

Abstract
Objectives:
Cholinergic projections to cerebral cortical and subcortical regions are decreased in Parkinson disease (PD), but not evaluated in the parkinsonian syndromes of multiple system atrophy (MSA-P) and progressive supranuclear palsy (PSP). We studied cholinergic innervation in these disorders as compared to age-appropriate normal control subjects.
Methods:
We used PET with [11C]PMP to measure acetylcholinesterase (AChE) activity in multiple cerebral cortical and subcortical regions. We studied 22 normal controls, 12 patients with PD, 13 patients with MSA-P, and 4 patients with PSP.
Results:
We found significantly decreased AChE activity in most cerebral cortical regions in PD and MSA-P, and a similar but nonsignificant decrease in PSP. No differences were found between PD and MSA-P. Significantly decreased AChE activity was found in PD in striatum, cerebellum, and thalamus, with a marginally significant decrease in mesencephalon and no change in pons. Significantly greater declines in AChE activity in all subcortical regions were seen in MSA-P and PSP vs in PD. Decreased AChE activity in brainstem and cerebellum of all 3 disorders correlated with disturbances of balance and gait.
Conclusions:
Cerebral cortical cholinergic activity is decreased to a similar level in Parkinson disease (PD), parkinsonian syndromes of multiple system atrophy (MSA-P), and progressive supranuclear palsy (PSP) as compared to normal controls. Subcortical cholinergic activity is significantly more decreased in MSA-P and PSP than in PD. The more substantial decrease reflects greater impairment in the pontine cholinergic group, which is important in motor activity, particularly gait. These differences may account for the greater gait disturbances in the early stages of MSA-P and PSP than in PD.
GLOSSARY
- AChE
= acetylcholinesterase;
- AD
= Alzheimer disease;
- LDT
= laterodorsal tegmental nuclei;
- MSA-P
= parkinsonian syndromes of multiple system atrophy;
- PD
= Parkinson disease;
- PDD
= Parkinson disease with dementia;
- PPT
= pedunculopontine tegmental nuclei;
- PSP
= progressive supranuclear palsy;
- ROI
= region of interest.
Deficits of cholinergic innervation of the cerebral cortex have been identified neuropathologically in Alzheimer disease (AD),1,2 Parkinson disease (PD), and Parkinson disease with dementia (PDD).3–5 Development of N-[11C]-methylpiperidin-4-yl proprionate ([11C]PMP), an acetylcholinesterase (AChE) substrate, with PET has enabled antemortem measurement of cholinergic innervation, permitting correlation with clinical functions.6–9 This approach verified the deficit of cholinergic innervation in AD and showed greater deficiency in advanced cases.10,11 [11C]PMP PET studies also demonstrated cerebral cortical cholinergic defects in PD that are more severe in PDD.11 Studies utilizing [11C]PMP PET in both PD and PDD demonstrated correlations between cerebral cortical AChE activity and tests of working memory, attention, and executive function.12
Deficits of cholinergic projections in AD and PD have been found also in multiple subcortical regions,9,13-15 but the functional correlates are unclear. Also, little information is available regarding cholinergic projections in the parkinsonian syndromes of multiple system atrophy (MSA-P) and progressive supranuclear palsy (PSP). We used [11C]PMP with PET to study cholinergic projections in MSA-P and PSP, and compared the results in normal controls and patients with PD. The rationale was to understand further the pathophysiology of these disorders, and to test the hypothesis that subcortical cholinergic projections, which originate principally in the pedunculopontine tegmental (PPT) and laterodorsal tegmental nuclei (LDT), would be more decreased in MSA-P and PSP than in PD.
METHODS
Patient selection and evaluation.
