

Abstract
For many biological investigations, groups of individuals are genetically sampled from several geographic locations. These sampling locations often do not reflect the genetic population structure. We describe a framework using marginal likelihoods to compare and order structured population models, such as testing whether the sampling locations belong to the same randomly mating population or comparing unidirectional and multidirectional gene flow models. In the context of inferences employing Markov chain Monte Carlo methods, the accuracy of the marginal likelihoods depends heavily on the approximation method used to calculate the marginal likelihood. Two methods, modified thermodynamic integration and a stabilized harmonic mean estimator, are compared. With finite Markov chain Monte Carlo run lengths, the harmonic mean estimator may not be consistent. Thermodynamic integration, in contrast, delivers considerably better estimates of the marginal likelihood. The choice of prior distributions does not influence the order and choice of the better models when the marginal likelihood is estimated using thermodynamic integration, whereas with the harmonic mean estimator the influence of the prior is pronounced and the order of the models changes. The approximation of marginal likelihood using thermodynamic integration in MIGRATE allows the evaluation of complex population genetic models, not only of whether sampling locations belong to a single panmictic population, but also of competing complex structured population models.
INVESTIGATIONS using genetic samples from individuals taken across a geographic or biological range—for example, water frogs caught at several ponds, blood samples of humans collected in several villages, or viruses collected from different host species that have the same disease—are common. Whether the individuals studied belong to a single population that is long-term randomly mating or to two or more populations that have varying degrees of genetic isolation from each other is an important concern. Because the geographic information about the locations often does not give a clear indication about the degree of genetic isolation of the individuals, we often use the genetic data themselves to calculate test statistics to suggest whether or not the locations belong to the same population. Many programs (Hudson et al. 1992b; Michalakis and Excoffier 1996; Rousset 1996; Neigel 2002; Weir and Hill 2002; Holsinger et al. 2002) use allele frequencies to calculate FST for pairs of locations or use Fisher's exact test to reject panmixia for the whole or subsets of the data (Raymond and Rousset 1995; Rousset 2008).
Several methods test explicitly whether two populations are or are not panmictic (for example, Hudson et al. 1992a; Rousset 1996). These methods are often applied to all pairs of a multiple-population data set. This is problematic, because both Beerli (2004) and Slatkin (2005) have shown that pairwise analyses can inflate the effective population size estimates, thereby confounding estimators of migration that use the effective number of migrants.
Alternatives to tests based on allele frequencies have been implemented, for example, in the programs STRUCTURE (Pritchard et al. 2000), BAPS (Corander et al. 2008), and STRUCTURAMA (Huelsenbeck and Andolfatto 2007). These methods allow the assignment of individuals to groups using the compatibility of their multilocus genotypes. They can thus be used to group locations into panmictic units on the basis of allele profiles and geography; this capability led to many advancements in landscape genetics and phylogeography. If we are interested in directionality of migration, however, this framework is often insufficient because the assignment methods offer only limited insight into population processes, such as migration, mutation, or fluctuation of population size, that underlie and account for the present genetic structure (Palsbøll et al. 2007).
We describe here another alternative, using Bayesian inference, that calculates probabilities of explicit population models using coalescence theory (a historical review is given by Kingman 2000). An extension of the original n-coalescent of Kingman to multiple populations with migration (Strobeck 1987; Hudson 1991) leads to probabilistic inference programs that consider potentially complex migration patterns among sampling locations (for example, Beerli and Felsenstein 2001; Beerli 2006; Kuhner 2006). The program MIGRATE (Beerli and Felsenstein 2001; Beerli 2006) allows the calculation of a likelihood ratio test (LRT) for nested population models, but these calculations only approximate the LRT (Beerli 2008), need a moderately complicated approach with several independent runs (Beerli 2009), or require time-consuming large-scale simulations (Carstens et al. 2005). In our approach, a Bayes factor (BF) takes the role of an LRT. BFs and LRTs are not equivalent, however: the BF is the ratio of the marginal likelihoods of two hypotheses M1 and M2, whereas the LRT measures support for one hypothesis over another at the maximum likelihood. BFs are better suited for model selection than LRTs because one can compare nonnested as well as nested models. In addition, the programming and the successful application of Bayesian inference programs are often simpler than maximum likelihood (Beerli 2006).
Here, we report on the effect of two different approximations of the marginal likelihood on BF and therefore on the support for specific population models. We provide examples of the use of these methods to extend our tool set for investigating whether sampling locations are part of a panmictic population or are parts of a more complex population structure. Our approach unifies the analysis of population models and allows a wide spectrum of comparisons, from simple tests of whether locations sampled are part of a single population to more complex questions, such as whether there are unambiguous migration directions among populations; it also calculates posterior distributions of parameters of these models.
MATERIALS AND METHODS
Our approach to population model selection uses a framework that allows inferring parameters using coalescence theory. The population models are simple structured coalescence models with possibly many parameters (Beerli and Felsenstein 2001).
Bayes Factor estimation:
In a typical Bayesian inference using Markov chain Monte Carlo (MCMC) methods we do not need to calculate the marginal likelihood to estimate the posterior probability distribution of the parameters of a specific model because the MCMC analysis depends only on likelihood ratios and not absolute likelihoods. Because BF is a ratio of marginal likelihoods of two models, however, calculation of these absolute likelihoods is essential. Because we use absolute likelihoods, we can now easily compare more than two models with the BF framework by choosing a reference model and comparing or ranking other candidate models with that.
We augmented the program MIGRATE (Beerli 2006) with a module to calculate the marginal likelihood
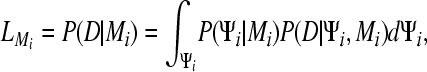 |
(1) |
which is the probability density of the data where the parameters, for example population sizes and migration rates, and nuisance parameters, for example genealogies Ψi, of the model Mi are integrated out using the prior distribution 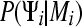 . The marginal likelihood is difficult to estimate with sufficient accuracy because not only the region around the mode, but also the tails of the distribution need to be explored. This is not straightforward in an MCMC context where we bias toward more likely solutions and so have a tendency to sample the tails of the distribution less frequently. The marginal likelihood is calculated in a Bayesian context and needs proper prior distributions to exist. Improper priors would lead to infinitely large tails that do not allow a consistent estimate of the marginal likelihood. We contrast two different methods to estimate the marginal likelihood: harmonic mean (Newton and Raftery 1994; Kass and Raftery 1995) and path sampling (Gelman and Meng 1998). Studies of path sampling have recently led to an alternative method of estimating marginal likelihoods (thermodynamic integration) (Gelman and Meng 1998; Lartillot and Philippe 2006; Friel and Pettitt 2005, 2008).
. The marginal likelihood is difficult to estimate with sufficient accuracy because not only the region around the mode, but also the tails of the distribution need to be explored. This is not straightforward in an MCMC context where we bias toward more likely solutions and so have a tendency to sample the tails of the distribution less frequently. The marginal likelihood is calculated in a Bayesian context and needs proper prior distributions to exist. Improper priors would lead to infinitely large tails that do not allow a consistent estimate of the marginal likelihood. We contrast two different methods to estimate the marginal likelihood: harmonic mean (Newton and Raftery 1994; Kass and Raftery 1995) and path sampling (Gelman and Meng 1998). Studies of path sampling have recently led to an alternative method of estimating marginal likelihoods (thermodynamic integration) (Gelman and Meng 1998; Lartillot and Philippe 2006; Friel and Pettitt 2005, 2008).
Harmonic mean estimator:
Newton and Raftery (1994) described an approximation of Equation 1 using a harmonic mean estimator. Our stabilized harmonic mean estimator is a natural adaptation of Newton and Raftery's harmonic mean estimator to problems that treat genealogies as nuisance parameters and summarize over all possible genealogies G using the Metropolis–Hastings algorithm (our MCMC sampler was described in detail by Beerli 1998, 2006 and Beerli and Felsenstein 1999, 2001). We approximate the marginal likelihood as
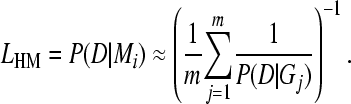 |
(2) |
The extension from single-locus to multilocus data is not straightforward even with unlinked loci. We developed a method for combining independently inferred marginal likelihoods that allows fast parallel computation of unlinked loci. The combined marginal likelihoods are the product of the independent marginal likelihoods for each locus and a scaling factor K for loci,
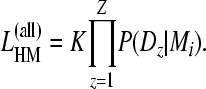 |
(3) |
The scaling factor
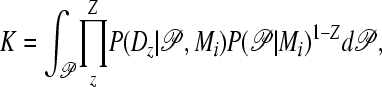 |
(4) |
where Z is the number of loci. We describe the scaling factor K in detail in the appendix. K can be approximated using prior, likelihood, and posterior values reported during the MCMC run (appendix). Our program MIGRATE version 3.1 calculates K and reports locus-specific and combined marginal-likelihood values when multiple loci are used.
