

Abstract
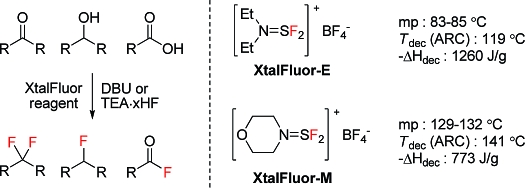
Diethylaminodifluorosulfinium tetrafluoroborate (XtalFluor-E) and morpholinodifluorosulfinium tetrafluoroborate (XtalFluor-M) are crystalline fluorinating agents that are more easily handled and significantly more stable than Deoxo-Fluor, DAST, and their analogues. These reagents can be prepared in a safer and more cost-efficient manner by avoiding the laborious and hazardous distillation of dialkylaminosulfur trifluorides. Unlike DAST, Deoxo-Fluor, and Fluolead, XtalFluor reagents do not generate highly corrosive free-HF and therefore can be used in standard borosilicate vessels. When used in conjunction with promoters such as Et3N·3HF, Et3N·2HF, or DBU, XtalFluor reagents effectively convert alcohols to alkyl fluorides and carbonyls to gem-difluorides. These reagents are typically more selective than DAST and Deoxo-Fluor and exhibit superior performance by providing significantly less elimination side products.
Introduction
It is well recognized that the introduction of a C−F structural feature within a bioactive molecule often imparts desirable physiological properties.(1) As a consequence, fluorinated biologically active compounds are increasingly common in the realm of pharmaceuticals and agrochemicals. This widespread interest and increasing demand for site selective fluorination of organic compounds has prompted the extensive development of novel specialized fluorination technologies and reagents capable of incorporating fluorine atoms in a safe, efficient and controlled fashion. Among the myriad of fluorination methodologies currently available, the direct replacement of oxygen by fluoride, commonly referred to as deoxofluorination, is one of the most effective and routinely used approaches that is particularly applicable to a wide variety of functional groups.(2)
Deoxofluorination using nucleophilic-type fluorinating reagents has a long development history, notably with the early recognition that SF4 could directly transform such groups as hydroxyl to fluoride, carbonyl to difluoromethylene, and carboxylic acid to trifluoromethyl.(3) Since this initial discovery, several reagents were developed, such as DAST (diethylaminosulfur trifluoride),(4) Deoxo-Fluor (bis(2-methoxyethyl)aminosulfur trifluoride),(5) Yarovenko’s reagent (N,N-diethyl-2-chloro-1,1,2-trifluoroethylamine),(6) Ishikawa’s reagent (N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine),(7) 1,1,2,2-tetrafluoroethyl-N,N-dimethylamine (TFEDMA),(8) 2,2-difluoro-1,3-dimethylimidazolidine (DFI),(9) and perfluoro-1-butanesulfonyl fluoride (PBSF).(10) DAST and Deoxo-Fluor are the most widely used deoxofluorinating agents and arguably the best in this class in terms of broad spectrum of applicability.(11)
From a historical perspective, dialkylaminosulfur trifluorides were developed as liquid alternatives to the parent reagent, i.e., SF4. The latter is a gaseous substance that is extremely toxic and corrosive. Its handling therefore necessitates extensive safety measures and specialized equipment. On the other hand, DAST is a more easily handled fluorinating agent and generally superior to SF4 in that the latter requires much higher temperatures (typically 100 °C) and leads to undesired side products. As such, DAST became widely used in deoxofluorinations and has found applications in a myriad of transformations. However, it was soon recognized that liquid DAST was thermally unstable and highly explosive.(12) Investigation of this phenomenon by thermal analyses has shown that the decomposition occurs in two distinct stages. The first event occurring at approximately 90 °C is a nonexothermic disproportionation of DAST leading to SF4 and bis(diethylamino)sulfur difluoride. Upon further heating, the latter undergoes detonation/explosion with generation of unidentified gases and black char.(13) The safety concerns over DAST prompted the development of a safer dialkylaminosulfur trifluoride reagent, namely bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor).(3) In this context, it has been shown by differential scanning calorimetry (DSC) that DAST and Deoxo-Fluor have the same decomposition temperature, but the latter degrades more slowly with somewhat lower heat evolution. In spite of these significant advances, the hazards associated with this class of reagent are considered unsafe for industrial use in a batch-type process. As a matter of fact, considerable efforts were deployed to develop continuous-flow processes to prevent reagent accumulation and circumvent the inherent problems associated with both DAST and Deoxo-Fluor.(14)
From their initial preparation to their final use, handling dialkylaminosulfur trifluoride reagents remains problematic in many aspects of their life cycle. First, purification of crude reagents by vacuum distillation is a hazardous process that requires extensive safety measures due to their explosiveness. Later, after manufacturing, dialkylaminosulfur trifluorides are subject to stringent shipping regulations. Then, in hand, great care must be exercised in handling these fuming liquids as they react extremely violently with water. Over time, these liquids are known to discolor, and a redistillation can be required to be used satisfactorily.(15) Upon use, DAST and Deoxo-Fluor generate free HF which is very volatile (bp 20 °C), highly toxic, extremely corrosive to skin and other tissues including bone, and readily etches glass.(16) Finally, deoxofluorinations using dialkylaminosulfur trifluorides are problematic in certain cases in that dehydration to an olefin often occurs. In light of these considerations, the development of safer deoxofluorinating reagents is warranted. In this context, enhanced thermal stability and crystallinity would be desirable attributes that would address most shortcomings of DAST and Deoxo-Fluor. A crystalline reagent would not only facilitate its isolation and purification but would also offer the convenience of handling a solid reagent.
In the quest to identify a safe deoxofluorinating reagent capable of incorporating fluorines in a controlled fashion, we surmised that dialkylaminodifluorosulfinium salts could act as surrogates for aminosulfur trifluorides and provide a safer alternative reagent. Preliminary investigations in our laboratories have shown that dialkylaminodifluorosulfinium salts were promising leads in that regard and warranted further investigation.(17) Herein, we report the results of our recent findings.
