

Abstract
Horizontal gene transfer (HGT) is believed to be a major source of genetic variation, particularly for prokaryotes. It is believed that horizontal gene transfer plays a major role in shaping bacterial genomes and is also believed to be responsible for the relatively rapid dissemination and acquisition of new, adaptive traits across bacterial strains. Despite the importance of horizontal gene transfer as a major source of genetic variation, the bulk of research on theoretical evolutionary dynamics and population genetics has focused on point mutations (sometimes coupled with gene duplication events) as the main engine of genomic change. Here, we seek to specifically model HGT processes in bacterial cells, by developing a mathematical model describing the influence that conjugation-mediated HGT has on the mutation–selection balance in an asexually reproducing population of unicellular, prokaryotic organisms. It is assumed that mutation–selection balance is reached in the presence of a fixed background concentration of antibiotic, to which the population must become resistant to survive. We find that HGT has a nontrivial effect on the mean fitness of the population. However, one of the central results that emerge from our analysis is that, at mutation–selection balance, conjugation-mediated HGT has a slightly deleterious effect on the mean fitness of a population. Therefore, we conclude that HGT does not confer a selection advantage in static environments. Rather, its advantage must lie in its ability to promote faster adaptation in dynamic environments, an interpretation that is consistent with the observation that HGT can be promoted by environmental stresses on a population.
HORIZONTAL gene transfer (HGT) is any form of direct transfer of genetic material between two organisms, where one organism is not the parent of the other (the latter case is known as vertical gene transfer) (Ochman et al. 2000; Brown 2003; Kurland et al. 2003; Gogarten and Townsend 2005). HGT has become a subject of great interest for both molecular and evolutionary biologists, because it is believed that HGT plays a large role in reshaping prokaryotic genomes (Ochman et al. 2000; Brown 2003; Kurland et al. 2003; Gogarten and Townsend 2005). HGT is believed to be primarily responsible for the rapid spread of antibiotic drug resistance in bacterial populations (Walsh 2000).
Currently, there are three known mechanisms by which HGT occurs (Ochman et al. 2000; Brown 2003; Kurland et al. 2003; Gogarten and Townsend 2005): (1) transformation, when an organism collects genetic material from its environment; (2) transduction, when a virus directly infiltrates a bacterium with genetic material; and (3) bacterial conjugation, when a bacterium transfers genetic information via intercellular contact with another bacterium.
Bacterial conjugation is believed to be the most important mechanism responsible for HGT (Ochman et al. 2000; Brown 2003; Kurland et al. 2003; Gogarten and Townsend 2005), and so we focus on developing mathematical models describing the role that conjugation-mediated HGT has on the mutation–selection balance of bacterial populations. Given the presumed importance that HGT has for the spread of antibiotic drug resistance in bacterial populations, the mathematical models we develop look at the influence of HGT on the mutation–selection balance in the presence of an antibiotic. This is not the most realistic setting in which to study HGT, since it is more relevant to look at the role that HGT plays in the evolution and spread of antibiotic drug resistance in an initially nonresistant population. Nevertheless, it is important to understand the mutation–selection balance first, since this serves as a starting point for modeling dynamics.
The best-characterized bacterial conjugation system is the F+/F− system (Russi et al. 2008). Here, a bacterium containing what is termed an F-plasmid fuses with a bacterium lacking the F-plasmid. The bacterium containing the F-plasmid is termed an F+ bacterium, while the bacterium that does not contain this plasmid is termed an F− bacterium. When the F+ bacterium meets an F− bacterium, it transfers one of the strands of the F-plasmid to the F− bacterium via a pilus. Once a strand of the F-plasmid has been transferred from the F+ bacterium to the F− bacterium, a copy of the plasmid in both cells is produced by daughter strand synthesis using the DNA template strands. The F− bacterium then becomes an F+ bacterium that transcribes its own pilus and is able to transfer the F+ plasmid to other bacteria in the population (Russi et al. 2008). This process is illustrated in Figure 1.
Figure 1.—

Illustration of the process of bacterial conjugation. In steps 1 and 2, an F+ bacterium containing the F-plasmid (blue) binds to an F− bacterium lacking the plasmid. One of the template strands from the F-plasmid then moves into the F− bacterium, as shown in step 3. In step 4, the complementary strands are synthesized to reform the complete F-plasmids in both bacteria. Both bacteria are now of the F+ type.
The F+/F− system is in some ways atypical for bacterial conjugation systems: The F-plasmid system studied in the K12 strain of Escherichia coli was permanently derepressed, which meant that conjugation between F+ and F− cells occurred at a significantly elevated rate. Generally, conjugative plasmids tend to be repressed, so that only a small fraction of the plasmid-bearing bacterial population is able to participate in conjugation at any given time (Ghigo 2001). It is believed that this reduces the metabolic costs associated with continuously maintaining the enzymatic machinery necessary for conjugation (Ghigo 2001). Indeed, it is known that mutant forms of the F-plasmid system are permanently derepressed (Ghigo 2001), so it is possible that these are the strains that were accidentally generated under the experimental conditions that the plasmids were being studied. Nevertheless, because the F-plasmid system is one of the best-characterized bacterial conjugation systems, and because it is representative of all known bacterial conjugation systems, we believe it makes sense to base our initial model for conjugation-mediated HGT on the F-plasmid system.
METHODS
We assume that the genome of each bacterium consists of two DNA molecules. The first DNA molecule contains all of the genes necessary for the proper growth and reproduction of the bacterium itself. It corresponds to the large, circular chromosome defining the bacterial genome. We assume that there exists a wild-type genome characterized by a “master” DNA sequence. It is assumed that a bacterium with the master genome has a wild-type fitness, or first-order growth rate constant, given by 1. Such a bacterium is termed viable. Furthermore, we assume that any mutation to the bacterial genome renders the genome defective, so that the bacterium then has a fitness of 0. Bacteria with defective genomes are termed nonviable. This is known as the single-fitness-peak approximation in quasispecies theory (Tannenbaum and Shakhnovich 2005).