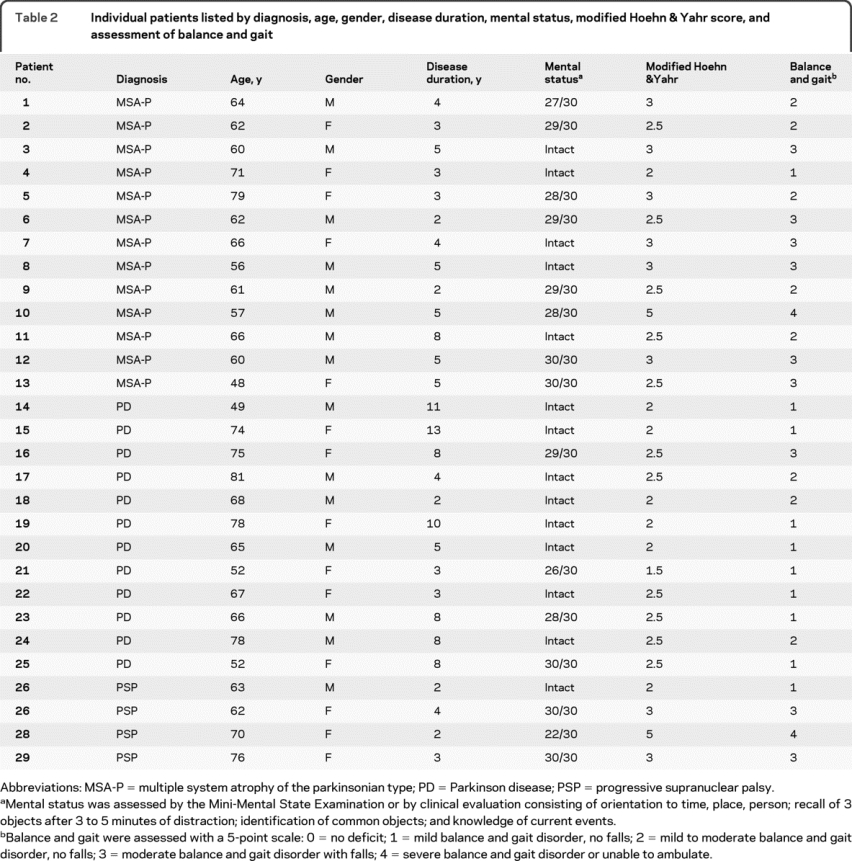
We used published criteria for the diagnosis of PD,16 MSA-P,17 and PSP.18 We studied 22 normal control subjects, 12 patients with PD, 13 patients with MSA-P, and 4 patients with PSP (table 1). In addition to neurologic examination and testing for orthostatic hypotension, the patients were tested for mental status by Mini-Mental State Examination or clinical assessment of cognition utilizing orientation to time, place, person; recall of 3 objects after 3 to 5 minutes of distraction; identification of common objects; and knowledge of current events (table 2). We also assessed the severity of the balance and gait disorder with a 5-point scale and determined the modified Hoehn & Yahr score (table 2). The normal controls were men and women in similar age groups as the patients and had no history of neurologic disorders, no neurologic complaints, and normal neurologic and general physical examinations.
Table 1 Number of patients, gender distribution, and mean age ± standard deviation for NC, PD, MSA-P, and PSP groups
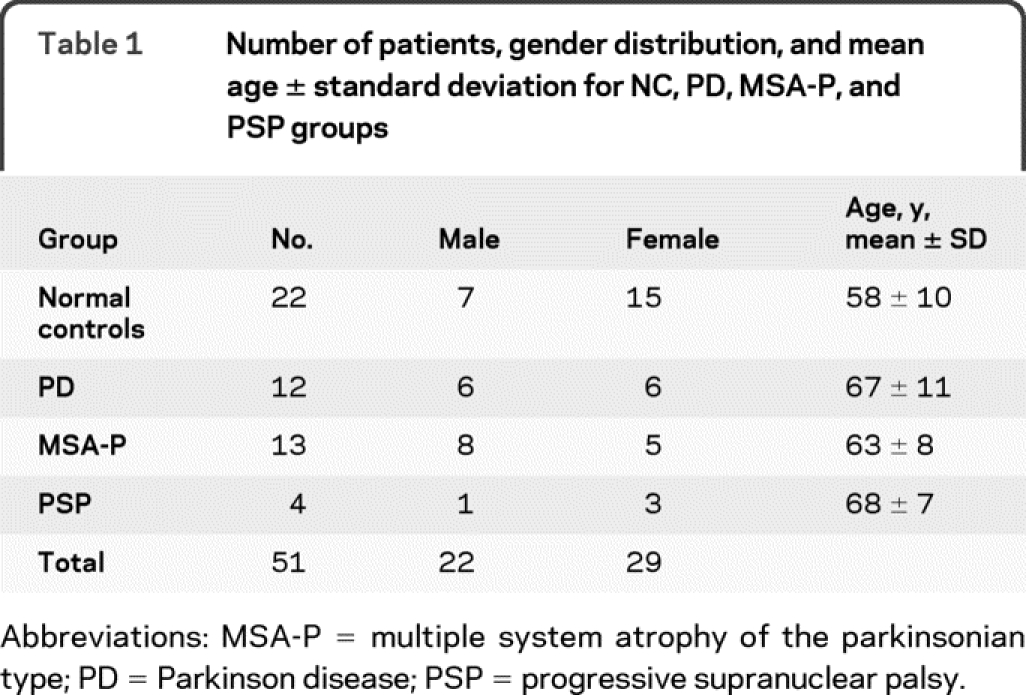
Table 2 Individual patients listed by diagnosis, age, gender, disease duration, mental status, modified Hoehn & Yahr score, and assessment of balance and gait
Standard protocol approvals, registrations, and patient consents.
We initiated the studies after protocol approval by the Institutional Review Board, the ethical standards committee on human experimentation, and the Radioactive Drugs Review Committee. We received written informed consent from all subjects studied.
PET methods.
