

Abstract
An expansion of structure-activity studies on a series of substituted 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine PDE4 inhibitors and the introduction of a related [1,2,4]triazolo[4,3-b]pyridazine based inhibitor of PDE4 is presented. The development of SAR included strategic incorporation of known substituents on the critical catachol diether moiety of the 6-phenyl appendage on each heterocyclic core. From these studies, (R)-3-(2,5-dimethoxyphenyl)-6-(4-methoxy-3-(tetrahydrofuran-3-yloxy)phenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (10) and (R)-3-(2,5-dimethoxyphenyl)-6-(4-methoxy-3-(tetrahydrofuran-3-yloxy)phenyl)-[1,2,4]triazolo[4,3-b]pyridazine (18) were identified as highly potent PDE4A inhibitors. Each of these analogues was submitted across a panel of 21 PDE family members and was shown to be highly selective for PDE4 isoforms (PDE4A, PDE4B, PDE4C, PDE4D). Both 10 and 18 were then evaluated in divergent cell-based assays to assess their relevant use as probes of PDE4 activity. Finally, docking studies with selective ligands (including 10 and 18) were undertaken to better understand this chemotypes ability to bind and inhibit PDE4 selectively.
The second messenger cyclic 3,5-adenosine monophosphate (cAMP) is a key regulator of numerous signaling cascades.1,2 As such, the production of cAMP by adenylate cyclase (AC) and the degradation of cAMP by phosphodiesterases (PDEs) are highly regulated. There are 11 primary families of PDEs (designated PDE1 - PDE11) expressed throughout the human body, with several families having multiple isoforms. Much effort has been devoted to understanding the physiological role of each PDE isoform, and the pharmacological inhibition of select PDE isoforms has proven therapeutically beneficial for several indications.2-4 The PDE4 family is comprised of 4 primary gene products (PDE4A, PDE4B, PDE4C, PDE4D) and is highly expressed in neutrophils and monocytes, CNS tissue and smooth muscles of the lung.2-4 Not surprisingly, the clinical utility of PDE4 inhibitors has focused on asthma, chronic obstructive pulmonary disease (COPD), memory enhancement and as a general modulator of inflammation.4
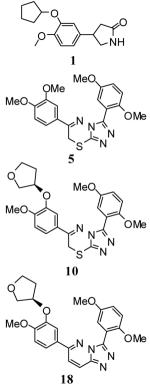
At present, there are numerous chemotypes known to inhibit PDE4 and several of these inhibitors are currently being evaluated in clinical settings.5 Reported molecules include rolipram (1), roflumilast (2) and cilomilast (3). It is notable that each of these small molecules contains a common catechol diether motif. Crystallographic studies of PDE4 bound to several of these derivatives have shown the catechol diether forming a key hydrogen bond with a conserved glutamine residue (Gln443 in PDE4B and Gln369 in PDE4D).4,6,7 The choice of alkyl, cycloalkyl and heterocyclic ether moieties is presumably driven by improvements to the potency of each molecule and selected DMPK properties. It is noteworthy that methoxy, cyclopentyloxy, cyclopropylmethoxy and 2-difluoromethoxy are repeatedly incorporated into known PDE4 inhibitor scaffolds. Another common ether substituent, an O-3-tetrahydrofuranyl such as in 4, is found in numerous published and patented chemotypes.8-10 The frequency with which these moieties are incorporated on small molecules intended to down-regulate PDE4 is compelling. However, there are few reports that directly compare these substituents on the same core chemotype to gain an appreciation of each moieties potential and limitations.
Recently, we introduced a series of substituted 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as potent PDE4 inhibitors including 5.11 Our initial 77 member matrix library revealed that the 3,4-catachol diether motif on the 6-phenyl appendage on the triazolothiadiazine ring system played a key role in defining this chemotype’s pharmacophore. Here, we systematically incorporate the methoxy, cyclopentyloxy, cyclopropylmethoxy, 2-difluoromethoxy and O-3-tetrahydrofuranyl moieties onto the 6-phenyl-triazolothiadiazine core in an attempt to improve potency, but also better understand these frequently utilized groups. The DMPK profiles of our initial triazolothiadiazines showed low microsomal stability [high intrinsic clearance and low t1/2 (rat) (data not shown)]. The removal/alteration of aromatic methoxy groups has been shown to positively affect microsomal stability in several instances given the propensity for demethylation and glucuronidation of this functional group. We were further interested in transposing the substitution patterns found on the triazolothiadiazine ring to the related triazolopyridazine ring system in order to remove the lone sulfur atom and eliminate S-oxidation as an additional mechanism of metabolism and clearance.