The second DNA molecule is the F-plasmid, which we assume consists of two regions. The first region comprises the various genes necessary for bacterial conjugation. The second region is assumed to encode for the various enzymes conferring resistance to a given antibiotic. As with the single-fitness-peak approximation made for the bacterial genome, we assume that there are master sequences for both the conjugation and the antibiotic drug resistance regions. If the region coding for bacterial conjugation corresponds to a given master sequence, then, assuming that the bacterium is also viable, the F-plasmid may be copied into another viable F− bacterium. Otherwise, we assume that the plasmid cannot be copied into another bacterium, in which case the bacterium is treated as an F− bacterium. Similarly, if the region coding for antibiotic drug resistance corresponds to a given master sequence, then we assume that the bacterium is resistant to the antibiotic. Otherwise, the bacterium is not resistant to the antibiotic and is assumed to die with a first-order rate constant κD. We assume that only viable bacteria interact with the antibiotic, since nonviable bacteria do not grow and so may be treated as dead.
A given viable genome may be characterized by a two-symbol sequence σ = ± ±, specifying the state of the conjugation and resistance portions of the plasmid, respectively. A “+” is taken to signify that the given genome region is identical to the corresponding master sequence, and a “−” is taken to signify that the given genome region differs from the corresponding master sequence. To develop the evolutionary dynamics equations governing this population, we let nσ denote the number of organisms in the population with genome σ. We wish to develop expressions for dnσ/dt for the various σ's. We do not consider nonviable genomes, since they do not reproduce or participate in the conjugation process and therefore do not contribute to the evolutionary dynamics of the population.
The semiconservative replication of the bacterial genome is not necessarily error free, so that there is a probability p, the replication fidelity, that a given template strand will produce a daughter genome that is identical to the original parent. Because our genome consists of three genome regions, we may define three such probabilities, denoted pv, pc, and pr, corresponding to the replication fidelities for the viability, conjugation, and resistance portions of the genome. If we assume that sequence lengths are long, then making an assumption known as the neglect of back mutations (Tannenbaum and Shakhnovich 2005), we assume that a template strand derived from a parent that differs from the master genome produces a daughter that differs from the master genome with probability 1.
We assume that conjugation occurs between a viable F+ bacterium and a viable F− bacterium. Thus, conjugation can occur only between a bacterium of type + ± and a bacterium of type − ±. This process is modeled as a second-order collision reaction with a rate constant γ. The conjugation process itself involves the transfer of one of the strands of the plasmid from the F+ bacterium to the F− bacterium, so that the full plasmid needs to be resynthesized in both bacteria via daughter strand synthesis. This introduces the possibility of replication errors in either one of the bacteria.
It should be emphasized that we are assuming for simplicity that all bacteria in the population contain exactly one plasmid. We also assume that, during conjugation, the plasmid transferred from the F+ bacterium replaces the plasmid in the F− bacterium. This is a simplifying assumption that will obviously have to be reexamined in future research, where we anticipate developing more accurate models that allow for variable plasmid numbers in the bacterial cell. The basis for this assumption derives from the observation that plasmids of similar compatibility classes cannot coexist in the same cell (Uhlin and Nordstrom 1975) and that bacteria can control the number of plasmids in the cell (Park et al. 2001).
Putting everything together, we obtain that the evolutionary dynamics equations are
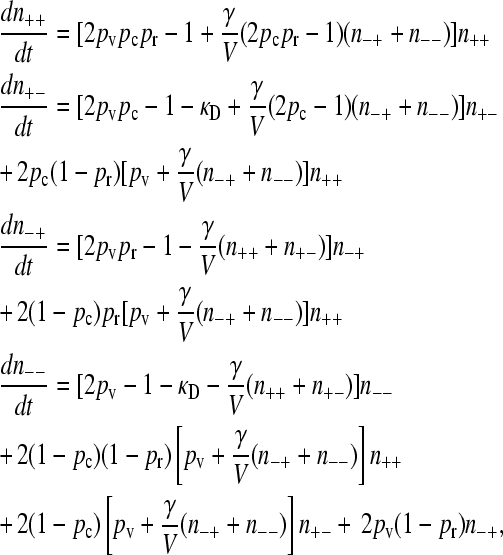 |
(1) |
where V is defined as the system volume. To put the equations into a form that makes the analysis of the mutation–selection balance possible, we define n to be the total population of organisms and then define population fractions xσ via xσ = nσ/n. We also define a population density ρ = n/V, and we assume that ρ is constant. The assumption of a constant ρ can be achieved if we assume that the system volume is not a constant, but rather grows with the population size in such a way to maintain a constant overall population density. The idea is that each cell takes up a certain amount of space, so that the total volume of the system is proportional to the total number of cells.
Converting from population numbers to population fractions, we obtain
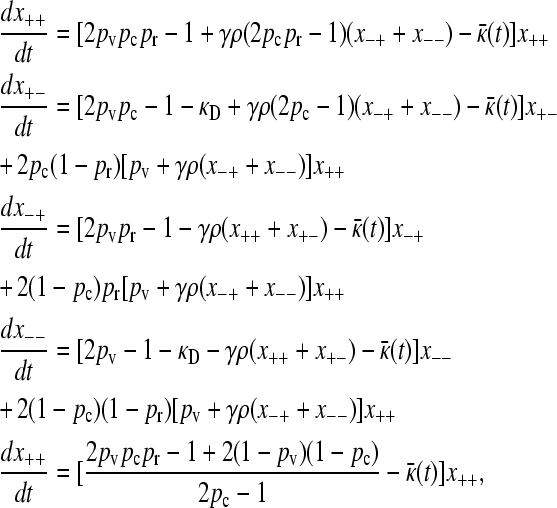 |
(2) |
where κ̄ (t)=(1/n)(dn/dt)=x+++x−++(1−κD)(x+−+x−−) is the mean fitness of the population or, equivalently, first-order growth constant of the population.
Another point to be noted from our equations is that, in their original formulation using absolute population numbers, the equations assume unrestricted exponential growth. However, when we change variables from population numbers to population fractions, then the form of the equations is identical to what would be obtained if we made a more realistic assumption that the population was growing in a chemostat (Tannenbaum and Shakhnovich 2005).
