

Abstract
Scar formation in the nervous system begins within hours after traumatic injury and is characterized primarily by reactive astrocytes depositing proteoglycans that inhibit regeneration. A fundamental question in CNS repair has been the identity of the initial molecular mediator that triggers glial scar formation. Here we show that the blood protein fibrinogen, which leaks into the CNS immediately after blood–brain barrier (BBB) disruption or vascular damage, serves as an early signal for the induction of glial scar formation via the TGF-β/Smad signaling pathway. Our studies revealed that fibrinogen is a carrier of latent TGF-β and induces phosphorylation of Smad2 in astrocytes that leads to inhibition of neurite outgrowth. Consistent with these findings, genetic or pharmacologic depletion of fibrinogen in mice reduces active TGF-β, Smad2 phosphorylation, glial cell activation, and neurocan deposition after cortical injury. Furthermore, stereotactic injection of fibrinogen into the mouse cortex is sufficient to induce astrogliosis. Inhibition of the TGF-β receptor pathway abolishes the fibrinogen-induced effects on glial scar formation in vivo and in vitro. These results identify fibrinogen as a primary astrocyte activation signal, provide evidence that deposition of inhibitory proteoglycans is induced by a blood protein that leaks in the CNS after vasculature rupture, and point to TGF-β as a molecular link between vascular permeability and scar formation.
Introduction
The identity of the molecule that triggers glial scar formation after injury or disease is a fundamental and unresolved question in CNS repair. Astrocytes become reactive in response to CNS traumatic injury or disease characterized by blood–brain barrier (BBB) breakdown, increased vascular permeability, edema formation, inflammatory responses, and neuronal damage, as in spinal cord injury (SCI), stroke, Alzheimer disease (AD), multiple sclerosis (MS), and brain trauma (Abbott et al., 2006). Reactive astrocytes demarcate the injury site from healthy tissue by forming a glial scar (Faulkner et al., 2004), which consists mainly of chondroitin sulfate proteoglycans (CSPGs), including neurocan and phosphacan, the major factors that inhibit axon regeneration after CNS injury (Fawcett and Asher, 1999; McKeon et al., 1999). CSPGs are upregulated by reactive astrocytes soon after injury (Jones et al., 2003; Silver and Miller, 2004). However, the early molecular events that trigger reactive astrogliosis and induce secretion of inhibitory proteoglycans are poorly understood.
One of the earliest events after brain trauma or SCI is leakage of blood components into brain parenchyma at areas that correlate with the formation of reactive astrocytes (Schnell et al., 1999; Preston et al., 2001). The soluble blood protein fibrinogen is converted to insoluble fibrin by the action of thrombin and is deposited in the nervous system promptly after vascular damage or BBB disruption (Akassoglou and Strickland, 2002; Adams et al., 2004, 2007a). Fibrinogen plays a causative role in nervous system disease as a regulator of inflammation (Akassoglou et al., 2004; Adams et al., 2007b; Paul et al., 2007), remyelination (Akassoglou et al., 2002), and neurodegeneration (Adhami et al., 2006; Schachtrup et al., 2007). Fibrinogen mediates functions in the nervous system as a ligand for cell-specific receptors. In microglia, fibrinogen induces activation of Akt and Rho via the CD11b/CD18 integrin receptor (complement receptor 3) (Adams et al., 2007b). In neurons, it induces phosphorylation of epidermal growth factor receptor (EGFR) via the αvβ3 integrin (Schachtrup et al., 2007). Given the potential of fibrinogen for signal transduction via a wide range of cellular receptors and its presence in the CNS microenvironment immediately after injury, we hypothesized that fibrinogen could be an early signal that triggers activation of astrocytes.
The present study reveals the unexpected finding that fibrinogen regulates TGF-β-mediated signal transduction within CNS tissues after vascular damage and induces reactive astrocytosis and deposition of CSPGs. Mice genetically or pharmacologically depleted of fibrinogen show a dramatic reduction in active TGF-β and reduced astrocytosis and neurocan deposition after injury. In primary astrocyte cultures, fibrinogen is a potent inducer of secretion of proteoglycans, and conditioned medium of fibrinogen-treated astrocytes inhibited neurite outgrowth. Active TGF-β was undetectable in fibrinogen solutions, but plasma-isolated fibrinogen coimmunoprecipitated with latent TGF-β, which is activated by astrocytes. These results identify fibrinogen-bound latent TGF-β as the molecular inducer of the inhibitory properties of the gliotic scar after vascular damage.
Materials and Methods
Mice.
C57BL/6J mice (Jackson Laboratory) and C57BL/6J-inbred mice deficient for fibrinogen (Fib −/−) (Suh et al., 1995) were used. All animal procedures were performed under the guidelines set by the University of California, San Francisco, Institutional Animal Care and Use Committee and are in accord with those set by the National Institutes of Health.
Cortical stab wound injury.
Cortical stab wound injury (SWI) was performed as described previously (Lin et al., 2005). Mice were anesthetized with avertin (0.2 ml of a 1.25% solution/10 g of body weight) and placed in a stereotaxic apparatus (David Kopf Instruments). A midline incision was made through the scalp, and the skin was retracted laterally. The periosteum was cleaned from the skull, and a hole was drilled over the right cerebral hemisphere, exposing the dura. A 30-gauge needle was inserted stereotaxically (anteroposterior [AP], −1.0 mm; mediolateral [ML], −1.0 mm; dorsoventral [DV], −1.70 mm from the bregma according to Paxinos and Watson) and left in place for 5 min. The needle was removed, the skin was sutured, and the mice were allowed to recover and returned to their cages. For stereotactic injection of active TGF-β and simultaneous SWI, TGF-β (R&D Systems) was dissolved in 4 mm HCl containing 1 mg/ml bovine serum albumin (BSA) and diluted to 0.04 mg/ml with artificial CSF (ACSF) composed of the following (in mm): 125 NaCl, 26.2 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 12.5 glucose. Active TGF-β or vehicle control (1 μl) diluted with ACSF was slowly injected (0.2 μl/min) into the cortex (at the same coordinates used for SWI) with a 10 μl Hamilton syringe attached to a 30-gauge needle.