Results and Discussion
Synthesis of Dialkylaminodifluorosulfinium Salts
The first examples of dialkylaminodifluorosulfinium salts were reported over three decades ago by Markovskii et al. In this account, the authors describe that DAST and the dimethylamino, piperidino and morpholino analogues all react with BF3·Et2O to form the corresponding dialkylaminodifluorosulfinium tetrafluoroborates.(18) In this context, although DAST possesses electron lone pairs, BF3 does not act as a Lewis acid but rather as an irreversible fluoride ion acceptor, and the resulting dialkylaminosulfinium ion is stabilized as the tetrafluoroborate.(19) Shortly thereafter, Cowley et al. found that dimethylaminosulfur trifluoride reacted with BF3, PF5, and AsF5 to form the corresponding dimethylaminodifluorosulfinium salts.(20) A year later, Mews and Henle also reported that dimethylaminosulfur trifluoride readily loses a fluoride to BF3.(21) Over a decade later, Pauer et al. determined the crystal structure of dimethylaminodifluorosulfinium hexafluoroarsenate.(22) More recently, Pashinnik et al. reported that morpholinosulfur trifluoride reacts with SeF4 to form morpholinodifluorosulfinium pentafluoroselenate.(23) Throughout these accounts, the chemical reactivity of isolated dialkylaminodifluorosulfinium salts has been scarcely studied. They were only employed in the reaction with trimethylsilylmorpholine to give tris(morpholino)sulfinium tetrafluoroborate, and to the best of our knowledge, the fluorination of an allylic alcohol in a prostaglandin represents the sole example of deoxofluorination using a dialkylaminodifluorosulfinium salt.(24) It is noteworthy that the presence of a dialkylaminosulfinium species in solution has been inferred in the fluorination of thiocarbonyls using Deoxo-Fluor in the presence of catalytic amounts of SbCl3.(25) In this account, the identification of an in situ generated dialkylaminosulfinium species by 19F NMR is described but the counterion was not characterized. A similar intermediate had been previously proposed for the reaction of DAST with ZnI2 or SbCl3 in the specific context of transforming sulfoxides to α-fluorothioethers.(26)
In order to further evaluate the thermal stability of dialkylaminodifluorosulfinium salts and their potential use as fluorinating reagents, we initially secured some diethylaminodifluorosulfinium tetrafluoroborate (XtalFluor-E, 1) and morpholinodifluorosulfinium tetrafluoroborate (XtalFluor-M, 2) by reproducing the original Markovskii procedure (Figure 1).(27) As we previously communicated, XtalFluor-E crystallizes directly out of solution upon the reaction of DAST and BF3·Et2O in diethyl ether, but the isolated solid was somewhat amorphous and highly hygroscopic. In the hopes of obtaining a less moisture-sensitive material, the forgoing salt was initially recrystallized in warm 1,2-dichloroethane (DCE), which upon rapid cooling led to needles melting at 72−76 °C, consistent with Markovskii’s published results (vide supra). A second crystallization trial at higher temperature did not lead to the same morphology, even when seeded with aforementioned needles. Instead, a denser and cleaner product with higher melting point was obtained. Powder X-ray diffraction (XPRD) of the latter confirmed the generation of a distinctly different polymorph, herein referred to as Type II polymorph. Whereas Markovskii reportedly obtained needles (herein referred to as Type I polymorph) melting at 74−76 °C, the new morphology consists of flakes with a melting point of 89.8 °C measured by DSC and 83−85 °C measured by capillary. The fusion enthalpy of the higher-melting type II polymorph was measured at 101.3 J/g, whereas the lower melting type I polymorph gave 86.4 J/g, confirming that the new polymorph is more stable. Moreover, the Type II polymorph is less moisture sensitive, exhibits superior handling properties and is storage stable. Similarly, the morpholinodifluorosulfinium tetrafluoroborate that we have obtained using a different procedure (vide infra) exhibited a higher mp of 133.2 °C measured by DSC and 129−132 °C measured by capillary as compared with the 104−106 °C mp previously disclosed by Markovskii. Although we were not able to reproduce the original prisms, the significantly higher melting crystals we have now obtained is indicative of a new and more stable polymorphic form.
Figure 1.

Structures of fluorinating reagents.
As mentioned above, the preparation of DAST has the disadvantage of requiring a distillation. This purification step is hazardous, requires extensive safety measures and specialized equipment, and is a major cost contributor to this relatively expensive reagent. In that regard, a crystalline reagent derived from DAST would also be desirable by allowing isolation via simple filtration. In this context, we envisaged using crude undistilled DAST in the preparation of XtalFluor-E, thereby eliminating the need for a time-consuming and hazardous distillation. Gratifyingly, addition of BF3·THF to crude DAST (prepared from N,N-diethyltrimethylsilylamine and SF4 in dichloromethane) directly led to crystalline XtalFluor-E in the desired polymophic form (Type II) with an isolated yield of 90% (Scheme 1).
Scheme 1. One-Pot Preparation of XtalFluor-E from N,N-Diethyltrimethylsilylamine.

All the previously reported dialkylaminodifluorosulfinium salts were prepared via fluoride transfer to Lewis acids such as BF3, PF5, AsF5, and SbF4, and the types of salts were limited to the corresponding counteranions. However, during the course of mechanistic investigations (vide infra), DAST was found to react exothermically with tetrafluoroboric acid and provides diethylaminodifluorosulfinium tetrafluoroborate with concomitant elimination of HF. In fact, the exothermic reaction proceeded nearly quantitatively with an isolated yield of 96% (Scheme 2). This finding constitutes a novel method for the preparation of dialkylaminodifluorosulfinium salts. Moreover, this unprecedented Brønsted acid exchange method complements the traditional fluoride transfer procedure, and now provides access to aminodifluorosulfinium species bearing other types of counteranions. For example, diethylaminodifluorosulfinium trifluoromethanesulfonate salt (3) can be readily prepared by reacting DAST with triflic acid and constitutes the first example of a triflate salt of a dialkylaminodifluorosulfinium. An additional benefit is that the foregoing triflate salt has a higher melting point (97−101 °C) as compared to the corresponding tetrafluoroborate.
Scheme 2. Novel Synthesis of Diethylaminodifluorosulfinium Salts Using Strong Brønsted Acids.

Fluorination of Alcohols with XtalFluor-E and XtalFluor-M Reagents
As mentioned above, Pashinnik et al. reported the deoxofluorination of an allylic alcohol using morpholinodifluorosulfinium tetrafluoroborate (2) in acetonitrile and claim an 85% yield of the corresponding fluoride as a mixture of epimers. This represents the sole record of a deoxofluorination reported in the literature using an aminodifluorosulfinium salt. We therefore set out to assess the potential scope of such salts from a broader perspective. Fluorinations of various alcohols using XtalFluor-M were initially attempted under conditions similar to those reported in the Pashinnik procedure, i.e., using acetonitrile as solvent. Unexpectedly, geraniol led to an intractable mixture, whereas hydrocinnamyl alcohol (4) provided acetamide 5 as the major product, presumably via a Ritter-type reaction with the solvent. These indications point to an incompatibility with acetonitrile. However by using dichloromethane as solvent, we found that XtalFluor-E (1) did convert hydrocinnamyl alcohol into the desired fluoride, albeit sluggishly. More specifically, all the starting material was consumed within 5 min and led to a complex reaction mixture from which fluoride 6, ether 7, and sulfinate 8 were isolated in 32%, 27%, and 9% yield, respectively (Scheme 3).
Scheme 3. Reaction of Hydrocinnamyl Alcohol with Diethylaminodifluorosulfinium Tetrafluoroborate.

As we previously communicated, the mechanistic pathways of deoxofluorinations with DAST and XtalFluor-E are similar in that they both involve a common dialkylaminodifluorosulfane intermediate.4,28 However, since XtalFluor-E releases tetrafluoroboric acid instead of the DAST-generated HF, the reactions with XtalFluor-E are fluoride-starved, and side reactions occur.(17)
In this context, formation of ether 7 would be ascribed to a nucleophilic displacement of the activated sulfane species by the alcohol instead of fluoride. To a lesser extent, ejection of diethylamine from the transient sulfane, followed by alcohol addition on the resulting sulfoxonium would account for the formation of the sulfinate 8. In light of the fast reaction rate and product distribution observed, the dialkylaminodifluorosulfinium salt is a potent electrophile that leads to a reactive alkoxy-N,N-dialkylaminodifluorosulfane species, but the reaction mixture is fluoride-starved. In this context, we surmised that an exogenous source of fluoride would be required to mitigate side reactions. Indeed, a cleaner conversion of hydrocinnamyl alcohol (4) to 1-fluoro-3-phenylpropane (6) could be achieved when the reaction was performed in the presence of Et3N·3HF as a promoter (Table 1, entry 3). The addition order was a key parameter in this reaction. In fact, adding the exogenous fluoride immediately after the substrate was contacted with XtalFluor-E led to essentially the same product distribution as a reaction without Et3N·3HF (Table 1, entries 1 and 4). In other words, coupling of XtalFluor-E with the alcohol and subsequent reactions of the adduct are quasi instantaneous, thereby exemplifying the highly reactive nature of the salt.