To determine the values for pv, pc, and pr, we assume that daughter strand synthesis has a per-base mismatch probability ɛ, which incorporates all DNA error-correction mechanisms such as proofreading and mismatch repair. Because we are assuming complementary double-stranded DNA molecules, we assume that all postreplication mismatches are corrected via various lesion repair mechanisms (e.g., nucleotide excision repair, NER). However, because at this stage there is no discrimination between parent and daughter strands, a mismatch either is correctly repaired with probability 1/2 or is fixed as a mutation in the genome with probability 1/2 (Voet et al. 2008). Thus, the net per-base mismatch probability is ɛ/2. If the total sequence length is L, then the probability of producing a mutation-free daughter from a given parent template strand is (1 − ɛ/2)L.
If we define μ = Lɛ, so that μ is the average number of mismatches per template strand per replication cycle, and if we assume that 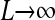 while μ is held constant, then we obtain that
while μ is held constant, then we obtain that 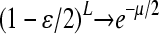 . For the case of the three-gene model we are considering, we let Lv, Lc, and Lr denote the lengths of the genome controlling viability, conjugation, and resistance, respectively. Defining L = Lv + Lc + Lr and αv = Lv/L, αc = Lc/L, and αr = Lr/L, we then obtain that
. For the case of the three-gene model we are considering, we let Lv, Lc, and Lr denote the lengths of the genome controlling viability, conjugation, and resistance, respectively. Defining L = Lv + Lc + Lr and αv = Lv/L, αc = Lc/L, and αr = Lr/L, we then obtain that
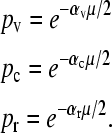 |
(3) |
It should be noted that holding μ constant in the limit of infinite genome length is equivalent to assuming a fixed per genome replication fidelity in the limit of long genomes.
RESULTS AND DISCUSSION
We present the mean fitness at mutation–selection balance, denoted by 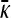 , for two different sets of parameter regimes: (1) arbitrary κD, but with
, for two different sets of parameter regimes: (1) arbitrary κD, but with 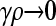 and
and 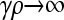 , and (2) arbitrary γρ, but with
, and (2) arbitrary γρ, but with 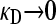 and
and 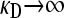 .
.
Details of the derivations of the various results are in the appendix.
Behavior of 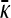 for arbitrary κD:
for arbitrary κD:
The steady-state mean fitnesses for arbitrary κD for the 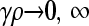 cases are provided in Table 1. We can show that
cases are provided in Table 1. We can show that 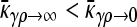 .
.
TABLE 1.
A summary of the steady-state mean fitness results obtained from our model
| Regime | Mean fitness |
|---|---|
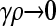 , arbitrary κD , arbitrary κD
|
max{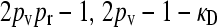 } } |
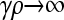 , arbitrary κD , arbitrary κD
|
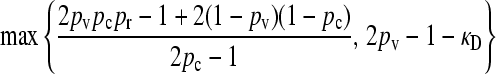 |
 |
 |
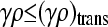 , , 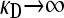
|
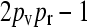 |
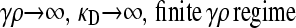 |
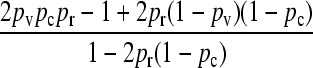 |
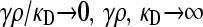 |
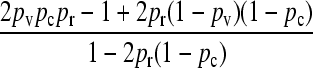 |
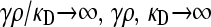 |
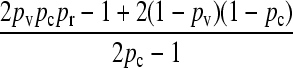 |
Figure 2 shows plots of 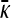 vs. μ for both the
vs. μ for both the 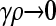 and the
and the 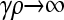 limits. Plots were obtained using both the analytical formulas obtained in this article, as well as via stochastic simulations of replicating organisms.
limits. Plots were obtained using both the analytical formulas obtained in this article, as well as via stochastic simulations of replicating organisms.
Figure 2.—

Plots of 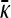 vs. μ for both the
vs. μ for both the 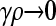 and the
and the 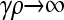 limits. The parameter values we took are αv = 0.6, αc = αr = 0.2, and κD = 10. We show both analytical results and results from stochastic simulations. The analytical results are plotted using thin solid lines, where the top curve corresponds to the γρ = 0 result, while the bottom curve corresponds to the γρ = ∞ result. The dotted line corresponds to the stochastic simulation for γρ = 0, and the dashed line corresponds to the stochastic simulation for γρ = ∞. Parameter values for the stochastic simulations were Lv = 30, Lc = Lr = 10, and a population size of 1000.
limits. The parameter values we took are αv = 0.6, αc = αr = 0.2, and κD = 10. We show both analytical results and results from stochastic simulations. The analytical results are plotted using thin solid lines, where the top curve corresponds to the γρ = 0 result, while the bottom curve corresponds to the γρ = ∞ result. The dotted line corresponds to the stochastic simulation for γρ = 0, and the dashed line corresponds to the stochastic simulation for γρ = ∞. Parameter values for the stochastic simulations were Lv = 30, Lc = Lr = 10, and a population size of 1000.
Behavior of 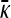 for arbitrary γρ:
for arbitrary γρ:
Now we consider the behavior of 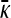 for arbitrary values of γρ, but where κD is either very small or very large. Combined with the results of the previous subsection, we may then piece together a qualitative sketch of how
for arbitrary values of γρ, but where κD is either very small or very large. Combined with the results of the previous subsection, we may then piece together a qualitative sketch of how 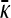 depends on κD and γρ.
depends on κD and γρ.
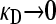 :
:
When 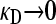 , there is no selective advantage for maintaining antibiotic drug resistance genes in the genome, and so we expect these genes to be lost to genetic drift. Thus, we expect, at mutation–selection balance, that x++ = x−+ = 0. From Table 1, we also have that
, there is no selective advantage for maintaining antibiotic drug resistance genes in the genome, and so we expect these genes to be lost to genetic drift. Thus, we expect, at mutation–selection balance, that x++ = x−+ = 0. From Table 1, we also have that 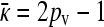 .
.
Furthermore, the fraction of viable conjugators, x++ + x+−, exhibits a transition as a function of γρ. For sufficiently small values of γρ, we have that x++ + x+− = 0, while for sufficiently large values of γρ, we have that
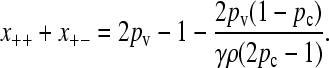 |
(4) |
The transition between the two regimes may be shown to occur at
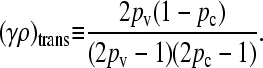 |
(5) |
It may be shown that the disappearance of the conjugators below the critical value of γρ corresponds to a localization to delocalization transition over the portion of the plasmid coding for conjugation, so that this transition is a conjugation-mediated HGT analog of the well-known error catastrophe from quasispecies theory (Tannenbaum and Shakhnovich 2005).