Stereotactic injection of fibrinogen.
Male C57BL/6 mice were used for stereotactic injection of fibrinogen; controls received ACSF. The mice were anesthetized with avertin (0.2 ml/10 g of body weight) and placed in the stereotaxic apparatus. Fibrinogen (Calbiochem) was dissolved in endotoxin-free distilled water, diluted to 5 mg/ml with ACSF, and kept at 37°C. Fibrinogen (1 μl of 5 mg/ml) or ACSF was slowly injected (0.2 μl/min) with a 10 μl Hamilton syringe attached to a 33-gauge needle into mouse cortex at the same coordinates used for SWI. Stereotactic injection of 1 μl of 5 mg/ml control protein, albumin, fibronectin, or laminin (all from Sigma) was performed as described above. Fibrinogen fractions I-2 and I-9 were isolated as described previously (Mosesson and Sherry, 1966; Mosesson et al., 1972; Galanakis et al., 2007). For TGF-β receptor type I inhibitor injection, the inhibitor SB431542 (Sigma) was dissolved in DMSO solution and diluted to 0.1 mm with ACSF (final DMSO concentration, 1%). TGF-β receptor type I inhibitor solution (3 μl of 0.1 mm) or 3 μl of ACSF containing 1% DMSO was slowly injected (0.2 μl/min) with a 10 μl syringe attached to a 33-gauge needle into the lateral ventricle (AP, −0.1 mm; ML, −1.0 mm, DV, −2.5 mm) 30 min before fibrinogen injection.
Immunohistochemistry.
Mice were transcardially perfused with cold saline followed by 4% paraformaldehyde under avertin anesthesia. Brain samples were removed, postfixed, cryoprotected, cut into 30 μm sections, and processed as described previously (Adams et al., 2007b; Schachtrup et al., 2007). The primary antibodies were rat anti-glial fibrillary acidic protein (GFAP) (1:1000; Zymed), sheep anti-human fibrinogen (1:200; U.S. Biological), rabbit anti-neurocan (1:500) (Milev et al., 1996), and mouse anti-active TGF-β (1:200; R&D Systems). Sections were permeabilized in 0.1% Triton X-100, blocked with 5% BSA, and incubated for 24 h at 4°C with primary antibodies. Sections were rinsed in PBS with 0.1% Triton X-100 and incubated with secondary antibodies conjugated with Alexa Fluor 488 or 594 (1:200; Invitrogen) for 1 h in the dark. After washing in PBS, sections were mounted on glass slides and coverslipped with Prolong Gold antifading agent (Invitrogen). For light microscopy, after primary antibody incubation sections were incubated with biotinylated secondary antibody (1:500; Vector Laboratories), followed by the ABC Elite system (1:500; Vector Laboratories), and developed in 3,3′-diaminobenzidine (Vector Laboratories). The sections were mounted on glass slides, air dried, dehydrated, and coverslipped with Permount (Fisher).
Systemic fibrinogen depletion.
Mice were depleted of fibrinogen with ancrod as described previously (Akassoglou et al., 2002, 2004; Adams et al., 2007b). Ancrod or buffer was administered by mini-pumps, which deliver 0.5 μl/h; thus, mice received 2.4 U of ancrod per day.
Culture of primary astrocytes.
Cortical astrocytes were isolated as described previously (McCarthy and de Vellis, 1980). Brain cortices were isolated at P3 and transferred into HBSS (Invitrogen). The dura was removed, and the cortices were minced with scissors. Four cortices were transferred into 21.5 ml of HBSS (Invitrogen), digested in 2.5 ml of trypsin-EDTA (2.5%, Invitrogen) and 1.0 ml of pancreatin (2.5%, ICN Biochemicals) for 30 min at 37°C with occasional shaking. Trypsin was deactivated by adding 7.5 ml of DMEM with 10% fetal bovine serum (Invitrogen) and 1.5% penicillin/streptomycin (Invitrogen). Digested cortices were collected by centrifugation at 500 × g for 5 min, resuspended in 20 ml of DMEM with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin, and plated onto poly-d-lysine-coated 75 cm2 tissue culture flasks. The media were changed on day 2 and every third day thereafter. After 10–14 d, when astrocytes had reached confluency, microglia and oligodendrocytes were removed by shaking the flasks at 240 rpm for 6 h. The adherent astrocyte cultures were rinsed with PBS and passaged using trypsin-EDTA (1:3 split). After astrocytes reached confluency, they were used for experiments.
RNA isolation and quantitative PCR.
RNA was isolated from primary astrocyte cultures with the RNAeasy kit (Qiagen), according to the manufacturer's instructions. RNA was reverse transcribed to cDNA with the GeneAmp RNA PCR Core kit (Applied Biosystems) and random hexamer primers. Real-time PCR analysis was performed with the Opticon DNA Engine 2 (MJ Research) and the Quantitect SYBR Green PCR kit (Qiagen) with 1.5 μl of cDNA template in a 25 μl reaction mixture. PCR efficiencies of the primers were calculated by serial dilution of template; no significant differences were found between the target genes and the housekeeping genes. Results were analyzed with the Opticon 2 Software and the comparative CT method as described previously (Livak and Schmittgen, 2001; Passino et al., 2007). Data are expressed as 2−ΔΔCT for the experimental gene of interest normalized to the housekeeping gene and presented as fold change relative to control. The following primers were used: neurocan: Fwd 5′-TGC AAC CAC GGC TAA GCT C-3′, Rev 5′-GGG GAT AAG CAG GCA ATG AC-3′; GAPDH: Fwd 5′-CAA GGC CGA GAA TGG GAA G-3′, Rev 5′-GGC CTC ACC CCA TTT GAT GT-3′.