Localized predominantly in cholinergic cell bodies and axons, acetylcholinesterase (AChE) is a marker of cerebral cholinergic pathways.10,19 AChE localizes to axons projecting from nucleus basalis to cerebral cortex and within intrinsic cerebral cortical neurons. In vivo AChE activity can be studied with a radiolabeled acetylcholine analog that enters the brain where it is metabolized by AChE, producing a metabolite that is trapped at the site of production.8 This method, utilizing [11C]PMP, has been used in humans to examine brain AChE activity.10,12 We prepared [11C]PMP in high radiochemical purity (95%) and specific activity greater than 3.70 × 107 MBq/mmol (1,000 Ci/mmol) by N-[11C]methylation of 4-piperidinyl propionate20 and injected it IV as a 666 MBq (18 mCi) dose. All scans were acquired in 3-dimensional mode on a Siemens ECAT EXACT-HR+ (Siemens Medical Systems Inc., Knoxville, TN). Image data were reconstructed using Fourier rebinning (FORE) followed by 2-D ordered subset expectation maximization (4 iterations, 16 subsets) resulting in spatial resolution of approximately 5.5 mm full width at half maximum both in-plane and axially. [11C]PMP-PET acquisition was accomplished with a dynamic sequence of 15 PET scans over 60 minutes. The fraction of unmetabolized [11C]PMP in plasma samples was determined using SEP-PAK C18 cartridge (Waters, Milford, MA) chromatography and an internal standard of [3H]PMP, a modification of a method described previously.21 Data were coregistered, reoriented, and nonlinearly warped using NeuroStat routines.22,23 A 2-tissue compartment model (PMP and hydrolyzed product) yielded estimates of K1 (transport into tissue), k2 (tissue clearance of authentic tracer back to blood), k3 (hydrolysis rate from [11C]PMP to acetate and choline, i.e., AChE activity), and blood volume for time activity curves generated from the dynamic data set. The rate constant k4 was fixed to zero, because the action of AChE on [11C]PMP is irreversible, and the distribution volume (K1/k2) was constrained. K1, k2, k3, and cerebral blood volume were estimated pixel-by-pixel from the entire cortical data set.10,19 The free-fraction of radiotracer in brain tissue (f2) was assumed to be (k2/K1), the inverse of the estimate of the distribution volume of free (unhydrolyzed) PMP in tissue. The k3 values reported here were corrected for the free fraction f2 (k3rep = k3est/f2). In our previous work19 k3 values corrected in this manner were identified as k3*. While theoretically the reported k3 values are unit-less, the f2 correction is obtained using K1/k2; hence the reported k3 values have the same units as an irreversible influx constant (mL tissue mL−1 blood min−1). This variable is not the irreversible influx constant for PMP, however, as this would be given by K1/k3/(k2+k3). The rationale for using this k3 index is not primarily to correct for the free-fraction in tissue, but to stabilize the estimate of AChE activity. The estimates of k2 and k3 are highly correlated, as noise propagates similarly into both rate constants; thus the estimate of an “f2-corrected” k3 has proven to be as stable as when the ratio K1/k2 is constrained, but does not require K1/k2 to be fixed to a constant value.
We assessed multiple cerebral cortical regions and subcortical sites. We examined bilaterally the anterior cingulate, lateral frontal, superior lateral parietal, inferior lateral parietal, superior lateral temporal, inferior lateral temporal, amygdala, hippocampus, and the whole cerebral cortex. Data from each side of the brain were averaged in all structures. We also examined bilaterally the caudate nucleus and putamen combined; cerebellar vermis and hemispheres combined; and pons, mesencephalon, and thalamus. [11C]PMP-PET data were not normalized, as the disease processes under study could affect AChE activity in all cerebral structures. Instead, quantitative values of [11C]PMP k3 were used directly as indices of AChE activity. We compared data between the normal control group and the individual patient groups and also compared data between PD and MSA-P groups.
Data analysis.
We computed the mean and SD of k3 values in each region of interest (ROI) for the 3 disease groups individually as well as the normal control group. The decrease in AChE activity of each disease group is reported as the percentage decline relative to the normal control group mean k3 value for each ROI, and an associated p value is calculated from the corresponding 2-sample t test. We calculated the p value threshold by controlling the false discovery rate among all tests for multiple ROIs across all groups. The p value threshold at which we can claim significance is 0.03, which corresponds to a false discovery rate no greater than 5%.24 With the original threshold at 0.05, the false discovery rate was less than 10%. Owing to the small sample size, the results involving the PSP group need to be interpreted with caution. We used Spearman rank correlations to evaluate the relationship between k3 values and measures of balance and gait.
RESULTS
Cerebral cortex.
