

SUMMARY
To investigate the emergence and current situation of terrestrial rabies in Cuba, a collection of rabies virus specimens was employed for genetic characterization. These data supported the monophyletic nature of all terrestrial rabies viruses presently circulating in Cuba but additionally delineated several distinct variants exhibiting limited spatial distribution which may reflect the history of rabies spread on the island. The strain of rabies currently circulating in Cuba, which emerged on the island in the early 20th century, has very close evolutionary ties to the Mexican dog type and is a member of the cosmopolitan lineage widely distributed during the colonial period. The Cuban rabies viruses, which circulate predominantly within the mongoose population, are phylogenetically distant from viruses circulating in mongooses in other parts of the world. These studies illustrate, at a global level, the adaptation of multiple strains of rabies to mongoose species which should be regarded as important wildlife hosts for rabies re-emergence. Given the recent emergence of human cases due to bat contact in Cuba, this study also included a single insectivorous bat specimen which was found to most closely resemble the rabies viruses known to circulate in Mexican vampire bats.
INTRODUCTION
The emergence of mongoose rabies in the Caribbean has been reviewed previously [1]. In an attempt at control of rodent populations in sugar-cane plantations, the small Indian mongoose (Herpestes auropunctatus) was introduced to many Caribbean islands during the latter part of the 19th century. The mongoose proved to be ineffective at rodent control but, due to its high reproductive rate and adaptability, this species flourished and has had significant environmental impact in all areas where it was introduced. Subsequently, this species has emerged as a rabies reservoir in some but not all introduced areas. Four Caribbean islands (Cuba, Grenada, Puerto Rico and Hispaniola, where reporting comes primarily from the Dominican Republic) currently report mongoose rabies to the WHO rabies database [2]. The original source of the viruses responsible for these outbreaks has been a matter of some debate. It had been speculated that rabies may have been introduced into the region with the mongoose host from India but limited sequence analysis of rabies viruses from Puerto Rico and the Dominican Republic suggested that rabies emerged from other reservoirs, such as the dog, independently on these two islands [3]. However, to date, there is no information on the evolutionary origins of the enzootic in Cuba.
In Cuba, the first indication of the presence of rabies in the mongoose population was in 1936, when a dog contracted the disease after contact with a mongoose, while the first case of human rabies was registered in 1948, again after documented mongoose contact [1]. However, confirmation of the disease in mongooses was reported only in 1956 and since then case numbers have climbed significantly. By 1976, 114 human cases had been recorded and significant numbers of cases were reported in domestic animals with rather lower numbers of confirmed rabid mongooses. Since 1980 an extensive national programme to control rabies in domestic animals by vaccination was implemented on the island and this, together with improved surveillance, led to reductions in total cases in domestic animals and an increase in the proportion of reported mongoose cases [4]. Despite these activities, rabies incidence in dogs presently remains significant in some regions of the country especially in suburban and rural areas; such occurrence is frequently associated with contacts with the mongoose population, leading to the generally accepted belief that the mongoose is currently the primary reservoir for rabies on the island although some dog-to-dog transmission cannot be discounted. Previous mongoose population control programmes employing poisoned eggs had limited success in controlling either mongoose populations or rabies in mongooses and they were subsequently discontinued [5]; there is no current activity to control rabies in mongooses in Cuba.
The most recent information from the RabNet database [6] covers a 9-year period, 1991–1999, during which time 1155 laboratory-confirmed animal rabies cases (mean of 128 per year) were reported in Cuba. Out of these cases, 280 were in dogs (24·2%), 279 were in other domestic species, including cats and livestock (24·2%), 584 (50·6%) were in wildlife, predominantly the mongoose, and bats, which are infrequently reported, comprised just 12 cases (1%) of the total. Notably eight of these reports in bats occurred in 1999, the last year for which comprehensive data are available. Although mongooses account for about 50% of all reported cases, these figures probably do not represent the actual relative importance of this species with respect to rabies incidence due to the inevitable reporting bias in favour of domestic species.
Human rabies is now infrequently reported on the island, in part due to good access to post-exposure prophylaxis (PEP), and indeed, throughout the country no case of human rabies transmitted by dogs has been reported since 1976. However, after a decade (1977–1987) without any human case reports, one case presented in 1988 following contact with a wild animal. Since then, a total of nine human cases have been reported; eight of these cases occurred following bites by non-haematophagous bats and the other victim was presumed to be infected via contact with a wild cat [4, 5].