Table 1. Effects of Additives on the Reaction Profiles of Hydrocinnamyl Alcohol with Diethylaminodifluorosulfinium Tetrafluoroboratea.
| yieldc (%) |
|||||||
|---|---|---|---|---|---|---|---|
| entry | additive | addition orderb | time (h) | 6 | 7 | 8 | 4 |
| 1 | none | n/a | 0.1 | 32 | 27 | 9 | 0 |
| 2 | TEA·3HF | A | 1.0 | 78 | 4 | 0 | 5 |
| 3 | TEA·3HF | B | 0.5 | 84 | 2 | 0 | 3 |
| 4 | TEA·3HF | C | 0.2 | 39 | 26 | 6 | 0 |
| 5 | DBU | A | 17 | 92 | 0 | 0 | 6 |
| 6 | DBU | B | 19 | 76 | 0 | 0 | 23 |
All reactions were performed at room temperature using 1.5 equiv of reagent and additive.
Method A: reagent was added to a mixture of alcohol and additive. Method B: alcohol was added to a mixture of reagent and additive. Method C: additive was added to a mixture of alcohol and reagent;.
Nonisolated HPLC yields using m-xylene as internal standard.
Unlike anhydrous hydrogen fluoride and Olah’s reagent, it is important to emphasize that Et3N·3HF is much less corrosive and can be handled in borosilicate glassware up to 150 °C without etching.(29) Moreover, Et3N·3HF is soluble in organic solvents, anhydrous, and inexpensive. The role of the latter is to serve as a source of HF, and it is unlikely that HF causes the dialkylaminodifluorosulfinium tetrafluoroborate to revert back to dialkylaminosulfur trifluoride and generate tetrafluoroboric acid. On the contrary, DAST reacts exothermically with tetrafluoroboric acid to provide dialkylaminodifluorosulfinium tetrafluoroborate and HF in 96% isolated yield. Moreover, 19F NMR analysis of a 1:1 mixture of XtalFluor-E and Et3N·3HF in deuterated dichloromethane did not show the characteristic DAST peak expected at 40.9 ppm.
While considering the mechanism at play, we surmised that the presence of a non-nucleophilic strong base could serve to deprotonate the putative alkoxy-N,N-dialkylaminodifluorosulfane species and prevent release of diethylamine. This would not only suppress the pathway leading to the sulfinate 8 byproduct but also now allow ejection of requisite fluoride anions. Validation of the concept was realized when DBU allowed formation of the desired fluoride, i.e., in the absence of exogenous fluoride (Table 1, entries 5, 6). Again, addition order was critical, and increased conversion was observed when the reagent was added last. Although the reaction rates with DBU were slower than those using Et3N·3HF, none of the ether 7 and sulfinate 8 side products were observed, thereby suggesting that the deprotonated alkoxy-N,N-dialkylaminodifluorosulfane adduct is more stable.
With these promising leads in hand, substrate scope was next assessed on a wide variety of alcohol substrates, including primary, secondary, tertiary, allylic, and anomeric alcohols (Table 2). In the case of hydrocinnamyl alcohol, greater conversion to the corresponding fluoride was achieved when the reactions were initiated at lower temperatures. Conversions using Et3N·3HF were initially more problematic in sec-alcohols. However, this problem was easily resolved through the use of Et3N·2HF which is considered a more nucleophilic and less basic fluoride ion source than Et3N·3HF.(30) Although not commercially available, this reagent is easily generated in situ from the appropriate amounts of Et3N·3HF and Et3N. Thus, in the fluorination of 1-phenyl-3-butanol (9) using Et3N·2HF in conjunction with XtalFluor-M, 4% of the starting material remained unreacted as compared with 17% and 31% using Et3N·3HF and Et3N·HF, respectively (Table 2, entries 5−7).
Table 2. Deoxofluorinations of Alcohols with XtalFluor-E and XtalFluor-M Reagents.
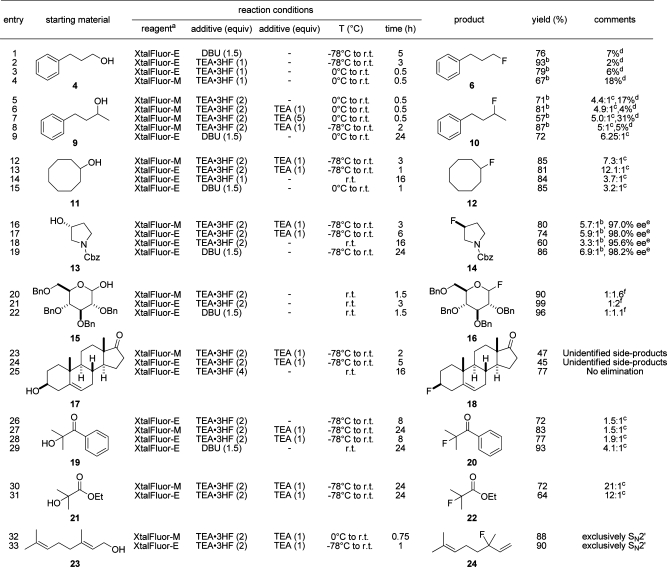 |
All reactions were performed using 1.5 equiv of XtalFluor reagent.
Nonisolated HPLC yields using m-xylene as internal standard.
Fluoro/alkene ratio calculated by 1H NMR on crude product.
Remaining alcohol estimated by HPLC.
ee of starting material: 99.9%.
α/β ratio calculated by 1H NMR.
Another important consideration is the amount of elimination side products which is typically observed in deoxofluorinations reactions using DAST and Deoxo-Fluor. Not only does the formation of these undesired products consume valuable reagent and negatively impact the yields, but the purification of the desired fluoride is oftentimes challenging due to similar physical properties between the two. The use of Et3N·2HF is also advantageous in that regard since less elimination is observed as compared with Et3N·3HF. As shown in entries 13 and 14, the XtalFluor-E-mediated deoxofluorination of cyclooctanol (11) using the latter as promoter provided a mixture of cyclooctyl fluoride (12) and cyclooctene in a 3.7:1 ratio. However, combination of both colder temperature and the use of Et3N·2HF markedly increased the selectivity to 12.1:1. These results compare favorably with those reported using DAST and Deoxo-Fluor (2.3:1 and 5.7:1, respectively).(31)
The foregoing method was next challenged on enantiopure (R)-benzyl 3-hydroxypyrrolidine-1-carboxylate (13) to determine the stereochemical course of the substitution. Thus, treatment of this reagent with XtalFluor-E and Et3N·2HF provided the inverted fluoride 14 in 98.0% ee admixed with benzyl 2,5-dihydro-1H-pyrrole-1-carboxylate in a 5.9:1 ratio, respectively. Even better results in terms of selectivity and stereochemical integrity were observed using the same fluorinating reagent and DBU (Table 2, entry 19). On the basis of these results, both the Et3N·2HF- and DBU-promoted fluorinations mainly proceed in an SN2 fashion with minimal loss of enantiopurity.