To understand this behavior, we note that plasmids with defective genes for conjugation nevertheless replicate due to the replication of the bacteria in which they reside. Thus, for plasmids with functional genes for conjugation to be preserved in the population, their additional growth rate due to conjugation must overcome the loss of functionality due to replication mistakes in the genes controlling conjugation. If the conjugation rate is too slow and unable to overcome this loss of functionality, then the fraction of conjugators in the population drops to zero.
Figure 3 illustrates the regimes, as a function of μ and γρ, where a positive fraction of conjugators exist at steady state and where the fraction of conjugators is zero. This is computed for the κD = 0 limit. Note that, as μ increases, γρ must be pushed to higher values so that there is a positive fraction of conjugators at steady state. As explained before, this increase in γρ is necessary to overcome the mutation-induced loss of functionality as μ increases.
Figure 3.—

Regimes of existence and nonexistence of conjugators as a function of μ and γρ, where κD = 0. The boundary between the two regimes was computed analytically.
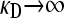 :
:
We now consider the case where 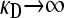 . In contrast to the case where
. In contrast to the case where 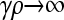 of the previous subsection, where we could solve for
of the previous subsection, where we could solve for 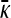 for arbitrary values of κD, here we cannot readily analytically solve for
for arbitrary values of κD, here we cannot readily analytically solve for 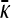 for arbitrary values of γρ. However, we can obtain analytical solutions for
for arbitrary values of γρ. However, we can obtain analytical solutions for 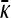 in certain limiting cases of γρ and then interpolate between the two solution regimes. As will be seen in the subsection comparing theory and simulation, this approach turns out to be fairly accurate.
in certain limiting cases of γρ and then interpolate between the two solution regimes. As will be seen in the subsection comparing theory and simulation, this approach turns out to be fairly accurate.
In the first limiting case, we assume that γρ remains finite in the limit that 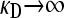 . This ensures that x+− = x−− = 0, since the rate of death due to the presence of antibiotics is so fast that no nonresistant genotypes are present in the population.
. This ensures that x+− = x−− = 0, since the rate of death due to the presence of antibiotics is so fast that no nonresistant genotypes are present in the population.
We then obtain either that 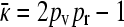 or that
or that 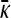 is the solution to the following equation:
is the solution to the following equation:
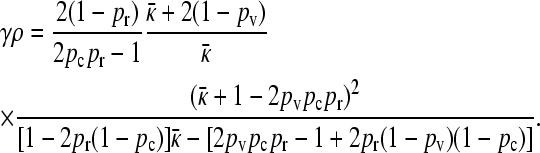 |
(6) |
In the first case, we have that x++ = 0, while in the second case we have that x++ > 0. The transition between the two regimes may be shown to occur at
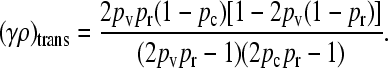 |
(7) |
where x++ = 0 for γρ ≤ (γρ)trans and x++ > 0 for γρ > (γρ)trans. We may show that this expression for (γρ)trans is larger than the corresponding expression for the κD = 0 case.
To understand the behavior of 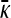 where γρ > (γρ)trans, we consider the asymptotic behavior of
where γρ > (γρ)trans, we consider the asymptotic behavior of 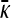 in the limit as
in the limit as 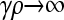 . In this case, Equation 6 reduces to
. In this case, Equation 6 reduces to
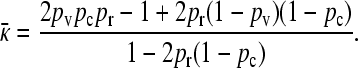 |
(8) |
We may show that this expression is smaller than the expression for 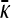 obtained in the arbitrary κD, infinite γρ case.
obtained in the arbitrary κD, infinite γρ case.
We now consider the second limiting case in the 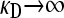 limit, specifically where γρ is itself infinite. Here, however, the ratio between κD and γρ plays an important role in the competition between death of nonresistant bacteria and their “rescue” by conjugation with resistant bacteria. Thus, here, we assume that both
limit, specifically where γρ is itself infinite. Here, however, the ratio between κD and γρ plays an important role in the competition between death of nonresistant bacteria and their “rescue” by conjugation with resistant bacteria. Thus, here, we assume that both 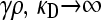 , but we take γρ/κD to have some given value in this limit.
, but we take γρ/κD to have some given value in this limit.
We may show that
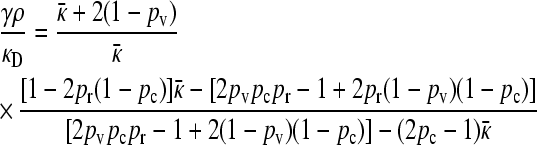 |
(9) |
and so obtain that
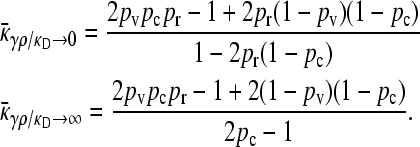 |
(10) |
Therefore, for large κD, we expect that 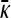 will initially be given by 2pvpr − 1 up to a critical value of γρ, after which it begins to decrease according to Equation 6. Once γρ becomes sufficiently large, we expect that the γρ/κD ratio is such that the functional form for
will initially be given by 2pvpr − 1 up to a critical value of γρ, after which it begins to decrease according to Equation 6. Once γρ becomes sufficiently large, we expect that the γρ/κD ratio is such that the functional form for 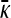 transitions from the finite γρ solution to the infinite γρ, fixed γρ/κD solution. To estimate the transition point between the two solution regimes, we equate the values for γρ as a function of
transitions from the finite γρ solution to the infinite γρ, fixed γρ/κD solution. To estimate the transition point between the two solution regimes, we equate the values for γρ as a function of 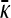 for the two solutions. This allows us to solve for
for the two solutions. This allows us to solve for 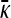 and thereby allows us to solve for γρ.
and thereby allows us to solve for γρ.
We then obtain that the transition point occurs at
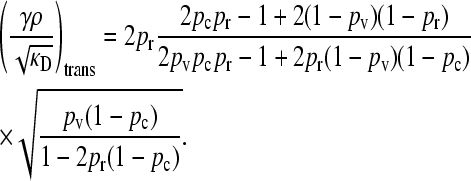 |
(11) |
Note that, as 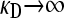 , we have that
, we have that 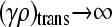 and
and 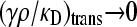 , so the assumptions that allowed us to make the calculation above are valid.