Path sampling or thermodynamic integration estimator:
MCMC sampling spends more time in areas of the search space proportional to the likelihood; as a result little attention is paid to regions with low likelihoods despite the fact that they may be large. Marginal likelihood is the integral over the whole search space and therefore may depend on accurate representation of these low-likelihood areas. Path sampling allows exploring these low-likelihood areas by distorting the acceptance ratio of the MCMC procedure with scaling factor τ ranging from zero to 1.0, where at τ = 0.0 the process samples from the prior distribution and at τ = 1.0 it samples from the distribution of interest. Thus, we calculate the log marginal likelihood using the expectation of the distribution of all coalescent genealogies G given the data D evaluated at scaling factor τ,
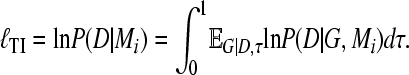 |
(5) |
We approximate this integral using the trapezoidal rule for the scaling factor τ, using a small number of scaling values τ0 = 0 < τ1 < … < τk < … < τn = 1 and the corresponding marginal likelihoods y0 … yn as
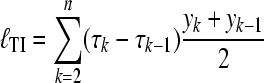 |
(6) |
with the average of log likelihoods, ln P(D | Gj, Mi), at a given scaling value τk,
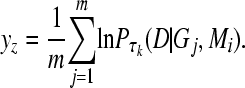 |
(7) |
For multiple unlinked loci we then use
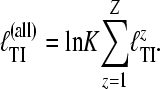 |
(8) |
The K is the same as the one in Equation 4. MIGRATE already used a scheme to run parallel MCMC chains to improve the exploration of search space using discrete scaling values τk that is based on the scheme proposed by Geyer and Thompson (1995): Metropolis coupled Markov chain Monte Carlo (MCMCMC). They formulated their method in terms of thermodynamic properties in which a chain that accepts always, with τ = 0.0, is the hottest chain with a temperature of 1/τ = ∞ because the chain bounces randomly in many different areas of the search space, and a chain with τ = 1.0 is cold because its movements are smaller. After each chain attempts a change of the genealogy, the system allows for swapping trees among neighboring MCMC chains with scaling factors τi and τi+1 to improve the parameter estimates. The swap ratio depends on the relative likelihood ratios of randomly chosen pairs of chains with different τ and is
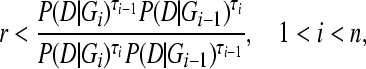 |
(9) |
where r is a uniform random number between 0 and 1, and n is the number of chains with different scaling factors τ. We use the term scaling classes to express the different discrete classes with different values of τ. One could express the same classes as temperature classes where the temperature Ti is 1/τi.
For the thermodynamic integration we record the likelihood values for each chain; these values are then used to calculate the averages yk, which are used to calculate the marginal likelihoods. This is a static variant of the step-stone method proposed by Wangang Xie, Ming-Hiu Chen, Yu Fan, Lynn Kuo, and Paul Lewis (L. Kuo and P. Lewis, personal communication in 2008).
Using discrete classes τk may be too simple for phylogenetic applications (cf. Lartillot and Philippe 2006), but results in consistent estimates even for few scaling classes (Figure 1), except that the magnitudes of the estimates of the marginal likelihood (the area under the curve) are correlated with the number of scaling classes. The calculation time for each scaling class is about the same, so a run with 4 scaling classes will be about eight times faster than a run with 32 scaling classes. In principle, the different chains can be run in parallel, but the gain in speed is limited because the chains run in lockstep and need to wait on the slowest chain. Because many simulations (not shown) revealed that the shape of the path sampling function (Figure 1) is very similar with different migration models, we propose a different treatment of the first (the hottest) interval, defined by the scaling factors τ0 and τ1 with log-likelihood values y0 and y1, respectively. We calculate the area of this first interval analytically, using a cubic Bézier spline with two additional control points c(0) and c(1) that are calculated using the first three points. A point is a pair of τi and log-likelihood yi and is defined as pi = (τi, yi). The additional control points are
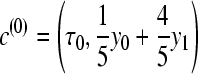 |
(10) |
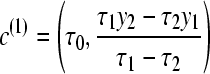 |
(11) |
so that we have four control points
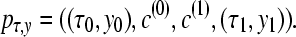 |
(12) |
The values of the y-axis of the additional control points were chosen so that the Bézier curve mimics the path sampling function estimated with many scaling classes. We calculate a point p(w) on the Bézier function using
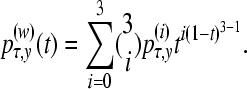 |
(13) |
The partial marginal likelihood by integrating the parametric function over the hottest interval is then
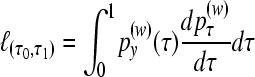 |
(14) |
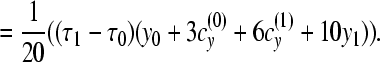 |
(15) |
This Bézier quadrature allows shorter run times than approaches with more scaling classes, an important fact because the estimation of large problems with many parameters can take a long time to run.
Figure 1.—

Comparison of integration accuracy. The shaded curve with shaded squares shows means of 32 chains at equally spaced intervals of τ. Solid squares mark the curve derived from 16 chains. The dashed line marks the curve from 4 chains. The dotted line is the cubic Bézier-spline approximation of the first interval of the 4-chain run.
Simulation studies to test the approximations to the marginal likelihood:
The quality of the two estimators 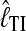 and
and 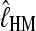 was tested using simulated data. These data sets were generated using a coalescence-based simulator (distributed from http://people.sc.fsu.edu/∼beerli/programs).
was tested using simulated data. These data sets were generated using a coalescence-based simulator (distributed from http://people.sc.fsu.edu/∼beerli/programs).
One- and two-population simulations:
The HM and TI approximations were compared with a standard test statistic based on allele frequencies (Hudson et al. 1992a), using two groups of simulated two-population data:
One hundred artificial DNA data sets containing 1000 sites for 10 individuals in each of two populations using a model with no immigration into population 2 with parameters Θ1 = 0.005, Θ2 = 0.01,
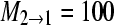 ,
, 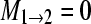 were analyzed with nine different models. Θ is the mutation-scaled effective population size, 4Neμ, and M is the ratio of the immigration rate m and the mutation rate μ per site and generation. The marginal likelihoods of eight alternative models were then compared with the marginal likelihood of the model used to simulate the data, the “true” model. This comparison of marginal likelihood ratios is equivalent to Bayes factors.
were analyzed with nine different models. Θ is the mutation-scaled effective population size, 4Neμ, and M is the ratio of the immigration rate m and the mutation rate μ per site and generation. The marginal likelihoods of eight alternative models were then compared with the marginal likelihood of the model used to simulate the data, the “true” model. This comparison of marginal likelihood ratios is equivalent to Bayes factors.Simulations of four sets of 100 single-locus data sets with different degrees of isolation from each other were used to compare the Bayes factor method against a traditional test based on frequencies. These four sets were simulated with (a) Θ = 0.01 and the 20 individuals randomly split into two groups; (b) Θ1 = Θ2 = 0.005,
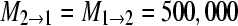 (this is equivalent to a total Nm = 1250); (c) Θ1 = Θ2 = 0.005,
(this is equivalent to a total Nm = 1250); (c) Θ1 = Θ2 = 0.005, 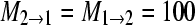 (this is equivalent to a total Nm = 0.25); and (d)
(this is equivalent to a total Nm = 0.25); and (d) 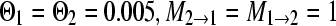 (this is equivalent to a total Nm = 0.0025). The analyses of these four sets were done for two models, a single-population model and a full two-population model.
(this is equivalent to a total Nm = 0.0025). The analyses of these four sets were done for two models, a single-population model and a full two-population model.
Large-scale population simulations:
Many real problems include many sampling locations for which the association of sampling locations and panmictic populations is unknown. We simulated data for 50 loci from 3 populations using a scenario as outlined in Figure 2A. This stepping-stone model has five parameters and for each locus 300 bp were simulated using these values: Θ1 = 0.003, Θ2 = 0.003, Θ3 = 0.004, 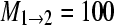 ,
, 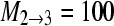 . The individuals (120, 120, and 160) in the 3 populations were then randomly grouped into 6, 6, and 8 sampling locations, respectively. The full data set contained 20 locations with 20 individuals each. These particular settings were chosen because they mimic potential data sets that use anonymous loci from the nuclear genome. A naive application of these data would ask for a 20-population analysis. With a default MIGRATE run we would need to estimate 20 population sizes and 380 migration parameters, a daunting task with few loci. A total of six potential migration models using different numbers of populations and different migration models were explored. The six cases presented use models with 1, 2, 3, and 20 populations with several candidate models (Table 1, Figure 2). Specific MIGRATE run conditions are described in supporting information, File S1.
. The individuals (120, 120, and 160) in the 3 populations were then randomly grouped into 6, 6, and 8 sampling locations, respectively. The full data set contained 20 locations with 20 individuals each. These particular settings were chosen because they mimic potential data sets that use anonymous loci from the nuclear genome. A naive application of these data would ask for a 20-population analysis. With a default MIGRATE run we would need to estimate 20 population sizes and 380 migration parameters, a daunting task with few loci. A total of six potential migration models using different numbers of populations and different migration models were explored. The six cases presented use models with 1, 2, 3, and 20 populations with several candidate models (Table 1, Figure 2). Specific MIGRATE run conditions are described in supporting information, File S1.