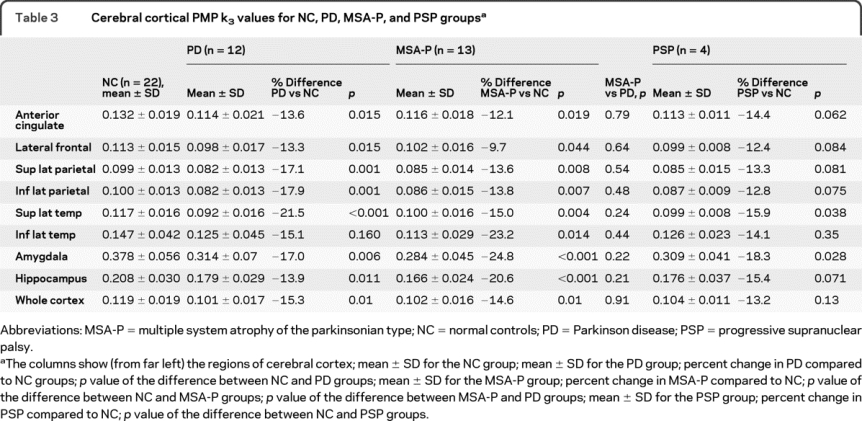
PMP k3 values are markedly decreased in all regions studied within the PD, MSA-P, and PSP groups as compared to the normal controls (table 3). The difference between the normal control group and the PD group is significant in all regions except for the inferior lateral temporal cortex. The difference between the normal control group and the MSA-P group is significant in all regions studied. There is no significant difference between the PD and the MSA-P groups for any of the cerebral cortical regions. Although the changes found in the PSP group in comparison to the normal control group are similar in magnitude to those in the PD and MSA-P groups, none of the regions is significantly different from the normal control group except for the superior lateral temporal region. The small number of patients with PSP studied probably accounts for the lack of significance in this group.
Table 3. Cerebral cortical PMP k3 values for NC, PD, MSA-P, and PSP groups
Subcortical structures.
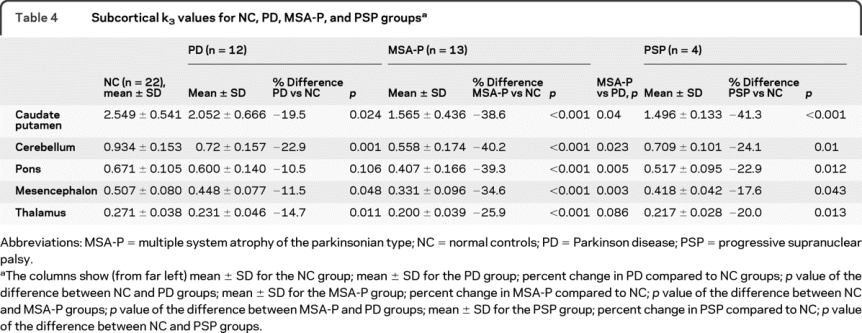
The figure illustrates PMP k3 values obtained from the levels of the thalamus, basal ganglia, mesencephalon, and pons/cerebellum in the normal control, PD, MSA-P, and PSP groups. In PD as compared to normal controls, k3 values are significantly decreased in caudate nucleus and putamen, cerebellum, and thalamus, with a marginally significant decrease in mesencephalon and no significant change in pons (table 4). In MSA-P as compared to normal controls, k3 values are significantly decreased in all regions studied. The decrease in k3 values between PD and MSA-P groups is significantly different for all regions except the thalamus owing to greater declines in MSA-P. In the PSP group, despite the small number of cases studied, significant differences in k3 values from normal controls are found in all subcortical structures examined.

Figure PMP k3 activity in 4 brain levels.
Maps of mean PMP k3 activity in the following groups: normal control (NC), Parkinson disease (PD), multiple system atrophy of the parkinsonian type (MSA-P), and progressive supranuclear palsy (PSP). The column on the far left shows the group average PMP map for the 22 normal controls for all 4 brain levels displayed to the same peak (3.0 min−1) (see the single long color bar to the left of this column). As using the same peak does not permit evaluation of changes within individual structures of interest owing to the large dynamic range of acetylcholinesterase (AChE) values in the brain, we scaled the PMP maps to suit individual regions (see the 4 color bars corresponding to thalamus, basal ganglia, mesencephalon, and pons/cerebellum). The 4 columns to the right of the color bars show regional AChE values from group averages in the 4 groups, from left to right NC, PD, MSA-P, and PSP. In the NC column, 1 arrow points to the thalamus, and a second arrow points to the mesencephalon. The individual images show that AChE activity is reduced in the thalamus in all 3 patient groups, with the greatest decrease seen in MSA-P; decreased in the striatum in all 3 groups, with greater declines in MSA-P and PSP than in PD; slightly decreased in the mesencephalon in PD and markedly decreased in MSA-P and PSP; markedly decreased in all 3 patient groups in the cerebellum, greatest in the MSA-P group; and unchanged in the pons in PD, but significantly decreased in MSA-P and PSP groups.
Table 4. Subcortical k3 values for NC, PD, MSA-P, and PSP groups
Balance and gait disorders.
We evaluated the relationship of balance and gait disturbances to k3 reductions in all cerebral cortical and subcortical structures studied. The results demonstrate a correlation between the severity of the balance and gait disorders for patients with PD, patients with MSA-P, and patients with PSP and reductions in k3 values in the midbrain (p < 0.025) and cerebellum (p < 0.013), but not for any other structure.