, so the assumptions that allowed us to make the calculation above are valid.
Figure 4 shows three plots of 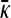 vs. γρ for κD = 10. One of the plots was obtained by numerically solving for the mutation–selection balance using fixed-point iteration. The other two plots correspond to the infinite κD, finite γρ and infinite κD, fixed γρ/κD expressions for
vs. γρ for κD = 10. One of the plots was obtained by numerically solving for the mutation–selection balance using fixed-point iteration. The other two plots correspond to the infinite κD, finite γρ and infinite κD, fixed γρ/κD expressions for 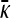 given in the preceding subsections. Note that already for κD = 10 the approximate analytical solutions capture the dependence of
given in the preceding subsections. Note that already for κD = 10 the approximate analytical solutions capture the dependence of 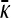 on γρ fairly accurately.
on γρ fairly accurately.
Figure 4.—

Plots of 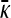 vs. γρ for κD = 10, μ = 0.4, αv = 0.6, αc = αr = 0.2. The plot marked with the solid line was obtained by numerically solving for
vs. γρ for κD = 10, μ = 0.4, αv = 0.6, αc = αr = 0.2. The plot marked with the solid line was obtained by numerically solving for 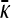 using fixed-point iteration. The dashed line was obtained by using the infinite κD, finite γρ expression for
using fixed-point iteration. The dashed line was obtained by using the infinite κD, finite γρ expression for 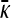 , while the dotted line was obtained by using the infinite κD, fixed γρ/κD expression for
, while the dotted line was obtained by using the infinite κD, fixed γρ/κD expression for 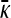 .
.
Conclusions:
We developed a mathematical model describing the role that conjugation-mediated HGT has on the mutation–selection balance of a unicellular, asexually reproducing, prokaryotic population. We found that, in a static environment at mutation–selection balance, conjugation actually reduces the mean fitness of the population. However, by studying the dependence of the mean fitness on γρ for large values of κD, the antibiotic-induced first-order death rate constant, we find that the behavior is somewhat more complicated: For small values of γρ, the mean fitness is constant, and the fraction of viable conjugators in the population is 0. At a critical value of γρ, the fraction of viable conjugators begins to increase, and the mean fitness decreases to its minimum value. After reaching its minimum, the mean fitness increases asymptotically to the 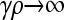 limit, which is nevertheless smaller than the small γρ value for the mean fitness. We developed approximate analytical solutions for the functional dependence of the mean fitness on γρ in the limit of large κD and found that these solutions agree well with simulation. Although the fitness variations as a function of γρ were fairly small for the parameter values studied, we believe that this is nontrivial behavior that is important to characterize.
limit, which is nevertheless smaller than the small γρ value for the mean fitness. We developed approximate analytical solutions for the functional dependence of the mean fitness on γρ in the limit of large κD and found that these solutions agree well with simulation. Although the fitness variations as a function of γρ were fairly small for the parameter values studied, we believe that this is nontrivial behavior that is important to characterize.
Although the results of our article are based on a highly simplified model, they nevertheless suggest that HGT does not provide a selective advantage in a static environment. This is likely due to the fact that, due to mutation, HGT can destroy antibiotic drug resistance in a previously resistant cell. While HGT can also confer resistance to a nonresistant cell, natural selection alone is sufficient to maximize the population mean fitness in a static environment. HGT simply has the net effect of destroying favorable genes, thereby lowering the mean fitness. This result may be viewed as an example of the “If it is not broken, do not fix it” principle.
Thus, on the basis of the results of this article, we argue that HGT likely has a selective advantage only in dynamic environments, where it would act to speed up rates of adaptation. While this result needs to be checked in future research, it is nevertheless consistent with the observation that bacteria can regulate their rates of HGT. For example, it is known that, in response to stress, bacteria can activate the SOS response (Beaber et al. 2004), which has the effect of increasing rates of HGT. It is also suspected that bacteria can increase their mutation rates in response to stress (Bjedov et al. 2003), which, coupled with the observation that mismatch-repair-deficient cells, or mutators, have significantly increased rates of recombination and HGT (Denamur et al. 2000), suggests that there is a strong correlation between HGT, stress, and adaptation. This is consistent with our results suggesting that HGT should be kept at a minimal level in static environments and increased in dynamic environments. It is also worth mentioning that while conjugation-mediated HGT has not been specifically modeled before in this manner (at least to our knowledge), other HGT-like models have been studied (Cohen et al. 2005; Park and Deem 2007), and these studies have found that HGT does indeed allow for faster adaptation in dynamic environments (Cohen et al. 2005).
It should be noted that we have obtained our conclusions by an analysis of the mean fitness of the population. Thus, our analysis is based on what is known as a group selection approach, whereby we assume that what is beneficial for the population as a whole dictates what will actually be observed. The group selection approach is known to have some serious drawbacks, specifically because it cannot explicitly account for selfish behavior such as defection from a cooperative strategy or independently replicating entities that act as parasites on a larger host system (the plasmids may be viewed as such an example).
However, in a certain sense, even analyses based on individual-selection models are themselves group selection approaches. For example, in analyzing the dynamics of defection and cooperation, one looks at the rate of growth of defectors vs. the rate of growth of cooperators. If, by group selection, one means that the mean fitness of the entire population is used to determine the structure of the mutation–selection balance, then indeed this approach will fail to give the correct results. However, if two mean fitnesses are used, one for the defectors and one for the cooperators, then the resulting analysis will capture the coevolutionary dynamics between cooperators and defectors in the correct manner. Similarly, when dealing with host–parasite interactions, one does not simply consider the mean fitness of the host and study the influence that the parasites have on the host fitness. Rather, one works with mathematical models that consider both the host and the parasite fitnesses, which again leads to a coevolutionary dynamics that can lead to correct results. This is the approach one must take when dealing with plasmid–bacteria systems or with viral–bacteria systems.