Further along the substrate−scope evaluation, 2,3,4,6-tetra-O-benzyl-d-glucose (15) provided glycosyl fluoride 16 in essentially quantitative yield as a mixture of anomers.(32) Deoxofluorination of geraniol using either XtalFluor-E or XtalFluor-M and Et3N·2HF proceeded smoothly and exclusively in a SN2′ fashion. Initial attempts to fluorinate a homoallylic alcohol, namely androstenolone (17) using the same reagent system were low yielding and more problematic. However, with further optimization with the former fluorinating reagent, Et3N·3HF gave superior results and provided 77% isolated yield of the 3β-isomer consistent with previous results obtained with DAST.(33) Finally, deoxofluorinations were tested on two tertiary alcohol substrates, i.e., 2-hydroxy-2-methyl-1-phenylpropan-1-one (19) and ethyl 2-hydroxy-2-methylpropanoate (21). On the former alcohol, XtalFluor-E and Et3N·2HF led to a mixture of desired fluoride 20 and elimination product in a modest 1.9:1 ratio, but a greater selectivity of 4.1:1 was observed using DBU as promoter. While the DBU/XtalFluor-E reagent combination was optimal in this case, the use of Et3N·2HF/XtalFluor-M on 2-hydroxy-2-methylpropanoate (21) gave a superior selectivity of 21:1. Thus, on the basis of these results, there are no general trends for the ideal combination of XtalFluor-E and XtalFluor-M fluorinating reagents with Et3N·2HF, Et3N·3HF, and DBU promoters. However, various reagent/promoter combinations are complementary to each other and provide opportunities for optimization.
Geminal Difluorination of Carbonyls with XtalFluor-E and XtalFluor-M Reagents
Prior to our original communication, the use of aminodifluorosulfinium salts for the geminal difluorination of carbonyls was unprecedented. In preliminary trials, we found that XtalFluor-E alone was incapable of performing such transformations. For example, when 4-tert-butylcyclohexanone (27) was treated with XtalFluor-E, no detectable conversion to 4-tert-butyl-1,1-difluorocyclohexane (28) was observed even after 4 days at room temperature. Interestingly, Merck chemists have also reported that 10 mol % of BF3·Et2O substantially retards the rate of reaction in the deoxofluorination of a ketone by Deoxo-Fluor.(34) These results are somewhat contradictory to previous claims to the effect that BF3·Et2O catalyzes the dialkylaminosulfur trifluoride mediated deoxofluorination of ketones.(31) All indications suggest that BF3·Et2O does not act as a Lewis acid but rather as an irreversible fluoride ion acceptor to form dialkylaminodifluorosulfinium tetrafluoroborate salts with attenuated reactivity.
Although not supported by conclusive experimental proof, the first step toward geminal difluorination is generally considered to be the addition of HF across the carbonyl group to provide a fluorohydrin, which then partakes in dehydroxyfluorination with the reagent. In the case of DAST and Deoxo-Fluor, the initial source of free HF arises by hydrolysis of the reagents with trace amounts of water or by the deliberate addition of ethanol. Likewise, exogenous HF would be required to initiate deoxofluorinations of carbonyls using dialkylaminodifluorosulfinium salts. Accordingly, we found that the deoxofluorination of hydrocinnamaldehyde (25) using either XtalFluor-E or XtalFluor-M was indeed promoted with Et3N·3HF and provided the geminal difluoride 26 with no detectable amounts of vinylfluoride side product (Table 3, entry 1, 2). Although no reaction was observed between XtalFluor-E and 4-tert-butylcyclohexanone, the addition of Et3N·2HF efficiently allowed the desired transformation to occur and provided a 91% yield of gem-difluoride 28 admixed with vinyl fluoride in a 62:1 ratio. This result favorably compares with the 5:1 and 2:1 ratios reportedly observed with Deoxo-Fluor and DAST, respectively. As was the case for alcohols, conversions were generally greater when Et3N·2HF was employed in lieu of Et3N·3HF. However, greater selectivities were generally obtained using the latter, but in this context, higher reaction temperatures, i.e., refluxing DCE, were required to achieve full conversion. For example, the selectivity of deoxofluorination on 4-(Boc-amino)cyclohexanone (29) could be upgraded from 1.8:1 to 5.1:1 by using Et3N·3HF instead of Et3N·2HF and by performing the reaction under more concentrated conditions in refluxing DCE. Likewise, high-yielding selectivities were obtained in the synthesis of ethyl 4,4-difluorocyclohexanecarboxylate (32), a key intermediate in the commercial drug Maraviroc.(35) In this case, XtalFluor-M gave a higher 15:1 selectivity than XtalFluor-E (13:1), results which again compare favorably to the 1:1 selectivity reported using DAST. Application of the foregoing procedure on cyclohexane-1,4-dione mono(ethylene glycol) acetal (33) provided the desired gem-difluoride (34) in a 9:1 selectivity, whereas Deoxofluor was reported to yield a 0.8:1 ratio.(16) Even more impressive results were obtained when 2 equiv of XtalFluor-M was employed which led to no detectable amounts of vinyl side product (Table 3, entries 8 and 9). Likewise, no elimination was observed in the fluorination of N-Cbz-protected 3-pyrrolidinone (35), whereas N-Cbz-protected 4-piperidinone (37) led to a 13:1 selectivity in favor of the desired gem-difluoride 38. Finally, both ethyl 2-oxo-2-phenylacetate (39) and 1-phenoxypropan-2-one (41) were readily converted to their corresponding difluorides using XtalFluor-E and Et3N·2HF. Throughout these examples, all reactions were essentially completed within 1 day, were generally high yielding, and consistently exhibited higher selectivities than those reported using DAST and Deoxo-Fluor.
Table 3. Deoxofluorinations of Aldehydes and Ketones with XtalFluor-E and XtalFluor-M Reagents.
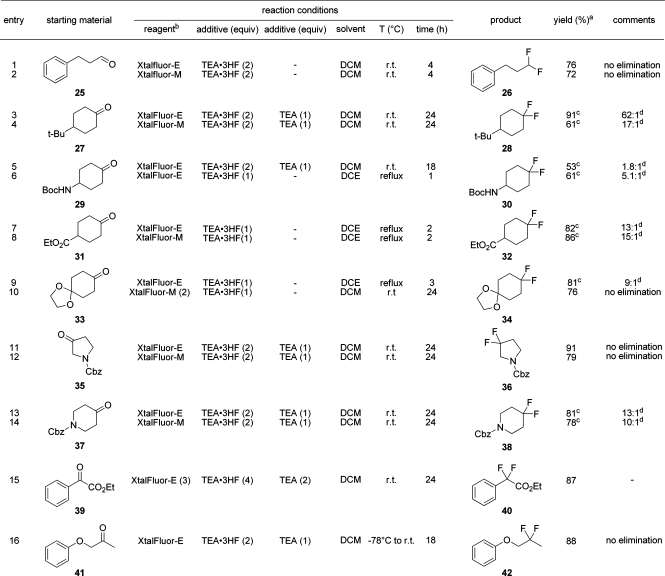 |
Isolated yields of products.
Unless otherwise noted, 1.5 equiv of reagent was employed.
Unseparated mixture of fluoro and alkene.
Fluoro/alkene ratio calculated by 1H NMR on crude product.