The mean age of the normal control subjects is approximately one decade younger than the patient groups (table 1). To adjust for this age effect, we checked all comparisons using the analysis of covariance method for each ROI with age as a continuous covariate. The k3 differences in most ROIs are attenuated with slightly different p values. The significant differences between normal controls and PD cases in the original analysis become marginally significant with p values around 0.05 (from 0.05 to 0.09) in anterior cingulate cortex, lateral frontal cortex, hippocampus, as well as whole cortex and thalamus. The results of all other comparisons, however, are not affected.
DISCUSSION
This study revealed similar patterns of diminished k3 values, which represent AChE activity, within cerebral cortex in PD and MSA-P as compared with normal controls. Direct comparison of PD with MSA-P showed no significant differences for any region studied. Differences between PSP and normal controls were generally similar to differences found in PD and MSA-P in comparison to normal controls, but the results in PSP were not significant, likely because this component of the study was insufficiently powered. In subcortical regions, AChE activity was decreased in PD as compared to normal controls in all regions studied, although the decline in pons was nonsignificant and the decrease in mesencephalon was marginally significant. Comparisons of both MSA-P and PSP with normal controls showed more severe declines in all regions studied than in PD. Comparison of MSA-P with PD verified the greater involvement of subcortical structures in MSA-P.
These results indicate that degenerative process in PD, MSA-P, and PSP affect the major cholinergic pathways of the CNS. Had the degenerative process spared the cholinergic connections, PMP k3 values likely would have been slightly increased, not decreased as we found, because the decline in tissue volume would have increased the density of AChE in surviving tissue.
The present data cannot answer the question of whether the cholinergic pathways are disproportionately affected compared to tissue loss as a whole. Nevertheless, our method, which utilizes ROIs centered on the peak of activity in an anatomic region, is relatively insensitive to atrophy, because the increasing compactness of surviving tissue compensates for activity lost due to degenerated tissue components. Given that most of our k3 values were 10%–40% lower than those in normal controls, we suspect that the degeneration in cholinergic pathways is out of proportion to degeneration in the tissue as a whole, especially in subcortical structures of MSA-P, where values were decreased by 26%–40% compared to normal controls. Our index for AChE activity, k3, has been corrected for the free tissue fraction f2. While this added stability to the fits, the K1/k2 correction term used as 1/f2 is sensitive to partial volume effects such as tissue atrophy. Estimates of K1/k2 were decreased in patient groups by only 2%–7% in cerebral cortical regions and thalamus, slightly over 10% in basal ganglia, about 20% in brainstem structures, and up to 30% in cerebellar hemispheres (in the MSA-P group). In all cases, decreases in the AChE activity index exceeded decreases in the atrophy-sensitive measure K1/k2.
It is intriguing that all 3 disorders, characterized by parkinsonism and known to have underlying involvement of the nigrostriatal dopaminergic pathway, also involve brain cholinergic pathways. The disorders differ widely in pathogenesis. PD is characterized by condensation of α-synuclein into intraneuronal Lewy bodies, MSA-P by condensation of α-synuclein into oligodendroglial inclusions, and PSP by tau aggregation into intraneuronal neurofibrillary tangles. We speculate that dopaminergic pathways and cholinergic pathways have features in common that make both susceptible to these varied degenerative processes. Alternatively, degeneration of dopaminergic pathways might secondarily lead to effects on cholinergic terminals.
Cholinergic projections to cerebral cortex originate in neurons of basal forebrain, including medial septum, nucleus of the diagonal band of Broca, and basal nucleus of Meynert. These neurons innervate the entire cerebral cortex, hippocampus, olfactory bulb, and basolateral amygdala. The projections constitute an important part of higher cognitive functions, including attention, learning, and memory. In PD, several specific cognitive disorders occur in the absence of dementia.25-31 These include impairment of executive function,30 perceptual motor dysfunction,31 and disorders of visuospatial functioning, including internal representation of visuospatial information and spatial memory.25-29 These abnormalities have some relationship to cholinergic innervation, as PD includes loss of cholinergic neurons in the nucleus basalis3–5,32-34 and reduced muscarinic binding and choline acetyltransferase activity in hippocampus and neocortex.13 Several investigations have also demonstrated cholinergic deficits in the cerebral cortex of patients with PD without dementia,9,14,15 although the deficits are more severe and widespread in PDD.11
A previous investigation utilized techniques similar to ours in examining cerebral cortical AChE activity in PD with normal cognition and in PDD as compared to normal controls.12 This investigation examined cerebral cortical regions analogous to those we reported, but did not present data from individual regions or study subcortical structures. The study showed that overall cerebral cortical AChE activity was reduced 12.7% in PD without dementia and 20.9% in PDD as compared to normal controls. The results in their PD group without dementia are comparable to our findings, which showed 15.3% reduction in whole brain cortex of PD as compared to normal controls. Another study utilizing [11C]MP4A and PET showed a significant reduction of AChE activity in the cerebral cortex of PD and a nonsignificant reduction in PSP.14 In contrast, there was a significant reduction of AChE activity in thalamus in PSP but no significant change in PD. These results are similar to ours; however, the severity of reduction in cerebral cortical AChE activity in PSP in our study suggests that we may have found a significant reduction if we had examined a larger number of cases. Moreover, we found a significant reduction of thalamic AChE activity in PD, but the degree was considerably less than in PSP.