The synthesis of substituted 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines was accomplished via previously reported methods (for full synthetic details associated with the synthesis of substituted 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines see the supporting information).11 The key condensation reaction between appropriately substituted 2-bromo-1-phenylethanone (ultimately the phenyl ring at the C6 position of the heterocycle) and appropriately substituted 4-amino-3-phenyl-1H-1,2,4-triazole-5(4H)-thione (ultimately the phenyl ring at the C3 position of the heterocycle) was accomplished in ethanol at elevated temperatures. Utilizing the known SAR from our previously reported matrix library, we opted to maintain the 2,5-dimethoxy substitution pattern on the 3-phenyl appendage on the heterocyclic core. To incorporate the cyclopentyloxy, cyclopropylmethoxy, 2-difluoromethoxy and O-3-tetrahydrofuranyl moieties unto the 2-bromo-1-phenylethanone precursor, we relied upon 1-(3-hydroxy-4-methoxyphenyl)ethanone as an orthogonally protected starting reagent. For the cyclopentyloxy and cyclopropylmethoxy substituents, we utilized a nucleophilic displacement of the corresponding alkyl bromides to ultimately provide the substitution pattern found in derivatives 6 and 7 (Table 1). Reaction of 1-(3-(cyclopentyloxy)-4-methoxyphenyl)ethanone with dodecane-1-thiol in sodium methoxide/DMF at 100 °C provided demethylation in a mild manner.12 Treatment of the resulting 1-(3-(cyclopentyloxy)-4-hydroxyphenyl)ethanone with sodium 2-chloro-2,2-difluoroacetate in DMF at 100 °C afforded the incorporation of the 2-difluoromethoxy functionality on the C4 position of the catachol moiety (found in derivative 8).13 Mitsonobu conditions were utilized to condense tetrahydrofuran-3-ol (both racemic and R) with 1-(3-hydroxy-4-methoxyphenyl)ethanone to provide analogues 9 and 10.
Table 1.
PDE4A inhibition by compounds 5-10, 17 and 18.
| Analogue # | R1 | R2 | PDE4A IC50 (nM) |
|
|---|---|---|---|---|
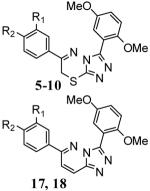
|
5 | −OCH3 | −OCH3 | 6.7 ± 0.4 |
| 6 | −OCypent | −OCH3 | 13 ± 0.8 | |
| 7 | −OCH2Cyprop | −OCH3 | 6.1 ± 0.9 | |
| 8 | −OCH2Cyprop | −OCHF2 | 11 ± 0.7 | |
| 9 | −O(3-THF)[rac] | −OCH3 | 3.4 ± 0.4 | |
| 10 | −O(3-THF)[R] | −OCH3 | 3.0 ± 0.2 | |
| 17 | −O(3-THF)[S] | −OCH3 | 7.3 ± 3.8 | |
| 18 | −O(3-THF)[R] | −OCH3 | 1.5 ± 0.7 |
data represents the results from three seperate experiments (SD provided). Definitions: OCH3 = methoxy, OCypent = cyclopentyloxy, OCH2Cyprop = cyclopropylmethoxy, OCHF2 = 2-difluoromethoxy, O(3-THF) = O-3-tetrahydrofuranyl [racemic, R or S enantiomers].
The synthesis of [1,2,4]triazolo[4,3-b]pyridazines was based upon the precedented works of Tisler and coworkers, Greenblatt and coworkers and Street and coworkers.14-16 The general method is outlined in Scheme 1 and begins with the coupling of commercially available 2,5-dimethoxybenzoic acid (11) and 3-chloro-6-hydrazinylpyridazine (12) to provide N’-(6-chloropyridazin-3-yl)-2,5-dimethoxybenzohydrazide (13) in good yields. Direct treatment of 13 with POCl3 at elevated temperature afforded the cyclization to core [1,2,4]triazolo[4,3-b]pyridazine ring system (analogue 14) and provided a common analogue for entry into the convergent syntheses of multiple products via end-stage Suzuki-Miyaura couplings. Here, we were primarily interested in examining the biochemical viability of this alternate heterocycle and limited our explorations to the appropriately substituted O-3-tetrahydrofuranyl derivatives. Boronic acids 15 and 16 were synthesized independently utilizing the aforementioned Mitsonobu protocols and displacement of an aryl bromide with boronic acid. Following purification of 15 and 16, standard Suzuki-Miyaura conditions with microwave irradiation produced the appropriately substituted [1,2,4]triazolo[4,3-b]pyridazines 17 and 18 in good yields.