Immunoblots and coimmunoprecipitation.
To detect astrocyte-secreted CSPG or neurocan, primary astrocytes were serum starved overnight and treated with 2.5 mg/ml fibrinogen for various times. Astrocytes were activated with epidermal growth factor (EGF) (Calbiochem) or TGF-β (R&D Systems). Astrocytes were pretreated with 50 nm PD168393 (Calbiochem), an inhibitor of EGFR phosphorylation, or 10 μm SB431542 (Sigma), an inhibitor of TGF-β receptor type I, 1 h before fibrinogen treatment. Supernatants were sampled, and CSPGs were digested for 3 h at 37°C with 0.01 U chondroitinase ABC enzyme (ChABC) (Sigma) per milliliter of astrocyte-conditioned medium. To inhibit active TGF-β we used 10 μg/ml of a pan-active TGF-β antibody (R&D Systems). For detection of latent TGF-β binding protein 1 (LTBP1) and latency-associated protein (LAP), we used human plasminogen-free fibrinogen samples isolated from plasma by ion-exchange chromatography, glycine precipitation, and gel filtration (Calbiochem) or as described previously (Mosesson and Sherry, 1966; Mosesson et al., 1972; Galanakis et al., 2007).
For coimmunoprecipitation, 100 μg of fibrinogen (Calbiochem) was incubated with rabbit anti-fibrinogen antibody (1:200) bound to A-agarose beads for 4 h at room temperature. After three washes, the beads were resuspended in sample buffer, boiled for 10 min, and centrifuged. Protein extracts were separated by electrophoresis on 4–12% gradient or 6% SDS-PAGE gels as described previously (Akassoglou et al., 2002) and probed with the following antibodies: neurocan (1:1000) (Milev et al., 1996), P-EGFR, EGFR, P-Smad2 (1:1000, Cell Signaling Technology), Smad2 (1:1000, Cell Signaling Technology), active TGF-β (1:1000, R&D Systems), GFAP (1:1000, Zymed), LTBP1 (1:1000, Santa Cruz Biotechnology), LTBP1 (1:500, R&D Systems), LAP1 (1:1000, R&D Systems), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000, Cell Signaling Technology). Blots were washed three times with TBS-T, incubated with peroxidase-labeled secondary antibodies (goat anti-rabbit IgG, Cell Signaling Technology, 1:5000 or goat anti-mouse IgG, Santa Cruz Biotechnology), diluted in 5% nonfat milk in TBS-T for 1 h at room temperature, and washed again, followed by detection with chemiluminescence (ECL, GE Healthcare).
Astrocyte-conditioned medium.
Astrocytes were plated in 24-well plates at 1 × 105 cells per well. Astrocytes were untreated or treated with 2.5 mg/ml fibrinogen for various times. All experiments were performed with astrocytes aged for 30 d in culture, unless otherwise noted. For neurite outgrowth assay studies, astrocytes were treated with fibrinogen overnight; the medium was changed, and astrocyte-conditioned medium was harvested 2 d later. Therefore, sampled astrocyte-conditioned medium did not contain fibrinogen. For inhibitor studies, astrocytes were pretreated with the TGF-β receptor I inhibitor SB431542 (10 μm; Sigma) 1 h before fibrinogen treatment. For digestion of CSPGs, astrocyte-conditioned medium was treated for 3 h at 37°C with 0.01 U of ChABC (Sigma) per milliliter of astrocyte-conditioned medium. Astrocyte-conditioned media were filtered through 0.45 μm filters and frozen at −80°C.
Neurite outgrowth assay.
Cortical neurons were isolated from E16 C57BL/6 mice as described previously (Kim et al., 2002), plated overnight at a density of 37,500 cells per well in eight-well Nunc plates coated with poly-d-lysine, cultured with 80% astrocyte-conditioned medium for 24 h, fixed with 4% paraformaldehyde, and stained with anti-β-tubulin (1:200, Sigma). Neurite outgrowth and length were quantified as described previously (Niederöst et al., 1999; Wang et al., 2002; Schachtrup et al., 2007). Neurite outgrowth was determined as the proportion of total cells bearing neurites longer than the diameter of the cell body, an indication of successful initiation of neurite outgrowth. Neurite length (micrometers) was measured from the portion of cells that successfully initiated neurite outgrowth. The number of neurite-bearing cells and neurite length was determined from 400–500 neurons per condition. Ten representative images per well were taken with an Axioplan II epifluorescence microscope (Carl Zeiss) with a dry Plan-Neofluar 40 × 0.75 NA objective and an Axiocam HRc CCD camera. Images were acquired and analyzed with Axiovision image analysis software. All experiments were repeated four times and were performed in triplicate.
TGF-β measurement.
The amounts of active and latent TGF-β in blood-isolated fibrinogen or in astrocyte supernatant were measured with an ELISA kit (R&D Systems), according to the manufacturer's instructions. Active TGF-β levels were measured in 2.5 mg/ml of fibrinogen isolated from blood and in the supernatant of serum-starved astrocytes treated for 1 h with 2.5 mg/ml of blood-isolated fibrinogen or TGF-β or left untreated. To determine levels of latent TGF-β, HCl was added to the samples to activate latent TGF-β to the immunoreactive form.
Immunocytochemistry.
Cells were rinsed with ice-cold PBS, fixed in 4% PFA for 30 min at 4°C, washed three times with PBS, blocked in PBS with 5% BSA and 0.1% Triton X-100 for 30 min at 4°C, and washed three times in PBS. The cells were then incubated with anti-GFAP (1:1000, Zymed), active TGF-β (1:1000, R&D Systems), or anti-β-tubulin (1:200, Sigma) in PBS with 1% BSA overnight. After three washes in PBS, the cells were incubated with secondary antibody (Jackson ImmunoResearch Laboratories, 1:200) for 45 min in PBS with 3% BSA, washed three times in PBS, and coverslipped with Slowfade Gold containing DAPI (Invitrogen).