Cholinergic projections to subcortical regions arise principally in the PPT and LDT nuclei.35-37 These nuclei project rostrally to thalamus, lateral hypothalamus, and basal forebrain, and caudally to cerebellum and pontine and medullary reticular formation. The nuclei also project to basal ganglia and substantia nigra pars compacta. The PPT/LDT have been implicated in the coordination of gait.35 The finding of more severe degeneration of the subcortical cholinergic projections in PSP and MSA-P than in PD may at least partially account for the greater disability in gait found in the latter 2 disorders than in PD in the earlier stages of the diseases. Connections of PPT/LDT to reticular activating system are important, as the system participates in arousal and wakefulness. Rostral projections to upper brainstem and thalamus may be responsible for the association of REM sleep with dreaming, while the caudal projections to reticular formation induce immobility during dreaming.37 Deficits in cholinergic projections to some of these subcortical sites have been described in PD.15 A recent report describes loss of cholinergic PPT/LDT neurons in postmortem examinations of MSA as compared to normal controls.38 These findings are in keeping with the current results.
The low k3 values in cerebellum may not reflect just denervation of the PPT/LDT, but also loss of cholinergic neurons and fibers intrinsic to cerebellum. The predominant fibers in cerebellum thought to utilize ACh are the ascending mossy fibers that innervate granule cells and neurons in the molecular layer of cerebellar cortex.39 Degeneration of these connections may contribute to the decreased cerebellar k3 values in our study.
A previous article evaluated cholinesterase activity in MSA-C.40 In contrast to our findings, this investigation demonstrated no change in k3 in cerebral cortex, but k3 was decreased in thalamus and posterior lobe of cerebellar cortex. The authors attributed the decreased thalamic k3 to loss of innervation from PPT and LDT nuclei, as we have. They attributed the low k3 values in the posterior lobe to degeneration of the cholinergic synapses in the molecular layer and not to loss of projections from PPT/LDT.
The current study was not designed to determine whether cholinergic denervation is a distinctive hitherto unrecognized signature feature of MSA-P and PSP rather than a reflection of nonspecific degeneration of brainstem, basal ganglia, and cortex as the disease progresses. Additional studies with ligands that portray other neurotransmitter systems would be needed to resolve the issue of selective vulnerability of cholinergic pathways.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Nan, C.-N. Wang, and X. Wang.
DISCLOSURE
Dr. Gilman served as Chair of the Safety Monitoring Committees for clinical trials conducted by Elan Corporation, Wyeth, and Transition Therapeutics; served as a consultant to the Food and Drug Administration; has served/serves on scientific advisory boards for GlaxoSmithKline, Adamas Pharmaceuticals, Johnson & Johnson, Solvay Pharmaceuticals, Inc., and Longitude Capital; has received funding for travel from GlaxoSmithKline; receives royalties from the publication of Contemporary Neurology Series (Oxford University Press, 1982), MedLink Neurology (MedLink Corporation, 1992), Experimental Neurology (Elsevier, 2003), and Neurobiology of Disease (Elsevier, 2005); and receives research support from the NIH (NIA P50 AG08671 [PI], U01-AG16976 [PI], and NINDS P01 NS15655 [PI]). Dr. Koeppe receives research support from Elan Corporation and from the NIH (UO1 AG024904-01 [Co-I], PO1 NS15655 [Co-I], RO1 HL079540 [Co-I], RO1 DA022520 [Co-I], and RO1 DA016423 [Co-I]). Dr. Nan serves as an Associate Editor of Statistics in Biosciences. C.-N. Wang and X. Wang report no disclosures. Dr. Junck has served on a scientific advisory board for Genentech, Inc.; has received travel expenses and/or honoraria for lectures or educational activities not funded by industry; and receives research support from Genentech, Inc. and the NIH (P01-CA85878 [Co-I] and NCI P01-CA59827 [Co-I]). Dr. Chervin serves on scientific advisory boards for Pavad Medical, Inc. and Sweet Dreamzzz, Inc.; has received research equipment from Respironics; his university received contributions from Respironics and Sepracor Inc. toward an endowed professorship eventually held by him; serves as an Associate Editor of Sleep, on the editorial boards of Sleep Medicine and the Journal of Clinical Sleep Medicine, and as a Section Editor for UpToDate; could potentially receive royalty payments for US Patent 7,190,995 B2, assigned to University of Michigan and Michigan Tech University (issued 2007): System and method for analysis of respiratory cycle-related EEG changes in sleep-disordered breathing; has filed a patent, assigned to University of Michigan and Michigan Tech University, regarding automated polysomnographic assessment for rapid eye movement sleep behavior; has filed a provisional patent, assigned to University of Michigan, regarding a new approach to continuous positive airway pressure for patients with sleep apnea; has received honoraria for speaking and/or educational activities from Accel Healthcare Communications, Physician's Weekly, and CME LLC; served in the past as a consultant for Guidepoint Global, Advantage Healthcare, Alexza Pharmaceuticals, OrbiMed Advisors, LLC, and Respironics; receives or received research support from the NIH (HL080941 [PI], RR000042 [PI for GCRC-supported study], HL083129 [Site PI], NS015655 [Co-I], HL087819 [Co-I], HL089918 [Co-I], HL03645 [Co-I], NS02009 [PI], HD38461 [PI], NS15655 [Co-I], and NS42698 [Co-I]), the Children's Memorial Hospital (Chicago, IL), the Michael J. Fox Foundation, and the Arthritis Foundation; and holds stock options in Pavad Medical, Inc. Dr. Consens serves on a scientific advisory board for GlaxoSmithKline; has received travel expenses and/or honoraria for lectures or educational activities not funded by industry; and has received/receives research support from the Michigan Clinical Research Unit (MO1-RR00042 [PI]) and from the NIH (NIA R01 AG 19360 [Co-I], 1PO1 GM 067189-01A2 [Co-I], and 5 P50 NS15655 [Co-I]. A. Bhaumik reports no disclosures.