Scheme 1.

a
aReagents and conditions: (i) CDI, DMF, rt 2h; (ii) POCl3,105 °C, 2h; (iii) 15 or 16, Pd(PPh3)4,2Maq.Na2CO3,DME, μwave, 90 °C 30 min.
PDE4A inhibition profile
With several new analogues now in hand, we evaluated their inhibitory potency against PDE4A via a previously reported, purified enzyme fluorescence polarization assay (IMAP; Molecular Devices, CA).11 The results for the newly synthesized 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines 5-10 and the novel [1,2,4]triazolo[4,3-b]pyridazines 17 and 18 are shown in Table 1. For 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines 5-10, each substitution pattern yielded a molecule with potency in the low nanomolar range. The enantiomerically pure O-(3-THF)[R] substitution of 10 showed the best potency with an IC50 value of 3.0 nM. As a result, the enantiomerically pure O-(3-THF)[R] and O-(3-THF)[S] substitutions were incorporated onto the [1,2,4]triazolo[4,3-b]pyridazine core structure and the resulting constructs were found to have excellent potencies for PDE4A inhibition (IC50 value of 7.3 ±3.8 nM for 17 and 1.5 ±0.7 nM for 18). Several analogues were also explored with varying substitutions on the phenyl ring attached to the C3 position of the 1,2,4-triazole ring system. Substitutions included methoxy, fluoro, chloro and trifluoromethyl groups on the ortho, meta and para positions of the phenyl ring (for synthetic details and characterization of these analogues see the Supporting Information section). Analysis of these analogues confirmed that various substitutions at one of the ortho positions were necessary to maintain potent PDE4A inhibition (see supporting information). Additional substitutions have been shown to be tolerated without effect on the inhibition of PDE4A.
Selectivity panel of PDE isoforms
Having arrived at several compounds with good potency profiles and divergent core heterocycles, it was of interest to confirm the selectivity of these agents against a panel of PDE isoforms. BPS Bioscience (San Diego, CA)17 has available a panel of 21 PDE isoforms from all 11 primary PDE families except PDE6 available for activity profiling. We submitted 5, 10, 18 and 1 for analysis across this panel and the resulting IC50 determinations are shown in Table 2.
Table 2.
PDE isoform selectivity data for 1, 5, 10 and 18.
| PDE isof orm* |
1 I C50 /% inh. |
5 I C50 /% inh. |
10 I C50 /% inh. |
18 I C50 /% inh. |
|
|---|---|---|---|---|---|
|
PDE1A | inactive | inactive | 36% | 32% |
| PDE1B | inactive | inactive | 52% | 56% | |
| PDE1C | inactive | 26% | 49% | 74% | |
| PDE2A | inactive | 41% | 68% | 54% | |
| PDE3A | inactive | 1.7 μM | 56% | 54% | |
| PDE3B | inactive | 720 nM | 4.6 μM | 2.3 μM | |
| PDE4A1A | 102 nM | 12.9 nM | 0.26 nM | 0.6 nM | |
| PDE4B1 | 901 nM | 48.2 nM | 2.3 nM | 4.1 nM | |
| PDE4B2 | 534 nM | 37.2 nM | 1.6 nM | 2.9 nM | |
| PDE4C1 | 40% | 452 nM | 46 nM | 106 nM | |
| PDE4D2 | 403 nM | 49.2 nM | 1.9 nM | 2.1 nM | |
| PDE5A1 | inactive | 60% | 58% | 51% | |
| PDE7A | inactive | 73% | 48% | 59% | |
| PDE7B | inactive | 33% | 43% | 35% | |
| PDE8A1 | inactive | 57% | inactive | inactive | |
| PDE9A2 | inactive | inactive | inactive | inactive | |
| PDE10A1 | inactive | 823 nM | 632 nM | 388 nM | |
| PDE11A4 | inactive | inactive | inactive | inactive |
data generated by BPS Biosciences (CA) [http://www.bpsbioscience.com]and represents either the IC50 value or the % inhibition at 10 μM of compound.
