

Abstract
In-depth, reproducible coverage of complex proteomes is challenging because the complexity of tryptic digests subjected to LC-MS/MS analysis frequently exceeds mass spectrometer analytical capacity, which results in undersampling of data. In this study, we used cancer cell lysates to systematically compare the commonly used GeLC-MS/MS (1-D protein + 1-D peptide separation) method using four repetitive injections (2-D/repetitive) with a 3-D method that included solution isoelectric focusing and involved an equal number of LC-MS/MS runs. The 3-D method detected substantially more unique peptides and proteins, including higher numbers of unique peptides from low-abundance proteins, demonstrating that additional fractionation at the protein level is more effective than repetitive analyses at overcoming LC-MS/MS undersampling. Importantly, more than 90% of the 2-D/repetitive protein identifications were found in the 3-D method data in a direct protein level comparison, and the reproducibility between data sets increased to greater than 96% when factors such as database redundancy and use of rigid scoring thresholds were considered. Hence, high reproducibility of complex proteomes, such as human cancer cell lysates, readily can be achieved when using multidimensional separation methods with good depth of analysis.
Keywords: Proteomics, Proteome coverage, Complex proteomes, Multidimensional, Reiterative LC-MS/MS, Proteome reproducibility, Proteome data analysis, Database limitations, Melanoma
Short abstract
Comparison of a GeLC-MS/MS proteome analysis utilizing repetitive injections (2-D/repetitive runs) and a 3-D method showed greater depth of analysis for the 3-D method when equal mass spectrometer time was utilized. Both methods minimized undersampling, and when stringent data filtering was used, both methods produced highly reproducible results with 90% overlap of protein names and greater than 96% overlap when relatively trivial database and data analysis factors were considered.
Introduction
Over the past decade, protein separation techniques and peptide analysis by mass spectrometry have greatly improved, and qualitative and quantitative proteome comparisons have become powerful tools for defining the composition of complex proteomes and the functions of protein complexes.(1) Current ion trap mass spectrometers, in particular, are very sensitive and have fast analysis times that enable the identification of hundreds of proteins in a single LC-MS/MS analysis.2−6 However, obtaining comprehensive protein profiles from very complex samples, such as biological fluids or mammalian cell or tissue lysates, remains challenging due to the large number of proteins present in the sample over wide concentration ranges. To improve proteome coverage, samples typically are fractionated at either the protein level or peptide level, or both. Regardless of the initial fractionation methods used, the final step in most strategies involves analysis of tryptic peptides by LC-MS/MS. However, “undersampling” will occur, that is, only a subset of the peptides will be identified, if the complexity of these tryptic digests exceeds the analytical capacity of the mass spectrometer (e.g., when more peptides elute from the HPLC column per unit time than can be analyzed, or low-abundance peptides are below the instrument detection limit).7,8
A common observation when complex proteomes are analyzed is that the lists of proteins identified are often quite variable, which has raised concerns about the general value of proteomics for most applications, if it actually does have poor reproducibility. Typically, the identifications of abundant proteins are reproducible, while most of the variability is observed in lower-abundance proteins. A major factor contributing to this low reproducibility is undersampling as discussed above. One strategy of achieving more comprehensive proteome coverage actually exploits this variability by simply performing repetitive LC-MS/MS analyses on each sample.9,10 Other strategies for increasing the number of proteins identified include prefractionating the proteome based on protein physical properties,11,12 improving separation of the peptide mixture at the HPLC separation step,13−16 modifying the ESI interface to enhance ionization,17,18 or adding an ion mobility spectrometry (FAIMS) interface before the ion trap.(19) Proteome coverage also can be improved at the data analysis stage by using high-mass-accuracy data(20) or improved data-process algorithms.21−23 Other strategies include a two-step method that reanalyzes the same sample with an inclusion list (eliminating redundant precursor selection in the second analysis)24,25 and a “replay run” that analyzes a sample twice from a single injection with targeted analysis of undersampled features in the replay run.(26) Although each of these methods can achieve increased sensitivity, it is difficult to assess the relative importance of strategies that have not been directly compared in a single study due to variations in samples, study design, mass spectrometry platforms, data-analysis strategies used, and other laboratory-to-laboratory variations. Consequently, a number of direct comparisons of alternative analysis strategies have been conducted to identify the best analysis approaches for specific types of samples such as serum and cell or tissue lysates.27,28
Factors that should be considered when comparing alternative analysis strategies are the amount of total protein required for the analysis and the total amount of mass spectrometry instrument time required by each method. It is now well-established that repetitive LC-MS/MS analyses, longer HPLC gradients, or additional fractionation—via either more fractions or more modes of separation of complex proteomes—will usually confer some benefit in terms of depth of analysis. The key factor is to determine the most time- and resource-effective strategies for achieving a high depth of analysis, as these factors directly affect sample throughput and proteome analysis cost.
In this study, we compared the effects of two commonly used strategies: GeLC-MS/MS with repetitive LC-MS/MS analysis and additional protein prefractionation and assessed their effects on depth of analysis and reproducibility using cancer cell lysates. Neither strategy requires expensive special hardware or reagents, and both have been integrated into diverse analyses of complex proteomes by multiple laboratories. Because of its simplicity, repetitive analysis often has been used to increase protein identifications after different fractionation schemes,29,30 most of which involve two dimensions of separation at either the peptide or protein levels, such as Mud-PIT or GeLC. Utilizing a cell extract of the human metastatic melanoma cell line 1205Lu,(31) the GeLC-MS/MS analysis with and without repetitive injections was compared and these data sets were compared to a 3-D method that included the prefractionation of the cell lysate using microscale solution isoelectrofocusing (MicroSol IEF) prior to the SDS-PAGE separation.32−34 To balance the total number of LC-MS/MS runs and total instrument time between the repetitive 2-D method and the 3-D method, the 20 fractions from the SDS-PAGE fractionation each were analyzed four times and the cumulative result was compared with the 80-fraction proteome analysis using the 3-D approach. Other factors such as HPLC gradient, mass spectrometry method, and data processing were kept uniform to demonstrate clearly the relative effects of repetitive analyses versus an additional prefractionation step at the protein level. These comparisons showed excellent reproducibility between the methods, and substantially improved proteome coverage using the 3-D method compared with the repetitive analysis of fractions from the 2-D method.
