM-1 immunostaining showed that there were no metaphyseal capillaries that ran parallel to the HC columns, as seen in the control mice. Instead, there were dilated vascular spaces running perpendicular to the HC (Fig. 9F,G). Bone formation was also affected. Primary trabeculae were not present, and the secondary trabeculae became a large mass of bone (Fig. 9H,I).
Fig. 9.

Inhibition of MMP activity inhibits residual endochondral ossification at the MMP-9−/− growth plate. (A–D) H&E-stained sections of metatarsals of 1-week-old MMP-9−/− mice treated for 1 week (A,B) or 2 weeks (C,D) with either vehicle or the MMP inhibitor GM6001, showing further lengthening of the HC (hc) zone in the GM6001-treated mice (B,D) compared with the vehicle-treated mice (A,C). (E) Bar graphs of the mean and standard deviation of the HC zone lengths of six control and GM6001-treated bones after 1 and 2 weeks of treatment; the differences between control and treated samples at each time point were statistically significant (P<0.01). (F,G) Immunostaining of tissue sections with PECAM-1 antibody showing metaphyseal vessels running parallel to hypertrophic chondrocyte columns in the vehicle-treated mice (F, arrows) and dilated vessels running perpendicular to the hypertrophic chondrocyte columns in the GM6001-treated mice (G, arrows). (H,I) H&E-stained sections showing primary (H, arrowheads) and secondary (H, arrows) trabeculae in the vehicle-treated mice, and a mass of bone instead of secondary trabeculae in the GM6001-treated mice (I, arrows). (J,K) TRAP staining of tissue sections showing the presence of TRAP+ cells at the cartilage-bone junction (arrows) and on trabecular bone surfaces (arrowheads) in both vehicle-treated (J) and GM6001-treated (K) mice. Bars, 400 μm (A–D); 100 μm (F–K).
Since the trabecular bone phenotype resulting from the inhibition of all MMPs may be because of inhibition of bone remodeling, we next determined whether the treated mice showed an abnormal osteoclast number by staining for TRAP activity, a marker of osteoclasts. We observed large numbers of osteoclasts both at the cartilage-bone junction and on the trabecular bone surfaces in the treated mice (Fig. 9J,K), indicating that the abnormal bone remodeling in the treated mice was probably due to an alteration in osteoclastic activity and not in osteoclast number.
DISCUSSION
In this study we explored the functional relationships between MMP-9, VEGF and osteoclasts at the growth plate during endochondral bone formation. Our results demonstrated that all of these components are necessary for normal endochondral ossification and that their functions are interdependent (Fig. 10). MMP-9 may degrade ECM components to regulate angiogenesis either by path clearing or by regulating the bioavailability of sequestered growth factors such as VEGF. VEGF not only regulates angiogenesis, but also regulates the recruitment of other crucial cells such as osteoclasts. Osteoclasts function by providing MMP-9 activity, as well as other MMPs, which contributes to HC turnover and bone ECM remodeling.
Fig. 10.
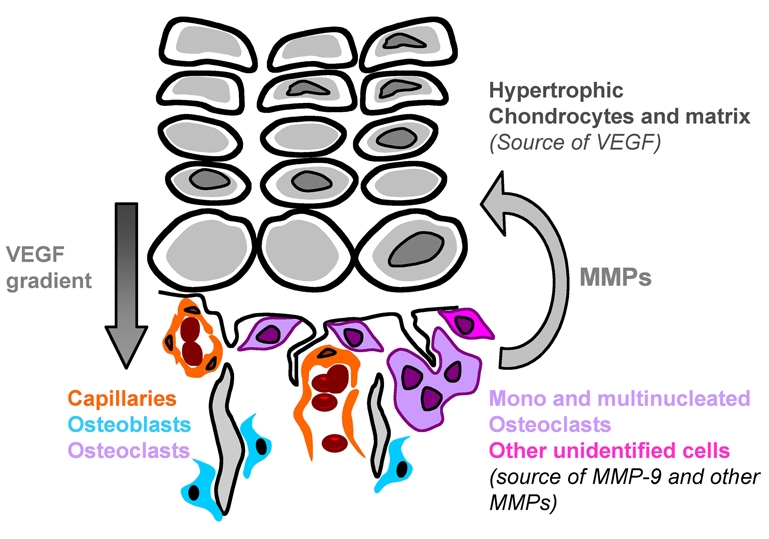
Complementary functions of VEGF, MMP-9 and osteoclasts at the growth plate. At the HC-bone junction, MMP-9 is secreted by mononucleated and multinucleated osteoclasts, as well as by other non-osteoclastic cells. MMP-9 can degrade the ECM leading to the release of VEGF sequestered in the HC. However, MMP-9 also has functions besides regulating VEGF bioavailability, such as ECM degradation and remodeling. VEGF can act on endothelial cells, as well as on mononucleated osteoclasts and osteoblasts. Besides MMP-9, osteoclasts also secrete other MMPs that can also cleave the ECM.