Miscellaneous Reactions using XtalFluor-E and XtalFluor-M Reagents
A brief survey of other types of oxygen-based functional groups was undertaken to further expand on the scope of fluorinating capabilities of XtalFluor salts and to compare with known reactions with DAST and Deoxo-Fluor (Table 4). Both aliphatic and aromatic carboxylic acids 43 and 45 were easily fluorinated to the corresponding acyl fluorides by either reagent. However, it is noteworthy that, even under forcing conditions, exhaustive fluorination to the corresponding trifluoromethyls was unsuccessful. This limitation could be ascribed to the fact that free-HF is not generated in the process.(36) Further along the substrate−scope studies, the well-precedented fluorination of sulfoxides to α-fluorothioethers was also successfully performed on methyl phenyl sulfoxide (47). Lastly, using XtalFluor-E, a one-pot process was developed where an O-trimethylsilyl cyanohydrin generated in situ from hydrocinnimalhehyde and TMSCN afforded the α-fluoronitrile 50 in excellent yields.(37)
Table 4. Miscellaneous Reactions Using XtalFluor-E and XtalFluor-M Reagents.
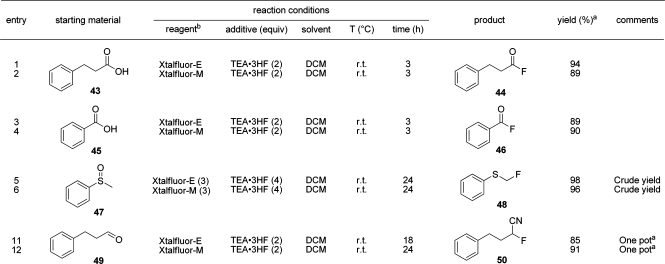 |
Isolated yields of products.
Unless otherwise noted, 1.5 equiv of reagent was employed.
Thermal Safety Assessments of XtalFluor-E and XtalFluor-M
A preliminary thermal screening was performed using differential scanning calorimetry (DSC) to establish if they are safe to handle from a thermal hazard safety perspective. In previously disclosed DSC values, DAST reportedly decomposed at 140 °C releasing 1700 J/g, whereas Deoxo-Fluor decomposed at 140 °C with 1100 J/g of energy.(5) However, recently published DSC data on DAST has shown strong exothermic activity from 131 to 163 °C with a Tmax at 162 °C and a release of 1252 J/g.(35b) Since the latter value is much lower than the one previously reported, we elected to retest DAST and Deoxo-Fluor using the same calibrated DSC instrument as the one to be used for assessing XtalFluor-E and XtalFluor-M (Figure 2). Thus, DAST exhibited a very sharp Tmax peak at 155 °C and a release of 1641 J/g, whereas Deoxo-Fluor was much broader with a Tmax at 158 °C and a release of 1031 J/g. For comparison, DSC analysis on XtalFluor-E showed a decomposition temperature (Tmax) at 205 °C with an exothermic heat (−ΔH) of 1260 J/g. In general, a higher decomposition temperature and a lower exothermic heat generated during decomposition are indicative of a more stable compound and provide greater safety. On the basis of these results, XtalFluor-E is significantly more stable as it decomposes at approximately at 50 °C above DAST and Deoxo-Fluor. Even superior results were observed with XtalFluor-M, which decomposed at an even higher temperature of 243 °C (Tmax) while releasing only 773 J/g. Moreover, isothermal DSC of both XtalFluor-E and XtalFluor-M set at 90 °C showed no observable degradation in the time frame tested, i.e., 5000 min. At the same temperature, DAST and Deoxo-Fluor were reported to degrade within 300 and 1800 min, respectively.(5)
Figure 2.

Overlapped DSC thermograms of DAST, Deoxo-Fluor, XtalFluor-E, and XtalFluor-M.
In order to accurately assess the thermal hazards and set the safety limits for process, storage, and transport temperatures, the onset temperature self-accelerated thermal decomposition must be identified by more reliable methods. Accelerated rate calorimetry (ARC), isothermal calorimetry (IC), and thermal screening unit (TSU) measurements provide more realistic estimates of the run-away temperature.(38) For example, previous hazard safety studies reported by Rossi et al. on Deoxo-Fluor showed that the onset of decomposition is around 70 °C using a Calvet calorimeter (C80), a value much lower than that detected by DSC (120 °C).(39) Likewise, DAST reportedly undergoes a very energetic decomposition between 50 and 60 °C.(40) It is noteworthy that these figures are somewhat lower than those previously obtained by ARC which showed onsets of decomposition at 85 °C for DAST and 100 °C Deoxo-Fluor.(5) Therefore, commercial samples of DAST and Deoxo-Fluor were reevaluated by ARC using the same unit model and similar sample loading as previously reported (Figure 3).(41) Thus, the onset of decomposition started at 60 °C for both DAST and Deoxo-Fluor, a value which is more aligned with recent reports. Furthermore, the maximum heat generation rate of Deoxo-Fluor occurred at 170 °C with a recorded 505 °C/min, a marked difference with the previously reported 10 °C/min (Figure 4).(5) These discrepancies could be attributed to the presence of impurities in the commercial samples, as it is well recognized that dialkylaminosulfur trifuorides discolor with aging, and several impurities were previously detected in commercial sources of Deoxo-Fluor.(42) These ARC results were next compared with those of XtalFluor-E and XtalFluor-M by using the same set of parameters. Thus, ARC testings on approximately 1 g samples showed decomposition onsets at 119 °C for XtalFluor-E and 141 °C for XtalFluor-M, a significant increase in margin safety over DAST and Deoxo-Fluor.
Figure 3.

ARC thermograms of DAST, Deoxo-Fluor, XtalFluor-E, and XtalFluor-M: temperature over time.
Figure 4.

ARC thermograms of DAST, Deoxo-Fluor, XtalFluor-E and XtalFluor-M: self-heat rate versus temperature.
Since ARC is performed under near adiabatic conditions, the information provided is generally considered as highly reliable and provides valuable heat and pressure generation rates.(43) In this context, a maximum pressure generation rate of 855 psi/min was observed at 149 °C for DAST, whereas Deoxo-Fluor decomposed at a marginally lower rate of 788 psi/min at 170 °C (Figure 5). In contrast, XtalFluor-E showed a maximum pressure generation rate of 357 psi/min at 191 °C while 85 psi/min at 246 °C was recorded for XtalFluor-M. Thus, the maximum rates of both XtalFluor salts occur at significantly higher temperatures than those of DAST and Deoxo-Fluor. Moreover, the maximum rate of pressure rise for XtalFluor-E is approximately half the rates of DAST and Deoxo-Fluor whereas XtalFluor-M is about 1 order of magnitude lower. A similar trend is also observed when comparing heat generation rates. Maximum rates of temperature rise of 310 °C/min and 50 °C/min were recorded for XtalFluor-E and M respectively, as compared to 711 °C/min and 505 °C/min for DAST and Deoxo-Fluor.
Figure 5.

ARC thermograms of DAST, Deoxo-Fluor, XtalFluor-E, and XtalFluor-M: pressure generation rate versus temperature.