Materials and Methods
Materials
RPMI-1640 cell culture medium, SYPRO Ruby stain, and NuPAGE precast gels were from Invitrogen Corporation, Carlsbad, CA. Ultra pure urea, thiourea, DTT, and CHAPS were from GE Healthcare, Ltd., Giles, U.K. The Bradford protein assay kit was from Thermo Fisher Scientific, Waltham, MA, and sequencing-grade porcine trypsin was from Promega Corporation, Madison, WI. The 1205Lu human melanoma cell line was a gift from Dr. Meenhard Herlyn at the Wistar Institute. All other reagents were from Sigma-Aldrich, St. Louis, MO.
Preparation of Cell Lysates
The human melanoma 1205Lu cell line was maintained in RPMI-1640 medium supplemented with 10% fetal calf serum. Cells were harvested on ice by scraping in cold phosphate-buffered saline (PBS) with inhibitors (0.3 mM PMSF, 2 mM Sodium orthovanadate, 50 mM sodium fluoride, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin) when 70−90% confluence was reached. The tissue culture plates then were washed three times in cold PBS with inhibitors; the washes were collected, combined with the initial scraped cells, collected by centrifugation, and the cell pellet was quick frozen in liquid nitrogen followed by storage at −80 °C. Cell lysates were prepared using 800 μL of lysis buffer (25 mM Tris, pH 8.0, 8 M urea, 2 M thiourea, 4% CHAPS, 1 mM DTT) per cell pellet collected from a 15-cm tissue culture dish. The lysate was sonicated with a probe sonicator on ice with a duty cycle setting of 50% and an output level <5%. The sample was chilled for 1 min for each 20-s sonication. Insoluble material was removed by centrifugation at 100 000g for 60 min and was estimated to be <1% of the total protein based on staining intensity of Colloidal Coomassie SDS gels. The supernatant was reduced using DTT and alkylated with N,N-dimethylacrylamide (DMA), as previously described.(11) Total protein in the supernatant was measured using the Bradford method.
MicroSol IEF Fractionation
Cell lysates (1.5 mg) were separated into four pH ranges by MicroSol IEF32−34 using a ZOOM IEF Fractionator (Invitrogen Corporation, Carlsbad, CA). For these separations, the fractionator contained five immobiline gel disks, each sustaining a pH of 3.0, 5.4, 6.2, 7.0, and 12, respectively. These disks delineated four separation chambers and the cell lysate in a sample buffer consisting of 8 M urea, 2 M thiourea, 4% CHAPS, 1% DTT, 1% pH 3−7 ZOOM focusing buffer, and 1% pH 7−12 ZOOM focusing buffer was loaded into the middle two separation chambers. The outer chambers were loaded with sample buffer only and standard electrode solutions were used. Samples were focused at a constant 750 V and a maximum of 1 mA for 3 h. The solution in each sample chamber was removed and measured. Each chamber was rinsed with a small volume of sample buffer so that, when combined with the original solution removed from that chamber, the final volume was 700 μL. Proteins trapped in the immobiline gel disks were recovered using two sequential 175 μL extractions for 30 min each using 10 mM Tris, pH 8, 1% SDS for 30 min.
1-D SDS-PAGE
The cell lysate and MicroSol IEF fractions were initially analytically separated on 10% NuPAGE minigels (with MES running buffer) using standard separation conditions. Preparative SDS-PAGE was run using unfractionated cell lysate and the four MicroSol IEF fractions in an analogous manner to the analytical run, except the separation distance was limited to 40 mm to minimize required numbers of fractions and gel volume per fraction. Typically, six lanes—two lanes from the GeLC (2-D) method containing 60 μg each of cell lysate and one lane each from the 3-D method—were sliced into 4 mm × 1 mm × 1 mm thick gel pieces. Two adjacent pieces in each lane were combined in a digestion well and were digested overnight with trypsin. Corresponding digestions from the duplicate 2-D method lanes were combined, generating a total of 20 digestions for the 2-D method. The 3-D method had four fractions of 20 digestions each, totaling 80 digestions.
LC-MS/MS
Trypsin digestions were injected into a 75 μm i.d. × 15 cm PicoFrit (New Objective, Inc., Woburn, MA) column packed with 5 μm Magic C18 resin and peptides were separated by nano-HPLC (Eksigent Technologies, Dublin, CA) interfaced with a LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). For each analysis, 8 μL of trypsin digest was loaded onto the column using solvent A (0.1% formic acid in Milli-Q [Millipore Corporation, Billerica, MA] water). Peptides were subsequently eluted using the following gradient conditions with 0.1% formic acid in acetonitrile as solvent B: 1−28% B over 42 min, 28−50% B over 25.5 min, 50−80% B over 5 min, and 80% B for 5 min. To minimize carryover, a 25 min blank injection was run between each sample. Twenty 2-D samples were analyzed first, followed by another replicated injection of the same 2-D samples. Then, 80 3-D samples were analyzed, followed by the third and fourth replicated injections of the 2-D samples. The LTQ mass spectrometer was operated with dynamic exclusion enabled for 30 s, full scans from 400−2000 m/z, and data-dependent MS/MS analysis on the six most intense ions.
Data Processing
MS2 data was extracted and searched using the SEQUEST algorithm (Ver. 28, rev. 13, University of Washington, Seattle, WA) in BioWorks (Ver. 3.1, Thermo Fisher Scientific, Waltham, MA), and the human UniRef 100 (Ver. May, 2007) protein database, which was downloaded from the Protein Information Resource at Georgetown University. To generate the decoy database, the protein amino acid sequence for each database entry was reversed and the entire reversed database was appended in front of the original forward sequences. The data were searched against the combined forward/reverse database using partial trypsin specificity with a 2.5 Da precursor mass tolerance and 1 Da fragment ion mass tolerance. Consensus protein lists were generated by DTASelect (Ver 1.9, licensed from Scripps Research Institute, La Jolla, CA) after applying the following filters: full tryptic boundaries, XCorr ≥ 1.8 (z = 1), 2.1 (z = 2), 3.25 (z = 3), ΔCn ≥ 0.05. Custom software was then used to ensure that each unique peptide sequence was used only once in assembling the protein list. To identify common and unique proteins found by 2-D and 3-D methods, protein and peptide data were put into a relational database (MySQL) and matched using custom software.