Microscopy and image-acquisition and analysis.
Images were acquired with an Axioplan II epifluorescence microscope (Carl Zeiss) equipped with dry Plan-Neofluar objectives (×10 0.3 NA, ×20 0.5 NA, or ×40 0.75 NA), an Axiocam HRc CCD camera, and Axiovision image analysis software. Quantitative image analysis for the immunostained mouse cortical sections was performed on three equally spaced sections through the level of the injection site (AP −1.0 mm). To maintain consistency between the selected sections, a rectangular box (200 × 1000 μm) was localized to the medial and lateral region 100 μm away from needle track in each image as described previously (Buffo et al., 2008). The digitized images were analyzed with ImageJ software (National Institutes of Health). The number of pixels per image with an intensity above a predetermined threshold level was quantified by measurement of the immunoreactive areas for GFAP, neurocan, and active TGF-β. Total immunoreactivity was calculated as percentage area density defined as the number of pixels (positively stained areas) divided by the total number of pixels (sum of positively and negatively stained area) in the imaged field (Pickford et al., 2008). All quantitative analyses were performed in a blinded manner.
Statistical analyses.
Statistical significance was determined with one-way ANOVA and Bonferroni's post-test (multiple comparisons). Statistical calculations were performed with GraphPad Prism. The data are presented as mean ± SEM.
Results
Genetic or pharmacologic depletion of fibrinogen reduces astrocytosis and neurocan expression after SWI
To identify a possible role for fibrinogen in astrocyte activation, we used SWI, a model of brain injury that induces reactive astrocytosis (Fig. 1 A) (Silver and Miller, 2004; Sofroniew, 2005). After SWI, we detected massive fibrin deposition, which colocalized with reactive astrocytes (Fig. 1 B). Fibrinogen was not detected in control uninjured brain (Fig. 1 B). To determine whether fibrinogen is required for astrocyte activation after SWI, we examined deposition of inhibitory CSPGs, which are secreted by astrocytes and are critical for inhibiting CNS repair, in mice genetically or pharmacologically depleted of fibrinogen. Fib −/− mice or mice treated with the fibrin-depleting agent ancrod showed a fourfold reduction in astrocyte activation and a threefold reduction in neurocan deposition than wild-type (WT) mice (Fig. 1 C–E).
Figure 1.
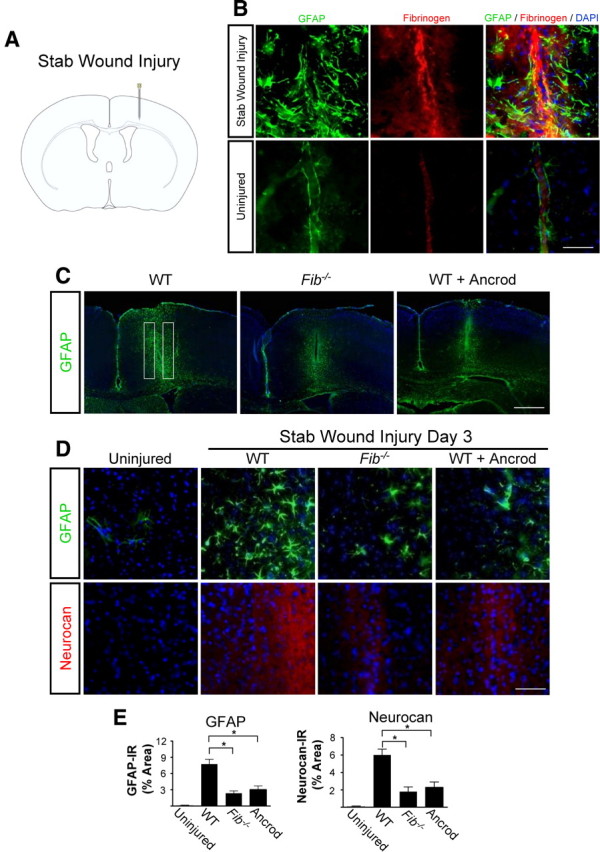
Fibrinogen depletion decreases astrogliosis and neurocan expression. A, SWI, a model of cortical trauma. B, Three days after SWI, immunolabeling for fibrin (red) and GFAP (green) revealed perivascular fibrin colocalizing with reactive astrocytes in brain coronal sections of mice (top). Uninjured mice show no fibrinogen deposition in the brain (bottom). C, Low-magnification images of Fib −/− and ancrod-depleted WT mice show reduced astrogliosis demonstrated by immunolabeling of GFAP astrocytes (green) 3 d after SWI. Box indicates area of quantification. D, Fib −/− and ancrod-treated WT mice show reduced astrogliosis and neurocan expression 3 d after SWI, shown by immunolabeling of astrocytes for GFAP (green) and neurocan (red). Uninjured WT mice served as controls. E, Quantification revealed lower levels of GFAP and neurocan in Fib −/− and ancrod-treated mice than in WT mice (n = 5 per group) after SWI. Values are mean ± SEM. *p < 0.001. Scale bars: B, 75 μm; C, 500 μm; D, 60 μm.
Fibrinogen induces neurocan upregulation in primary astrocytes
To examine whether fibrinogen directly regulates astrocyte functions, we treated primary astrocytes in vitro. Fibrinogen induced a fivefold increase in neurocan RNA (Fig. 2 A) and robust neurocan protein secretion in astrocyte supernatants (Fig. 2 B,C). Fibrinogen induced neurocan secretion in astrocyte supernatants as early as 1 d after treatment (Fig. 2 B), and the effects of fibrinogen were sustained up to 7 d after treatment (Fig. 2 C). Fibrinogen showed sustained induction of neurocan in astrocytes using TGF-β (Fig. 2 B) or EGF as positive controls (Fig. 2 C). Fibrinogen did not affect astrocyte proliferation, indicating that alterations in cell division are not a contributor to the effects of fibrinogen on inducing neurocan expression (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Induction of neurocan by fibrinogen was concentration dependent (Fig. 2 D). Interestingly, fibrinogen even at a concentration of 0.3 mg/ml, which is 10% of the physiological level of fibrinogen in the blood (Mosesson et al., 2001), was sufficient to induce neurocan protein expression (Fig. 2 D).