Concluding Remarks
Our investigations on the physicochemical properties of dialkylaminodifluorosulfinium salts have led to important findings, including a novel and complementary preparative method using strong acids and the discovery of a novel and more stable polymorphic form of diethylaminodifluorosulfinium tetrafluoroborate. Detailed safety hazard assessments have demonstrated the enhanced thermal stability of XtalFluor-E and XtalFluor-M over Deoxo-Fluor and DAST. Moreover, a new preparative method of XtalFluor-E has eliminated the need to perform the hazardous distillations of DAST, a major cost contributor in the preparation of the latter. Although XtalFluor-E and XtalFluor-M were poor fluorinating agents of alcohols and found totally inert toward carbonyls, they excelled when used in conjunction with promoters, such as Et3N·3HF, Et3N·2HF, or DBU. A set of general reaction conditions has been developed and the fluorinations were generally high yielding and significantly more selective in providing less elimination side products as compared to Deoxo-Fluor and DAST. Contrary to the latter reagents, XtalFluor-E and XtalFluor-M are easily handled free-flowing solid which do not generate highly toxic and corrosive free HF, and are therefore compatible with borosilicate glassware and common multipurpose industrial equipment. The aforementioned desirable attributes address most of the shortcomings associated with the use of Deoxo-Fluor and DAST. Above and beyond handling, safety, and selectivity considerations, XtalFluor-E and XtalFluor-M are easily prepared and cost-effective reagents.
Experimental Section
Preparation of XtalFluor-E (1) Using DAST and BF3·THF
To a solution of diethylaminosulfur trifluoride (8.2 mL, 62 mmol) in anhydrous 1,2-dichloroethane (150 mL) at room temperature was added, dropwise and under nitrogen, neat BF3·THF (6.8 mL, 62 mmol) over a period of 45 min while keeping the reaction temperature below 30 °C. The suspension was stirred for an additional 30 min and then filtered under a blanket of nitrogen. The solid material was rinsed twice with diethyl ether (2 × 50 mL) and then dried under vacuum to provide 1 (12.1 g, 85%) as colorless crystals: mp 83−85 °C (lit.(18) mp 74−76 °C); 1H NMR (CD3CN, 300 MHz) δ 3.87 (m, 4H), 1.35 (t, J = 7.2 Hz, 6H); 19F NMR (CD3CN, 282 MHz) δ 12.9 (m, 2F), −151.1 (s, 4F); 13C NMR (CD3CN, 75 MHz) δ 45.5, 12.6.
One-Pot Preparation of XtalFluor-E Using N,N-Diethyltrimethylsilylamine, SF4, and BF3·THF
To a 5 L flange necked flask fitted with magnetic stirrer, temperature probe, bubbler, and nitrogen inlet was added dichloromethane (150 mL) and then the flask cooled to −78 °C. Sulfur tetrafluoride (70 g, 0.65 mmol) was subsurfaced while keeping the temperature below −65 °C. To the resulting pale purple solution was added dropwise a solution of diethylaminotrimethylsilane (90 g, 0.62 mol) in dichloromethane (42 mL) while keeping the temperature below −60 °C. The resulting solution was allowed to slowly warm to room temperature and stirred overnight. To the resulting dark amber solution was added dichloromethane (558 mL) followed by boron trifluoride tetrahydrofuran complex (68 mL, 0.61 mol) dropwise over 30 min keeping the temperature between 15 and 25 °C. The suspension was stirred for an additional 60 min and then filtered under a blanket of nitrogen. The solid material was rinsed with diethyl ether (3 × 150 mL)and then dried under vacuum to provide 1 (126 g, 89%) as off-white crystals: mp 83−85 °C; the NMR spectra were identical in all respects to those reported above.
Preparation of XtalFluor-M (2) Using Morpho-DAST and BF3·THF
To a solution of morpholinosulfur trifluoride (13.8 mL, 114 mmol) in anhydrous 1,2-dichloroethane (200 mL) at room temperature was added, dropwise and under nitrogen, a solution of BF3·THF (12.6 mL, 114 mmol) in anhydrous 1,2-dichloroethane (100 mL) over a period of 45 min, while keeping the reaction temperature below 25 °C. The suspension was stirred for an additional 30 min and then filtered under a blanket of nitrogen. The solid material was rinsed twice with anhydrous 1,2-dichloroethane (2 × 25 mL) and then dried under vacuum to provide morpholinodifluorosulfinium tetrafluoroborate (25.8 g, 94%) as colorless crystals: mp 129−132 °C (lit.(18) mp 104−106 °C); 1H NMR (CD3CN, 300 MHz) δ 3.90−3.85 (m, 8H); 19F NMR (CD3CN, 282 MHz) δ 10.2 (s, 2F), −151.3 (s, 4F); 13C NMR (CD3CN, 75 MHz) δ 65.7, 48.3 (br).
Representative Procedure Using an XtalFluor Reagent and TEA·3HF in DCM: Preparation of 1,1-Difluoro-3-phenylpropane (26)(17)
To a solution of triethylamine trihydrofluoride (326 μL, 2.0 mmol) in dichloromethane (3.0 mL) at room temperature were successively added XtalFluor-E (344 mg, 1.5 mmol) and 3-phenylpropionaldehyde (132 μL, 1.0 mmol). After 2 h, the reaction mixture was quenched at room temperature with a 5% aqueous sodium bicarbonate solution and stirred for 15 min, and the resulting mixture was extracted twice using dichloromethane. The organic phases were combined, dried over magnesium sulfate, and filtered through a pad of silica gel. Solvents were evaporated, and the resulting crude material was purified by silica gel flash chromatography using pentane to provide the title compound (119 mg, 76%) as a clear oil: 1H NMR (CDCl3, 300 MHz) δ 7.38−7.22 (m, 5H), 5.65 (tt, 2JH−F = 56.7 Hz, 3JH−H = 4.4 Hz, 1H), 2.82 (t, J = 7.7 Hz, 2H), 2.20 (m, 2H). 19F NMR (CDCl3, 282 MHz) δ −117.5 (dt, 2JH−F = 56.7 Hz, 3JH−F = 16.9 Hz, 1F); 13C NMR (CDCl3, 75 MHz) δ 140.2, 128.9, 128.6, 126.7, 117.0 (t, 1JC−F = 238.9 Hz), 35.9 (t, 2JC−F = 20.5 Hz), 28.7 (t, 3JC−F = 6.1 Hz).
Representative Procedure Using an XtalFluor Reagent and TEA·3HF in DCE: Preparation of 4-Carbethoxy-1,1-difluorocyclohexane (32).(35)
To a solution of triethylamine trihydrofluoride (163 μL, 1.0 mmol) in 1,2-dichloroethane (2.0 mL) was added at room temperature XtalFluor-M (362 mg, 1.5 mmol) followed by 4-carbethoxycyclohexanone (159 μL, 1.0 mmol), and the reaction mixture was heated to reflux. After 2 h, the reaction mixture was cooled to room temperature, quenched with a 5% aqueous sodium bicarbonate solution, and stirred for 15 min, and the resulting mixture was extracted twice using dichloromethane. The organic phases were combined, dried over magnesium sulfate, and filtered through a pad of silica gel. Solvents were evaporated, and the resulting crude material was purified by silica gel flash chromatography using pentane to provide the title compound (166 mg, 86%) admixed with 4-carbethoxy-1-fluorocyclohex-1-ene (15:1 ratio respectively) as a clear oil. Major compound: 1H NMR (CDCl3, 300 MHz) δ 4.11 (q, J = 7.0 Hz, 2H), 2.53−1.61 (m, 9H), 1.23 (t, J = 7.0 Hz, 3H); 19F NMR (CDCl3, 282 MHz) δ −94.3 (d, 2JF−F = 237.5 Hz, 1F), −99.8 (d, 2JF−F = 237.4 Hz, 1F); 13C NMR (CDCl3, 75 MHz) δ 174.2, 127.2 (t, 1JC−F = 241.6 Hz), 60.5, 40.5, 32.5 (t, 2JC−F = 24.3 Hz), 25.0, 14.1.