Results
Experimental Strategy
The strategy used to compare (1) the conventional GeLC-MS/MS method (protein separation by SDS gel + reverse-phase peptide separation), (2) GeLC-MS/MS with repetitive injections, and (3) a 3-D method that used MicroSol IEF fractionation prior to the SDS-PAGE step (protein IEF + SDS gel + reverse-phase peptide separation) is summarized in Figure 1. To directly compare the relative impact on proteome coverage of repetitive injections versus an additional protein separation step, equal amounts of the same cell lysate were fractionated and an equal number of LC-MS/MS analyses were used. Similarly, all other experimental parameters were maintained as constant as possible.
Figure 1.

Experiment outline. A cell lysate of 1205Lu cell line was processed in such a manner that each common step was comparable. Details of the methods are described in the Materials and Methods. The 2-D method consisted of SDS-PAGE and LC-MS/MS. The same samples were reanalyzed another three times as a repetitive analysis strategy. The 3-D method consisted of MicroSol IEF, SDS-PAGE, and LC-MS/MS. Subsequently, the final minimum consensus protein lists from the three data sets were compared to evaluate the ability of enhancing sensitivity of repetitive analysis and additional fractionation strategies.
MicroSol IEF Prefractionation
MicroSol IEF initially was developed to prefractionate complex proteomes prior to narrow pH range 2-D gels(33) and was subsequently used prior to 1-D SDS-PAGE as part of a 4-D strategy for fractionating serum or plasma.(11) In this study, we evaluated its utility for prefractionating cell lysates prior to GeLC-MS/MS, since this separation mode is orthogonal to SDS-PAGE and does not suffer from many of the limitations of 2-D gels. Because the dynamic range of protein concentrations in cell lysates is substantially less than in plasma or serum, the MicroSol IEF separation was designed to yield four final fractions rather than the larger number of fractions typically used for plasma. The immobiline membrane disks between chambers were extracted because, as shown previously, proteins with pI values close to the pH of the disk remain in the disk but can be recovered in high yield.32−34 Typical results for fractionation of a melanoma cell lysate are shown in Figure 2. Extracts from disks with extreme pH values (3.0 and 12) showed only trace amount of proteins, as few proteins have pI’s near these values. Extractions from disks located between separation chambers contained a mixture of proteins unique to that disk and proteins present in adjacent chambers as previously observed.32−34 When similar samples were separated by MicroSol IEF and analyzed on 2-D gels, comparisons of solution fractions and membrane fractions showed that most proteins recovered from membrane disks were either unique to the membrane or had pI’s slightly lower than the pH of the membrane (data not shown). Since the goal in this experiment was to fractionate the entire proteome into a small number of fractions at the MicroSol IEF step, disk extracts were combined with the fraction to its right to maximize recovery (Figure 2). The extraction from the pH 12 disk was discarded as it contained a negligible amount of protein. Thus, after MicroSol IEF fractionation, the cancer cell proteome was divided into four fractions of roughly similar complexity and with a simpler protein content.
Figure 2.

Separation of the melanoma 1205Lu cell extract by MicroSol IEF and pooling to produce four fractions. Equal portions of each fraction and separation membrane disk extract were analyzed by SDS-PAGE to evaluate separation and relative amounts of total protein in each fraction. The solution recovered from individual chambers was pooled with the adjacent membrane disk extract on the low pH side of the pool, as shown at the bottom of the gel, to produce four fractions. The extract from the pH 12 membrane disk had negligible protein and was not used further.
GeLC-MS/MS Analysis
For the conventional GeLC-MS/MS method, 60 μg of the original cell lysate, which was close to a maximum load while avoiding band distortion, was loaded into each of two lanes of a NuPAGE gel (Figure 3A). Proteins were separated until the tracking dye migrated 40 mm. After staining the gel with Colloidal Coomassie, the 40-mm lane was divided into 20 equal fractions and digested with trypsin. Digests from corresponding positions in the two replicate lanes were combined to yield sufficient volume for four 8 μL injections. Hence, 120 μg of total cell lysate was fractionated into 20 fractions.
Figure 3.

SDS-PAGE separation of melanoma 1205Lu cell lysate and MicroSol fractions for proteome analysis. Samples were electrophoresed until the tracking dye migrated 4 cm; gels were stained with Colloidal Coomassie and individual lanes were cut into 20 equal-sized slices, as shown. (A) For the 2-D method, the unfractionated lysate of the 1205Lu cells was separated in two lanes (60 μg/lane). The supernatants from corresponding slices in the two lanes were combined after trypsin digestion. (B) MicroSol IEF fractions derived from 120 μg of cell lysate were separated for the 3-D method.
For the 3-D method, the amount of each fraction loaded onto the preparative gel was the protein recovered from 120 μg of total cell lysate (Figure 3B). Hence, the total amount of protein in the four gel lanes in Figure 3B should be close to the amount of protein in the two lanes in Figure 3A, provided sample losses during the MicroSol IEF procedure were low, as has been previously demonstrated.32−34 As noted above, the gel lanes from the MicroSol fractions were cut and digested in the same manner as the gel slices from Figure 3A, except in this case there were no duplicate gel lanes to be combined. HPLC and mass spectrometer performance were carefully monitored to ensure consistent performance through these experiments. To further minimize effects of minor instrument performance variations, samples were injected in an order described in Materials and Methods. Consistent performance of the autoinjector was monitored by weighing each sample vial before and after injection. There was no significant difference in the injection amount between the two methods (t-test p > 0.05).
Comparison of Peptide and Protein Counts between the 2-D, 2-D/Repetitive Runs, and 3-D Methods
All data were consistently analyzed and filtered as described in Materials and Methods, which resulted in estimated false-positive rates (FPR) calculated by dividing reverse-hit peptide counts by forward-hit peptide counts of 1.6% and 1.4% for 2-D and 3-D data, respectively. Figure 4 shows the nonredundant peptide and protein counts for the 3-D data set as well as differing numbers of replicates for the 2-D sample set. Similar numbers of peptide and protein identifications were obtained for all individual 2-D data sets (data not shown). As expected, the total number of nonredundant peptides increased moderately as additional replicate data sets were combined, and the incremental increase diminished as each new replicate was added. It was evident from the curve shown in Figure 4A that the data set resulting from combining four replicate runs was approaching a plateau. Parallel trends were observed for the peptide data sets defined by proteins identified by ≥3, 2, or 1 peptide(s). A total of 25 641 nonredundant peptides were identified after combining all four replicates of the 2-D analysis, which was 26% less than the 32 216 peptides identified by the 3-D method.