It is apparent that both 10 and 18 represent novel inhibitors of five isoforms of PDE4 with sub-nanomolar potencies found for PDE4A1A. The modest activities found for PDE3B and PDE10A1 represent a sizeable enough difference in potency as to confirm the cell-based utility of these agents as PDE4 inhibitors Interestingly, PDE10 is considered to be an important modulator of cGMP regulation in the brain. Additionally, 10 and 18 may represent good starting points for optimizing compounds with potency for PDE10. This PDE family is considered to be an important regulator of cGMP in the brain, and to data few reports of potent and selective PDE10 inhibitors have surfaced.
The identification of novel enzyme inhibitors via screening in purified enzyme assays provides an important means for the identification of small molecules with a known mechanism of action. However, not all active small molecules found in such screens find utility in cell-based assays for a myriad of reasons. To evaluate whether the substantial in vitro inhibition of PDE4 by 10 and 18 could be replicated in living cells we pursued two divergent, cell-based assays of PDE4 activity.
Cyclic-nucleotide gated ion channel cell-based assay
The first cell-based analysis of PDE4 activity utilized a recently reported assay based on the coupling of a constitutively activated G-protein coupled receptor (GPCR) and cyclic-nucleotide gated (CNG) ion channel coexpressed in HEK293 cells.18 The read-out for this assay is based on measurement of membrane electrical potential by a potential-sensitive fluorophore (ACTOne™ dye kit). Inhibitors of PDE4 will interfere with the native enzymatic conversion of cAMP to AMP resulting in increased intracellular levels of the cyclic nucleotide due to constitutive activity of the GPCR. In response to increased amounts of cAMP, the CNG ion channel opens resulting in membrane polarization. The dye reacts to this alteration in membrane polarity with an increase in fluorescence detectable by fluorescence spectroscopy of whole cells read on a fluorescence microtitre plate reader.
Compounds 5, 10 and 18 were used in this analysis as along with the common PDE4 inhibitor 1, and the results are shown in Figure 2. In this assay, 1 was noted to produce an effective response (EC50) (registered as % activity) of 131.5 nM. In comparison, the triazolothiadiazine based inhibitors were found to be more potent in this cell-based assay with 5 and 10 registering an EC50 value of 18.7 and 2.3 nM, respectively. The EC50 of the lone triazolopyridazine 18 was 34.2 nM.
Figure 2. Cell-based analysis of novel PDE4 inhibitors.

Effect of PDE4 inhibition by 1, 5, 10 and 18 in a cell-based cyclic nucleotide-gated cation channel biosensor assay. Specific EC50 values are as follows; (A) EC50 for 1 = 131.5 nM. (B) EC50 for 5 = 18.7 nM. (C) EC50 for 10 = 2.3 nM. (D) EC50 for 18 = 34.2 nM. The data represents the results from four separate experiments.
Protein-fragment Complementation (PCA) cell-based assay
PCAs take advantage of the ability of well-engineered protein fragments to form a functional monomer with measurable enzymatic activity when brought into suitable proximity by interacting proteins to which the fragments are fused.20,21 For our purposes, we utilized a previously reported PCA based on reporter enzyme Renilla reniformis luciferase (Rluc) N- and C-terminal fragments of Rluc fused to the catalytic subunits (Cat) and inhibiting regulatory subunits (Reg) of protein kinase A (PKA).19 The signaling cascades initiated by GPCR activation is mediated by cAMP production and activation of numerous protein kinases.1,20 Negative regulation of these events is the sole domain of the phosphodiesterase class of enzymes.2 One ubiquitous pathway is activated when cAMP triggers the disassociation of the PKA catalytic and regulatory subunits, which in turn, enables numerous signaling events. In the Rluc PCA PKA reporter, the regulatory subunit II beta cDNA is fused through a sequence coding for a flexible polypeptide linker of ten amino acids (Gly.Gly.Gly.Gly.Ser)2 to the N-terminal fragment (Rluc F[1]) [amino acids 1-110 of Rluc] and the cDNA of the PKA catalytic subunit alpha is fused through the same flexible linker to the C-terminal fragment (Rluc F[2] )[amino acids 111-311 of Rluc]. The resulting constructs are designated Reg-F[1] and Cat-F[2] and reconstitute enzymatic activity of Rluc in the absence of cAMP It has been recently demonstrated that this assay could be used to detect the effects of PDE4 inhibition on PKA activation downstream of of basal β-2 adrenergic receptor (β2 AR) activities.19
Here, we evaluated the effects of 1, 10 and 18 in HEK293 cells stably expressing the β2AR and transiently transfected with the required PKA-Rluc fragments [Reg-F[1] and CatF[2]]. It was confirmed that isoproterenol (19) activation of the β2AR is able to reduce luminescence (indicating dissociation of the Rluc biosensor complex and consequent activation of PKA catalytic activity) (Figure 3A). Further, pretreatment with the selective β2AR inverse agonist IC118551 (20; decrease of basal β2AR activity) was capable of preventing the effects of 19 as was previously shown.19 These important controls confirm that alterations of the luminescence signal are primarily mediated through the actions of the β2AR signaling to PKA. Further, the effect of 1 confirms the responsiveness of the assay to PDE4 inhibition. Treatment with 10 and 18 at 100 μM and 10 μM concentrations displayed marked loss of luminescence highly suggesting a β2AR mediated increase of cAMP due to inhibition of PDE4 (Figure 3B). Next, we examined the real-time kinetics of PKA subunit dissociation by administering 10 at a 10 μM concentration. The shown real-time kinetics are normalized on the control experiment of administering 10 following pretreatment with 1 μM of the inverse β2AR agonist 20. In four independent experiments, the presence of 10 reduced the luminescence of the cell-based system by 25% to 50% within 2 minutes of administration (Figure 3C).