Figure 2.—

Population structures used to generate artificial data sets. (A) The true population model, where population 3 receives immigrants from population 2, which receives immigrants from population 1. Each solid disk represents a random sample from the population and therefore represents an arbitrary sample subdivision (location sample) of the panmictic population 1, or 2, or 3. (A–C) show alternative partitionings used of the true population model. (B) The 20 location samples are lumped into 2 populations, combining populations 2 and 3 (see A). (C) An alternative 2-population model. (D) A naive 20-population model assuming each sampling location represents a population; arrows in A, B, and lines in D represent potential migration routes; migration routes in C are bidirectional from dark to light locations.
TABLE 1.
List of migration models used for simulation study
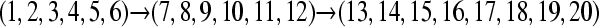
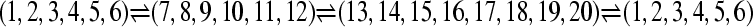
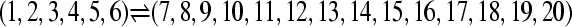
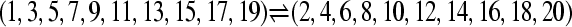
Effect of prior choice and prior range:
We explored the effect of the choice of the prior distribution on the marginal likelihood by using simulated multilocation single-locus data. We compared two exponential and two uniform prior distributions: narrow uniform prior distribution for Θ and M with a minimum of 0.00001, 0.0 and a maximum of 0.1, 5000, respectively; a wide uniform distribution with a maximum for Θ and M of 0.5 and 50,000; a narrow exponential distribution with the same minimum and maximum as the narrow uniform but a mean of 0.01 and 100 for Θ and M, respectively; and a wide exponential distribution with minimum and maximum of the wide uniform, but with a mean of 0.1 and 1000, respectively. Specific MIGRATE run conditions are described in File S1.
Model selection:
Model choice probabilities si were calculated as suggested by Kass and Raftery (1995) by
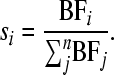 |
(16) |
Example data set:
Our example problem reanalyzes part of a data set of humpback whales from four sampling locations in the Southern Atlantic collected by Engel et al. (2008): near Brazil, Antarctica 1 (west of the Antarctic peninsula), Antarctica 2 (east of the Antarctic peninsula), and Colombia (Figure 1 in Engel et al. 2008). The data were analyzed using several different migration models. We used three subsamples of the original data, two with 10 and one with 30 randomly selected individuals from each location. We also ran one of the data sets twice for all example models to assess the effect of the Markov chain Monte Carlo error. We established a most likely mutation model within the constraints for MIGRATE by using PAUP* (Swofford 2003) to estimate parameters for site rate variation and transition/transversion ratio.
RESULTS
Comparison of approximations of the marginal likelihood:
In all but trivial situations we cannot calculate LM or its log value, lM, analytically. Using simulated data, we compared the two different methods for approximating LM: the thermodynamically estimated 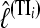 using coupled scaling classes TIi and the harmonic mean HM estimated as
using coupled scaling classes TIi and the harmonic mean HM estimated as 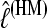 . In the context of coalescent simulations the artificial data Di simulated from a set of true parameter values still include considerable variability, so we do not expect a particular
. In the context of coalescent simulations the artificial data Di simulated from a set of true parameter values still include considerable variability, so we do not expect a particular 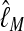 (for short:
(for short: 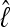 ) from all data sets. Nevertheless, we expect that the different approximations will result in the same
) from all data sets. Nevertheless, we expect that the different approximations will result in the same 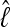 for a specific data set. Figure 3 shows a comparison of the two different approximations of lM. The relative magnitude of
for a specific data set. Figure 3 shows a comparison of the two different approximations of lM. The relative magnitude of 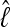 among the different data sets is the same: a data set that shows low
among the different data sets is the same: a data set that shows low 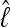 with the HM estimator also shows low values for the different TI schemes.
with the HM estimator also shows low values for the different TI schemes. 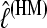 is little affected by the number of scaling classes, whereas the number of scaling classes affects the absolute value of the
is little affected by the number of scaling classes, whereas the number of scaling classes affects the absolute value of the 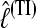 . When the results of a specific data set are compared, the TI4 method delivers lower
. When the results of a specific data set are compared, the TI4 method delivers lower 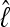 than the HM4, HM16, HM32, TI16, and TI32 methods. The thermodynamically estimated
than the HM4, HM16, HM32, TI16, and TI32 methods. The thermodynamically estimated 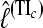 using independent scaling classes is identical to the coupled scaling classes (data not shown).
using independent scaling classes is identical to the coupled scaling classes (data not shown).
Figure 3.—

Comparison of the log marginal likelihood lM inferred by harmonic mean (HM) and thermodynamic integration (TI), using 4, 16, and 32 scaling classes of 10 independent data sets (sorted by magnitude of the likelihood of the TI runs with four scaling classes). HM values for the 16- and 32-chain runs were so similar that the squares overlap on the y-axis scaling used, but are different up to 5 log-likelihood units. The simulated data were generated using model 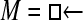 ▪.
▪.
Bayes factor estimation:
Instead of reporting BF, we report its log-equivalent LBF, which is 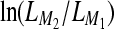 or
or 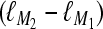 . The log marginal-likelihood values
. The log marginal-likelihood values 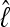 are dependent on the approximation, and the LBF depends on the difference of the log marginal likelihoods
are dependent on the approximation, and the LBF depends on the difference of the log marginal likelihoods 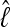 and therefore the relative difference among models is more important than the unbiased recovery of
and therefore the relative difference among models is more important than the unbiased recovery of 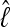 (Figure 3). Figure 4 compares the dependency of the approximations on the length of the run. The shortest run took only 5 sec with four chains, visiting 30,000 states and discarding the first 10,000; the longest four-chain run took 5 min 21 sec, visiting and discarding 256 times more states. The thermodynamic integration approximation results in LBFs with high repeatability and little variance even with only short runs, whereas the LBFs using the HM estimator are unstable even for long runs, and it appears that MCMCMC searches with many chains result in reduced reliability of the HM estimators.
(Figure 3). Figure 4 compares the dependency of the approximations on the length of the run. The shortest run took only 5 sec with four chains, visiting 30,000 states and discarding the first 10,000; the longest four-chain run took 5 min 21 sec, visiting and discarding 256 times more states. The thermodynamic integration approximation results in LBFs with high repeatability and little variance even with only short runs, whereas the LBFs using the HM estimator are unstable even for long runs, and it appears that MCMCMC searches with many chains result in reduced reliability of the HM estimators.
Figure 4.—

Comparison of the LBF (ln BF) values for different run lengths of the MCMC chain. The squares and circles are LBF values using the average marginal likelihoods from five replicated runs. The vertical bars mark the range between the largest and the smallest LBF value from five replicated runs. (A) LBF approximated using the harmonic mean; (B) LBF approximated by thermodynamic integration. The simulated data were generated using model 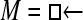 ▪ and LBF =
▪ and LBF = 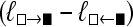 . LBF scales in A and B are very different.
. LBF scales in A and B are very different.
Numerous artificial single-locus data sets from a model with two populations of unequal size, in which only one population receives migrants from the other, were generated; this model has three parameters that are free to vary: population sizes 1 and 2 and immigration rate from population 2 to population 1, which is 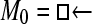 ▪. Populations are indicated by squares. Two open squares indicate populations constrained to have the same size; one open and one solid square indicate population sizes are not constrained. Arrows indicate allowed migration direction [from population 1 to 2, from 2 to 1, or in both directions; arrows with two heads indicate symmetric migration rate parameters (M = m/μ)]. These data sets were analyzed with all nine possible simple models (one parameter, □; two parameters,
▪. Populations are indicated by squares. Two open squares indicate populations constrained to have the same size; one open and one solid square indicate population sizes are not constrained. Arrows indicate allowed migration direction [from population 1 to 2, from 2 to 1, or in both directions; arrows with two heads indicate symmetric migration rate parameters (M = m/μ)]. These data sets were analyzed with all nine possible simple models (one parameter, □; two parameters, 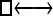 ▪,
▪, 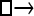 ▪,
▪, 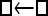 ; three parameters,
; three parameters, 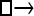 ▪,
▪, 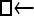 ▪,
▪, 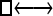 ▪, □⇆□; four parameters, □⇆▪); models that exclude gene flow among the populations were omitted. We calculated log Bayes factors,
▪, □⇆□; four parameters, □⇆▪); models that exclude gene flow among the populations were omitted. We calculated log Bayes factors, 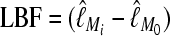 . These report the chance of accepting Mi over M0. In Table 2 LBF using TI16 rejects models that have more parameters than the true model or that disregard unidirectional migration with high frequency. Very simple models and asymmetric models are often accepted as plausible models. LBF using HM is indecisive, even with models that are very different from the true model, such as
. These report the chance of accepting Mi over M0. In Table 2 LBF using TI16 rejects models that have more parameters than the true model or that disregard unidirectional migration with high frequency. Very simple models and asymmetric models are often accepted as plausible models. LBF using HM is indecisive, even with models that are very different from the true model, such as 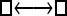 . Overall, the estimates from TI deliver a clearer guide about which models to prefer than the highly variable HM estimates (see File S1), which, on average, are less decisive.