Modern tools of viral characterization, including both antigenic [7] and genetic [8] methods of analysis, have been extensively applied to field isolates of rabies virus. As the type virus of the Lyssavirus genus, within which seven distinct viral genotypes are currently recognized [9, 10], the classical rabies virus that constitutes genotype 1 has been most extensively studied. Of the five coding regions contained by the non-segmented negative sense viral genome [11], three genes (N, P and G) have been targeted for global studies on viral variation [12–14]. Regardless of the target sequence employed, similar conclusions regarding the phylogeny and evolution of rabies viruses are evident. In a study of P gene diversity, Nadin-Davis et al. [14] showed that genotype 1 rabies viruses segregate into two main clades, one of which is restricted in its distribution to the American continent (designated as American indigenous) while the other clade is globally distributed and includes many strains that also circulate in the Americas. The majority of the viruses of the American indigenous clade circulate in chiropteran hosts together with a limited number of strains of terrestrial carnivores, including those associated with skunks from the southern United States, raccoons in the United States and Canada, and skunks from certain regions in Mexico [15]. In contrast, the other major grouping of rabies viruses comprises strains which are exclusively maintained in terrestrial carnivores and includes a particular subgroup now known as the cosmopolitan lineage. This lineage, believed to have originated in Europe, was widely disseminated around the world as a consequence of colonial activities that took place from the 16th to 19th centuries [3, 16].
This report is the first to describe a comprehensive study of the nature of the viruses that currently circulate in Cuba. The study uses phylogenetic methods to explore the evolutionary origins of these viruses and explores the utility of genetic and antigenic methods as future epidemiological tools.
METHODS
Viral isolates
All viral isolations were performed either at the Rabies Laboratory of Hygiene and Epidemiology or at the National Reference Laboratory for Rabies at Pedro Kouri Institute (IPK) in Havana, Cuba, using suckling Balb/c mice inoculated intracerebrally. Rabies diagnosis was performed by a direct immunofluorescent antibody test (FAT) according to Dean et al. [17]. Table 1 presents a complete listing of the isolates employed in this study.
Table 1.
Summary of isolates employed in study
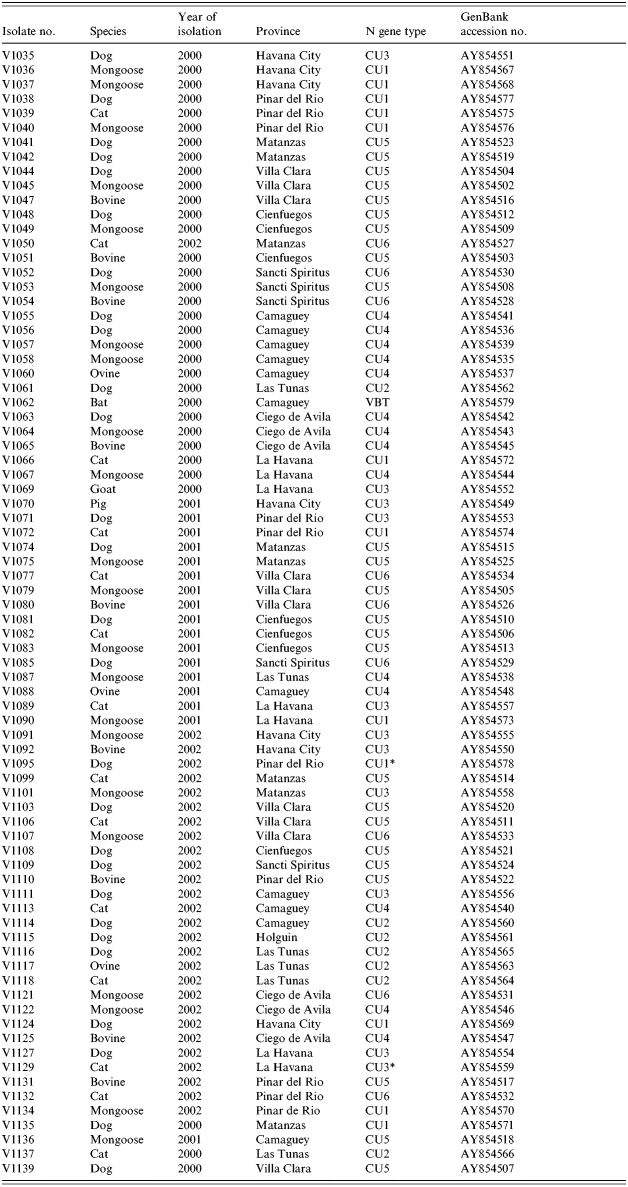
VBT, Vampire bat strain.
Support for the association of this isolate with the indicated group was modest.
RNA extraction
Total RNA was recovered from 0·1 g of infected brain tissue using TRIzol as indicated by the supplier (Invitrogen, Burlington, ON, Canada). RNA pellets were dissolved in ribonuclease-free sterile water and RNA concentration was determined spectrophotometrically.