Figure 3. Effect of PDE inhibition on β2AR regulated PKA activities in vivo.

Bioluminescence in A-C was detected from stable β2AR-HEK293 (see reference 19) cells transiently transfected with the PKA reporter Reg-F[1]:Cat-F[2]2 (from three independent experiments). (A) Effect of combinations of pretreatment with the selective β2AR-antagonist 20 (1 μM) and treatment with 1 (100 μM; 30 min) or 19 (1 μM, 30 min) on the on the association of Reg-F[1]:Cat-F[2] (mean ± s.d. from independent triplicates). (B) Concentration dependent effect of 18, 10 and a related triazolothiadiazine that possesses no PDE4 inhibition on the association of Reg-F[1]:Cat-F[2] (30 min, mean ± s.d. from independent triplicates). Percentage of PKA activation is normalized based upon 20 (1 μM) pretreated cells. (C) Real-time kinetics (normalized on the control experiment = pretreatment with 1 μM 20) of Reg-F[1]:Cat-F[2] dissociation in response to 10 treatment (10 μM, four independent samples).
Docking of 10 at PDE4B
Given the potency, selectivity and cell-based PDE4 inhibition results for this chemotype, it was of interest to examine its binding modality to the PDE4 structure. The PDE classes of enzymes are comprised of an N-terminal domain, a catalytic domain and a C-terminal domain. Crystallographic analyses of several PDE isozymes have aided researchers in understanding the divergent activities and pharmacology of this class of proteins.6,23 Importantly, structures of PDE4 have been reported in complex to AMP and several small molecule inhibitors.24-28 Through these efforts it was shown that the three domains of PDE4 are coordinated through interactions with two metal cations (Zn2+ and Mg2+).7 The residues that coordinate these metals are highly conserved across the PDE family. Both the Zn2+ and Mg2+ play important roles in the catalytic mechanism of cAMP hydrolysis by coordinating the phosphate moiety. Other important insights include the recognition of a conserved glutamine residue (Q443 in PDE4B) that serves as an important binding residue for the purine motif of cAMP and cGMP.
From the myriad of known PDE4 inhibitors, one common structural motif that continues to be exploited are catachol diether-based small molecules.4 Indeed, the structure of 1 formed the basis for numerous catachol diether based inhibitors of PDE4 and the visualization of 1 bound to PDE4B provided the rational for its potent inhibition profile. From numerous crystallographic analyses and modeling efforts it is clear that the catachol diether based inhibitors bind to the catalytic domain of PDE4 through specific hydrogen bonds with the conserved glutamine residue. Our initial SAR explorations of triazolothiadiazine based PDE4 inhibitors confirmed that a 3,4-dimethoxy phenyl moiety linked to the C6 position of the 3,6-dihydro-2H-1,3,4-thiadiazine ring was a crucial substitution pattern for potent PDE4 inhibition.11 Interestingly, the phenyl ring attached to the C3 position of the 1,2,4-triazole ring system was found to be more amendable to random substitutions without loss of function. This formed the basis for our supposition that these novel PDE4 inhibitors were binding in a similar pose to that of 1.