Figure 2.
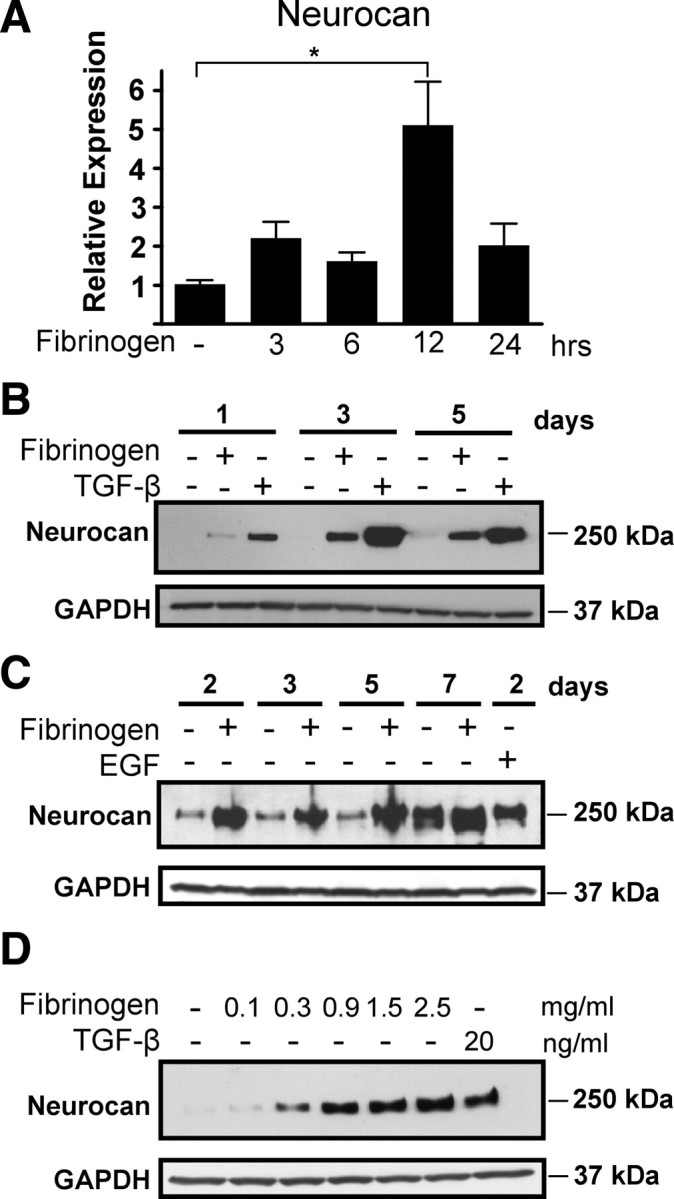
Fibrinogen regulates neurocan expression in primary astrocytes. A, Increased expression of neurocan mRNA in primary astrocytes after fibrinogen treatment as determined by quantitative PCR and normalized to GAPDH. Results are from three independent experiments performed in duplicate. B, C, Potent and rapid fibrinogen-induced neurocan protein secretion by primary astrocytes with TGF-β (B) or EGF (C) as positive controls. D, Fibrinogen-induced neurocan expression is concentration-dependent. Primary astrocytes were treated for 2 d with fibrinogen (0.1–2.5 mg/ml). TGF-β served as a positive control. Representative blots are shown from three independent experiments. Values are mean ± SEM of three independent experiments. *p < 0.01.
Fibrinogen induces astrocyte-mediated inhibition of neurite outgrowth via the TGF-β receptor pathway
CSPG and neurocan expression by astrocytes is regulated by the TGF-β receptor and EGFR pathways (Smith and Strunz, 2005). Inhibition of EGFR did not prevent fibrinogen-induced CSPG expression (Fig. 3 A), and fibrinogen did not induce phosphorylation of EGFR in astrocytes (Fig. 3 B), suggesting that the EGFR pathway is not involved in fibrinogen-induced neurocan upregulation. In contrast, a TGF-β receptor inhibitor abolished fibrinogen-induced neurocan secretion (Fig. 3 C). Moreover, in primary astrocytes fibrinogen induced phosphorylation of Smad2, which is the downstream effector of the TGF-β receptor pathway (Fig. 3 D). Inhibiting TGF-β receptor abolished fibrinogen-induced Smad2 phosphorylation (Fig. 3 D).
Figure 3.

Fibrinogen regulates astrocyte-induced formation of inhibitory ECM through the TGF-β receptor pathway in vitro. A, Fibrinogen-induced inhibitory CSPG expression by primary astrocytes is not regulated through the EGFR pathway. Conditioned medium from fibrinogen-treated and untreated astrocytes was blotted with anti-CSPG antibody. Astrocytes were incubated with EGFR inhibitor 1 h before stimulation with fibrinogen. B, Fibrinogen does not induce phosphorylation of the EGFR in astrocytes. Serum-starved astrocytes were treated with fibrinogen for the indicated times or left untreated, and lysates were blotted for P-EGFR. EGF served as a positive control. C, Treatment of primary astrocytes with a TGF-β receptor type I inhibitor blocked fibrinogen-induced neurocan expression. TGF-β served as a positive control. D, TGF-β receptor type I inhibitor blocks fibrinogen-induced Smad2 phosphorylation. E, Neurite outgrowth assay with astrocyte-conditioned medium. Conditioned medium (CM) from fibrinogen-treated primary astrocytes aged for 30 d inhibited neurite outgrowth of cortical neurons. This inhibitory effect was abolished by pretreatment with TGF-β receptor type I inhibitor. F, ChABC digestion of conditioned medium from fibrinogen-treated primary astrocytes aged for 40 d in culture reduced the inhibition of neurite outgrowth of cortical neurons. G, Quantification of neurite outgrowth and neurite length revealed an inhibitory effect of fibrinogen-treated astrocyte-CM in astrocytes aged for 30 d. The TGF-β receptor inhibitor abolished this effect. Values are mean ± SEM of at least three different experiments. A minimum of 900–1000 neurons per condition were analyzed. *p < 0.001. Representative blots are shown from three independent experiments. Scale bar, 40 μm.