Address correspondence and reprint requests to Dr. Sid Gilman, Department of Neurology, University of Michigan, 300 N. Ingalls 3D15, Ann Arbor, MI 48109-5489 sgilman@med.umich.edu
Editorial, page 1406
Study funding: Supported by the NIH (NS044233, NS15655, AG08671, and UL1RR024986).
Disclosure: Author disclosures are provided at the end of the article.
Received August 2, 2009. Accepted in final form January 6, 2010.
REFERENCES
- 1.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 1976;2:1403. [DOI] [PubMed] [Google Scholar]
- 2.Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 1983;219:1184–1190. [DOI] [PubMed] [Google Scholar]
- 3.Whitehouse PJ, Hedreen JC, White CL 3rd, Price DL. Basal forebrain neurons in the dementia of Parkinson disease. Ann Neurol 1983;13:243–248. [DOI] [PubMed] [Google Scholar]
- 4.Candy JM, Perry RH, Perry EK, et al. Pathological changes in the nucleus of Meynert in Alzheimer's and Parkinson's diseases. J Neurol Sci 1983;59:277–289. [DOI] [PubMed] [Google Scholar]
- 5.Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer's disease, paralysis agitans and Korsakoff's Disease. Acta Neuropathol 1983;61:101–108. [DOI] [PubMed] [Google Scholar]
- 6.Irie T, Fukushi K, Akimoto Y, Tamagami H, Nozaki T. Design and evaluation of radioactive acetylcholine analogs for mapping brain acetylcholinesterase (AChE) in vivo. Nucl Med Biol 1994;21:801–808. [DOI] [PubMed] [Google Scholar]
- 7.Irie T, Fukushi K, Namba H, et al. Brain acetylcholinesterase activity: validation of a PET tracer in a rat model of Alzheimer's disease. J Nucl Med 1996;37:649–655. [PubMed] [Google Scholar]
- 8.Kilbourn MR, Snyder SE, Sherman PS, Kuhl DE. In vivo studies of acetylcholinesterase activity using a labeled substrate, N-[11C]methylpiperdin-4-yl propionate ([11C]PMP). Synapse 1996;22:123–131. [DOI] [PubMed] [Google Scholar]
- 9.Kuhl DE, Minoshima S, Fessler JA, et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer's disease, and Parkinson's disease. Ann Neurol 1996;40:399–410. [DOI] [PubMed] [Google Scholar]
- 10.Kuhl DE, Koeppe RA, Minoshima S, et al. In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer's disease. Neurology 1999;52:691–699. [DOI] [PubMed] [Google Scholar]
- 11.Bohnen NI, Kaufer DI, Ivanco LS, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 2003;60:1745–1748. [DOI] [PubMed] [Google Scholar]
- 12.Bohnen NI, Kaufer DI, Hendrickson R, et al. Cognitive correlates of cortical cholinergic denervation in Parkinson's disease and parkinsonian dementia. J Neurol 2006;253:242–247. [DOI] [PubMed] [Google Scholar]
- 13.Lange KW, Wells FR, Jenner P, Marsden CD. Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson's disease. J Neurochem 1993;60:197–203. [DOI] [PubMed] [Google Scholar]
- 14.Shinotoh H, Namba H, Yamaguchi M, et al. Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson's disease and progressive supranuclear palsy. Ann Neurol 1999;46:62–69. [PubMed] [Google Scholar]
- 15.Fujita M, Ichise M, Zoghbi SS, et al. Widespread decrease of nicotinic acetylcholine receptors in Parkinson's disease. Ann Neurol 2006;59:174–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol 1999;56:33–39. [DOI] [PubMed] [Google Scholar]
- 17.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 19.Koeppe RA, Frey KA, Snyder SE, Meyer P, Kilbourn MR, Kuhl DE. Kinetic modeling of N-[11C]methylpiperidin-4-yl propionate: alternatives for analysis of an irreversible positron emission tomography trace for measurement of acetylcholinesterase activity in human brain. J Cereb Blood Flow Metab 1999;19:1150–1163. [DOI] [PubMed] [Google Scholar]