To explore this hypothesis, we conducted docking simulations using the AutoDock software.29 We first retrieved the three-dimensional coordinates for PDE4B from the Protein Data Bank (PDB ID: 1XMY). Protein and ligand structures were prepared in AutoDock29 and previously reported PDE4-inhibitor complexes were taken into account when preparing the active site grid box. Flexibility was granted to the active site glutamine and the ligand(s). Following multiple docking simulations the most favorable binding conformations were extracted based upon calculated binding constants (reported as Ki values and found to be in the low nanomolar range for favorable docking orientations). The primary docking modality for 10 is shown in Figure 4. Importantly, this docking orientation is consistent with the idea that the catachol diether forms an integral hydrogen bond with Q443 (right panel) and the aromatic moiety is clearly positioned between the conserved isoleucine (I410) and phenylalanine (F446). The remainder of the molecule is shown to extend into the catalytic domain in close proximity to both the Zn2+ and Mg2+ cations. Such an orientation would block the approach of cAMP to the catalytic domain and forms the basis for inhibiting PDE4. This docking orientation is consistent with the known SAR for this chemotype whereby the 3,4-dimethoxy phenyl moiety at the C6 position of the 3,6-dihydro-2H-1,3,4-thiadiazine ring is crucial for maintaining inhibition in the low nanomolar range whereas the opposite phenyl ring is more amendable to change without significant loss of potency. It also demonstrates that alterations of the core heterocycle from the general triazolothiadiazine structure to the triazolopyridazine structure will have limited affect on the inhibitory profile of these reagents. These docking poses may allow future SAR explorations to focus on improved pharmacokinetic and drug metabolism properties without compromising potency, efficacy and selectivity for this chemotype.
Figure 4. Model of PDE4B and Docking of 10.

The left panel details the entire PDE4B structure (N-terminal domain, a catalytic domain and a C-terminal domain) bound to 10. The right panel shows the catalytic domain bound to 10 including interactions with conserved glutamine (Q443) isoleucine (I410) and phenylalanine (F446) and the Zn2+ (grey) and Mg2+ (green) cations.
Numerous chemotypes exist for the potent, selective inhibition of PDE4. Here, we expand upon our discovery of substituted 6-(3,4-dialkoxyphenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and introduce 6-(3,4-dialkoxyphenyl)-[1,2,4]triazolo[4,3-b]pyridazines as novel inhibitors of PDE4. In this study, we examined the common structural substitutions found on other catachol diether based inhibitors of PDE4 and several of the resulting analogues are among the most potent inhibitors of this important cellular target. These include methoxy, cyclopentyloxy, cyclopropylmethoxy, 2-difluoromethoxy and O-3-tetrahydrofuranyl moieties. It was found that the chirally pure R-O-3-tetrahydrofuranyl substitution maintained the best potency in this study. Further, these reagents possess impressive selectivity for PDE4 versus other PDE family members. However, these chemotypes, like others, do not posses subtype selectivity across PDE4 isozymes.4
Our initial analysis of the compounds and their activity was based upon an in vitro analysis using purified PDE4 protein. It is critical to examine chemical probes discovered via purified-protein assays within cell-based contexts to confirm activity and establish that they are relevant for cell-based experimentation. Here, we examine selected analogues (5, 10 and 18) in two different cell-based assays. One assay is based upon the ability of PDE4 to reduce cAMP levels in a CNG cell line while the other utilizes a PCA reporter for PKA activity. Both analyses demonstrated the utility of these novel reagents as cell-based chemical probes of PDE4 activity.
Finally, with the myriad of structural data surrounding PDE4 and PDE4 complexes with selected inhibitors, it was important to explore the binding modality of these compounds. Docking studies demonstrated that these agents utilize the conserved binding mode whereby the catachol diether functionality forms a strong interaction with a conserved glutamine residue. This docking orientation further provides a roadmap for additional SAR around the seemingly modifiable phenyl ring attached to the 1,2,4-triazole moiety of the core heterocycle. This key aspect of these reagents may be of importance during attempts to modify ADME properties of these compounds without altering the affinity or selectivity for PDE4.
PDE4 inhibitors are highly sought after as probes of selected cell signalling pathways and as potential therapeutics in diverse areas including memory enhancement and COPD. Here, we expand on the potential of substituted triazolothiadiazines and introduce triazolopyridazines as potent and selective inhibitors of this important cellular target. Not only are selected analogues of this novel chemotype capable of down-regulating purified isozymes of PDE4, but they maintain excellent cell-based activity as well. Their binding modality is predicted to mimic known catachol diether based inhibitors of PDE4. Importantly, both computational and structure activity studies suggest that the phenyl ring at the C3 position of the 1,2,4-triazole ring system could be modified providing a mechanism for advanced SAR considerations.