We then examined the functional consequences of fibrinogen-mediated regulation of the TGF-β receptor pathway in astrocytes. Neurocan secretion by activated astrocytes is inhibitory to neurite outgrowth (Friedlander et al., 1994; Asher et al., 2000). Indeed, conditioned medium from fibrinogen-treated astrocytes significantly decreased both neurite length and the percentage of cells showing neurite outgrowth (Fig. 3 E–G), suggesting that fibrinogen renders astrocytes inhibitory to neurite outgrowth. As astrocytes age in vitro, secretion of proteoglycan increases. Treatment of cortical cultures with supernatants from untreated astrocytes that had aged in culture for 40 d (Fig. 3 F) decreased the percentage of cells with neurite outgrowth to a greater extent than supernatants from astrocytes aged 30 d (Fig. 3 G). However, regardless of whether astrocytes were aged in culture for 40 d (Fig. 3 F) or 30 d (Fig. 3 G), conditioned media from fibrinogen-treated astrocytes decreased in parallel both the percentage of cells showing neurite outgrowth and the neurite length relative to control conditioned media from untreated astrocytes. ChABC effectively removes the inhibitory properties of CSPGs and promotes axonal regrowth after SCI (Bradbury et al., 2002; Silver and Miller, 2004). Conditioned medium of fibrinogen-treated astrocytes treated with ChABC no longer inhibited neurite outgrowth, suggesting that fibrinogen-induced decrease of neurite outgrowth was, indeed, mediated by CSPGs (Fig. 3 F). Importantly, conditioned medium of astrocytes treated with fibrinogen in the presence of TGF-β receptor inhibitor did not inhibit neurite outgrowth (Fig. 3 E,G). This represents the first evidence that fibrinogen induces biological responses via the TGF-β receptor pathway.
Fibrinogen-bound latent TGF-β becomes activated by astrocytes
Consistent with its role in inflammatory and reparative processes after injury, fibrinogen binds a diverse array of cell-associated proteins, such as integrin and nonintegrin receptors, matrix components like fibronectin, and soluble factors, including cytokines, and growth factors, such as interleukin-1β, fibroblast growth factor-2, and vascular endothelial growth factor (Sahni and Francis, 2000; Sahni et al., 2003, 2004). We hypothesized that fibrinogen might have a novel function as a soluble or matrix-associated carrier for TGF-β and regulator of its bioavailability. Fibrinogen-induced Smad2 phosphorylation was abolished by a pan anti-TGF-β blocking antibody (Fig. 4 A), suggesting that active TGF-β is necessary for the induction of Smad2 phosphorylation by fibrinogen. However, active isoforms of TGF-β1, TGF-β2, and TGF-β3 were undetectable in fibrinogen isolated from plasma as measured by ELISA (Fig. 4 B). By binding to LAP, TGF-β exists in a functionally inactive latent form that cannot interact with cellular TGF-β receptors (Attisano et al., 1994). While active TGF-β was undetectable, latent TGF-β isoforms, particularly latent TGF-β1 and TGF-β2, were detected at high levels in plasma fibrinogen (Fig. 4 B) (2.4 pg of TGF-β per 10 μg of fibrinogen, or 3.45 μm TGF-β/1 m fibrinogen). According to our ELISA data 2.5 mg/ml of fibrinogen contain 0.6 ng/ml of latent TGF-β. Treatment of astrocytes with 2.5 mg/ml of fibrinogen revealed a delay in Smad2 phosphorylation compared with active 2 ng/ml TGF-β treatment (Fig. 4 C). These results show that active TGF-β is undetectable, whereas latent inactive TGF-β is present in plasma-isolated fibrinogen.
Figure 4.