. Overall, the estimates from TI deliver a clearer guide about which models to prefer than the highly variable HM estimates (see File S1), which, on average, are less decisive.
TABLE 2.
Summary of support for specific models using LBF approximated with harmonic mean (HM) and thermodynamic integration (TI) using 16 chains with different scalers
Evidence (“true” 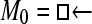 ▪) ▪) |
Counts (based on LBFTI and LBFHM) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nparam | 4 | 3 | 3 | 3 | 2 | 2 | 2 | 1 | ||||||||
| Model | □⇆▪ |
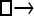 ▪ ▪ |
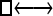 ▪ ▪ |
□⇆□ | 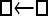 |
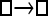 |
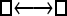 |
 |
||||||||
| Approximation | TI | HM | TI | HM | TI | HM | TI | HM | TI | HM | TI | HM | TI | HM | TI | HM |
| Against M0 | 0 | 46 | 28 | 36 | 0 | 48 | 0 | 57 | 70 | 50 | 54 | 35 | 0 | 59 | 11 | 40 |
| Against Mi | 100 | 54 | 72 | 64 | 100 | 52 | 100 | 43 | 30 | 50 | 46 | 65 | 100 | 41 | 89 | 60 |
One hundred single-locus data sets were analyzed, each with a total of 20 DNA sequences simulated using a three-parameter model with two different population sizes and unidirectional migration from population 2 to population 1 (model abbreviation is 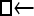 ▪). All other models 1–8 (Mi), such as the full model (□⇆▪) or the minimal model (
▪). All other models 1–8 (Mi), such as the full model (□⇆▪) or the minimal model (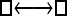 ), are compared with this “true” model (
), are compared with this “true” model (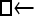 ▪), which represents the M0 hypothesis. nparam: number of parameter estimated.
▪), which represents the M0 hypothesis. nparam: number of parameter estimated.
A comparison with different strengths of migration rate among two populations (Table 3) shows that the 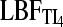 is more variable than the
is more variable than the 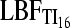 but the numbers of acceptances or rejections of a hypothesis (Table 3, Table 2S in File S1) are very similar between TI16 and TI4. In contrast, the LBFHM has a higher variability of outcomes.
but the numbers of acceptances or rejections of a hypothesis (Table 3, Table 2S in File S1) are very similar between TI16 and TI4. In contrast, the LBFHM has a higher variability of outcomes.
TABLE 3.
Comparison of the influence of the approximation on the power of LBF for simple models with different migration schemes
| Evidence | Counts (based on 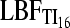 , , 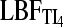 , and LBFHM) , and LBFHM) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | 1a | 1b | 2a | 2b | ||||||||
| Nm | ∞ | 1250 | 0.25 | 0.0025 | ||||||||
| Approximation | 16 | 4 | H | 16 | 4 | H | 16 | 4 | H | 16 | 4 | H |
| Against M0 | 0 | 5 | 26 | 0 | 8 | 29 | 70 | 49 | 53 | 100 | 100 | 78 |
| Against M1 | 100 | 94 | 73 | 100 | 92 | 71 | 30 | 51 | 47 | 0 | 0 | 22 |
LBF compared a full model (model M1 = □⇆▪) with a panmictic population (model M0 =  ). Models used to simulate the data were as follows: 1a, a single population, the sampled individuals split randomly into two sets (
). Models used to simulate the data were as follows: 1a, a single population, the sampled individuals split randomly into two sets (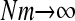 ); 1b, two populations exchanging many migrants (Nm = 1250); 2a, two populations exchanging a moderate number of migrants (Nm = 0.25); and 2b, two populations with very low migration rate (Nm = 0.0025). The marginal likelihoods used in the LBF were approximated with thermodynamic integration (TI) with 16 and 4 scaler bins and with the harmonic mean (HM4).
); 1b, two populations exchanging many migrants (Nm = 1250); 2a, two populations exchanging a moderate number of migrants (Nm = 0.25); and 2b, two populations with very low migration rate (Nm = 0.0025). The marginal likelihoods used in the LBF were approximated with thermodynamic integration (TI) with 16 and 4 scaler bins and with the harmonic mean (HM4).
Comparison with a panmixia test method:
Currently, coalescence-based inference programs do not test whether the sampling locations are in separate populations or not. Therefore, summary statistics such as FST, Fisher's exact test, or population genetic clustering programs (Pritchard et al. 2000; Evanno et al. 2005; Huelsenbeck and Andolfatto 2007; Manel et al. 2007; Guillot 2008) are being used to establish groups of individuals or sampling locations that most likely form panmictic populations. Waples and Gaggiotti (2006) showed that contingency table permutation methods work well. Hudson, Boos, and Kaplan (Hudson et al. 1992a) developed a permutation test (HBK) that has great potential but seems to be little used despite its power to establish panmixia. For our comparison we used four scenarios: (1a) a single population was sampled and then the sample was randomly partitioned into two “populations”, (1b) two populations exchanging 1250 migrants per generation, (2a) two populations exchanging 1 migrant every 4 generations, and (2b) two populations exchanging 1 migrant every 400 generations. Table 3 reveals that for a real panmictic population (1a), 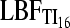 ,
, 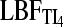 , and LBFHM detect panmixia in 100, 94, and 73 of the data sets, respectively, whereas HBK finds that all 100 data sets are panmictic. Recognition of panmixia in scenario 1b was 100, 92, and 71 for
, and LBFHM detect panmixia in 100, 94, and 73 of the data sets, respectively, whereas HBK finds that all 100 data sets are panmictic. Recognition of panmixia in scenario 1b was 100, 92, and 71 for 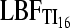 ,
, 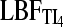 , and LBFHM, respectively, whereas the HBK method marks all data sets panmictic. With LBFTI, all data sets from scenario 2b fit a two-population model; with 2a the acceptance of a two-population model shrank to 70, 49, and 53 of 100, signaling considerable uncertainty about finding the correct population model. HBK declares all data sets under scenario 2 to contain two populations. LBFHM shows, for all scenarios, much larger variability in acceptance and rejection of panmixia (see File S1), resulting in a lower total acceptance of the correct model.
, and LBFHM, respectively, whereas the HBK method marks all data sets panmictic. With LBFTI, all data sets from scenario 2b fit a two-population model; with 2a the acceptance of a two-population model shrank to 70, 49, and 53 of 100, signaling considerable uncertainty about finding the correct population model. HBK declares all data sets under scenario 2 to contain two populations. LBFHM shows, for all scenarios, much larger variability in acceptance and rejection of panmixia (see File S1), resulting in a lower total acceptance of the correct model.
Effect of loci and model complexity:
Table 4 shows the LBFs for six migration models (Table 1). The thermodynamic integration method consistently chooses the true model as the best model. Differences for the other models depend on the haphazard choice of the order of the loci. Because only 50 loci were simulated for all runs, the first locus is shared among all runs, and the second locus is shared among all runs except the 1-locus runs, etc. We expect that with many loci a clear order of models is achieved. The model order for the 50-locus run is 1, 2, 3, 6, 5, and 4. Runs with many loci (>10) suggest that the one-population model (5) is superior to the two-population model that combines the locations in an intermixed pattern (4) and also suggest that the 400-parameter (6) analysis is preferable over analyses with wrongly combined locations. Runs with only few loci may suffer because there are not enough data to correctly rank incorrect models 3, 4, 5, and 6. The reported Bayes factor values suggest that model 1 should be picked with probability 1.0 over the five alternatives. More loci increase this certainty considerably: the difference between the first and the second best model is already very large for a single locus. The number of loci and the BF differences are positively correlated. The results for the harmonic mean estimator suggest that the preferred model is the 9-parameter model (2) and not the model that was used to simulate the data (1).
TABLE 4.
Comparison of log Bayes factors (marginal log-likelihood differences) approximated by thermodynamic integration and harmonic mean estimator, for different models and different numbers of loci
| LBF for modela |
Rank of model |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1st | 2nd | 3rd | 4th | 5th | 6th | |
| Thermodynamic integration | ||||||||||||
| Loci | ||||||||||||
| 1 | 0 | −10 | −86 | −583 | −594 | −253 | 1 | 2 | 3 | 6 | 4 | 5 |
| 2 | 0 | −552 | −1,946 | −3,167 | −3,338 | −467 | 1 | 6 | 2 | 3 | 4 | 5 |
| 5 | 0 | −697 | −2,432 | −4,757 | −4,826 | −542 | 1 | 6 | 2 | 3 | 4 | 5 |
| 10 | 0 | −1,136 | −4,266 | −8,566 | −8,352 | −2,328 | 1 | 2 | 6 | 3 | 5 | 4 |
| 20 | 0 | −2,072 | −5,914 | −12,913 | −12,379 | −4,835 | 1 | 2 | 6 | 3 | 5 | 4 |
| 50 | 0 | −4,829 | −14,683 | −30,147 | −28,439 | −15,245 | 1 | 2 | 3 | 6 | 5 | 4 |
| Harmonic mean estimator | ||||||||||||
| 1 | 0 | −7 | −20 | −29 | −28 | −35 | 1 | 2 | 3 | 5 | 4 | 6 |
| 2 | 0 | −76 | −83 | −123 | −70 | −168 | 1 | 5 | 2 | 3 | 4 | 6 |
| 5 | 0 | −124 | −133 | −215 | −160 | −308 | 1 | 2 | 3 | 5 | 4 | 6 |
| 10 | 0 | −236 | −201 | −430 | −161 | −420 | 1 | 5 | 3 | 2 | 6 | 4 |
| 20 | 0 | −438 | −565 | −1,085 | −453 | −943 | 1 | 2 | 5 | 3 | 6 | 4 |
| 50 | 0 | −819 | −1,266 | −2,613 | −1,266 | −2,723 | 1 | 2 | 5 | 3 | 4 | 6 |
Model 1 was used to simulate the data and is the reference model.