Supplementary Material
Figure 1.

Structures of several known PDE4 inhibitors.
Acknowledgments
We thank Ms. Allison Peck for critical reading of this manuscript. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research, the Intramural Research Program of the National Human Genome Research Institute and the National Institute of Dental and Craniofacial Research.
Footnotes
Supporting Information. Experimental methods for the cyclic nucleotide-gated cation channel assay, protein-fragmentation complementation assays and molecular docking and the full synthetic procedures and characterization of reported compounds are detailed. This material is available free of charge via the Internet.
References and Notes
- (1).Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat. Rev. Mol. Cell. Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- (2).Bender AT, Beavo JA. Cyclic Nuleotides Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- (3).McKenna JM, Muller GW. In: Cyclic Nucleotide Phosphodiesterases in Health and Disease. Beavo JA, Francis SH, Houslay MD, editors. CRC Press; 2006. p. 667. [Google Scholar]
- (4).Zhang KYJ, Ibrahim PN, Gillette S, Bollag G. Phosphodiesterase-4 as a potential drug target. Expert Opin. Ther. Targets. 2005;9:1283–1305. doi: 10.1517/14728222.9.6.1283. [DOI] [PubMed] [Google Scholar]
- (5).Huang Z, Ducharme Y, Macdonald D, Robichaud A. The next generation of PDE4 inhibitors. Curr. Opin. Chem Biol. 2001;5:432–438. doi: 10.1016/s1367-5931(00)00224-6. [DOI] [PubMed] [Google Scholar]
- (6).Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Notle RT. Crystal Structures of the Catalytic Domain of Phosphordiesterase 4B Complexed with AMP, 8-Br-AMP and Rolipram. J. Mol. Biol. 2004;337:355–365. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- (7).Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim S-H, Schlessinger J, Zhang KYJ. Structural Basis for the Activity of Drugs that Inhibit Phosphodiesterases. Structure. 2004;12:2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- (8).Dunn RF, Kuester EM, Conticello RD, Hopper AT. Preparation of (hetero)aryl pyrazole derivatives as phosphodiesterase 4 inhibitors. PCT Int. Appl. 2007 WO 2007123953. [Google Scholar]
- (9).Napoletano M, Norcini G, Pellacini F, Marchini F, Morazzoni G, Ferlenga P, Pradella L. The Synthesis and Biological Evaluation of a Novel Series of Phthalazine PDE4 Inhibitors I. Bioorg. Med. Chem. Lett. 2000;10:2235–2238. doi: 10.1016/s0960-894x(00)00449-2. [DOI] [PubMed] [Google Scholar]
- (10).Andrés JI, Alonso JM, Diaz A, Fernández J, Iturrino L, Martinez P, Matesanz E, Freyne EJ, Deroose F, Boeckx G, Petit D, Diels G, Megens A, Somers M, Van Wauwe J, Stoppie P, Cools M, De Clerck F, Peeters D, de Chaffoy D. Synthesis and Biological Evaluation of Imidazol-2-one and 2-Cyanoiminoimidazole Derivatives: Novel Series of PDE4 Inhibitors. Bioorg. Med. Chem. Lett. 2002;12:653–658. doi: 10.1016/s0960-894x(01)00817-4. [DOI] [PubMed] [Google Scholar]
- (11).Skoumbourdis AP, Huang R, Southall N, Leister W, Guo V, Cho M-H, Inglese J, Nirenberg M, Austin CP, Xia M, Thomas CJ. Identification of a potent new chemotype for the selective inhibition of PDE4. Bioorg. Med. Chem. Lett. 2008;18:1297–1303. doi: 10.1016/j.bmcl.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Katoh T, Awasaguchi K.-i., Mori D, Kimura H, Kajimoto T, Node M. Lipase-Catalyzed Asymmetric Dealkoxylcarbonylation of σ-Symmetrical β-Ketodiesters and Its Application to the Synthesis of (-)-Podocarpic Acid. Synlett. 2005;19:2919–2922. [Google Scholar]