Figure 4.

Comparison of peptide and protein coverage for the 2-D and 3-D methods. (A) Nonredundant peptide counts from a single 2-D analysis, combined data from increasing numbers of replicate analyses, and the 3-D method. (B) Corresponding nonredundant protein counts for the same data sets as shown in panel A. The total number of LC-MS/MS runs that each data set contains is shown at the bottom of the figure.
Protein counts showed a similar trend as the peptide counts (Figure 4B), that is, adding a second replicate increased the number of nonredundant proteins more markedly than adding a third and fourth replicate. For example, the number of proteins identified by two or more peptides increased by 594, 305, and 162 when two, three, and four replicates were combined. The most appropriate comparison of the 3-D and 2-D methods is between the 3-D data set and four replicates of the 2-D samples because they both involve a total of 80 LC-MS/MS runs using equal amounts of initial cell lysate. When protein identifications based on two or more peptides were counted, the 3-D method identified 3486 proteins compared with 2850 proteins for the 2-D method. These 636 additional proteins (22.3% increase) indicate a clear advantage of the 3-D method compared with the 2-D/replicate run method, even when equal amounts of mass spectrometer time are utilized.
Comparisons of the Proteomes Identified by the 2-D and 3-D Methods
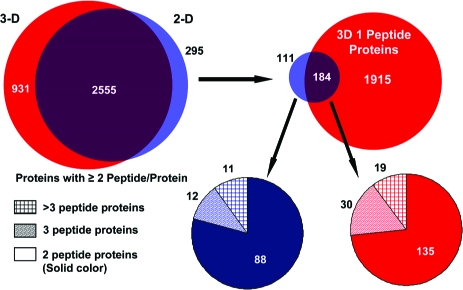
The proteomes produced by the 2-D/four repetitive-run and the 3-D methods were compared using the lists of proteins identified by two or more peptides. This comparison was facilitated by the fact that both data sets were searched using the same database and that DTASelect lists all protein names identified in the search. If a protein identification carried one protein name that could be found in the other data set, we deduced that both data sets had identified the same protein. This comparison at the protein name level showed extensive overlap between the two data sets even at this first level comparison (Figure 5). Specifically, the two data sets shared 2555 proteins, which corresponded to 90% of the smaller 2-D/repetitive-run proteome. The 295 proteins that appeared to be unique to the less in-depth 2-D/replicate run data set were further investigated to determine the basis for this apparent lack of reproducibility between in-depth analyses of the same biological sample. Further analysis showed that 184 of these proteins were present in the 3-D data set as single-hit proteins. A majority (135) of these 184 proteins were two-peptide proteins in the 2-D data, indicating that the 3-D method identified these proteins by one less peptide. When the unfiltered data for the 3-D proteome was examined using randomly selected proteins in this group, we found that a substantial number of these proteins had been identified by the same second peptide as in the 2-D data set, but one or more of the filtering parameters was slightly lower than the cutoff values selected for global data filtering. As a result, these moderate changes in scoring parameters reduced the two- or three-hit protein identification in the 2-D data set to a 1-hit protein in the 3-D data set. Three examples are shown in Table 1. We also further evaluated the 111 proteins found exclusively in the 2-D method. More than three-quarters (88) of them were two-peptide proteins. Among these identifications, the 11 proteins identified by more than three unique peptide sequences were a particular concern, as the number of peptide identifications suggested that these proteins were not near the detection threshold and should have been identified in the more in-depth 3-D proteome. We found that the majority of the peptide sequences associated with these 11 proteins in the 2-D data set also existed in the 3-D data set, but they were assigned to highly homologous proteins (Supplemental Table 1). Although a large number of common sequences for these proteins were observed in both data sets, a small number of unique sequences led the DTASelect program to assign these peptides to different proteins for each data set. Of the 385 peptides assigned to the 111 proteins unique to the 2-D/replicate run data set, 226 peptides were present in the filtered 3-D data.
Figure 5.

Overlap of identified proteins between the 2-D repetitive and 3-D methods. The two methods identified 2555 common proteins with two or more peptides per protein (90% of the total proteins identified in the smaller 2-D/replicate-run data set). Of the 295 apparently unique proteins in the 2-D method, 184 proteins were identified in the 3-D data set by one peptide. The pie charts in the lower panels show the number of peptide hits for the 2-D method for the proteins that were not directly identified in the 3-D data set (111 proteins or 3.9% of the proteins in the 2-D data set) and those identified by a single peptide in the 3-D data set (183 proteins or 6.5% of the proteins in the 2-D data set).
Table 1. Examples of Proteins Identified in the 2-D/Replicate Data Set by ≥2 Peptides but Not in the 3-D Data Set.
| 2-D methodb |
3-D methodbc |
|||||||
|---|---|---|---|---|---|---|---|---|
| IDa | Name | Sequence | Xcorr | ΔCn | z | Xcorr | ΔCn | z |
| Q53FT3 | Uncharacterized protein C11orf73 | R.LVQTAAQQVAEDK.F | 4.74 | 0.35 | 2 | 4.62 | 0.35 | 2 |
| R.LAQNPLFWK.T |
2.69 | 0.185 | 2 | 2.11 | 0.046 | 2 | ||
| Q8IW35 | Leucine-rich repeat and IQ domain-containing protein 2 | K.AGLLPCPEPTIISAILK.D | 3.39 | 0.129 | 3 | 2.94 | 0.314 | 2 |
| K.LPMILTQR.S |
2.57 | 0.051 | 2 | 1.98 | 0.091 | 2 | ||
| O95563 | Brain protein 44 | R.TVFFWAPIMK.W | 2.87 | 0.254 | 2 | 3.08 | 0.263 | 2 |
| K.LRPLYNHPAGPR.T |
3.54 | 0.283 | 3 | 3.12 | 0.328 | 3 | ||
| K.WGLVCAGLADMAR.P | 2.87 | 0.254 | 2 | - | - | - | ||
These proteins, which were identified by two or three peptides in the 2-D/replicate data set, were identified by a single peptide in the 3-D data set. In each case, a second peptide was detected in the 3-D data set but failed to pass the data filter cutoff due to slightly lower values for Xcorr or ΔCn (bolded values in table).
The filtering cutoff values used were Xcorr ≥ 1.8 (+1); 2.1 (+2); 3.25 (+3), ΔCn ≥ 0.05.