Representative Procedure Using an XtalFluor Reagent and TEA·2HF: Preparation of N-Cbz-3,3-difluoropyrrolidine (36)(44)
To a solution of triethylamine trihydrofluoride (326 μL, 2.0 mmol) and triethylamine (139 μL, 1.0 mmol) in dichloromethane (3.0 mL) at room temperature were successively added XtalFluor-E (344 mg, 1.5 mmol) and N-Cbz-pyrrolidin-3-one (219 mg, 1.0 mmol). After 24 h, the reaction mixture was quenched at room temperature with a 5% aqueous sodium bicarbonate solution and stirred for 15 min, and the resulting mixture was extracted twice using dichloromethane. The organic phases were combined, dried over magnesium sulfate, and filtered through a pad of silica gel. Solvents were evaporated, and the resulting crude material was purified by silica gel flash chromatography using hexanes/EtOAc (9/1) to provide the title compound (220 mg, 91%) as a clear oil: 1H NMR (CDCl3, 300 MHz) δ 7.31−7.26 (m, 5H), 5.08 (s, 2H), 3.73−3.53 (m, 4H), 2.29−2.21 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ −102.7 (m, 2F); 13C NMR (CDCl3, 75 MHz) δ 154.6, 136.5, 127.6 (t, 1JC−F = 249.2 Hz), 127.0 (t, 1JC−F = 248.7 Hz), 128.7, 128.4, 128.2, 67.4, 53.0 (t, 2JC−F = 32.4 Hz), 52.9 (t, 2JC−F = 32.9 Hz), 43.9, 34.3 (t, 2JC−F = 24.4 Hz), 33.7 (t, 2JC−F = 23.9 Hz).
Representative Procedure Using an XtalFluor Reagent and DBU: Preparation of (S)-N-Cbz-3-fluoropyrrolidine (14)(44)
To a solution of (R)-N-Cbz-3-hydroxypyrrolidine (221 mg, 1.0 mmol, ee >99.9%) in dichloromethane (3.0 mL) cooled at −78 °C were successively added DBU (224 μL, 1.5 mmol) and XtalFluor-E (344 mg, 1.5 mmol). After being stirred under nitrogen for 30 min, the reaction mixture was allowed to warm to room temperature and stirred for 24 h. The reaction mixture was quenched with a 5% aqueous sodium bicarbonate solution and stirred for 15 min, and the resulting mixture was extracted twice with dichloromethane. The organic phases were combined, dried over magnesium sulfate, and filtered through a pad of silica gel. Solvents were evaporated, and the resulting crude material was purified by silica gel flash chromatography using hexanes/EtOAc (3/1) to afford the title compound (192 mg, 86%) admixed with N-Cbz-2,5-dihydropyrrole (6.9:1 ratio respectively) as a clear oil. Major product: 1H NMR (CDCl3, 300 MHz) δ 7.37−7.26 (m, 5H), 5.15 (d, 2JH−F = 52.5 Hz, 1H), 5.08 (s, 2H), 3.79−3.46 (m, 4H), 2.24−1.91 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ −177.8 (m, 1F); 13C NMR (CDCl3, 75 MHz) δ 154.9, 136.9, 128.7, 128.2, 128.1, 93.0 (d, 1JC−F = 176.8 Hz), 92.2 (d, 1JC−F = 176.2 Hz), 67.1, 53.0 (d, 2JC−F = 27.1 Hz), 52.7 (d, 2JC−F = 27.1 Hz), 44.2, 43.8, 32.4 (d, 2JC−F = 57.6 Hz), 32.1 (d, 2JC−F = 57.6 Hz). Enantiomeric excess was determined to be 98.2% ee (HPLC on a Chiralcel OJ 250 × 4.6 mm column, 215 nm, hexanes/ethanol/2-propanol/TFA 1000:20:5:1, 25 °C, 1.2 mL/min, tR = 22.00 min (major), tR = 27.06 min (minor)).
Acknowledgments
We are grateful to Dr. Richard Barnhart (Pfizer) for acting as a consultant for the thermal experimental screenings. We also thank Professor Pierre Deslongchamps (Université de Sherbrooke), Professor Paul Brassard (Université Laval, retired), and Ms. Marie-Marthe Leroux for proofreading the manuscript.
Supporting Information Available
Alternative experimental methods for the preparation of 1; experimental procedures, compound characterization data for all compounds, including references to the known compounds; copies of 1H NMR, 13C NMR, and 19F NMR spectra for all compounds; DSC and ARC thermograms of 1, 2, DAST, and DeoxoFluor; XPRD spectra of 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
Dedicated to Professor Paul Brassard on the occasion of his 80th birthday.
Patent pending.
Supplementary Material
References
- a Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320. [DOI] [PubMed] [Google Scholar]; b Hagmann W. K. J. Med. Chem. 2008, 51, 4359. [DOI] [PubMed] [Google Scholar]; c Müller K.; Faeh C.; Diederich F. Science 2007, 317, 1881. [DOI] [PubMed] [Google Scholar]
- For a recent review on fluorination methods and strategies employed in medicinal chemistry, see: Kirk K. L. Org. Process Res. Dev. 2008, 12, 305. [Google Scholar]
- Hasek W. R.; Smith W. C.; Engelhardt V. A. J. Am. Chem. Soc. 1960, 82, 543. [Google Scholar]
- Middleton W. J. J. Org. Chem. 1975, 40, 574. [Google Scholar]
- a Lal G. S.; Pez G. P.; Pesaresi R. J.; Prozonic F. M.; Cheng H. J. Org. Chem. 1999, 71, 7048. [Google Scholar]; b Lal G. S.; Pez G. P.; Pesaresi R. J.; Prozonic F. M. J. Chem. Soc., Chem. Commun. 1999, 215. [Google Scholar]
- Yarovenko N. N.; Raksha M. A. Zh. Obshch. Khim. 1959, 29, 2159. [Google Scholar]
- Takaoka A.; Iwakiri H.; Ishikawa N. Bull. Chem. Soc. Jpn. 1979, 52, 3377. [Google Scholar]
- Petrov V. A.; Swearingen S.; Hong W.; Petersen W. C. J. Fluorine Chem. 2001, 109, 25. [Google Scholar]
- Hayashi H.; Sonoda H.; Fukumura K.; Nagata T. J. Chem. Soc., Chem. Commun. 2002, 1618. [DOI] [PubMed] [Google Scholar]
- a Bennua-Skalmowski B.; Vorbrüggen H. Tetrahedron Lett. 1995, 36, 2611. [Google Scholar]; b Savu P. M.; Snustad D. U.S. Patent 6 248 889, 2001.