Reverse transcription–polymerase chain reaction (RT–PCR) and sequencing
Up to 2 μg of total brain RNA was employed for cDNA synthesis as previously described [18] using a positive sense rabies-specific oligonucleotide (Nseq0) [19] for priming. A 10 μl aliquot of cDNA was used in a 50 μl PCR using RabN5 oligonucleotide as the reverse sense primer [18]. Production of the expected 1478 bp product was evaluated by electrophoresis of aliquots of the PCRs through standard agarose gels. For those isolates for which no product could be detected, a nested PCR was undertaken using the same cycling conditions and internal primers RabNfor and RabNrev [18] to generate a 762 bp product. Amplicons were purified using a Wizard PCR purification system according to the manufacturer's instructions (Promega, Madison, WI, USA) prior to sequencing.
Nucleotide sequencing and phylogenetic analysis
Consensus sequences were determined from purified PCR products using a Li-Cor (Lincoln, NE, USA) 4200L two-dye automated sequencing system. Sequences were generated using IR 700 or IR 800 dye-labelled primers with sequences corresponding to those of the RabNfor and RabNrev primers and a Thermosequenase cycle sequencing kit (Amersham Biosciences, Baie d'Urfé, QC, Canada). Sequences were manually edited using Eseq v. 2 software (Li-Cor) and exported in fasta format for alignment in the Clustal X (v. 1.8) programme [20]. The phylip 3.6 software package [21] was used for phylogenetic analysis of the alignment, using either the neighbour-joining (NJ) algorithm as detailed elsewhere [14] or the maximum parsimony (MP) algorithm implemented with the dnapars programme. treeview [22] was used to generate graphical outputs of the trees. Partial N gene sequences determined for all Cuban isolates characterized in this study have been submitted to GenBank and assigned accession numbers AY854502-79. Translation of these nucleotide sequences to predict the partial encoded nucleoprotein was performed using the DNAsis software package (Helixx Technologies Inc., Scarborough, ON, Canada).
Antigenic typing
For antigenic analysis, selected Cuban isolates were propagated in neuroblastoma cell culture. A collection of 373 anti-N monoclonal antibodies (mAbs) was available for rabies virus typing as detailed previously [23]. Undiluted tissue culture supernatant of hybridoma cultures were used in indirect FAT assays [24] on acetone-fixed infected neuroblastoma cultures in Terasaki plates. Fluorescence was scored from negative (−) to weakly (+) or strongly (+++) positive.
RESULTS
Phylogenetic evaluation
The 78 isolates of this study included 22 from mongooses, 27 from dogs, 14 from cats, 14 from various livestock species and one of bat origin (see Table 1). All regions of the main island of Cuba were represented (Fig. 1) apart from the three most southerly provinces, Granma, Santiago de Cuba and Guantanamo, that do not currently report any rabies cases. All 78 specimens generated a PCR product that was used to determine a consensus sequence of a 530 base segment of the N gene, corresponding to bases 159–688 of the challenge virus standard (CVS) reference strain (GenBank accession no. D42112). A NJ algorithm, applied to the complete alignment of these data using the CVS strain as reference, generated the phylogeny represented in Figure 2.
Fig. 1.

Map of Cuba showing the administrative provincial organization of the island as referred to in the text.
Fig. 2.

Phylogenetic tree of all Cuban isolates sequenced in this study. The tree was generated by a neighbour-joining algorithm as described using partial rabies virus N gene sequences (530 bp). The CVS strain was used as an outgroup for the terrestrial isolates although the single bat specimen (V1062) was found to be the most divergent isolate within the sample set. Bootstrap values (out of 1000 data replicates) are indicated on many of the main branches and the major clades described in the text are illustrated to the right of the tree. A genetic scale is shown at bottom; the dashed line connecting V1062 indicates that this branch is not to scale.