Given the discussion in the above paragraph, it may then appear that there is something contradictory between the analysis we stated should be done on bacteria–plasmid systems and the analysis that we actually carried out. To resolve this, we must emphasize that the purpose of this article is not to characterize the coevolutionary dynamics of bacteria and plasmids. Rather, the purpose is to determine what effect the presence of plasmids and, in particular, the presence of plasmids that confer some selective advantage to the bacterial hosts (e.g., drug resistance) has on the overall fitness of a population of bacteria in a static environment. As stated, we find that the mean fitness has nontrivial aspects to its behavior, but the central result is that plasmids capable of moving between bacteria via conjugation actually have a deleterious effect on fitness in a static environment.
This in no way suggests that plasmids will therefore not undergo conjugation, since, as selfish genetic elements residing within bacterial cells, they are under a selection pressure to evolve a conjugation ability even if they negatively affect bacterial fitness (the case of viruses in the lytic phase makes this point very obvious). However, to the extent that bacteria can control rates of conjugation, the results of this article suggest that, in a static environment at mutation–selection balance, bacteria have an “interest” in keeping rates of conjugation as low as possible, as long as the costs associated with such control mechanisms are not prohibitive. This is consistent with the observation that HGT can become significantly elevated in response to stress.
There are several extensions of the model that should be considered in future research: First, we need to model the role that HGT plays in adaptive dynamics. Second, we need to develop more realistic models for the conjugation process itself. This includes moving away from the single-plasmid-per-bacterium model considered here and to actually model plasmid compatibility classes and the regulation of copy number inside bacterial cells. This also includes properly modeling the repression/derepression dynamics associated with the activation of the conjugation process itself. Third, our current model assumes that all plasmids have identical characteristics. This does not take into account that, in many models of recombination, a modifier allele that allows for cell-specific recombination rates is often considered and that the evolution of the recombination rate itself is often modeled. Such models will be useful to determine the optimal level of horizontal gene transfer in various environments and to understand the distribution of horizontal gene transfer rates in bacterial populations.
Finally, and perhaps most importantly, it is important to carry out experimental studies to see if the qualitative predictions made as to how the mean fitness of the population varies as a function of conjugation rate are correct. We believe that the existing model may be relevant for plasmid systems that have low copy number in their host cells. Recent work (Barrick et al. 2009) points to the kinds of experimental studies in this area that are desired.
APPENDIX: DERIVATION DETAILS OF THE ANALYTICAL RESULTS
Derivation of 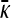 for arbitrary κD and
for arbitrary κD and 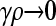 :
:
Due to the nature of exponential growth, for the population fractions to converge to a stable steady state we must have that 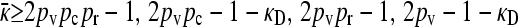 . Because 2pvpcpr − 1 < 2pvpr − 1 and 2pvpc − 1 − κD < 2pv − 1 − κD, it follows that
. Because 2pvpcpr − 1 < 2pvpr − 1 and 2pvpc − 1 − κD < 2pv − 1 − κD, it follows that 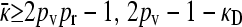 . However, if we then look at the steady-state version of Equation 2, obtained by setting the time derivatives to 0, we then obtain that x++ = x+− = 0. If x−+ > 0, then the third equation gives us that
. However, if we then look at the steady-state version of Equation 2, obtained by setting the time derivatives to 0, we then obtain that x++ = x+− = 0. If x−+ > 0, then the third equation gives us that 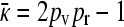 ; otherwise the fourth equation gives us
; otherwise the fourth equation gives us 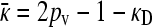 .
.
So, we have shown that 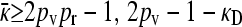 , and yet
, and yet 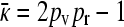 or 2pv − 1 − κD. These two requirements imply that
or 2pv − 1 − κD. These two requirements imply that 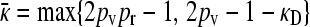 . Note that we have also shown that x++ + x+− = 0, so that our claim that conjugation is lost due to genetic drift has also been proven.
. Note that we have also shown that x++ + x+− = 0, so that our claim that conjugation is lost due to genetic drift has also been proven.
Derivation of 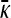 for arbitrary κD and
for arbitrary κD and 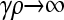 :
:
In the limit where 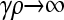 , we have that x−+ = x− − = 0. However, γρx−+ and γρx−− may converge to positive values. So, we define z−+ = γρx−+ and z−− = γρx−−.
, we have that x−+ = x− − = 0. However, γρx−+ and γρx−− may converge to positive values. So, we define z−+ = γρx−+ and z−− = γρx−−.
Because x−+ = x− − = 0, we also have that dx−+/dt = dx−−/dt = 0, and so from Equation 2 we have that
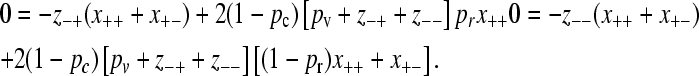 |
(A1) |
Summing these two equations and solving for z−+ + z−− gives
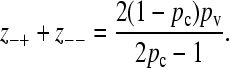 |
(A2) |
Substituting into the expressions for dx++/dt and dx+−/dt from Equation 2 we obtain, after some manipulation,
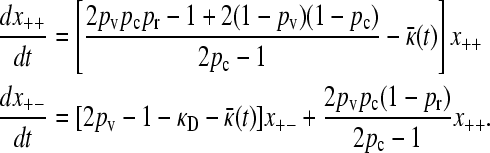 |
(A3) |
Following a similar argument to the  case, we obtain the expression for
case, we obtain the expression for  given in the main text.
given in the main text.
To prove that  , we need only show that
, we need only show that
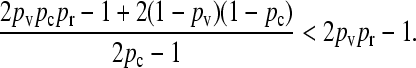 |
(A4) |
After some manipulation, it may be shown that this inequality is equivalent to pr < 1, which clearly holds, thereby proving the claim.
Derivation of 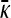 for κD→0 and arbitrary γρ:
for κD→0 and arbitrary γρ:
We can add the first two equations from Equation 2, and also the third and fourth equations, to obtain the pair of equations
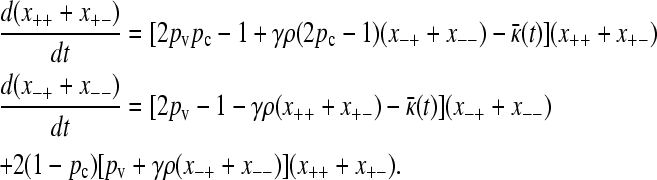 |
(A5) |
Summing these two equations then gives
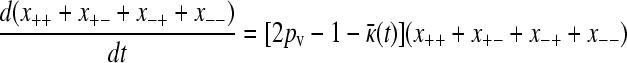 |
(A6) |
from which it follows that 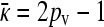 at steady state.
at steady state.