The functions of VEGF and MMP-9 in endochondral ossification do not completely overlap
Previous studies have suggested that the function of MMP-9 in endochondral ossification is to release VEGF that is sequestered in the ECM. The accumulation of HC is seen in mice that are deficient in MMP-9, treated with a VEGF inhibitor, or with conditional deletion of the Vegf gene in cartilage (Gerber et al., 1999; Haigh et al., 2000; Vu et al., 1998). In addition, MMP-9-deficient HC shows a delayed recruitment of capillaries in vitro (Vu et al., 1998). Our data now show that the functions of MMP-9 and VEGF, while synergistic, do not completely overlap, and thus the MMP-9−/− phenotype results from more than a defect in VEGF release. VEGF mRNA and protein are abundant in the MMP-9−/− HC. VEGF release from the MMP-9−/− HC is not impaired in vitro, and there is VEGF activity contributing to the vascularization and ossification of the MMP-9−/− growth plates. In addition, administration of exogenous recombinant VEGF to MMP-9−/−mice only partially rescues the phenotype. Thus, although VEGF bioavailability may be limiting in MMP-9 deficiency, our data suggest that MMP-9 is also necessary for other processes unrelated to the release of VEGF.
Osteoclasts are essential for normal endochondral ossification and are regulated differently by MMP-9 and VEGF
Osteoclasts are well-known matrix-resorbing cells and their presence at the chondro-osseous junction suggests that they contribute to endochondral ossification by secreting ECM-degrading enzymes. However, cells that express MMP-9, but that are TRAP negative, are also present at the chondro-osseous junction and may also contribute to ECM degradation. We predicted that, in the absence of osteoclasts, non-osteoclastic cells expressing MMP-9 would contribute to growth plate remodeling. Consistent with this model, we found that mice lacking osteoclasts showed MMP-9 expression in non-osteoclastic cells and display milder defects in growth plate ossification than those seen in MMP-9 deficiency. These non-osteoclastic MMP-9-expressing cells may function as the TRAP-negative ‘chondroclasts’. The four different genetically osteopetrotic mice point to primarily osteoblasts and their progenitors as the non-osteoclast MMP-9-expressing cells: Rank−/− mice have no osteoclasts but still have mononuclear phagocytes; Fos−/− mice have no osteoclasts but have an excess of macrophages and monocytes; Csf1op/op mice have no osteoclasts and very few monocytes and macrophages; and PU.1 −/− mice lack myeloid cells (Dougall et al., 1999; Grigoriadis et al., 1994; Li et al., 2000; Marks and Lane, 1976; McKercher et al., 1996; Tondravi et al., 1997; Yoshida et al., 1990). From their location, the TRAP-negative cells that express MMP-9 are presumably osteoblasts, their progenitors, or other mesenchymal cells that can express MMP-9 (Heissig et al., 2002). However, since HC does accumulate in osteopetrotic mice in the presence of MMP-9 expression, it is clear that MMP-9 produced by these TRAP-negative ‘chondroclasts’ is not sufficient for normal growth plate ossification. Whether this owes to a defect in MMP-9 activation or to other products made by osteoclasts remains to be determined. Clearly other MMPs, which are produced by osteoclasts, contribute, as further inhibition of MMPs in MMP-9−/− growth plates resulted in complete inhibition of endochondral ossification.