- Singh R. P.; Shreeve J. M. Synthesis 2002, 17, 2561. [Google Scholar]
- a Cochran J. Chem. Eng. News 1979, 57 (12), 74. [Google Scholar]; b Middleton W. J. Chem. Eng. News 1979, 57 (21), 43. [Google Scholar]
- Messina P. A.; Mange K. C.; Middleton W. J. J. Fluorine Chem. 1989, 42, 137. [Google Scholar]
- a Negi D. S.; Köppling L.; Lovis K.; Abdallah R.; Geisler J.; Budde U. Org. Process Res. Dev. 2008, 12, 345. [Google Scholar]; b Baumann M.; Baxendale I. R.; Martin L. J.; Ley S. V. Tetrahedron 2009, 65, 6611. [Google Scholar]
- Fauq A. H.; Singh R. P.; Meshri D. T. In Handbook of Reagents for Organic Synthesis − Fluorine Containing Reagents; Paquette L. A., Ed.; John Wiley & Sons Ltd.: England, 2007; pp 180−194. [Google Scholar]
- Yin J.; Zarkowsky D. S.; Thomas D. W.; Zhao M. M.; Huffman M. A. Org. Lett. 2004, 6, 1465. [DOI] [PubMed] [Google Scholar]
- Beaulieu F.; Beauregard L.-P.; Courchesne G.; Couturier M.; LaFlamme F.; L’Heureux A. Org. Lett. 2009, 11, 5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovskii L. N.; Pashinnik V. E.; Saenko E. P. Zh. Org. Khim. 1977, 13, 1116. [Google Scholar]
- Minkwitz R.; Molsbeck W.; Oberhammer H.; Weiss I. Inorg. Chem. 1992, 31, 2104. [Google Scholar]
- Cowley A. H.; Pagel D. J.; Walker M. L. J. Am. Chem. Soc. 1978, 100, 7065. [Google Scholar]
- Mews R.; Henle H. J. Fluorine Chem. 1979, 14, 495. [Google Scholar]
- Pauer F.; Erhart M.; Mews R.; Stalke D. Z. Naturforsch., B: Chem. Sci. 1990, 45, 271. [Google Scholar]
- Pashinnik V. E.; Martynyuk E. G.; Shermolovich Y. G. Ukr. Khim. Zh. (Russ. Ed.) 2002, 68, 83. [Google Scholar]
- Bezuglov V. V.; Pashinnik V. E.; Tovstenko V. I.; Markovskii L. N.; Freimanis Y. A.; Serkov I. V. Russ. J. Bioorg. Chem. 1996, 22, 688. [PubMed] [Google Scholar]
- Lal G. S.; Lobach E.; Evans A. J. Org. Chem. 2000, 65, 4830. [DOI] [PubMed] [Google Scholar]
- a McCarthy J. R.; Peet N. P.; LeTourneau M. E.; Inbasekaran M. J. Am. Chem. Soc. 1985, 107, 735. [Google Scholar]; b Wnuk S. F.; Robins M. J. J. Org. Chem. 1990, 55, 4757. [Google Scholar]
- XtalFluor-E (1) and XtalFluor-M (2) can be obtained from the Aldrich Chemical Co. (catalogue nos. 719439 and 719447, respectively) and Manchester Organics Ltd. (catalogue nos. J11026 and K11027, respectively).
- a Tewson T. J.; Welch M. J. J. Org. Chem. 1978, 43, 1090. [Google Scholar]; b Sutherland A.; Vederas J. C. Chem. Commun. 1999, 1739. [Google Scholar]
- a Haufe G. J. Prakt. Chem. 1996, 338, 99. [Google Scholar]; b McClinton M. A. Aldrichim. Acta 1995, 28, 31. [Google Scholar]; c Gatner K. Pol. J. Chem. 1993, 67, 1155. [Google Scholar]; d Franz R. J. Fluorine Chem. 1980, 15, 423. [Google Scholar]
- a Giudicelli M. B.; Picq D.; Veyron B. Tetrahedron Lett. 1990, 31, 6527. [Google Scholar]; b Yin J. Y.; Zarkowsky D. S.; Thomas D. W.; Zhao M. M.; Huffman M. A. Org. Lett. 2004, 6, 1465. [DOI] [PubMed] [Google Scholar]
- Lal G. S.; Pez G. P. US patent 6,222,064 B1, 2001.
- Kovac P.; Yeh H. J. C.; Jung G. L. J. Carbohydr. Chem. 1987, 6, 423. [Google Scholar]
- Rozen S.; Faust Y.; Ben-Yakov H. Tetrahedron Lett. 1979, 20, 1823. [Google Scholar]
- Mase T.; Houpis I. N.; Akao A.; Dorzoitis I.; Emerson K.; Hoang T.; Iida T.; Itoh T.; Kamei K.; Kato S.; Kato Y.; Kawasaki M.; Lang F.; Lee J.; Lunch J.; Maligres P.; Molina A.; Nemoto T.; Okada S.; Reamer R.; Song J. Z.; Tschaen D.; Wada T.; Zewge D.; Volante R. P.; Reider P. J.; Tomimoto K. J. Org. Chem. 2001, 66, 6775. [DOI] [PubMed] [Google Scholar]
- a Price D. A.; Gayton S.; Selby M. D.; Ahman J.; Haycock-Lewandowski S.; Stammen B. L.; Warren A. Tetrahedron Lett. 2005, 46, 5005. [Google Scholar]; b Haycock-Lewandowski S. J.; Wilder A.; Ahman J. Org. Process Res. Dev. 2008, 12, 1094. [Google Scholar]; c Ahman J.; Birch M.; Haycock-Lewandowski S. J.; Long J.; Wilder A. Org. Process Res. Dev. 2008, 12, 1104. [Google Scholar]
- Although there are a few examples of perfluoration of carboxylic acids using DAST and Deoxo-Fluor, these potentially hazardous reactions are conducted neat at high temperatures and are of very limited scope; see ref (5).
- a Effenberger F.; Osswald S. Tetrahedron: Asymmetry 2001, 12, 279. [Google Scholar]; b LeTourneau M. E.; McCarthy J. R. Tetrahedron Lett. 1984, 25, 5227. [Google Scholar]
- Dale D. J. Org. Process Res. Dev. 2002, 6, 933. [Google Scholar]
- Rossi F.; Corcella F.; Caldarelli F. S.; Heidempergher F.; Marchionni C.; Auguadro M.; Cattaneo M.; Ceriani L.; Visentin G.; Ventrella G.; Pinciroli V.; Ramella G.; Candiani I.; Bedeschi A.; Tomasi A.; Kline B. J.; Martinez C. A.; Yazbeck D.; Kucera D. J. Org. Process Res. Dev. 2008, 12, 322. [Google Scholar]
- DAST decomposition data: Bretherick’s Handbook of Reactive Chemical Hazards, 6th ed.; Urben P. G., Ed.; Butterworth-Heinemann: Oxford, Boston; 1999; Vol. 1, p 560. [Google Scholar]
- The ARC tests were conducted under a “heat−wait−search” mode; i.e., the sample is heated to a predetermined temperature, and the instrument waits and searches for exothermic activity generated by self-heating of the sample. The tests were done between the range of 30 to 300 °C with heating at 5 °C/min, 10 °C steps, a 20 min wait time, and a detection limit of 0.02 °C/min.
- Laali K. K.; Borodkin G. I. J. Fluorine Chem. 2002, 115, 169. [Google Scholar]
- Information on heat and pressure generation rates are particularly important in a manufacturing plant setting as it allows the determination of required cooling capacities and the size of emergency pressure-relief valves on process reactors.
- Giardina G.; Dondino G.; Grugni M. Synlett 1995, 55. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.