Model numbers are specified in Table 1.
Effects of prior distribution on the marginal likelihood:
Table 5 reveals that the marginal likelihoods depend on the prior distribution: the LBF values are different for different prior distributions. For the thermodynamic integration method, however, the order of the models is identical among the narrow and wide prior distributions, respectively, suggesting that most likely the runs were rather short for the wide-prior models. The harmonic mean estimator of the marginal likelihood is similarly affected by the choice of prior distributions. Using the harmonic mean estimator, the models are ranked differently for each of the different priors.
TABLE 5.
Log Bayes factors (LBF) estimated by thermodynamic integration and by the harmonic mean using different prior distributions
| Thermodynamic integration |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LBF for modela | Rank of model | |||||||||||
| Prior | 1 | 2 | 3 | 4 | 5 | 6 | 1st | 2nd | 3rd | 4th | 5th | 6th |
| Uniform narrow | 0 | −77 | −108 | −487 | −540 | −254 | 1 | 2 | 3 | 6 | 4 | 5 |
| Uniform wide | 0 | −232 | −165 | −409 | −277 | −364 | 1 | 3 | 2 | 5 | 6 | 4 |
| Exponential narrow | 0 | −17 | −84 | −531 | −542 | −255 | 1 | 2 | 3 | 6 | 4 | 5 |
| Exponential wide | 0 | −170 | −158 | −461 | −270 | −394 | 1 | 3 | 2 | 5 | 6 | 4 |
| Harmonic mean estimator |
||||||||||||
| LBF for modela |
Rank of model |
|||||||||||
| Prior | 1 | 2 | 3 | 4 | 5 | 6 | 1st | 2nd | 3rd | 4th | 5th | 6th |
| Uniform narrow | 0 | −3 | −29 | −23 | −4 | −55 | 1 | 2 | 5 | 4 | 3 | 6 |
| Uniform wide | 0 | −2 | −18 | −25 | −25 | −29 | 1 | 2 | 3 | 5b | 4b | 6 |
| Exponential narrow | 0 | −8 | −30 | −23 | −8 | −37 | 1 | 5b | 2b | 4 | 3 | 6 |
| Exponential wide | 0 | −8 | −23 | −36 | −18 | −55 | 1 | 2 | 5 | 3 | 4 | 6 |
Model 1 was used to simulate the data and is also the reference model.
Model numbers are specified in Table 1.
Tied.
Example analysis of migration patterns among humpback whales sampling locations in the south seas:
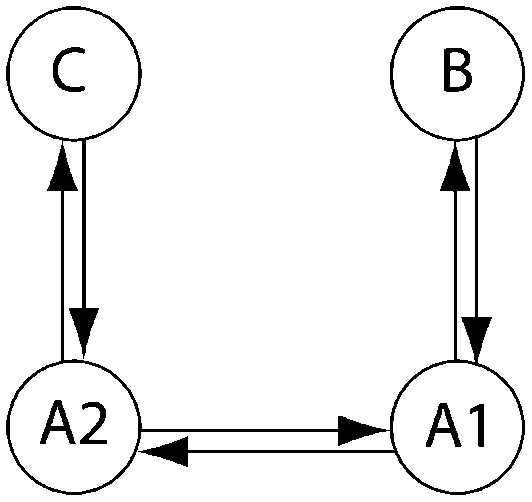
Olavarría et al. (2007) and Engel et al. (2008) described the interaction of several humpback whale populations (sampling locations). We use parts of their data to showcase how BF can inform the discussion of whether whales from these sampling locations belong to the same genetic population or not and whether some population models provide more appropriate descriptions than others. Our analysis does not completely resolve the complex population interactions of humpback whales, but it shows ways in which our method is more useful than current methods for model comparisons. Engel et al.(2008), using pairwise FST estimates, suggested that Antarctic locations A1 and A2 appear panmictic; they used additional sighting data to suggest that the individuals sampled near the Brazilian coast probably do not move to the presumed feeding grounds in the Antarctic but instead aggregate at some unknown location. We chose a subset of models to investigate (1) whether the regions Antarctica 1 and 2 belong to a single population and (2) whether the Brazilian individuals and Antarctic individuals belong to the same population. Table 6 shows the 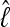 for each model tested for three subsets of the full data set. Model 6, which allows for structure between A1 and A2 and reduced gene flow between Antarctica and Brazil, has the highest marginal likelihood. This model was used as the reference in LBF to compare all models. Our analysis confirms the conclusion of Engel et al. (2008) that the connectivity between the Brazilian and Antarctic locations is reduced (model 6), but, unlike models 7–9, does not suggest complete isolation of the Brazilian individuals from the other locations. Model 2 is the second best model; it shares almost all features of model 6 except that the migration rates between Antarctica and Brazil are bidirectional. Models that suggest A1 and A2 are part of a panmictic population (models 3–5) have lower LBF values than models 2 and 6, but model 3 is superior to model 1. This suggests that A1 and A2 are probably not part of a panmictic population, but the data do not support a complex model with many parameters (model 1). Our current understanding of the population structuring is based on a single locus (mtDNA). These data are insufficient to resolve the complex interactions among southern Atlantic humpback whales.
for each model tested for three subsets of the full data set. Model 6, which allows for structure between A1 and A2 and reduced gene flow between Antarctica and Brazil, has the highest marginal likelihood. This model was used as the reference in LBF to compare all models. Our analysis confirms the conclusion of Engel et al. (2008) that the connectivity between the Brazilian and Antarctic locations is reduced (model 6), but, unlike models 7–9, does not suggest complete isolation of the Brazilian individuals from the other locations. Model 2 is the second best model; it shares almost all features of model 6 except that the migration rates between Antarctica and Brazil are bidirectional. Models that suggest A1 and A2 are part of a panmictic population (models 3–5) have lower LBF values than models 2 and 6, but model 3 is superior to model 1. This suggests that A1 and A2 are probably not part of a panmictic population, but the data do not support a complex model with many parameters (model 1). Our current understanding of the population structuring is based on a single locus (mtDNA). These data are insufficient to resolve the complex interactions among southern Atlantic humpback whales.
TABLE 6.