The bold values indicate they are below cutoff value. “-” indicates “not found.”
Sequence Coverage Comparisons of Putative Low-Abundance Proteins
To a first approximation, those proteins identified in the 2-D/repetitive-runs data set by only two or three peptides can be regarded as likely low-abundance proteins in the sample analyzed, since sequence coverage is usually a rough indicator of protein abundance level. The relative depth of coverage for these putative low-abundance proteins was compared in the 2-D/repetitive-run and the 3-D methods. As shown in Figure 6, for those proteins identified in both data sets, the majority showed greater sequence coverage in the 3-D data set. That is, of the 491 common proteins identified by two peptides in the 2-D data, the 3-D method found more peptides for 317 proteins (64.6%) and the remaining proteins in this group were identified with an equal number of peptides. Furthermore, in 114 cases, the 3-D method found at least three additional unique peptides, indicating a substantially greater depth of analysis. Of the 394 common proteins identified by the 2-D method with three peptides, 325 proteins (82.5%) were identified by an equal or larger number of peptides in the 3-D method. These data clearly indicate that, in most cases, the 3-D method had the ability to detect more peptides for low-abundance proteins than did the 2-D method.
Figure 6.

Comparison of the number of peptides identified in the 2-D/repetitive and 3-D methods. (A) Among proteins common to both data sets, 491 proteins were identified by two peptides in the 2-D/repetitive data set. The 3-D method found an equal number of peptides for 174 proteins and more peptides for 317 proteins. (B) Among 394 proteins identified with three peptides by the 2-D method, the 3-D method found one less peptide for 69 proteins (i.e., +1 for 2-D), an equal number of peptides for 99 proteins, and more peptides for 226 proteins.
Discussion
In this study, we systematically evaluated the relative merits of repetitive LC-MS/MS runs compared with introduction of an additional protein level separation step for increasing proteome coverage of cancer cell lysates. Factors that affect apparent reproducibility between proteome analyses performed on the same sample also were evaluated. The basic analysis platform used for repetitive analyses was the commonly utilized GeLC-MS/MS method, which can be considered to be a 2-D proteomics method as it involves two dimensions of separation, that is, protein separation using SDS-PAGE and reverse-phase HPLC separation of tryptic peptides. This method was compared to a 3-D method consisting of solution IEF at the protein level followed by the GeLC-MS/MS method.
It is important when comparing alternative analysis platforms to consider the total number of LC-MS/MS runs per proteome, because improved proteome coverage typically can be achieved by lengthening the HPLC gradient or by repeating LC-MS/MS analysis of complex samples.9,10 Similarly, many separation modes prior to LC-MS/MS can be at least incrementally improved by simply increasing the number of fractions collected, provided that the resolution of the separation method exceeds the initial fraction size used. But in some cases, increases in protein coverage may be too small to be considered advantageous when total analysis time per proteome is considered. Hence, evaluation of the merits of greater depth of analysis, particularly small improvements, must be constantly weighted relative to overall throughput. Furthermore, total mass spectrometer instrument time frequently is the limiting resource, and when fractionation prior to the LC-MS/MS step is used, it is the rate-limiting step in proteome analysis throughput. Hence, the most meaningful comparisons are those where the total mass spectrometer analysis time per proteome is held constant. In this study, we used a consistent gradient time and 80 LC-MS/MS runs for both the 3-D method and the 2-D/repetitive-run method (four repeat injections). Similarly, all other experimental variables were held as constant as possible, including use of replicate aliquots of a single cell lysate preparation, gel separation lengths, gel volumes per trypsin digestion reaction, instrument tuning, and data-analysis methods. Our goal was to determine, quantitatively, which method represents the more efficient utilization of mass spectrometer time when analyzing complex proteomes.
Robust proteome analysis methods should be reproducible in addition to identifying the majority of proteins present in a biological sample. One major cause of variations in proteins identified in replicate analyses of the same proteome is undersampling in the mass spectrometer, as discussed above. Therefore, high proteome coverage should be linked to good reproducibility of proteome analysis results because extensive proteome coverage will occur only if undersampling is minimized. A second factor that will contribute to poor reproducibility between proteome protein lists is use of data filtering conditions that result in high false-peptide and protein-identification rates, since false positives usually are random. Hence, data filtering stringency is another tradeoff that must be considered when selecting a proteome analysis strategy. While low stringency filters contribute to noise and low reproducibility, excessively stringent filters will greatly diminish the number of protein identifications and hence the value of the experiment. In the current study, data filters were used that yielded peptide false-positive rates between 1 and 2% as estimated using a decoy reverse database, thereby minimizing apparent poor reproducibility between data sets. This level of stringency results in very few false positives for proteins identified by two or more peptides and, while there are some false positives within the one-hit protein list, a majority of these identifications are correct.
The repetitive analyses of 2-D data showed increased peptide and protein counts (Figure 4) indicative of undersampling in the basic GeLC-MS/MS method used here. The overall gain from four repetitive analyses of proteins identified by two or more peptides was 1061 (59%) compared to the initial single analysis. A similar increase (61%) was observed in repetitive MudPIT using nine analyses.(10) As expected, the greatest positive impact on proteome coverage was use of a second replicate run, which increased the number of proteins identified by two or more peptides by 33% while doubling instrument time. In contrast, adding a third and fourth replicate only increased protein coverage by 13% and 6%, respectively. These data indicate that performing a second analysis of each fraction when using GeLC-MS/MS would be a positive tradeoff between instrument time and protein coverage. However, further doubling of instrument time by performing four repetitive runs is unlikely to represent optimal use of instrument time for most types of experiments.