VEGF regulates osteoclast recruitment during formation of the primary ossification site, as well as during growth plate ossification (Engsig et al., 2000; Gerber et al., 1999; Maes et al., 2002; Zelzer et al., 2002). It also regulates osteoclastic differentiation and function (Aldridge et al., 2005; Niida et al., 2005; Zelzer and Olsen, 2005). Similar to VEGF, MMP-9 positively regulates osteoclast recruitment during primary ossification (Engsig et al., 2000). Surprisingly, MMP-9 deficiency resulted in an increased number of osteoclasts at the chondro-osseous junction. Whether this is due to a compensatory mechanism that increases the recruitment of cells with ECM-degrading activities to overcome the MMP-9 deficiency or to negative regulation of osteoclast recruitment during growth plate ossification by MMP-9 remains to be elucidated. It is also possible that MMP-9 is required for osteoclast invasion of HC, either by degrading the ECM or by regulating the bioavailability of osteoclast chemotatic factors, and thus its deficiency causes osteoclasts to accumulate at the chondro-osseous junction.
Interestingly, administration of exogenous VEGF did not recruit more osteoclasts, but rather resulted in a reduction in their numbers, at the MMP-9−/− chondro-osseous junction. The additional VEGF appeared to act differentially on the recruitment of osteoclast progenitors, because exogenous VEGF decreased the number of mononuclear TRAP-positive cells to a greater extent than the multinucleated population. The improved angiogenesis and ossification by exogenous VEGF may negate the need for compensatory recruitment of ECM-resorbing cells. However, VEGF also affects the formation and/or function of osteoblasts (Hiltunen et al., 2003; Maes et al., 2002; Street et al., 2002; Zelzer et al., 2002), which are known to modulate osteoclast formation. Thus, exogenous VEGF may also reduce osteoclast number through its effects on osteoblasts. We conclude that the effect of VEGF on osteoclasts is not simply to increase recruitment, but varies depending on the complex interplay of many different factors.
MMPs and VEGF have different effects on bone turnover
In contrast to the synergistic control of cartilage vascularization, the functions of VEGF and MMPs differ in the regulation of bone homeostasis. Inhibition of VEGF activity led to fewer primary trabeculae, and thick and shortened secondary trabeculae, suggesting defects in bone formation. This is consistent with previous studies showing that VEGF regulates osteoblast differentiation and function (Hiltunen et al., 2003; Maes et al., 2002; Midy and Plouet, 1994; Street et al., 2002; Zelzer et al., 2002). However, inhibition of all MMPs resulted in both the absence of primary trabeculae and a thick mass of secondary trabeculae that seemed to have fused together; this suggests defects in both bone formation and resorption. MMP activity may be necessary for bone formation by having a direct effect on osteoblast function or through its ECM-degrading activity that may release factors such as VEGF or transforming growth factor (TGF)-β, which in turn act on osteoclasts and osteoblasts. Significant numbers of osteoclasts are present on the bone surfaces of GM6001-treated mice, suggesting that the defect in bone resorption is the result of inhibition of secreted MMP activity and not because of a lack of osteoclasts.
METHODS
Animal studies
MMP-9−/− mice have been described previously (Vu et al., 1998). The treatment of mice was by daily intraperitoneal injection. The following doses were used: recombinant human VEGF165 (Genentech, South San Francisco, CA) in PBS at 10 μg/mouse; mFlt(1–3)-IgG (Genentech) or control IgG in PBS at 25 μg/g; GM6001, which was made as a slurry in 4% carboxymethyl cellulose, at 100 μg/g. The GM6001 was a gift from Dr Richard Galardy. We are grateful to Dr Adrian Erlebacher for providing samples from c-Fos−/− mice and their WT littermates (Erlebacher et al., 1998); Dr S. R. McKercher for providing PU.1 −/− mice; and Dr Jacques Peschon for providing Rank−/− mice (Dougall et al., 1999; McKercher et al., 1996). The Csf1op/op mice were purchased from Jackson Labs.
Histological analysis, TRAP staining and immunohistochemistry
After euthanasia, the metatarsals were dissected free from the skin and fixed in 4% paraformaldehyde at 4°C overnight. The tissues were then washed in PBS and decalcified in 0.5 M EDTA (pH 7.4) for 7–10 days at 4°C prior to processing and embedding in paraffin. Sections (5 μm) were stained with hematoxylin and eosin, or with hematoxylin/Fast Green/Safranin-O. For quantification, the HC length was measured from control and treated samples (n=6). Data are presented as mean ± standard deviation (S.D.). Statistical analyses were performed using the Student’s t-test.