- 20.Snyder SE, Tluczek L, Jewett DM, Nguyen TB, Kuhl DE, Kilbourn MR. Synthesis of 1-[11C]methylpiperidin-4-yl propionate ([11C]PMP) for in vivo measurements of acetylcholinesterase activity. Nucl Med Biol 1998;25:751–754. [DOI] [PubMed] [Google Scholar]
- 21.Zubieta JK, Koeppe RA, Mulholland GK, Kuhl DE, Frey KA. Quantification of muscarinic cholinergic receptors with [11C]NMPB and positron emission tomography: method development and differentiation of tracer delivery from receptor binding. J Cereb Blood Flow Metab 1998;18:619–631. [DOI] [PubMed] [Google Scholar]
- 22.Minoshima S, Koeppe RA, Mintun MA, et al. Automated detection of the intercommissural line for stereotactic localization of functional brain images. J Nucl Med 1993;34:322–329. [PubMed] [Google Scholar]
- 23.Minoshima S, Koeppe RA, Frey KA, Kuhl DE. Anatomic standardization: linear scaling and nonlinear warping of functional brain images. J Nucl Med 1994;35:1528–1537. [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995;57:289–300. [Google Scholar]
- 25.Lees AJ, Smith E. Cognitive deficits in the early stages of Parkinson's disease. Brain 1983;106:257–270. [DOI] [PubMed] [Google Scholar]
- 26.Cooper JA, Sagar HJ, Jordan N, Harvey NS, Sullivan EV. Cognitive impairment in early, untreated Parkinson's disease and its relationship to motor disability. Brain 1991;114:2095–2122. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs DM, Marder K, Cote LJ, Sano M, Stern Y, Mayeux R. Neuropsychological characteristics of preclinical dementia in Parkinson's disease. Neurology 1995;45:1691–1696. [DOI] [PubMed] [Google Scholar]
- 28.Troster AI, Stalp LD, Paolo AM, Fields JA, Koller WC. Neuropsychological impairment in Parkinson's disease with and without depression. Arch Neurol 1995;52:1164–1169. [DOI] [PubMed] [Google Scholar]
- 29.Dubois B, Pillon B. Cognitive deficits in Parkinson's disease. J Neurol 1997;244:2–8. [DOI] [PubMed] [Google Scholar]
- 30.Gotham AM, Brown RG, Marsden CD. ‘Frontal’ cognitive function in patients with Parkinson's disease ‘on’ and ‘off’ levodopa. Brain 1988;111:299–321. [DOI] [PubMed] [Google Scholar]
- 31.Stern Y, Mayeux R, Rosen J, Ilson J. Perceptual motor dysfunction in Parkinson's disease: a deficit in sequential and predictive voluntary movement. J Neurol Neurosurg Psychiatry 1983;46:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano I, Hirano A. Parkinson's disease: neuron loss in the nucleus basalis without concomitant Alzheimer's disease. Ann Neurol 1984;15:415–418. [DOI] [PubMed] [Google Scholar]
- 33.Tagliavini F, Pilleri G, Bouras C, Constantinidis J. The basal nucleus of Meynert in idiopathic Parkinson's disease. Acta Neurol Scand 1984;70:20–28. [DOI] [PubMed] [Google Scholar]
- 34.Rogers JD, Brogan D, Mirra SS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol 1985;17:163–170. [DOI] [PubMed] [Google Scholar]
- 35.Jenkinson N, Nandi D, Muthusamy K, et al. Anatomy, physiology, and pathophysiology of the pedunculopontine nucleus. Mov Disord 2009;24:319–328. [DOI] [PubMed] [Google Scholar]
- 36.Winn P. How best to consider the structure and function of the pedunculopontine tegmental nucleus: evidence from animal studies. J Neurol Sci 2006;248:234–250. [DOI] [PubMed] [Google Scholar]
- 37.Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130:2770–2788. [DOI] [PubMed] [Google Scholar]
- 38.Schmeichel AM, Buchhalter LC, Low PA, et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology 2008;70:368–373. [DOI] [PubMed] [Google Scholar]
- 39.Jaarsma D, Ruigrok TJ, Caffe R, et al. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res 1997;114:67–96. [DOI] [PubMed] [Google Scholar]
- 40.Hirano S, Shinotoh H, Arai K, et al. PET study of brain acetylcholinesterase in cerebellar degenerative disorders. Mov Disord 2008;23:1154–1160. [DOI] [PubMed] [Google Scholar]