Of course, an alternative to performing a repetitive analysis of gel fractions would be to obtain more slices per gel lane. In analogous experiments where gel lanes were divided into 40 or 60 fractions, we observed increases in the number of proteins identified that were similar to those obtained in this study for duplicate and triplicate analyses of the 20 fractions per gel lane (data not shown). Although producing more fractions per gel lane increases the number of in-gel digestions, the overall increase in total analysis time for a proteome is minor. Hence, we generally prefer to use more fractions per lane rather than to replicate analyses when greater depth of analysis is desired using GeLC-MS/MS experiments. Longer gels and larger numbers of fractions per lane were not used in the current study because we wanted to keep total mass spectrometer time per proteome (approximately 160 h per proteome) within practical limits while simultaneously matching gel lengths, gel volumes, and other parameters. That is, extrapolating from other 2-D and 3-D experiments that we have performed, we expect that the total number of proteins for each data set (2-D, 2-D/repetitive runs, and 3-D) would have increased moderately if we would have used 40 or 60 slices per gel lane for all samples. But use of 40 or 60 fractions per gel lane would have increased total instrument time to about 320 and 480 h per proteome, which represents an impractically low throughput for most studies. Interestingly, as we increase the number of fractions per gel lane to 40 or 60 fractions, the incremental increases in new proteins identified diminish, analogous to the diminishing benefits of adding each additional replicate in the repetitive-run approach (Figure 4A). Although similar trends are observed for these two approaches, the mechanisms for increasing protein coverage are quite different. That is, using a larger number of gel fractions increases protein separation and simplifies the mixture of proteins present in each fraction, while repetitive runs exploit subtle variations in peptide separations in replicate HPLC runs and subtle variations in data-dependent selection of low-level ions for MS/MS fragmentation and analysis.
The 3-D method clearly provided superior protein and peptide coverage compared with the 2-D/repetitive method, which indicates that adding an additional protein separation step represents a more efficient use of mass spectrometer instrument time. This method identified 3486 proteins with two or more peptides, which is 22% more than the 2-D/repetitive method that used equal instrument time. At the peptide level, the 3-D method identified 30 385 high-confidence, nonredundant peptides, which is nearly 2.5 times more than that found in a single survey using the 2-D method (12 160) and 28% more than the cumulative count in four repetitive analyses (23 648). Furthermore, more unique peptides were found for most low-abundance proteins in the 3-D method data compared with the cumulative 2-D method data (Figure 6).
It is not surprising that adding solution IEF as an additional orthogonal protein separation step to a GeLC-MS/MS method is an efficient strategy for increasing proteome coverage and sequence coverage of lower-abundance proteins. MicroSol IEF separates the proteins that would normally be in a single gel slice into four gel slices (see Figure 3). Consequently, full-scan spectra were simplified, thereby minimizing undersampling. In addition, the simpler samples should decrease ion suppression effects and reduce dynamic range within each digest. Finally, in some cases, improved scores for MS2 spectra in the 3-D method probably resulted from a lower probability of interfering ions being isolated with the target ion for fragmentation. Thus, more peptides passed the data-filtering criteria as true positive identifications. While the repetitive analysis strategy also improved proteome coverage, it had neither a built-in mechanism to reduce repeated sampling of abundant ions between replicates, nor could it explore the ions below the MS2 triggering threshold. An alterative technique that has sometimes been used to improve replicate runs is to scan different mass ranges in each replicate. However, pilot experiments suggested that this approach was less productive than the simple repetitive analysis method used here.
One frequent criticism of proteomics methods is that the proteins identified on repeat analyses often are not very reproducible. A recent study suggested good reproducibility was achievable across 27 laboratories on a simple 20-protein mixture after uniform data processing was used.(35) But this simple sample of abundant proteins at the same concentration does not reflect real biological complexity. Hence, in the current study, we compared the reproducibility between different analysis methods using a very complex sample of biological interest, that is, a human cancer cell lysate. Among four replicate analyses of the 2-D samples, 1500−1600 proteins were shared between them (Supplemental Figure 1). This indicated at least 76% of the proteins observed in one analysis were reproducibly detected despite significant undersampling. More importantly, at least 90% of the proteins observed in the 2-D/four-replicate data set based on two or more peptides directly matched to a corresponding protein 3-D data set, and most of the apparent mismatches were caused by trivial data analysis issues.
A more rigorous comparison of the two comprehensive data sets showed that greater than 96% of the proteins identified in the 2-D/repetitive-run proteome were actually observed within the complete 3-D data set. One reason for the initially apparent, lower reproducibility when protein names were compared was slight variations in peptides scores together with use of rigid data filter cutoff values (see above and Figure 5). That is, the 10% of proteins that were apparently unique to the 2-D/repetitive-run data set included 184 proteins (6.4% of the 2-D/repetitive protein list) that were identified by a single peptide in the 3-D data set. Reasons why these proteins were only identified by a single peptide in the 3-D data set include run-to-run variations in automated selection of low-abundance signals for MS/MS and run-to-run variations in SEQUEST scores coupled with use of rigid data filters. The latter case appears to occur frequently as described above. A second contributing factor to the initially apparent, lower reproducibility at the protein list level is database redundancy and limitations of current software for consistently producing consensus protein lists from identified peptides. Of the 111 proteins apparently unique to the 2-D/repetitive-run data set and not identified by a single-hit protein in the 3-D data set, most were highly homologous to proteins identified in the 3-D data set (see Results and Supplemental Table 1). Among the 385 peptides belonging to the 111 unique proteins in the 2-D data, only 159 peptides were not found in the 3-D filtered data. Although these 111 unique proteins comprised 4% of all proteins identified, the 159 unique peptides were only 0.7% of all 2-D filtered peptides. This illustrates that very small variations in identified peptides can have a proportionally higher “apparent” impact on variations in identified proteins. Since we used each unique peptide a single time during assembly of consensus protein lists, the common sequences were assigned to the protein with the most unique sequences. Consequently, for a group of proteins that have high sequence identity, one or two unique peptides could determine which protein in the protein family emerged in the final consensus protein list. This illustrates that better software tools are needed for identifying and displaying putative unique proteins within protein families. Similarly, improved databases with uniform names or other labels that clearly indicate membership within a protein family would be beneficial.
In conclusion, additional prefractionation with MicroSol IEF substantially increased proteome coverage and sequence coverage compared with a GeLC-MS/MS-repetitive run method that utilized an equal amount of mass spectrometer time. Furthermore, the reproducibility of protein lists between the two methods was quite high because undersampling during data acquisition had been minimized. Most of the apparent differences in protein identifications were due to limitations of current sequence databases and protein naming conventions, as well as software limitations for filtering database search results and building consensus protein lists.
Acknowledgments
This work was supported by National Institutes of Health Grants CA120393 and CA94360, as well as institutional grants to the Wistar Institute, including an NCI Cancer Core Grant (CA10815), and grants from the Pennsylvania Department of Health. We thank Ms. Mea Fuller for her assistance in preparing the manuscript.