For tartrate-resistant acidic phosphatase (TRAP) staining, sections were deparaffinized and stained for TRAP activity using a leukocyte acid phosphatase kit with Fast Red Violet as a substrate (Sigma, St Louis, MO). After incubation with the TRAP solution at 37°C for 1 hour, sections were washed in distilled water and counterstained with either hematoxylin or Methyl Green (Sigma). Mononucleated and multinucleated TRAP-positive cells were counted under the microscope at a 40× magnification and expressed as the total number of TRAP-positive cells along the chondro-osseous junction. The counting was performed on a minimum of three mice per group, using both hind limbs, and using three to six different sections, with three metatarsals per section, for each hind limb. The results are expressed as the mean ± the S.E.M. Statistical analyses were carried out using the Student’s t-test. GraphPad calculator was used to perform the tests.
For immunohistochemistry, the sections were deparaffinized, washed in PBS, quenched in either PBS and 3% H2O2 for 5 minutes or in 0.3% H2O2 in methanol for 30 minutes, then washed in PBS, and treated with 0.1% trypsin in PBS for 30 minutes. For MMP-9 immunohistochemistry, the sections were treated with ficin (Zymed Laboratories, Inc., South San Francisco, CA) for 15 minutes at ambient temperature. The slides were then treated with 0.1 M glycine in PBS for 30 minutes, washed in PBS, and blocked in PBS with 3% BSA for 30 minutes, then washed in PBS, and blocked in PBS with 5% normal serum for 2 hours. The sections were incubated with primary antibody diluted in PBS with 1 mg/ml of BSA at 4°C overnight. The slides were then washed in PBS, blocked in PBS with 5% normal serum for 30 minutes, then incubated with biotinylated secondary antibody for 1 hour, washed in PBS, and incubated with Vector Elite ABC reagent (Vector Laboratory, Burlingame, CA) for 1 hour, washed in PBS, and developed using DAB substrate. Rat monoclonal anti-mouse CD31 antibody (clone MEC 13.3, Pharmingen, San Diego, CA) was used at a dilution of 1:100; the rabbit anti-cleaved collagen II epitope antibody (rabbit anti-Col 3/4 polyclonal antibody, Hdm Diagnostics and Imaging, Toronto, Canada) was used at 1:800; the MMP-9 rabbit polyclonal antibody was used at 1:100 (Vu et al., 1998); and the rabbit anti-human VEGF antibody (Ab1, Neomarkers, Fremont, CA) was used at 1:100 on frozen sections with tyramide amplification (NEN, Boston, MA) and True Blue HRP substrate (KPL, Gaithersburg, MD).
Labeling of proliferative cells in vivo with BrdU
Mice were injected intraperitoneally with 10 mg/ml of BrdU (Sigma), at doses of 0.01 ml/g, 1 hour prior to sacrifice. Bones were fixed and processed for paraffin sections as above. Sections were immunostained with an antibody against BrdU using a kit from Zymed.
In situ hybridization
Sections were deparaffinized, treated with proteinase K (20 μg/ml) for 5 minutes at ambient temperature, and hybridized with 35S-labeled antisense riboprobes in hybridization buffer (50% deionized formamide, 300 mM NaCl, 20 mM Tris-HCl pH 8.0, 5 mM EDTA, 0.5 mg/ml yeast tRNA, 10% dextran sulfate and 1×Denhardt’s solution) in a humidified chamber at 55°C overnight. Following hybridization, the slides were treated with RNase A, washed to a final stringency of 50% formamide, 2×SSC at 60°C, then dipped in emulsion, exposed for 1–2 weeks, developed, and counterstained with hematoxylin and eosin.
VEGF western blotting and ELISA
Growth plate HC was dissected, with or without perichondrium, from 14-day-old WT and MMP-9−/− mice and homogenized in RIPA buffer. The protein was quantified using a micro BCA assay (Pierce, Rockford, IL) and 40 μg of total protein of each sample was separated on a 10% Bis-Tris gel in MOPS running buffer (Novex Invitrogen, Carlsbad, CA) under non-reducing conditions, transferred onto a PVDF membrane, and blotted with an anti-mouse VEGF antibody recognizing all isoforms of VEGF (R&D Systems, Minneapolis, MN). For the measurement of VEGF by ELISA, growth plates from 14-day-old WT and MMP-9−/− mice were dissected, and the ossification fronts with the last rows of hypertrophic chondrocytes were isolated and incubated in DMEM/F12 under 5% CO2 for 48 hours. Cultures were kept in 24-well plates, with 20 pieces of cartilage per well in 500 μl of culture medium. After 48 hours, the conditioned media were centrifuged and the VEGF content was quantified by using a murine VEGF ELISA kit (R&D Systems), according to the manufacturer’s instructions.