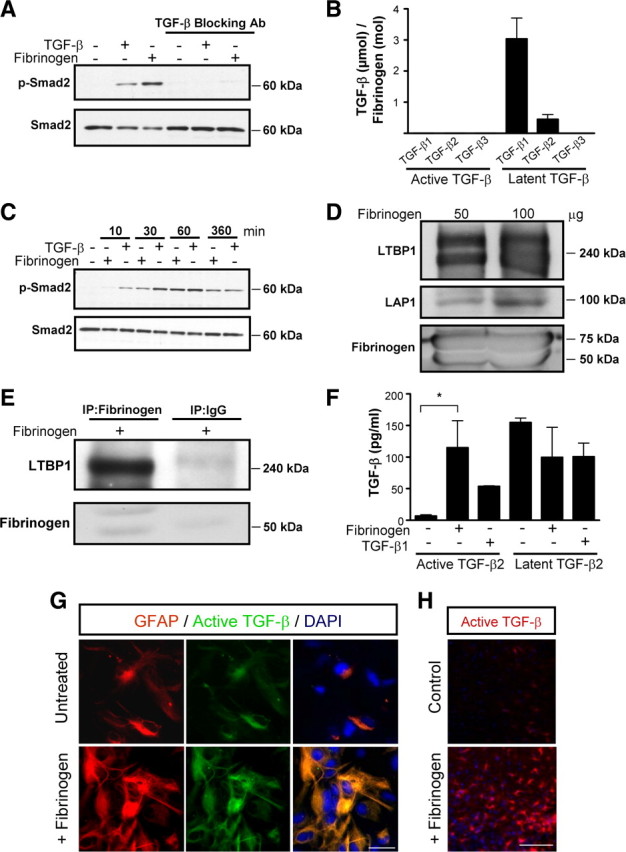
Fibrinogen-bound latent TGF-β is activated by astrocytes. A, Fibrinogen-induced Smad2 phosphorylation is blocked by anti-TGF-β neutralizing antibody. B, Plasma-isolated fibrinogen was tested for active and latent TGF-β with TGF-β isoform-specific ELISAs. C, Delayed fibrinogen-induced Smad2 phosphorylation in primary astrocytes. Astrocytes were treated with fibrinogen or TGF-β (positive control) for the indicated times or left untreated, and lysates were blotted for P-Smad2. TGF-β induces Smad2 phosphorylation (P-Smad2) as early as 10 min, whereas fibrinogen took 30 min. D, Western blotting of fibrinogen isolated from plasma reveals LAP1 and latent LTBP1. E, Plasma-isolated fibrinogen was immunoprecipitated with an antibody against human fibrinogen and immunoblotted with an antibody against LTBP1, revealing a complex of LTBP1 with fibrinogen. F, TGF-β ELISA revealed active TGF-β in supernatants of primary astrocytes treated with fibrinogen for 1 h. Results are from three independent experiments performed in duplicate. G, Immunolabeling for active TGF-β (green) and GFAP (red) revealed active TGF-β formation in astrocytes 1 h after fibrinogen treatment. H, Stereotactic injection of fibrinogen in mouse cortex results in active TGF-β formation, compared with control ACSF injection. Representative immunostaining is shown (n = 5 mice per group). Values are mean ± SEM. *p < 0.05. Scale bars: G, 13.5 μm; H, 90 μm.
Latent TGF-β consists of dimeric TGF-β covalently linked to LAP (Gentry and Nash, 1990). This complex is bound by LTBP1 (Miyazono et al., 1988), which contains N-terminal domains that bind to extracellular matrix (ECM) proteins to regulate the bioavailability of TGF-β within tissues (Unsöld et al., 2001). We therefore examined the possibility that the LTBP1-LAP complex binds fibrinogen. We performed Western blots on commercially available fibrinogen and on plasma fibrinogen isolated by the glycine isolation procedure, which removes potential coprecipitating plasma contaminants that do not bind directly to fibrinogen (Mosesson and Sherry, 1966; Mosesson et al., 1972; Galanakis et al., 2007). Western blotting showed LTBP1 and LAP proteins in plasma-isolated fibrinogen (Fig. 4 D). We further confirmed the presence of LTBP1 and LAP in our own preparation of the intact fibrinogen fraction I-2 isolated from human plasma that is a similar fraction with the commercially available fibrinogen (Mosesson and Sherry, 1966; Mosesson et al., 1972; Galanakis et al., 2007) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The presence of LTBP1 in two different preparations of fibrinogen was confirmed with both monoclonal and polyclonal antibodies against LTBP1 (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Coimmunoprecipitation with an antibody against fibrinogen showed the presence of LTBP1 (Fig. 4 E), suggesting that fibrinogen forms a complex with LTBP1.
Liberation of biologically active TGF-β requires its dissociation from the latent complex (Annes et al., 2003, 2004), which can be achieved through proteolytic cleavage of the large latent complex or conformational changes induced by engagement of latent TGF-β by specific cellular integrin receptors (Munger et al., 1999; Mu et al., 2002; Yang et al., 2007; Wipff and Hinz, 2008). Indeed, αvβ8 integrin binding to latent TGF-β is a major mechanism of TGF-β activation in astrocytes (Cambier et al., 2005). To test whether fibrinogen-bound latent TGF-β is activated by primary astrocytes, we measured active TGF-β in supernatants of fibrinogen-treated astrocytes. Within 1 h after treatment, the levels of active TGF-β2, the major TGF-β isoform expressed by astrocytes after injury (Lagord et al., 2002), had increased 25-fold (Fig. 4 F). Moreover, immunocytochemistry revealed active TGF-β formation in primary astrocyte cultures 1 h after fibrinogen treatment (Fig. 4 G). Consistent with these in vitro findings, stereotactic injection of fibrinogen into the cortex strongly induced formation of active TGF-β (Fig. 4 H). Active TGF-β immunoreactivity was 10-fold greater in fibrinogen-injected mice than in ACSF-injected controls (n = 5 mice per group, p < 0.001). These results suggest that plasma fibrinogen is a carrier of latent TGF-β, which becomes activated by astrocytes.
Fibrinogen regulates formation of active TGF-β by astrocytes after brain injury
To determine whether fibrinogen is required for active TGF-β formation, we measured active TGF-β after SWI in mice genetically or pharmacologically depleted of fibrinogen. As shown by immunolabeling for active TGF-β and GFAP, astrocytes were the major cell type positive for active TGF-β after injury (Fig. 5 A), and active TGF-β levels were dramatically reduced in Fib −/− and ancrod-treated mice compared with WT mice (Fig. 5 B,C). Furthermore, Western blotting of brain samples for the TGF-β receptor downstream effector Smad2 revealed a dramatic decrease in Smad2 phosphorylation in Fib −/− and ancrod-treated mice than in WT mice after SWI (Fig. 5 D). These results suggest that fibrinogen is necessary for generation of active TGF-β after brain injury.
Figure 5.