Supporting Information Available
Venn diagram comparing reproducibility in 2-D samples; table identifying closely related protein isoforms; list of proteins identified by two or more peptides using the 2-D/repetitive method; and list of proteins identified by two or more peptides using the 3-D method. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cravatt B. F.; Simon G. M.; Yates J. R. III. The biological impact of mass-spectrometry-based proteomics. Nature 2007, 450 (7172), 991–1000. [DOI] [PubMed] [Google Scholar]
- Pasini E. M.; Kirkegaard M.; Salerno D.; Mortensen P.; Mann M.; Thomas A. W. Deep coverage mouse red blood cell proteome: a first comparison with the human red blood cell. Mol. Cell. Proteomics 2008, 7 (7), 1317–30. [DOI] [PubMed] [Google Scholar]
- Dieguez-Acuna F. J.; Gerber S. A.; Kodama S.; Elias J. E.; Beausoleil S. A.; Faustman D.; Gygi S. P. Characterization of mouse spleen cells by subtractive proteomics. Mol. Cell. Proteomics 2005, 4 (10), 1459–70. [DOI] [PubMed] [Google Scholar]
- Omenn G. S.; States D. J.; Adamski M.; Blackwell T. W.; Menon R.; Hermjakob H.; Apweiler R.; Haab B. B.; Simpson R. J.; Eddes J. S.; Kapp E. A.; Moritz R. L.; Chan D. W.; Rai A. J.; Admon A.; Aebersold R.; Eng J.; Hancock W. S.; Hefta S. A.; Meyer H.; Paik Y. K.; Yoo J. S.; Ping P.; Pounds J.; Adkins J.; Qian X.; Wang R.; Wasinger V.; Wu C. Y.; Zhao X.; Zeng R.; Archakov A.; Tsugita A.; Beer I.; Pandey A.; Pisano M.; Andrews P.; Tammen H.; Speicher D. W.; Hanash S. M. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005, 5 (13), 3226–45. [DOI] [PubMed] [Google Scholar]
- Chen E. I.; Hewel J.; Felding-Habermann B.; Yates J. R. III. Large scale protein profiling by combination of protein fractionation and multidimensional protein identification technology (MudPIT). Mol. Cell. Proteomics 2006, 5 (1), 53–6. [DOI] [PubMed] [Google Scholar]
- Shi R.; Kumar C.; Zougman A.; Zhang Y.; Podtelejnikov A.; Cox J.; Wisniewski J. R.; Mann M. Analysis of the mouse liver proteome using advanced mass spectrometry. J. Proteome Res. 2007, 6 (8), 2963–72. [DOI] [PubMed] [Google Scholar]
- States D. J.; Omenn G. S.; Blackwell T. W.; Fermin D.; Eng J.; Speicher D. W.; Hanash S. M. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat. Biotechnol. 2006, 24 (3), 333–8. [DOI] [PubMed] [Google Scholar]
- Malmstrom J.; Lee H.; Aebersold R. Advances in proteomic workflows for systems biology. Curr. Opin. Biotechnol. 2007, 18 (4), 378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Jacobs J. M.; Camp D. G. II; Fang R.; Moore R. J.; Smith R. D.; Xiao W.; Davis R. W.; Tompkins R. G. Ultra-high-efficiency strong cation exchange LC/RPLC/MS/MS for high dynamic range characterization of the human plasma proteome. Anal. Chem. 2004, 76 (4), 1134–44. [DOI] [PubMed] [Google Scholar]
- Liu H.; Sadygov R. G.; Yates J. R. III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76 (14), 4193–201. [DOI] [PubMed] [Google Scholar]
- Tang H. Y.; Ali-Khan N.; Echan L. A.; Levenkova N.; Rux J. J.; Speicher D. W. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 2005, 5 (13), 3329–42. [DOI] [PubMed] [Google Scholar]
- Nissum M.; Kuhfuss S.; Hauptmann M.; Obermaier C.; Sukop U.; Wildgruber R.; Weber G.; Eckerskorn C.; Malmstrom J. Two-dimensional separation of human plasma proteins using iterative free-flow electrophoresis. Proteomics 2007, 7 (23), 4218–27. [DOI] [PubMed] [Google Scholar]
- Washburn M. P.; Wolters D.; Yates J. R. III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19 (3), 242–7. [DOI] [PubMed] [Google Scholar]
- Motoyama A.; Xu T.; Ruse C. I.; Wohlschlegel J. A.; Yates J. R. III. Anion and cation mixed-bed ion exchange for enhanced multidimensional separations of peptides and phosphopeptides. Anal. Chem. 2007, 79 (10), 3623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte N.; Lasaosa M.; Tholey A.; Heinzle E.; Huber C. G. Two-dimensional reversed-phase × ion-pair reversed-phase HPLC: an alternative approach to high-resolution peptide separation for shotgun proteome analysis. J. Proteome Res. 2007, 6 (11), 4363–73. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Kuromitsu J.; Oda Y. Evaluation of comprehensive multidimensional separations using reversed-phase, reversed-phase liquid chromatography/mass spectrometry for shotgun proteomics. J. Proteome Res. 2008, 7 (3), 1007–11. [DOI] [PubMed] [Google Scholar]
- Kelly R. T.; Page J. S.; Zhao R.; Qian W. J.; Mottaz H. M.; Tang K.; Smith R. D. Capillary-based multi nanoelectrospray emitters: improvements in ion transmission efficiency and implementation with capillary reversed-phase LC-ESI-MS. Anal. Chem. 2008, 80 (1), 143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page J. S.; Tang K.; Kelly R. T.; Smith R. D. Subambient pressure ionization with nanoelectrospray source and interface for improved sensitivity in mass spectrometry. Anal. Chem. 2008, 80 (5), 1800–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canterbury J. D.; Yi X.; Hoopmann M. R.; MacCoss M. J. Assessing the dynamic range and peak capacity of nanoflow LC-FAIMS-MS on an ion trap mass spectrometer for proteomics. Anal. Chem. 2008, 80 (18), 6888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas W.; Faherty B. K.; Gerber S. A.; Elias J. E.; Beausoleil S. A.; Bakalarski C. E.; Li X.; Villen J.; Gygi S. P. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell. Proteomics 2006, 5 (7), 1326–37. [DOI] [PubMed] [Google Scholar]