The bat-associated virus (V1062) clearly formed a distant outlier to the rest of the group, including the CVS strain, and is considered separately below while all remaining Cuban samples clustered as a group that was further subdivided into six main clades designated CU1–CU6. The CU1 cluster has moderate bootstrap support (value of 720) with an outlying specimen, V1095, associating with this group with only modest support (bootstrap value of 651). This cluster can be subdivided into two further groups comprising three specimens from Pinar del Rio (CU1a) and eight specimens (CU1b) originating from the neighbouring regions of Pinar del Rio, Havana City, La Havana, and Matanzas. The highly homogeneous and very strongly supported CU2 cluster (bootstrap value of 998) consisted of five specimens that originated from Las Tunas and one each from Camaguey and Holguin provinces. Ten specimens form a highly supported cluster CU3 (bootstrap value of 993) which is represented by specimens from La Havana, including Havana city, Pinar del Rio, Matanzas and one from Camaguey; isolate V1129 associates with this clade as an outlier with modest support (bootstrap value of 540). Clade CU4 (bootstrap value of 809), was divided into two well supported subgroups comprising viruses from Camaguey and Las Tunas (CU4a) and from Ciego de Avila and La Havana (CU4b). The CU5 cluster (bootstrap value of 971), comprised isolates from the central and northern regions of the island including Matanzas, Villa Clara, Cienfuegos, Sancti Spiritus, Pinar del Rio and one from Camaguey. The remaining nine specimens formed a cluster (CU6) far less well supported (bootstrap value of 541) and which originated from Villa Clara, Sancti Spiritus, Matanzas, Ciego de Avila and Pinar del Rio, a geographical distribution rather similar to that of CU5.
Another phylogenetic analysis, performed on these same data using the MP algorithm, supported a most similar tree topology (data not shown) and the bootstrap values (out of 100 replicates) for each clade are indicated (in parentheses) as follows: CU1 (63·4) with greater support for groups CU1a (98·7) and CU1b (73·6), CU2 (98·5), CU3 (100), again with V1129 as an outlying specimen (42·5), CU4 (73·7), CU5 (74·1) and CU6 (52·9). In this tree V1095 clustered with all other terrestrial viruses with a bootstrap value of 81·7 but did not cluster closely with any specific clade.
Nature of the nucleoprotein encoded by these viral variants
To explore the degree of variation conferred upon the rabies virus nucleoprotein as a result of the genetic variation at the N gene locus, all 78 partial N gene sequences were translated to protein. The resulting amino-acid sequence spanned from residues 40–215 of the nucleoprotein of the CVS reference sequence. An alignment of these sequences (data not shown) illustrated that specimen V1035, used as the reference Cuban sequence, differed from the CVS strain at just four positions (residues 102, 106, 136 and 214), mostly due to conservative amino-acid substitutions, while the single bat-associated isolate V1062 differed at a total of nine positions. Overall, the protein sequence was very highly conserved in all terrestrial Cuban viruses with only sporadic single amino-acid replacements observed for individual isolates. There was clearly no correlation of amino-acid substitution with the host species of origin. However, all members of clade CU6, as well as isolate V1062, exhibited an aspartic acid (Asp) at residue 110; all other viruses maintained glutamic acid (Glu) at this position. This observation suggested it might be worthwhile exploring antigenic methods of variant discrimination.
Antigenic analysis
A panel of anti-N monoclonal antibodies (mAbs) was evaluated by indirect FAT assay for reactivity to eight selected isolates representing the main genetic groupings as identified in the phylogenetic tree. While the majority of these antibodies reacted similarly to most of these viruses, a small group did exhibit differential reactivity. Out of this group the reactivity profiles of seven mAbs are summarized in Table 2. It is apparent that the bat-associated virus (V1062) was the only specimen to retain both binding sites for mAbs M1507 and M1512 and it was the only virus to react with mAb M1338; indeed the only mAb of this panel that did not react to this isolate was M866. With respect to the terrestrial viruses, the following patterns are evident. M866 exhibited a strong reaction with only V1036 (CU1 variant) although it did react weakly with two other specimens (V1115 and V1060 of different phylogenetic groupings). M878 bound to all isolates except for those of type CU2 (V1115 and V1061). M1495 and M1512 both reacted to the isolates representing variants CU4 and CU6 only while M1501 discriminated between these two viruses by reacting with V1121 only. M1507 reacted only to the two isolates of CU2 type. This small mAb panel would thus appear to be potentially useful in discriminating between most of the genetic variants of Cuban rabies viruses identified in this study with the exception of variants CU3 and CU5; however, further studies examining the reactivity patterns of a larger sampling of viruses will need to be undertaken to confirm the panel's utility.
Table 2.
Reactivity of selected monoclonal antibodies to representatives of the Cuban rabies virus variants
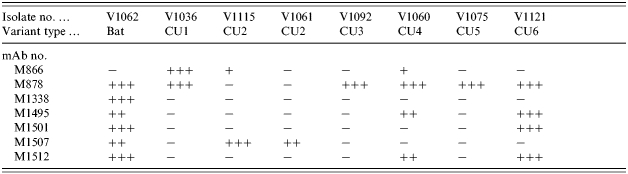
An indirect FAT applied to virus grown in cell culture was used to score mAb reactivity from − (no staining) to + (weak staining) to ++/+++ (strong to very strong staining).