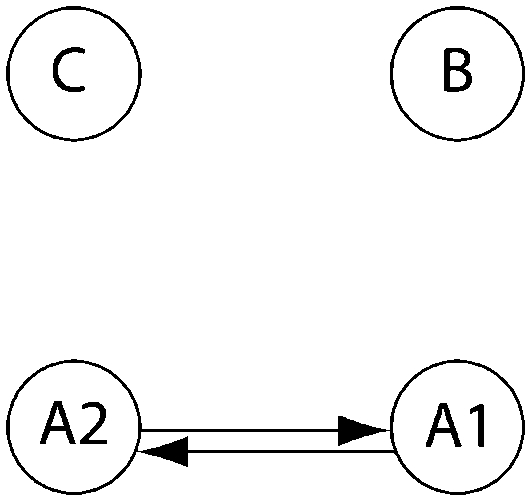
Log Bayes factor (LBF) using thermodynamic integration of different gene flow models Mi compared with model 6 for four sampling locations of humpback whales (C, Colombia; B, Brazil; A1, Antarctica east of the Antarctic peninsula; A2, Antarctica west of the Antarctic peninsula)
| LBF of nine models compared against M6 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| Method | Samples | 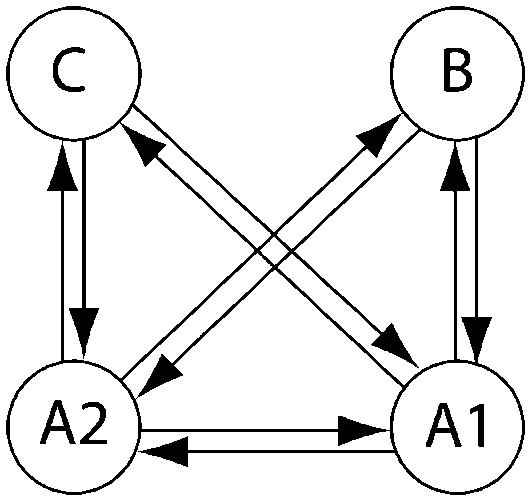 |
 |
 |
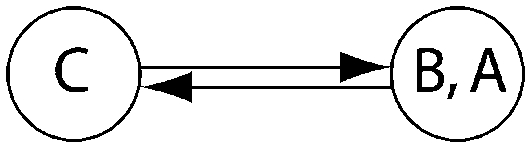 |
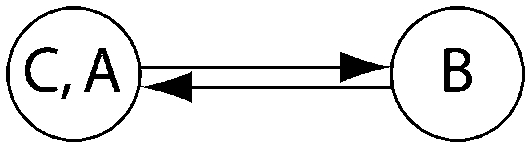 |
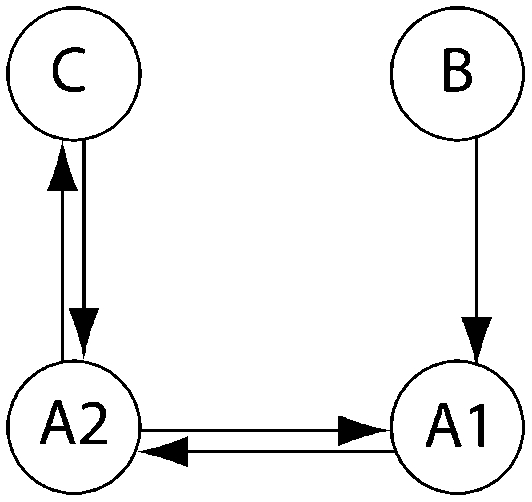 |
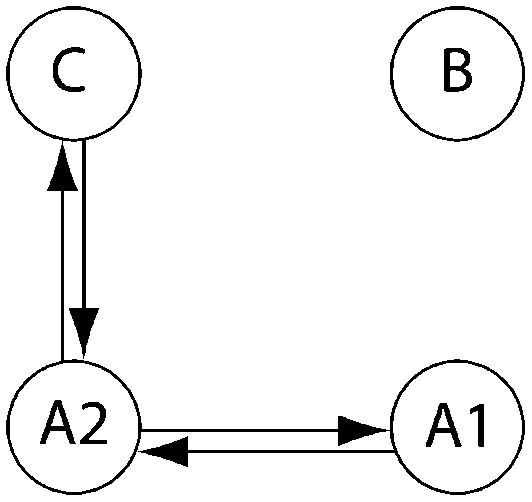 |
 |
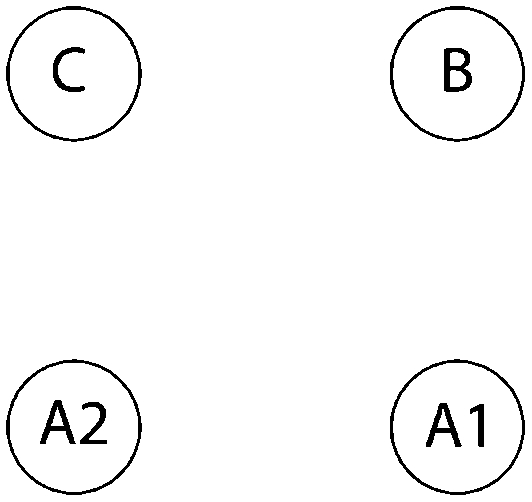 |
| 1 | 10a | −16.9 | −6.1* | −21.2 | −39.9 | 0.0*** | −198.8 | −241.7 | −531.4 | |
| 10a | −17.0 | −6.1* | −14.7 | −39.3 | −39.3 | 0.0*** | −217.0 | −194.1 | −484.5 | |
| 10 | −16.6 | −4.5** | −13.8 | −20.7 | −36.8 | 0.0*** | −237.3 | −241.9 | −523.8 | |
| 30 | −91.3 | −32.0 | −84.4 | −105.8 | −227.9 | 0.0*** | −291.4 | −167.8 | −605.5 | |
| 2 | 10a | −15.1 | −9.5 | −13.9 | −17.0 | −29.0 | 0.0*** | −180.0 | −222.6 | −494.3 |
| 10a | −14.7 | −9.3 | −13.5 | −16.3 | −28.8 | 0.0*** | −184.3 | −193.8 | −458.0 | |
| 10 | −14.6 | −7.8* | −12.4 | −15.4 | −25.2 | 0.0*** | −276.2 | −244.9 | −501.8 | |
| 30 | −75.0 | −30.5 | −77.0 | −83.1 | −164.6 | 0.0*** | −189.4 | −207.4 | −663.2 | |
Method 1, using 4 chains and Bézier approximation; method 2, using 32 chains. Model probabilities: *0.01 < si < 0.05; **0.05 < si < 0.10; and ***0.9 < si < 1.0.
Same data, but different start values of the MCMC run.
The data sets were analyzed using TI with 32 chains and 4 chains. The Bézier-corrected 4-chain marginal likelihoods result in LBF of the same magnitude as the 32-chain runs, despite the greatly reduced run time.
DISCUSSION
The approximation of lM using the harmonic mean estimator is concordant with the thermodynamic integration method, although the HM estimate is always higher than the TI estimator. Paul Lewis (P. Lewis, personal communication, 2009) has shown that this is an artifact of MCMC runs in which the HM estimator is biased toward the high probability regions of the parameter space. TI, in contrast, estimates very similar magnitudes of 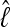 over replicated runs of the same data and run parameters. Nevertheless, the magnitude of
over replicated runs of the same data and run parameters. Nevertheless, the magnitude of 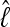 using the thermodynamic method is correlated with the number of classes, although the relative difference among models persists independently of the absolute magnitude. Using the Bézier quadrature with a low number of chains at different scalers removes this difference. The run time is dependent on the number of chains, so the use of the Bézier quadrature may be preferable for large data sets and large population models because running many MCMC chains requires more time than is usually available in computer time budgets.
using the thermodynamic method is correlated with the number of classes, although the relative difference among models persists independently of the absolute magnitude. Using the Bézier quadrature with a low number of chains at different scalers removes this difference. The run time is dependent on the number of chains, so the use of the Bézier quadrature may be preferable for large data sets and large population models because running many MCMC chains requires more time than is usually available in computer time budgets.
Analyses with different run lengths showed that the Bayes factor based on the harmonic mean estimator is more variable than that based on the thermodynamic integration estimator. Most disconcerting are the results with many chains because multiple LBF estimates based on the HM estimates show a wide range for the same data set, suggesting that an appropriate MCMC search results in unreliable HM estimates. The path of the MCMC chain influences the HM-based BF considerably because, for a good estimate of 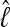 , the chain needs to explore areas of the solution space that have low probability. Once a low value is recorded, it affects the harmonic mean disproportionately. Runs that rarely visit such low values will report an
, the chain needs to explore areas of the solution space that have low probability. Once a low value is recorded, it affects the harmonic mean disproportionately. Runs that rarely visit such low values will report an 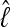 that is inflated. Using such values in the LBFHM leads to high variance because the low values are not visited in the correct proportions. Our results corroborate the work of other authors (for example, Lartillot and Philippe 2006) who consider the HM inferior to the TI method.
that is inflated. Using such values in the LBFHM leads to high variance because the low values are not visited in the correct proportions. Our results corroborate the work of other authors (for example, Lartillot and Philippe 2006) who consider the HM inferior to the TI method.
The LBF usually supports the correct model independent of the number of chains used in the thermodynamic approximation method. In the comparison in Table 2, several models were weakly supported. This is interesting because these alternative models (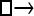 ▪,
▪, 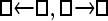 ), which are models with strong unidirectional gene flow, are viable competitors for the real model (
), which are models with strong unidirectional gene flow, are viable competitors for the real model (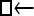 ▪) given the small sample size (20 individuals) and the large variance in coalescent simulations. Without multiple loci it is particularly difficult to estimate the migration direction from genetic data that often differ only in the frequency of alleles. The multilocus runs show the same general pattern as the analyses with few loci, but in the larger analyses the certainty of the order of models increases. The HM estimator is less certain for all scenarios than the TI estimator, corroborating the problems visible in Figure 4, suggesting again that the HM estimator should not be used.
▪) given the small sample size (20 individuals) and the large variance in coalescent simulations. Without multiple loci it is particularly difficult to estimate the migration direction from genetic data that often differ only in the frequency of alleles. The multilocus runs show the same general pattern as the analyses with few loci, but in the larger analyses the certainty of the order of models increases. The HM estimator is less certain for all scenarios than the TI estimator, corroborating the problems visible in Figure 4, suggesting again that the HM estimator should not be used.
LBF is relatively powerful for identifying appropriate models for samples from panmictic populations and well isolated populations, but showed a high variance for structured populations with moderate immigration rates (Table 3). In contrast, the Hudson–Boos–Kaplan estimator, using a permutation test, clearly suggested two populations for all analyzed data sets that were generated from models with reduced immigration rates. Because this test does not incorporate the uncertainty of the mutation model and the coalescence, however, it may overconfidently reject simpler (panmictic) interpretations.