- Jones A. R.; Siepen J. A.; Hubbard S. J.; Paton N. W. Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics 2009, 9 (5), 1220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen C. Y.; Russell S.; Mendoza A. M.; Meyer-Arendt K.; Sun S.; Cios K. J.; Ahn N. G.; Resing K. A. Improving sensitivity in shotgun proteomics using a peptide-centric database with reduced complexity: protease cleavage and SCX elution rules from data mining of MS/MS spectra. Anal. Chem. 2006, 78 (4), 1071–84. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Ma J.; Dou L.; Wu S.; Qian X.; Xie H.; Zhu Y.; He F. Bayesian nonparametric model for the validation of peptide identification in shotgun proteomics. Mol. Cell. Proteomics 2009, 8 (3), 547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A.; Gehlenborg N.; Bodenmiller B.; Mueller L. N.; Campbell D.; Mueller M.; Aebersold R.; Domon B. An integrated, directed mass spectrometric approach for in-depth characterization of complex peptide mixtures. Mol. Cell. Proteomics 2008, 7 (11), 2138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann M. R.; Merrihew G. E.; von Haller P. D.; MacCoss M. J. Post analysis data acquisition for the iterative MS/MS sampling of proteomics mixtures. J. Proteome Res. 2009, 8 (4), 1870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waanders L. F.; Almeida R.; Prosser S.; Cox J.; Eikel D.; Allen M. H.; Schultz G. A.; Mann M. A novel chromatographic method allows on-line reanalysis of the proteome. Mol. Cell. Proteomics 2008, 7 (8), 1452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker J. R.; Zhang H.; Eng J. K.; Fang R.; Piening B. D.; Feng L. C.; Lorentzen T. D.; Schoenherr R. M.; Keane J. F.; Holzman T.; Fitzgibbon M.; Lin C.; Cooke K.; Liu T.; Camp D. G. II; Anderson L.; Watts J.; Smith R. D.; McIntosh M. W.; Paulovich A. G. Head-to-head comparison of serum fractionation techniques. J. Proteome Res. 2007, 6 (2), 828–36. [DOI] [PubMed] [Google Scholar]
- Slebos R. J.; Brock J. W.; Winters N. F.; Stuart S. R.; Martinez M. A.; Li M.; Chambers M. C.; Zimmerman L. J.; Ham A. J.; Tabb D. L.; Liebler D. C. Evaluation of strong cation exchange versus isoelectric focusing of peptides for multidimensional liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2008, 7 (12), 5286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faca V.; Pitteri S. J.; Newcomb L.; Glukhova V.; Phanstiel D.; Krasnoselsky A.; Zhang Q.; Struthers J.; Wang H.; Eng J.; Fitzgibbon M.; McIntosh M.; Hanash S. Contribution of protein fractionation to depth of analysis of the serum and plasma proteomes. J. Proteome Res. 2007, 6 (9), 3558–65. [DOI] [PubMed] [Google Scholar]
- Chen M.; Ying W.; Song Y.; Liu X.; Yang B.; Wu S.; Jiang Y.; Cai Y.; He F.; Qian X. Analysis of human liver proteome using replicate shotgun strategy. Proteomics 2007, 7 (14), 2479–88. [DOI] [PubMed] [Google Scholar]
- Juhasz I.; Albelda S. M.; Elder D. E.; Murphy G. F.; Adachi K.; Herlyn D.; Valyi-Nagy I. T.; Herlyn M. Growth and invasion of human melanomas in human skin grafted to immunodeficient mice. Am. J. Pathol. 1993, 143 (2), 528–37. [PMC free article] [PubMed] [Google Scholar]
- Zuo X.; Speicher D. W. Comprehensive analysis of complex proteomes using microscale solution isoelectrofocusing prior to narrow pH range two-dimensional electrophoresis. Proteomics 2002, 2 (1), 58–68. [PubMed] [Google Scholar]
- Zuo X.; Speicher D. W. A method for global analysis of complex proteomes using sample prefractionation by solution isoelectrofocusing prior to two-dimensional electrophoresis. Anal. Biochem. 2000, 284 (2), 266–78. [DOI] [PubMed] [Google Scholar]
- Zuo X.; Echan L.; Hembach P.; Tang H. Y.; Speicher K. D.; Santoli D.; Speicher D. W. Towards global analysis of mammalian proteomes using sample prefractionation prior to narrow pH range two-dimensional gels and using one-dimensional gels for insoluble and large proteins. Electrophoresis 2001, 22 (9), 1603–15. [DOI] [PubMed] [Google Scholar]
- Bell A. W.; Deutsch E. W.; Au C. E.; Kearney R. E.; Beavis R.; Sechi S.; Nilsson T.; Bergeron J. J.; HUPO Test Sample Working Group; Beardslee T. A.; Chappell T.; Meredith G.; Sheffield P.; Gray P.; Hajivandi M.; Pope M.; Predki P.; Kullolli M.; Hincapie M.; Hancock W. S.; Jia W.; Song L.; Li L.; Wei J.; Yang B.; Wang J.; Ying W.; Zhang Y.; Cai Y.; Qian X.; He F.; Meyer H. E.; Stephan C.; Eisenacher M.; Marcus K.; Langenfeld E.; May C.; Carr S. A.; Ahmad R.; Zhu W.; Smith J. W.; Hanash S. M.; Struthers J. J.; Wang H.; Zhang Q.; An Y.; Goldman R.; Carlsohn E.; van der Post S.; Hung K. E.; Sarracino D. A.; Parker K.; Krastins B.; Kucherlapati R.; Bourassa S.; Poirier G. G.; Kapp E.; Patsiouras H.; Moritz R.; Simpson R.; Houle B.; LaBoissiere S.; Metalnikov P.; Nguyen V.; Pawson T.; Wong C. C.; Cociorva D.; Yates III J. R.; Ellison M. J.; Lopez-Campistrous A.; Semchuk P.; Wang Y.; Ping P.; Elia G.; Dunn M. J.; Wynne K.; Walker A. K.; Strahler J. R.; Andrews P. C.; Hood B. L.; Bigbee W. L.; Conrads T. P.; Smith D.; Borchers C. H.; Lajoie G. A.; Bendall S. C.; Speicher K. D.; Speicher D. W.; Fujimoto M.; Nakamura K.; Paik Y. K.; Cho S. Y.; Kwon M. S.; Lee H. J.; Jeong S. K.; Chung A. S.; Miller C. A.; Grimm R.; Williams K.; Dorschel C.; Falkner J. A.; Martens L.; Vizcaíno J. A. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat. Methods 2009, 6 (6), 423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.