Placement of the Cuban viruses in a global context
To examine the placement of the Cuban rabies specimens within a global context, representatives of all Cuban rabies variants, including the single bat isolate, were compared to other rabies viruses representative of many of the strains circulating around the world. Using a sample set of 69 partial rabies virus N gene sequences, together with corresponding sequence from a European Bat lyssavirus 2 (EBLV-2) specimen (9018HOL) employed as an outgroup (see virus list in Table 3), the phylogeny presented in Figure 3 was generated. Within this tree all Cuban terrestrial isolates again group as a single lineage which is very closely linked to the urban rabies cycle in Mexico (bootstrap value of 993). This cluster forms a branch of the cosmopolitan lineage that also includes isolates from Europe, the middle-East and many parts of Africa. It is notable that other mongoose isolates from South Africa (isolate 1500AFS) and from Sri Lanka (1077SRL) clustered to distinct lineages of the tree quite separate from each other and from the Cuban isolates. The single bat specimen from Cuba (V1062) was included in the American indigenous clade, specifically within the branch representative of those strains associated with vampire bats of Mexico, Trinidad, Brazil and Guyana.
Table 3.
Origin of rabies virus sequences employed in global comparison
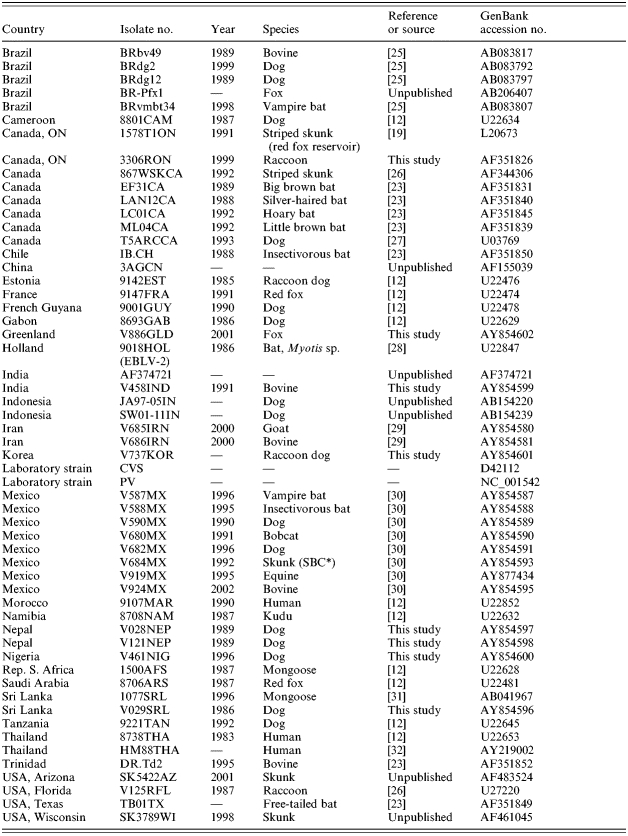
SBC, South Baja California.
Fig. 3.

Phylogenetic tree illustrating the placement of the Cuban rabies viruses in a global context. Partial N gene sequences (630 bp) for selected Cuban isolates were aligned with corresponding sequences representative of many rabies virus strains worldwide (see list in Table 3) together with an EBLV-2 sequence as outgroup; a tree was generated using a neighbour-joining algorithm. Bootstrap values (out of 1000 replicates) are shown on all main branches and major rabies strain groupings are illustrated to the right of the tree using the following abbreviations: CD, canid type; HP, herpestid type; SBC SK, South Baja California skunk variant; US SC SK, United States south central skunk variant. Many of these groups have been described previously [14]. A genetic scale is shown at bottom; the dashed line connecting the EBLV-2 sequence (9018HOL) indicates that this branch is not to scale.