It has been known for a while now through recent examples (Beerli and Felsenstein 1999; Felsenstein 2005; Heled and Drummond 2008) that the number of unlinked loci increases the accuracy of the coalescent estimators considerably; our comparison of the effect of multiple loci is no exception. Rejection of incorrect models became stronger with more loci when the marginal likelihood was approximated with thermodynamic integration. The harmonic mean estimator preferred a more complicated model with increased certainty, corroborating our findings with the two-population models (Tables 2 and 3) that the harmonic mean estimator should be avoided.
The Bayes factor framework demands proper priors, formally, priors that integrate to one. In our framework all priors are proper, although some may not be optimal: for example, uniform prior distributions over a very large range are wasteful because the posterior distribution covers only a small range of values and forces very long runs for accurate estimates. Our experimentation with different prior distributions shows that suboptimal priors can often result in long run times before convergence. The effect on the marginal likelihoods, however, seems small and the effect of such suboptimal priors on model choice seems negligible. In contrast, misspecification of the prior distribution, for example, choosing too narrow a prior distribution range, has a detrimental effect on the estimation of the posterior distribution of the parameters of the model and results in incorrect marginal likelihoods.
Our example (Table 6) confirms that, in a coalescence framework, a small sample per location has almost as much power as a large sample (cf. Felsenstein 2005) because not only is the LBF of a replicated run the same with the same sample, but also different randomly sampled sets of the same and larger size return the same ranking among the models. The Bézier-spline approximation of 4 chains gives LBF values that are equivalent to runs using 32 chains, but the run time is about one-eighth as long. This suggests that we are able to estimate LBF values of very large data sets in a reasonable time with good accuracy without the need to use a large number of chains or the reversible-jump MCMC (Green 1995) method that has recently been proposed by Lartillot (Lartillot and Philippe 2006) in a phylogenetic context. Our approach asks for independent runs for each model, in contrast to model selection approaches that use reversible-jump MCMC. This may look inelegant, but we believe that our method is preferable both because each run pays full attention to a single model and because the effort does not depend on the particular model-sampling algorithm and therefore is independent of the geometry of the complex solution space. In any study, the number of models depends on the number of populations and increases at a superexponential rate, so it is unlikely to evaluate all possible models, in contrast to mutation models, all of which are able to be evaluated (Huelsenbeck and Ronquist 2005). In addition, our scheme can be run in parallel without problems and without further programming.
The simulation study clearly shows that BFs are capable of distinguishing between different models and allow us to retrieve the model that was used to simulate the test data with high certainty when the true parameters produced a clear scenario. Single-locus data will often not be sufficient to retrieve a fairly complex model unambiguously, so that when available data are few, we should prefer simple models. Of course, multilocus data sets increase the certainty about the models considerably (Beerli and Felsenstein 1999; Heled and Drummond 2008).
We do not believe that our method should replace assignment- or allele-frequency-based methods, because for large problems the demand for large computer resources may make the analysis difficult or very time consuming. Our method does, however, add another tool for the researcher interested in natural population structures.
Our methods are available in the program MIGRATE from our website http://popgen.sc.fsu.edu. Simulated data sets and humpback whale example data sets are available at (http://people.sc.fsu.edu/∼beerli/data) or upon request.
Acknowledgments
We thank Thomas Uzzell, Kathleen Lotterhos, and two anonymous reviewers for their critical comments of the text; Joseph Felsenstein and Anuj Srivastava for discussions, and Fred W. Huffer for checking our proofs for combining independently evaluated posterior distributions and marginal likelihoods. We also acknowledge the generous use of the High Performance Computing facility at Florida State University. This work was supported by the joint National Science Foundation (NSF)/National Institute of General Medical Sciences Mathematical Biology program under National Institutes of Health grant R01 GM 078985 and by NSF grant DEB 0822626.
APPENDIX
Independent calculation of marginal likelihoods:
Maximum-likelihood inference for a multilocus data set can be run concurrently because, assuming the loci are independent, the calculation for each locus can be easily parallelized, and the final result is a simple combination of the individual results. In Bayesian inference, the independent calculation of the posterior distribution for each locus is simple. In contrast to the combination of maximum-likelihood estimates over loci, however, the product of these posterior distributions leads to an overuse of the prior. Correction for this overuse allows us to calculate the posterior distributions independently on different computers or CPU cores, therefore improving the speed of analysis considerably. The calculation of marginal likelihoods of a multilocus data set with independent calculations for each locus is difficult because the individual marginal likelihoods cannot be simply combined as in the maximum-likelihood analysis: there are interdependencies among the prior and the posterior distributions. Therefore, a scaling factor is needed for the combination of the locus-specific marginal likelihoods. Here we show how to correct for the overuse of priors and how to evaluate the multilocus marginal likelihoods that are generated from these independent posterior evaluations.
The combination of posteriors over multiple loci was done naively in our program MIGRATE (Beerli 2006); we overused the priors. This resulted in biases when the priors are highly skewed and do not match the posterior distribution. Analyses with uniform priors or single-locus analysis with any prior were not biased toward the prior mode.
Theorem 1. The posterior
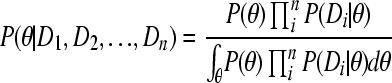 |
(A1) |
with independent locus data D1, D2, …, Dn, and a set of parameters θ can be calculated by
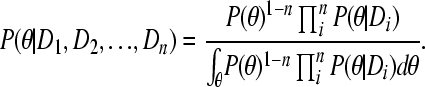 |
(A2) |
Proof. Expanding P(θ | Di) in (A2) leads to
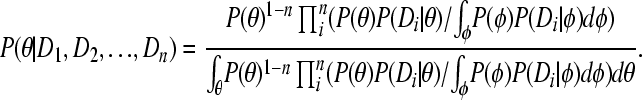 |
(A3) |
The integrals over φ cancel, so that
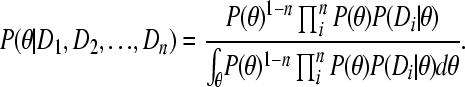 |
(A4) |
Moving the P(θ) in (A4) out of the products results in equivalence of (A1) and (A2).▪
The denominator in (A2) can be built up during the MCMC run. The main difference between (A1) and (A2) is that the latter allows completely independent calculation for the unlinked loci and therefore allows easy distribution of the inference on a computer cluster or even computer grids, facilitating the analysis of data sets with many unlinked loci.
The Bayesian inference offers a convenient tool for comparing different population models without requiring that models be nested. The marginal likelihoods are normally not computed during an MCMC run because these normalizing weights cancel in comparisons during the run. They need to be computed and recorded, however, when the combined marginal likelihoods need to be calculated; to do that we must evaluate the denominator of (A1):
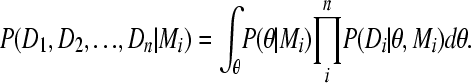 |
(A5) |
Theorem 2. The combined marginal likelihoods over all independent data blocks can be calculated as a product of independently calculated marginal likelihoods for each data block and a constant.
Proof. The combined estimator of the posterior distribution is
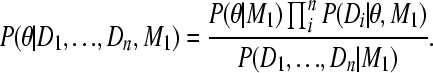 |
(A6) |
Converting the likelihoods using posteriors on the right,
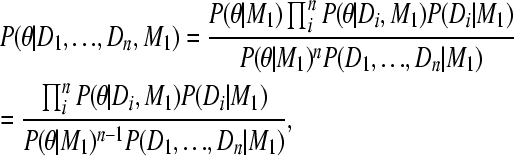 |
(A7) |
and moving P(D1, …, Dn | M1) to the left and P(θ | D1, … , Dn, M1) to the right results in
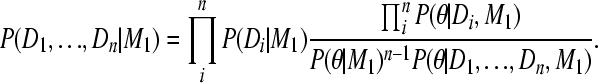 |
(A8) |
The fraction has to be a constant with respect to θ because both the product of the individual marginal likelihoods and the combined marginal likelihood on the left are also constants with respect to θ:
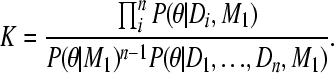 |
(A9) |
Moving the combined posterior and integrating both sides with θ leads to a reexpression of K,
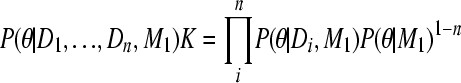 |
(A10) |
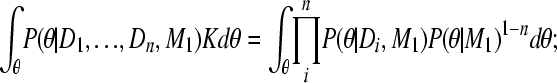 |
(A11) |
and because
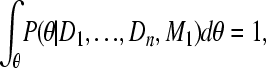 |
(A12) |
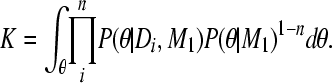 |
(A13) |
This allows the calculation of the combined marginal likelihood using independent inferences
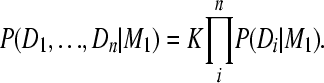 |
(A14) |
▪
The denominator in (A2) is equivalent to K and has already been calculated during the MCMC run; it can be reused to calculate the combined marginal likelihoods.