Substituting this value for 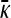 into the steady-state version of Equation A5, we obtain
into the steady-state version of Equation A5, we obtain
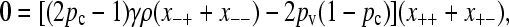 |
(A7) |
which gives either that x++ + x+− = 0 or that x−+ + x− − = 2pv(1 − pc)/[γρ(2pc − 1)]. If the second case holds, then since 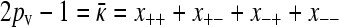 , we obtain that
, we obtain that
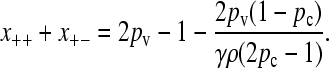 |
(A8) |
Now, for large values of γρ, we expect that the population will consist of a nonzero fraction of conjugators, so that x++ + x+− > 0. However, because x++ + x+− cannot be negative, we must have that
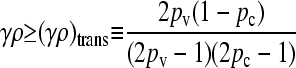 |
(A9) |
for x++ + x+− ≥ 0. Therefore, by continuity, we expect that x++ + x+− = 0 for γρ ≤ (γρ)trans and 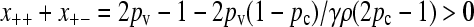 for
for 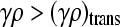 .
.
Derivation of 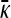 for κD→∞ and finite γρ:
for κD→∞ and finite γρ:
In this limiting case, although x+− = x−− = 0, it is possible that y+− ≡ κDx+− and y−− ≡ κDx−− have nonzero, finite values in the limit as 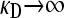 , and so we need to consider the effect of these quantities in our analysis. We then have that the steady-state version of Equation 2 reads
, and so we need to consider the effect of these quantities in our analysis. We then have that the steady-state version of Equation 2 reads
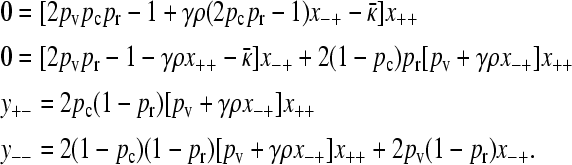 |
(A10) |
If x++ = 0 at steady state, then 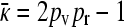 . So, let us consider the case where x++ > 0. Summing the first two equations from Equation A10 gives
. So, let us consider the case where x++ > 0. Summing the first two equations from Equation A10 gives
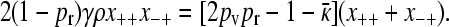 |
(A11) |
Summing the last two equations from Equation A10 then gives
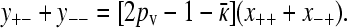 |
(A12) |
Now, in the limiting case being considered here, we have that 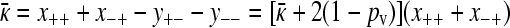 , and so,
, and so,
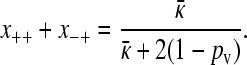 |
(A13) |
Since x++ > 0, the first equation from Equation A10 gives
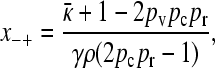 |
(A14) |
and so,
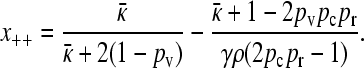 |
(A15) |
Substituting into Equation A11 gives the following nonlinear equation that 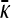 must satisfy,
must satisfy,
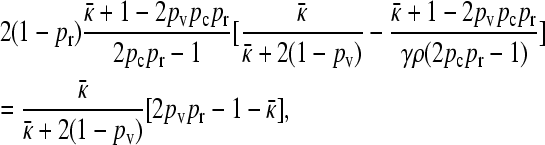 |
(A16) |
which, after some manipulation, may be shown to be equivalent to Equation 6.
To determine the critical value for the transition between the x++ = 0 and the x++ > 0 regimes, we note that if x++ is continuous at this transition, then we must have that x++ = 0 using the expression in Equation A15, which gives that 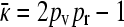 from Equation A16, so that
from Equation A16, so that 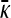 is also continuous at this transition. Solving for the critical value of γρ then gives
is also continuous at this transition. Solving for the critical value of γρ then gives
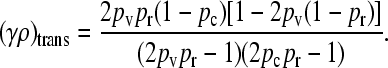 |
(A17) |
So, for γρ ≤ (γρ)trans, we have that x++ = 0 and 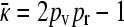 , while for γρ > (γρ)trans we have that x++ > 0 and
, while for γρ > (γρ)trans we have that x++ > 0 and 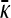 is given by the solution to Equation 8 or, equivalently, Equation A16.
is given by the solution to Equation 8 or, equivalently, Equation A16.
To show that this value for (γρ)trans is larger than the corresponding value obtained for κD = 0, we need to show that
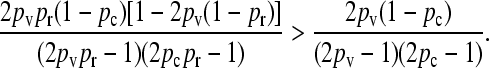 |
(A18) |
After some manipulation, this inequality may be shown to be equivalent to
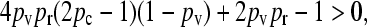 |
(A19) |
which clearly holds, and so the inequality is established.
Finally, to show that the value of 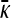 as
as 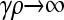 is smaller than the value of
is smaller than the value of 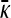 obtained in the arbitrary κD,
obtained in the arbitrary κD, 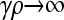 limit, we need to show that
limit, we need to show that
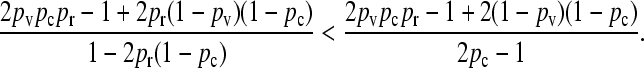 |
(A20) |
After some manipulation, this condition may be shown to be equivalent to
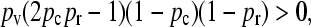 |
(A21) |
which establishes the inequality.
Derivation of 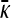 for
for 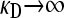 and fixed value of
and fixed value of 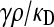 :
:
Because γρ is infinite, we expect that x−+ = x− − = 0, although z−+ ≡ γρx−+ and z−− ≡ γρx−− may converge to positive, though finite, values. Also, because the +− genomes, as conjugators, cannot be “rescued” by conjugators themselves, we expect that x+− = 0 in the limit as 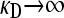 , although again it is possible that y+− ≡ κDx+− converges to a positive value. We expect only x++ > 0, since the ++ genomes are both conjugators and resistant to the antibiotic and so are not destroyed by conjugation or by antibiotic-induced death.
, although again it is possible that y+− ≡ κDx+− converges to a positive value. We expect only x++ > 0, since the ++ genomes are both conjugators and resistant to the antibiotic and so are not destroyed by conjugation or by antibiotic-induced death.