DISCUSSION
The observation reported here, that all recent rabies isolates from terrestrial species of Cuba form a single, well-supported monophyletic clade, argues in favour of a single introduction of rabies to the island and that all variants that presently circulate in Cuba are derived from this progenitor. The phylogeny predicted for the Cuban rabies viruses identified six principal groups that, based upon their branching pattern within the NJ tree of Figure 2, appear to have emerged in the order CU1, CU2, CU3, CU4, CU6 and CU5. Based on the sample set analysed in this study these variants exhibit some degree of regional distribution quite independent of the species involved. The CU1 variant was restricted to the more western regions of the island, specifically the provinces of La Havana, including Havana city, Pinar del Rio and Matanzas. The existence of the most ancient clade in this area might reflect the introduction of rabies into this region, a strong possibility given the role of Havana city as a major port. The existence of clade CU2 that was recovered only from the eastern provinces of Las Tunas, Camaguey and Holguin, suggests there may have been a translocation event that allowed the disease to spread rapidly into this area of the country. The CU3 clade was also located principally in the western provinces but included a single isolate from Camaguey as well – it is possible that the range of this variant is more extensive than suggested by our sampling or it is also possible that the Camaguey isolate represents a recent translocation event that may be confounding delineation of the natural spread of these viral variants. Variant CU4 was, with the exception of one sample divided into two well supported subclades that exhibited strong spatial clustering with CU4a found only in Camaguey and neighboring Las Tunas, while all but one sample from CU4b originated from Ciego de Avila province. The single sample (V1067) within clade CU4b that came from La Havana clearly departs from this trend and we suggest that this may also be due to a recent translocation event. The well represented CU5 clade was distributed over much of the island, suggesting the widespread movement of this more recently evolved viral variant. Clade CU6, which was distributed across the western half of the island, was not strongly supported by either NJ or MP analysis. However, our observation that it was predicted by both analyses suggests that these viruses do indeed group together, a conclusion strengthened by the presence of an amino-acid substitution observed exclusively in all members of this group when all terrestrial viral isolates were compared.
A small panel of mAbs that discriminated between representatives of most of these viral variants was identified after the reactivity of a large collection of anti-N mAbs was examined. The limited sequence data obtained for the nucleoprotein of these viruses suggests that residue 110 might be important for the binding of mAb M1501 that bound exclusively to members of clade CU6 and the single bat isolate in agreement with the observation that only these viruses of the collection maintained an Asp residue at this position. However, this was the only amino-acid substitution found that might explain the differential mAb binding patterns observed and it is presumed that other uncharacterized regions of the protein confer discriminatory binding properties to the other mAbs of the panel; in particular residues 376–379, contained within a linear antigenic site of certain rabies virus strains, have frequently been suggested as an important site for antigenic methods of strain discrimination [15]. While the data presented in Table 2 would suggest that this mAb panel might be useful for further epidemiological analysis, the trends seen with the small number of representative viruses tested in this study will need to be confirmed on a larger sample set.
The Cuban isolates are clearly placed within the cosmopolitan lineage as illustrated in Figure 3. This lineage is believed to have emerged from Western Europe and been distributed to many regions of the world, including parts of Africa, the Americas and Asia, during colonial times [16]. Indeed, dog variants that circulated in most of Latin America in the early 20th century [33] and which still persist in many parts of the continent fall within this lineage. Unfortunately a direct comparison between the Cuban rabies viruses and those from other Caribbean islands, notably Puerto Rico and the Domincan Republic [3], was not possible in this study due to the unavailability of nucleotide sequence data of the appropriate sequence window for viruses from these countries. However, it would appear likely that the Cuban viruses do not cluster closely with these other Caribbean rabies variants since Smith et al. [3] reported that both the Dominican Republican and Puerto Rican viruses grouped with other South American strains and rather distantly from Mexico and Central American viruses. This is in contrast to our findings which clearly indicate strong evolutionary connections between terrestrial rabies viruses of Cuba and Mexico and support the emergence of Cuban rabies from a progenitor that also evolved into urban rabies in Mexico. The Cuban cluster was clearly well separated from a group of Brazilian viruses that circulate in dogs and foxes (see BRAZIL CD group in Fig. 3). In contrast, the viruses that circulate in other mongoose reservoirs, in both Africa (represented by isolate 1500AFS) and Asia (isolate 1077SRL) are located in distinct branches of the rabies phylogeny and are clearly evolutionarily very different both from each other and from the Cuban viruses. The mongoose isolate in Sri Lanka (see ref. [31]) was closely related to the viruses that circulate in dogs on that island while African mongoose rabies viruses form a heterogeneous lineage with very distinct origins from other African canid viruses [34].
The single Cuban bat isolate examined in these studies was from a specimen of the species Eumops glaucinus, an insectivorous species that ranges from southern Florida, Mexico and into several central and South American countries; these populations also migrate to some Caribbean islands from the mainland and is not infrequently found throughout Cuba [35]. The virus, however, segregated within the American indigenous clade of rabies viruses and was closely related to previously characterized viruses that circulate in vampire bats of Mexico, Trinidad and South America [23]. The closest match to this Cuban bat rabies sample with a bootstrap value of 944 (data not shown), was to a Mexican isolate (V919MX), typical of vampire bat rabies, which was obtained from an equine in the state of Chiapas in south-eastern Mexico, an area of relatively close proximity to Cuba. The most likely explanation for this case is that this E. glaucinus bat was infected by spillover from the vampire bat reservoir on the American mainland prior to its migration to Cuba; vampire bats are not considered to be indigenous to Cuba and E. glaucinus has not previously been identified as a rabies reservoir. However, given the number of recent human cases that have been ascribed to exposure through bat contact in Cuba (M. Guzman et al., unpublished data), improved surveillance of bats on the island should be undertaken to investigate the prevalence of rabies in various species of indigenous and migratory bat populations.