Acknowledgments
We thank Helen Capili for her outstanding efforts in sectioning of the bone specimens, Andrew Tauscher for assistance with electron microscopy, and Dani Behonick for helping with the Rank−/− mouse experiments and staining of sections. This study was supported by grants from the National Institutes of Health (AR046238 to T.H.V. and Z.W.) and from the UCSF Program for Breakthrough Biomedical Research (to T.H.V). N.O. was a Michael Geisman Research Fellow of the Osteogenesis Imperfecta Foundation. Deposited in PMC for release after 12 months.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
N.O. conceived, designed and performed experiments; analyzed data; and prepared the manuscript. K.W. performed experiments and reviewed the manuscript. N.F. contributed reagents and reviewed the manuscript. Z.W. conceived and designed experiments and edited the manuscript. T.H.V. conceived, designed and performed experiments; analyzed data; and prepared and edited the manuscript.
REFERENCES
- Aldridge SE, Lennard TW, Williams JR, Birch MA. (2005). Vascular endothelial growth factor receptors in osteoclast differentiation and function. Biochem Biophys Res Commun. 335, 793–798 [DOI] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. (1999). Perlecan is essential for cartilage and cephalic development. Nat Genet. 23, 354–358 [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, et al. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H, et al. (1997). Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest 99, 1534–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof P, Truong K, Campana A. (2003). Regulation of trophoblastic gelatinases by proto-oncogenes. Placenta 24, 155–163 [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr (2000a). Merging the old skeletal biology with the new. I. Intramembranous ossification, endochondral ossification, ectopic bone, secondary cartilage, and pathologic considerations. J Craniofac Genet Dev Biol. 20, 84–93 [PubMed] [Google Scholar]
- Cohen MM., Jr (2000b). Merging the old skeletal biology with the new. II. Molecular aspects of bone formation and bone growth. J Craniofac Genet Dev Biol. 20, 94–106 [PubMed] [Google Scholar]
- Cohen MM., Jr (2006). The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet A 140, 2646–2706 [DOI] [PubMed] [Google Scholar]
- Colnot C. (2005). Cellular and molecular interactions regulating skeletogenesis. J Cell Biochem. 95, 688–697 [DOI] [PubMed] [Google Scholar]
- Daci E, Everts V, Torrekens S, Van Herck E, Tigchelaar-Gutterr W, Bouillon R, Carmeliet G. (2003). Increased bone formation in mice lacking plasminogen activators. J Bone Miner Res. 18, 1167–1176 [DOI] [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al. (1999). RANK is essential for osteoclast and lymph node development. Genes Dev. 13, 2412–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Petritsch C, Lu K, Liu P, Haller A, Ganss R, Song H, Vandenberg S, Bergers G. (2008). Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro Oncol. 10, 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Shen HC, Behonick DJ, Wilmes L, Eichten A, Korets LV, Kheradmand F, Werb Z, Coussens LM. (2007). Type I collagen is a genetic modifier of matrix metalloproteinase 2 in murine skeletal development. Dev Dyn. 236, 1683–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR, Henriksen K, Lenhard T, Foged NT, Werb Z, et al. (2000). Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 151, 879–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlebacher A, Filvaroff EH, Ye JQ, Derynck R. (1998). Osteoblastic responses to TGF-beta during bone remodeling. Mol Biol Cell 9, 1903–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. (1999). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 5, 623–628 [DOI] [PubMed] [Google Scholar]
- Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. (1994). c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 266, 443–448 [DOI] [PubMed] [Google Scholar]
- Gustafsson E, Aszodi A, Ortega N, Hunziker EB, Denker HW, Werb Z, Fassler R. (2003). Role of collagen type II and perlecan in skeletal development. Ann N Y Acad Sci. 995, 140–150 [DOI] [PubMed] [Google Scholar]
- Hackel C, Radig K, Rose I, Roessner A. (1995). The urokinase plasminogen activator (u-PA) and its inhibitor (PAI-1) in embryo-fetal bone formation in the human: an immunohistochemical study. Anat Embryol (Berl) 192, 363–368 [DOI] [PubMed] [Google Scholar]
- Haigh JJ, Gerber HP, Ferrara N, Wagner EF. (2000). Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development 127, 1445–1453 [DOI] [PubMed] [Google Scholar]
- Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA. (2002). Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109, 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen MO, Ruuskanen M, Huuskonen J, Mahonen AJ, Ahonen M, Rutanen J, Kosma VM, Mahonen A, Kroger H, Yla-Herttuala S. (2003). Adenovirus-mediated VEGF-A gene transfer induces bone formation in vivo. FASEB J. 17, 1147–1149 [DOI] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, et al. (1999). MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81–92 [DOI] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Chrysovergis K, Yamada S, Birkedal-Hansen H. (2003). MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J Cell Biol. 163, 661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM. (2004). Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci USA 101, 17192–17197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, et al. (2006). A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 281, 33814–33824 [DOI] [PubMed] [Google Scholar]
- Karsenty G, Wagner EF. (2002). Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2, 389–406 [DOI] [PubMed] [Google Scholar]
- Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, et al. (2000). RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA 97, 1566–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SW, Prockop DJ, Helminen H, Fassler R, Lapvetelainen T, Kiraly K, Peltarri A, Arokoski J, Lui H, Arita M, et al. (1995). Transgenic mice with targeted inactivation of the Col2 alpha 1 gene for collagen II develop a skeleton with membranous and periosteal bone but no endochondral bone. Genes Dev. 9, 2821–2830 [DOI] [PubMed] [Google Scholar]
- Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. (2002). Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 111, 61–73 [DOI] [PubMed] [Google Scholar]
- Maes C, Stockmans I, Moermans K, Van Looveren R, Smets N, Carmeliet P, Bouillon R, Carmeliet G. (2004). Soluble VEGF isoforms are essential for establishing epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest. 113, 188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks SC, Jr, Lane PW. (1976). Osteopetrosis, a new recessive skeletal mutation on chromosome 12 of the mouse. J Hered. 67, 11–18 [DOI] [PubMed] [Google Scholar]
- Martignetti JA, Aqeel AA, Sewairi WA, Boumah CE, Kambouris M, Mayouf SA, Sheth KV, Eid WA, Dowling O, Harris J, et al. (2001). Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 28, 261–265 [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, et al. (1996). Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 15, 5647–5658 [PMC free article] [PubMed] [Google Scholar]
- Midy V, Plouet J. (1994). Vasculotropin/vascular endothelial growth factor induces differentiation in cultured osteoblasts. Biochem Biophys Res Commun. 199, 380–386 [DOI] [PubMed] [Google Scholar]
- Mosig RA, Dowling O, DiFeo A, Ramirez MC, Parker IC, Abe E, Diouri J, Aqeel AA, Wylie JD, Oblander SA, et al. (2007). Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 16, 1113–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, Ikeda K, Shibuya M. (2005). VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci USA 102, 14016–14021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen BR, Reginato AM, Wang W. (2000). Bone development. Annu Rev Cell Dev Biol. 16, 191–220 [DOI] [PubMed] [Google Scholar]
- Ortega N, Behonick DJ, Werb Z. (2004). Matrix remodeling during endochondral ossification. Trends Cell Biol. 14, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega N, Behonick DJ, Colnot C, Cooper DN, Werb Z. (2005). Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol Biol Cell 16, 3028–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provot S, Schipani E. (2005). Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun. 328, 658–665 [DOI] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. (2001). Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 15, 2865–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. (2004). Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131, 5883–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street J, Bao M, deGuzman L, Bunting S, Peale FV, Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, et al. (2002). Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci USA 99, 9656–9661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL. (1997). Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 386, 81–84 [DOI] [PubMed] [Google Scholar]
- Tuysuz B, Mosig R, Altun G, Sancak S, Glucksman MJ, Martignetti JA. (2009). A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 17, 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD. (1990). The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 345, 442–444 [DOI] [PubMed] [Google Scholar]
- Zelzer E, Olsen BR. (2005). Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr Top Dev Biol. 65, 169–187 [DOI] [PubMed] [Google Scholar]
- Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D’Amore PA, Olsen BR. (2002). Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development 129, 1893–1904 [DOI] [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. (2004). VEGFA is necessary for chondrocyte survival during bone development. Development 131, 2161–2171 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. (2000). Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA 97, 4052–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]