- (13).Hall A, Brown SH, Chessell IP, Chowdhury A, Clayton NM, Coleman T, Giblin GMP, Hammond B, Healy MP, Johnson MR, Metcalf A, Michel AD, Naylor A, Novelli R, Spalding DJ, Sweeting J, Winyard L. 1,5-Biaryl pyrrole derivatives as EP1 receptor antagonists. Structure-activity relationships of 6-substituted and 5,6-disubstituted benzoic acid derivatives. Bioorg. Med. Chem. Lett. 2007;17:916–920. doi: 10.1016/j.bmcl.2006.11.059. [DOI] [PubMed] [Google Scholar]
- (14).Pollak A, Tišler M. Synthesis of Pyridazine Derivatives V: Formation of striazolo-(4,3-b)-pyridazines and bis-s-triazolo-(4,3-b,3′,4′-f)-pyridazines. Tetrahedron. 1966;22:2073–2079. [Google Scholar]
- (15).Albright JD, Moran DB, Wright WB, Jr., Collins JB, Beer B, Lippa AS, Greenblatt EN. Synthesis and Anxiolytic Activity of 6-(Substituted-pheyny)-1,2,4-triazolo[4,3-b]pyridazines. J. Med. Chem. 1981;24:592–600. doi: 10.1021/jm00137a020. [DOI] [PubMed] [Google Scholar]
- (16).Carling RW, Madin A, Guiblin A, Russell MGN, Moore KW, Mitchinson A, Sohal B, Pike A, Cook SM, Ragan IC, McKernan RM, Quirk K, Ferris P, Marshall G, Thompson SA, Wafford KA, Dawson GR, Atack JR, Harrison T, Castro JL, Street LJ. 7-(1,1-Dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl)-1,2-4-triazolo[4,3-b]pyridazine: A Functionally Selective γ-Aminobutyric AcidA (GABAA) α2/α3-Substype Selective Agonist That Exhibits Potent Anxiolytic activity but is Not Sedating in Animal Models. J. Med. Chem. 2005;48:7089–7092. doi: 10.1021/jm058034a. [DOI] [PubMed] [Google Scholar]
- (17).BPS Bioscience Inc. 11526 Sorrento Valley Rd. Ste. A2; San Diego, CA 92121. www.bpsbioscience.com.
- (18).Titus SA, Xiao L, Southall N, Lu J, Inglese J, Brasch M, Austin CP, Zheng W. A Cell-Based PDE4 Assay in 1536-Well Plate Format for High-Throughput Screening. J. Biomol. Screening. 2008;13:609–618. doi: 10.1177/1087057108319977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16916–16921. doi: 10.1073/pnas.0704257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nature Rev. Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- (21).Remy I, Michnick SW. Application of protein-fragment complementation assays in cell biology. BioTechniques. 2007;42:137–145. doi: 10.2144/000112396. [DOI] [PubMed] [Google Scholar]
- (22).Pierce KL, Premont RT, Lefkowitz RJ. Seven-Transmembrane Receptors Nat. Rev. Mol. Cell. Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- (23).Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT. Atomic Structure of PDE4: Insight into Phosphodiesterase Mechanism and Specificity. Science. 2000;288:1822–1825. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]
- (24).Lee ME, Markowitz J, Lee J-O, Lee H. Crystal structure of phosphodiesterase 4D and inhibitor complex. FEBS Lett. 2002;530:53–58. doi: 10.1016/s0014-5793(02)03396-3. [DOI] [PubMed] [Google Scholar]
- (25).Huai Q, Colicelli J, Ke H. The Crystal Structure of AMP-Bound PDE4 Suggests a Mechanism for Phosphodiesterase Catalysis. Biochemistry. 2003;42:13220–13226. doi: 10.1021/bi034653e. [DOI] [PubMed] [Google Scholar]
- (26).Huai Q, Wang H, Sun Y, Kim H-Y, Liu Y, Ke H. Three-Dimensional Structures of PDE4D in Complex with Roliprams and Implication on Inhibitor Selectivity. Structure. 2003;11:865–873. doi: 10.1016/s0969-2126(03)00123-0. [DOI] [PubMed] [Google Scholar]
- (27).Huai Q, Wang H, Zhang W, Colman RW, Robinson H, Ke H. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc. Nat. Acad. Sci. U.S.A. 2004;101:9624–9629. doi: 10.1073/pnas.0401120101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huai Q, Liu Y, Francis SH, Corbin JD, Ke H. Crystal Structures of Phosphodiesterases 4 and 5 in Complex with Inhibitor 3-Isobutyl-1-methylxanthine Suggests a conformational Determinant of Inhibitor Selectivity. J. Biol. Chem. 2004;279:13095–13101. doi: 10.1074/jbc.M311556200. [DOI] [PubMed] [Google Scholar]
- (29).Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1622. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.