Calculation of the scaling factor K in MIGRATE:
In a Bayesian inference run of MIGRATE, K is calculated from the recorded posterior probabilities P(θ | Di) and the prior P(θ) for a particular model M where θ are all the parameters of the model and Di is the data for each unlinked locus. For example, in a simple one-parameter scenario, θ = α, we record α and its prior during the MCMC run. Then we construct a histogram of the α-values that represents the posterior distribution P(α | D). The prior distribution is also calculated at the values of the histogram columns. Summing over the histogram corrected for the overuse of the prior approximates the integral and calculates K. With a single locus, K = 1 and the “combined” marginal likelihood is the same as the single-locus marginal likelihood. With multiple parameters the integral will be multidimensional. If we assume that the parameters are independent of each other, the integration can be simplified. If we believe that the parameters are correlated, then we would need to calculate a multidimensional histogram; this is more tedious but certainly doable. MIGRATE uses the assumptions that parameters are independent because in our experience mutation-scaled migration rates and mutation-scaled population sizes are almost uncorrelated.
Specification of population models when some populations are isolated:
MIGRATE uses two options to specify particular population models. The connection matrix allows the specification of directionality of gene flow, such as symmetric numbers of immigrants, symmetric immigration rates, average immigration rates, and immigration rates fixed to particular constants, for example, zero. If constants other than zero are used, then the start parameter settings need to be used in addition to the connection matrix to specify the values. This system allows approximating models where the populations are isolated from each other (Table 6) by inserting immigration rates that are very close to zero. For the humpback whale example we fixed all immigration rates to an isolated population as 100 times smaller than the mutation rate.
Run time considerations:
The run time of MCMC programs is often difficult to predict because little automatic control can be given to users to check whether the MCMC chain has converged and enough samples from the desired distribution have been taken. Almost all applications have a tendency to sample too few steps along the MCMC chain. The faster a single step in the chain can be evaluated, the more steps can be sampled in the same time. The runs in this work all used similar run–parameter options (data sets and parameters are available in File S1). The run time values in Fig. 3 for data set 10 were 18 min using 4 different scaling classes, 70 min using 16 scalers, and 152 min using 32 scalers. In MIGRATE a deliberate decision was made not to farm out the Metropolis-coupled Markov chain sampling that is used for the thermodynamic integration; the run time increases proportionally with the number of chains. The expected run times based on the shortest run with 4 concurrent chains took 18 min (4 × 4.5 min), for 16 concurrent chains × 4.5 = 72 min, and for 32 × 4.5 = 144 min; the actual run times (18, 72, and 152 min) fit these values well. The run time is dependent on the number of sampling locations and the number of individuals in each location. For large data sets these values change to hours or days. Except for the 20-location example, we deliberately ran only small data sets for which we could easily establish convergence of the MCMC chain. Different Bayes factor runs are independent of each other and therefore one can run many models at the same time on a cluster. Many analyses in this work were run, in fact, on the high-performance cluster at Florida State University.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.112532/DC1.
References
- Beerli, P., 1998. Estimation of migration rates and population sizes in geographically structured populations, pp. 39–53 in Advances in Molecular Ecology (NATO Science Series A: Life Sciences, Vol. 306), edited by G. Carvalho. IOS Press, Amsterdam,.
- Beerli, P., 2004. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol. Ecol. 13 827–836. [DOI] [PubMed] [Google Scholar]
- Beerli, P., 2006. Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics 22 341–345. [DOI] [PubMed] [Google Scholar]
- Beerli, P., 2008. MIGRATE documentation (version 3.0). Technical Report. http://popgen.sc.fsu.edu.
- Beerli, P., 2009. How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use? pp. 42–79 in Population Genetics for Animal Conservation (Conservation Biology, Vol. 17), edited by G. Bertorelle, M. W. Bruford, H. C. Hauffe, A. Rizzoli and C. Vernesi. Cambridge University Press, Cambridge, UK.
- Beerli, P., and J. Felsenstein, 1999. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, P., and J. Felsenstein, 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens, B., A. Bankhead, P. Joyce and J. Sullivan, 2005. Testing population genetic structure using parametric bootstrapping and MIGRATE-N. Genetica 124 71–75. [DOI] [PubMed] [Google Scholar]
- Corander, J., P. Marttinen, J. Sirén and J. Tang, 2008. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, M. H., N. J. R. Fagundes, H. C. Rosenbaum, M. S. Leslie, P. H. Ott et al., 2008. Mitochondrial DNA diversity of the Southwestern Atlantic humpback whale (Megaptera novaeangliae) breeding area off Brazil, and the potential connections to Antarctic feeding areas. Conserv. Genet. 9 1253–1262. [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14 2611–2620. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J., 2005. Accuracy of coalescent likelihood estimates: Do we need more sites, more sequences, or more loci? Mol. Biol. Evol. 23 691–700. [DOI] [PubMed] [Google Scholar]
- Friel, N., and A. Pettitt, 2005. Marginal likelihood estimation via power posteriors. Technical Report 05-10, Department of Statistics, University of Glasgow, Glasgow, UK.
- Friel, N., and A. Pettitt, 2008. Marginal likelihood estimation via power posteriors. J. R. Stat. Soc. Ser. B 70 589–607. [Google Scholar]
- Gelman, A., and X.-L. Meng, 1998. Simulating normalizing constants: from importance sampling to bridge sampling to path sampling. Stat. Sci. 13 163–185. [Google Scholar]
- Geyer, C. J., and E. A. Thompson, 1995. Annealing Markov-chain Monte-Carlo with applications to ancestral inference. J. Am. Stat. Assoc. 90 909–920. [Google Scholar]
- Green, P. J., 1995. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika 82 711–732. [Google Scholar]
- Guillot, G., 2008. Inference of structure in subdivided populations at low levels of genetic differentiation—the correlated allele frequencies model revisited. Bioinformatics 24 2222–2228. [DOI] [PubMed] [Google Scholar]
- Heled, J., and A. J. Drummond, 2008. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger, K. E., P. O. Lewis and D. K. Dey, 2002. A Bayesian approach to inferring population structure from dominant markers. Mol. Ecol. 11 1157–1164. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1991. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 7 1–44. [Google Scholar]
- Hudson, R. R., D. D. Boos and N. L. Kaplan, 1992. a A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9 138–151. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Slatkin and W. P. Maddison, 1992. b Estimation of levels of gene flow from DNA sequence data. Genetics 132 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck, J., and P. Andolfatto, 2007. Inference of population structure under a Dirichlet process model. Genetics 175 1787–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck, J. P., and F. Ronquist, 2005. Bayesian analysis of molecular evolution using MrBayes, pp. 183–232 in Statistical Methods in Molecular Evolution, edited by R. Nielsen. Springer, New York.
- Kass, R. E., and A. E. Raftery, 1995. Bayes factors. J. Am. Stat. Assoc. 90 773–795. [Google Scholar]
- Kingman, J. F. C., 2000. Origins of the coalescent: 1974–1982. Genetics 156 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner, M., 2006. Lamarc 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22 768–770. [DOI] [PubMed] [Google Scholar]
- Lartillot, N., and H. Philippe, 2006. Computing Bayes factors using thermodynamic integration. Syst. Biol. 55 195–207. [DOI] [PubMed] [Google Scholar]
- Manel, S., F. Berthoud, E. Bellemain, M. Gaudeul, J. E. Swenson et al., 2007. A new individual-based geographic approach for identifying genetic discontinuities. Mol. Ecol. 16 2031–2043. [DOI] [PubMed] [Google Scholar]
- Michalakis, Y., and L. Excoffier, 1996. A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142 1061–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigel, J. E., 2002. Is FST obsolete? Conserv. Genet. 3 167–173. [Google Scholar]
- Newton, M. A., and A. E. Raftery, 1994. Approximate Bayesian inference with the weighted likelihood bootstrap. J. R. Stat. Soc. Ser. B (Methodol.) 56 3–48. [Google Scholar]
- Olavarría, C., C. Baker, C. Garrigue, M. Poole, N. Hauser et al., 2007. Population structure of South Pacific humpback whales and the origin of the eastern Polynesian breeding grounds. Mar. Ecol. Prog. Ser. 330 257–268. [Google Scholar]
- Palsbøll, P. J., M. Bérubé and F. Allendorf, 2007. Identification of management units using population genetic data. Trends Ecol. Evol. 22 11–16. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multi-locus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, M., and F. Rousset, 1995. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86 248–249. [Google Scholar]
- Rousset, F., 1996. Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F., 2008. GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Res. 8 103–106. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 2005. Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol. Ecol. 14 67–73. [DOI] [PubMed] [Google Scholar]
- Strobeck, C., 1987. Average number of nucleotide differences in a sample from a single subpopulation: a test for population subdivision. Genetics 117 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L., 2003. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts.
- Waples, R., and O. E. Gaggiotti, 2006. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 15 1419–1439. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., and W. G. Hill, 2002. Estimating F-statistics. Annu. Rev. Genet. 36 721–750. [DOI] [PubMed] [Google Scholar]