ACKNOWLEDGEMENTS
We express our gratitude to all staff from the Rabies Laboratory of Hygiene and Epidemiology, Havana City and the National Reference Laboratory at Pedro Kouri Institute for their collaboration in this study. We also thank the Pan American Health Organisation (PAHO) for providing funds in support of the visit of Dr Gisset Torres to the Ottawa Laboratory (Fallowfield) in Canada thereby facilitating the initiation of this project. S. Craig and J. Armstrong are acknowledged for their excellent technical support and we thank E. Loza-Rubio (INIFAP, Mexico City) for supplying the Mexican isolates employed in this report. Finally we also thank two anonymous reviewers for their helpful suggestions.
DECLARATION OF INTEREST
None.
REFERENCES
- 1.Everard COR, Everard JD. Mongoose Rabies in the Caribbean. Annals of the New York Academy of Science. 1992;653:356–366. doi: 10.1111/j.1749-6632.1992.tb19662.x. [DOI] [PubMed] [Google Scholar]
- 2.WHO Rabies Database. http://www.who.int/rabies/resources/en. http://www.who.int/rabies/resources/en ). Accessed 26 January 2006.
- 3.Smith JS et al. Epidemiologic and historical relationships among 87 rabies virus isolates as determined by limited sequence analysis. Journal of Infectious Diseases. 1992;166:296–307. doi: 10.1093/infdis/166.2.296. [DOI] [PubMed] [Google Scholar]
- 4.Programa Nacional de Prevention y Control de la Rabia 1998. . Republica de Cuba. MINSAP,
- 5.Cruz de la Paz p. 23. . Situation of rabies in Cuba. Presented at the XV International Meeting Rabies in the Americas held in Santo Domingo, Dominican Republic, 31 October to 4 November 2004, pp.
- 6.WHO Rabies Database. http://www.who.int/globalatlas/defaults.asp. http://www.who.int/globalatlas/defaults.asp ). Accessed 26 January 2006.
- 7.Rupprecht CE, Baer GM The Natural History of Rabies. 2nd ed. Boca Raton: CRC Press; 1991. Antigenic relationships of lyssaviruses; pp. 69–100. , pp. [Google Scholar]
- 8.Smith JS, Jackson AC, Wunner WH. Rabies. San Diego: Academic Press; 2002. Molecular epidemiology; pp. 79–111. , pp. [Google Scholar]
- 9.Bourhy H, Bachir K, Tordo N. Molecular diversity of the Lyssavirus genus. Virology. 1993;194:70–81. doi: 10.1006/viro.1993.1236. [DOI] [PubMed] [Google Scholar]
- 10.Guyatt KJ et al. A molecular epidemiological study of Australian bat lyssavirus. Journal of General Virology. 2003;84:485–496. doi: 10.1099/vir.0.18652-0. [DOI] [PubMed] [Google Scholar]
- 11.Tordo N, Meslin FX, Kaplan MM, Koprowski H. Laboratory Techniques in Rabies. 4th ed. Geneva: WHO; 1996. Characteristics and molecular biology of the rabies virus; pp. 28–51. , pp. [Google Scholar]
- 12.Kissi B, Tordo N, Bourhy H. Genetic polymorphism in the rabies virus nucleoprotein gene. Virology. 1995;209:526–537. doi: 10.1006/viro.1995.1285. [DOI] [PubMed] [Google Scholar]
- 13.Badrane H, Tordo N. Host-switching in Lyssavirus history from the chiroptera to the carnivora orders. Journal of Virology. 2001;75:8096–8104. doi: 10.1128/JVI.75.17.8096-8104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadin-Davis SA et al. Lyssavirus P gene characterisation provides insights into the phylogeny of the genus and identifies structural similarities and diversity within the encoded phosphoprotein. Virology. 2002;298:286–305. doi: 10.1006/viro.2002.1492. [DOI] [PubMed] [Google Scholar]
- 15.Velasco-Villa A et al. Molecular epizootiology of rabies associated with terrestrial carnivores in Mexico. Virus Research. 2005;111:13–27. doi: 10.1016/j.virusres.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Nadin-Davis SA, Bingham J, King AA, Fooks AR, Aubert M, Wandeler AI. Historical Perspective of Rabies in Europe and the Mediterranean Basin. Paris: OIE; 2004. Europe as a source of rabies for the rest of the world; pp. 259–280. , pp. [Google Scholar]
- 17.Dean DJ, Ableseth MK, Atanasiu P, Meslin FX, Kaplan MM, Koprowski H. Laboratory Techniques in Rabies. 4th ed. Geneva: WHO; 1996. The fluorescent antibody test; pp. 88–95. , pp. [Google Scholar]