The steady-state equations then become
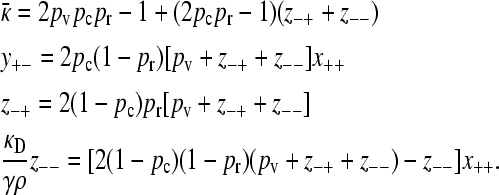 |
(A22) |
From the first equation we have that 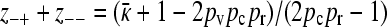 . We therefore have that
. We therefore have that
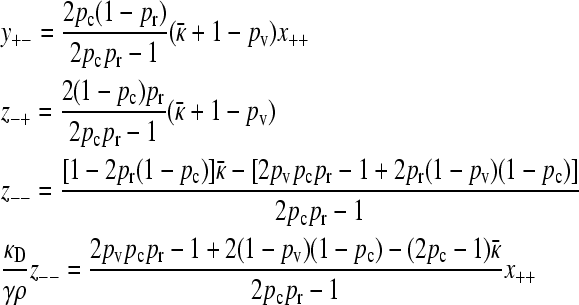 |
(A23) |
and we also have in this limit that 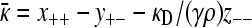 . Substituting in the expressions for y+− and κD/(γρ)z− −, we obtain
. Substituting in the expressions for y+− and κD/(γρ)z− −, we obtain
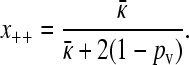 |
(A24) |
Substituting this expression into the last equality of Equation A23, and using the expression for z−−, gives us Equation 9.
Derivation of the transition point between the two functional forms for 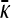 for
for 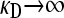 :
:
Equating the finite γρ with the infinite γρ expressions for 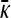 , we obtain that the transition point occurs where
, we obtain that the transition point occurs where
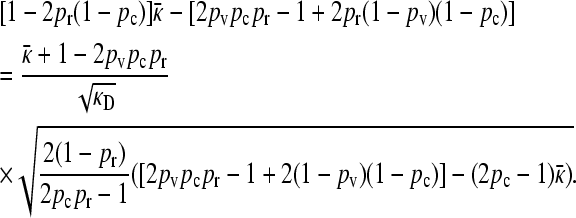 |
(A25) |
Since 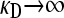 , we then obtain that the transition point occurs where the left-hand side is zero, so that
, we then obtain that the transition point occurs where the left-hand side is zero, so that 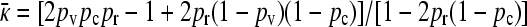 . To estimate the value of γρ where this transition occurs in the limit of large κD, we substitute the expression for
. To estimate the value of γρ where this transition occurs in the limit of large κD, we substitute the expression for 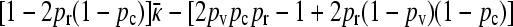 given in Equation A25 into Equation 6 and then substitute the value of
given in Equation A25 into Equation 6 and then substitute the value of 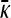 that we obtained for the transition. After some manipulation, we obtain the expression given by Equation 11.
that we obtained for the transition. After some manipulation, we obtain the expression given by Equation 11.
References
- Barrick, J. E., D. S. Yu, S. H. Yoon, H. Jeong, T. K. Oh et al., 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461 1243–1247. [DOI] [PubMed] [Google Scholar]
- Beaber, J. W., B. Hochhut and M. K. Waldor, 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427 72–74. [DOI] [PubMed] [Google Scholar]
- Bjedov, I., O. Tenaillon, B. Gerard, V. Souza, E. Denamur et al., 2003. Stress-induced mutagenesis in bacteria. Science 300 1404–1409. [DOI] [PubMed] [Google Scholar]
- Brown, J. R., 2003. Ancient horizontal gene transfer. Nat. Rev. Genet. 4 121–132. [DOI] [PubMed] [Google Scholar]
- Cohen, E., D. A. Kessler and H. Levine, 2005. Recombination dramatically speeds up evolution of finite populations. Phys. Rev. Lett. 94 098102. [DOI] [PubMed] [Google Scholar]
- Denamur, E., G. Lecointre, P. Darlu, O. Tenaillon, C. Acquaviva et al., 2000. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103 711–721. [DOI] [PubMed] [Google Scholar]
- Ghigo, J. M., 2001. Natural conjugative plasmids induce bacterial biofilm development. Nature (Lond.) 412 442–445. [DOI] [PubMed] [Google Scholar]
- Gogarten, J. P., and J. P. Townsend, 2005. Horizontal gene transfer, genome innovation, and evolution. Nat. Rev. Microbiol. 3 679–687. [DOI] [PubMed] [Google Scholar]
- Kurland, C. G., B. Canback and O. G. Berg, 2003. Horizontal gene transfer: A critical view. Proc. Natl. Acad. Sci. USA 100 9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman, H., J. G. Lawrence and E. A. Groisman, 2000. Lateral gene transfer and the nature of bacterial innovation. Nature (Lond.) 405 299–304. [DOI] [PubMed] [Google Scholar]
- Park, J. M., and M. W. Deem, 2007. Phase diagrams of quasispecies theory with recombination and horizontal gene transfer. Phys. Rev. Lett. 98 058101. [DOI] [PubMed] [Google Scholar]
- Park, K., E. Han, J. Paulsson and D. K. Chattoraj, 2001. Origin pairing (“handcuffing”) as a mode of negative control of P1 plasmid copy number. EMBO J. 20 7323–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russi, S., R. Boera and M. Coll, 2008. Molecular machinery for DNA translocation in bacterial conjugation, pp. 183–214 in Plasmids: Current Research and Future Trends, edited by G. Lipps. Caister Academic Press, Cambridge, UK.
- Tannenbaum, E., and E. I. Shakhnovich, 2005. Semiconservative replication, genetic repair, and many-gened genomes: extending the quasispecies paradigm to living systems. Phys. Life Rev. 2 290–317. [Google Scholar]
- Uhlin, B. E., and K. Nordstrom, 1975. Plasmid incompatibility and control of replication: copy mutants of the R-factor R1 in Escherichia coli K-12. J. Bacteriol. 124 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet, D., J. G. Voet and C. W. Pratt, 2008. Fundamentals of Biochemistry: Life at the Molecular Level, Ed. 3. Wiley, New York.
- Walsh, C., 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature (Lond.) 406 775–781. [DOI] [PubMed] [Google Scholar]