- 18.Nadin-Davis SA. Polymerase chain reaction protocols for rabies virus discrimination. Journal of Virological Methods. 1998;75:1–8. doi: 10.1016/s0166-0934(98)00106-2. [DOI] [PubMed] [Google Scholar]
- 19.Nadin-Davis SA, Casey GA, Wandeler A. Identification of regional variants of the rabies virus within the Canadian province of Ontario. Journal of General Virology. 1993;74:829–837. doi: 10.1099/0022-1317-74-5-829. [DOI] [PubMed] [Google Scholar]
- 20.Thompson JD et al. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Felsenstein J. PHYLIP: phylogeny inference package (Version 3.52c) Seattle, Washington: University of Washington; 1993. [Google Scholar]
- 22.Page RDM. TREEVIEW: an application to display phylogenetic trees on personal computers. Computer Applications in the Biosciences. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 23.Nadin-Davis SA et al. Antigenic and genetic divergence of rabies viruses from bat species indigenous to Canada. Virus Research. 2001;74:139–156. doi: 10.1016/s0168-1702(00)00259-8. [DOI] [PubMed] [Google Scholar]
- 24.Daoust P, Wandeler AI, Casey GA. Cluster of rabies cases of probable bat origin among red foxes in Prince Edward Island, Canada. Journal of Wildlife Diseases. 1996;32:403–406. doi: 10.7589/0090-3558-32.2.403. [DOI] [PubMed] [Google Scholar]
- 25.Ito M et al. Discrimination between dog-related and vampire-bat-related rabies viruses in Brazil by strain-specific reverse transcriptase–polymerase chain reaction and restriction fragment length polymorphism analysis. Journal of Clinical Virology. 2003;26:317–330. doi: 10.1016/s1386-6532(02)00048-3. [DOI] [PubMed] [Google Scholar]
- 26.Nadin-Davis SA, Huang W, Wandeler AI. Polymorphism of rabies viruses within the phosphoprotein and matrix protein genes. Archives of Virology. 1997;142:1–14. doi: 10.1007/s007050050133. [DOI] [PubMed] [Google Scholar]
- 27.Nadin-Davis SA, Casey GA, Wandeler AI. A molecular epidemiological study of rabies virus in central Ontario and western Quebec. Journal of General Virology. 1994;75:2575–2583. doi: 10.1099/0022-1317-75-10-2575. [DOI] [PubMed] [Google Scholar]
- 28.Bourhy H et al. Antigenic and molecular characterisation of bat virus in Europe. Journal of Clinical Microbiology. 1992;30:2419–2426. doi: 10.1128/jcm.30.9.2419-2426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nadin-Davis SA et al. Molecular and antigenic characterization of rabies viruses from Iran identifies variants with distinct epidemiological origins. Epidemiology and Infection. 2003;131:777–790. doi: 10.1017/s095026880300863x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loza-Rubio E et al. Detection of multiple strains of rabies virus RNA using primers designed to target Mexican vampire bat variants. Epidemiology and Infection. 2005;133:927–934. doi: 10.1017/S095026880500405X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arai YT et al. Characterization of Sri Lanka rabies virus isolates using nucleotide sequence analysis of nucleoprotein gene. Acta Virologica. 2001;45:327–333. [PubMed] [Google Scholar]
- 32.Hemachudha T et al. Sequence analysis of rabies virus in humans exhibiting encephalitic or paralytic rabies. Journal of Infectious Diseases. 2003;188:960–966. doi: 10.1086/378415. [DOI] [PubMed] [Google Scholar]
- 33.Acha PN. A review of rabies prevention and control in the Americas, 1970–1980. Over-all status of rabies. Bulletin de l'Office International des Epizooties. 1981;93:9–52. [Google Scholar]
- 34.Von Teichman BF et al. Molecular epidemiology of rabies virus in South Africa: evidence for two distinct virus groups. Journal of General Virology. 1995;76:73–82. doi: 10.1099/0022-1317-76-1-73. [DOI] [PubMed] [Google Scholar]
- 35.Taboada GS. Biografia Historica: Colonizacion. Habana, Cuba: Academic; 1979. The bats of Cuba; pp. 360–373. , pp. [Google Scholar]