Fibrinogen is necessary for active TGF-β formation and signaling in the CNS. A, Immunolabeling for GFAP (green) and active TGF-β (red) revealed astrocyte-specific expression of active TGF-β after SWI (yellow). B, Decreased levels of active TGF-β immunolabeling (red) in Fib −/− and ancrod-treated mice 3 d after SWI. Uninjured WT brain served as a negative control. C, Lower levels of active TGF-β after SWI in Fib −/− and ancrod-treated mice than in WT controls (n = 5 per group). D, Brain lysates of Fib −/− and ancrod-treated WT mice show reduced Smad2 phosphorylation 3 d after SWI. Brain lysates of uninjured WT mice do not show P-Smad2 activation. Lysates from two mice per experimental treatment are shown. Values are mean ± SEM. *p < 0.001. Scale bar, 60 μm.
Next, we investigated potential synergism between fibrinogen and active TGF-β. Stereotactic injection of purified active TGF-β after SWI potentiated the effect of SWI on GFAP and neurocan expression in WT mice (Fig. 6 A). However, Fib −/− mice injected with active TGF-β after SWI showed significantly decreased GFAP and neurocan immunoreactivity (Fig. 6 B). Notably, the activity of the injected TGF-β was not impaired in Fib −/− mice, because active TGF-β induces a similar threefold increase in both WT and Fib −/− mice (Fig. 6 B). Thus, as expected, active TGF-β can induce scar formation in the absence of fibrinogen, but fibrinogen is needed to obtain the full effect of endogenous TGF-β activation.
Figure 6.

Synergism between fibrinogen and active TGF-β in the induction of astrogliosis and neurocan deposition. A, Fib −/− mice show reduced astrogliosis and neurocan expression demonstrated by immunostaining for GFAP and neurocan, 3 d after TGF-β injection followed by SWI. Uninjured WT mice served as controls. Stereotactic injection of TGF-β induced a similar increase in astrogliosis and neurocan expression in Fib −/− and WT controls injected with ACSF. B, Quantification revealed significant lower levels of GFAP and neurocan in Fib −/− mice than in WT mice (n = 4 per group) after TGF-β injection and SWI. Values are mean ± SEM. *p < 0.001. Scale bar, 90 μm.
Stereotactic fibrinogen injection in the cortex induces scar formation via activation of the TGF-β receptor pathway
To determine whether fibrinogen can activate astrocytes in vivo, we injected fibrinogen stereotactically into the cortex (Fig. 7 A). Because a smaller needle is used for stereotactic injection than for SWI, injection of ACSF did not result in fibrinogen leakage into the cortex (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). GFAP immunoreactivity in the cortex was 3.4-fold greater after fibrinogen injection than after ACSF injection (Fig. 7 B,C). Fibrinogen injection resulted in less diffuse fibrinogen immunoreactivity than SWI (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Among other blood and ECM proteins, only fibrinogen increased GFAP immunoreactivity: stereotactic injection of albumin, laminin, or fibronectin did not affect astrocyte activation (Fig. 7 B,C). Even with no other injury, GFAP immunoreactivity and neurocan deposition in the cortex increased markedly after a single stereotactic fibrinogen injection (Fig. 7 D). Importantly, injecting TGF-β receptor inhibitor before fibrinogen significantly reduced fibrinogen-induced GFAP and neurocan immunoreactivity (Fig. 7 E).
Figure 7.
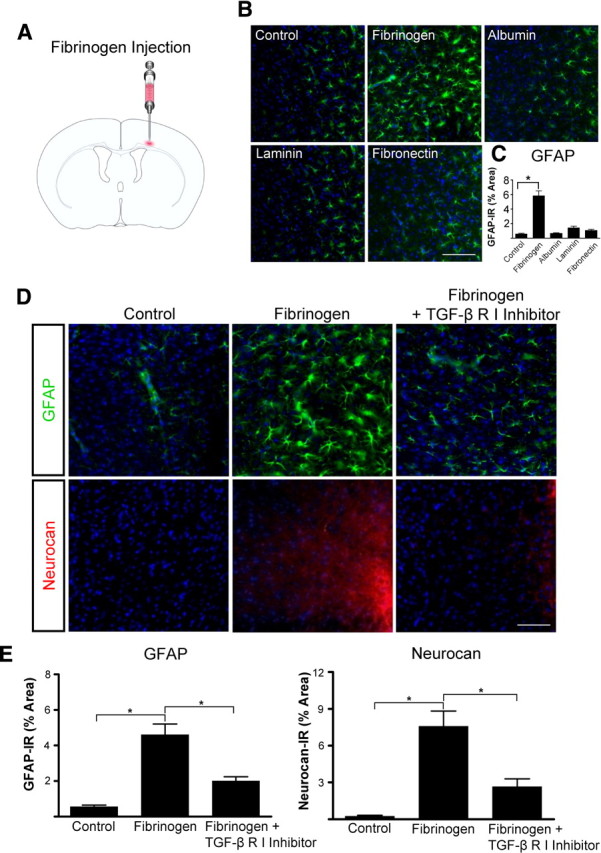
Fibrinogen regulates astrocyte-induced scar formation through the TGF-β receptor pathway in vivo. A, Stereotactic injection of fibrinogen into the cortex. B, Astrocyte immunolabeling (green) revealed reactive astrocytosis in fibrinogen-injected mice; injection of albumin, laminin, and fibronectin had no effect. C, Quantification revealed a 3.4-fold increase in astrogliosis after fibrinogen injection. n = 5 mice per condition. D, TGF-β receptor inhibitor injection in mice abolished fibrinogen-induced astrogliosis and neurocan deposition demonstrated by immunostaining for GFAP (green, top row) and neurocan (red, bottom row). E, Quantification revealed a sevenfold increase in GFAP and a tenfold increase in neurocan immunoreactivity in fibrinogen-injected mice compared with controls (n = 5 per group). TGF-β receptor inhibitor in fibrinogen-injected mice (n = 5) revealed a 2.5-fold decrease in GFAP immunoreactivity and a threefold decrease in neurocan immunoreactivity compared with fibrinogen-injected mice (n = 5). Values are mean ± SEM. *p < 0.001. Scale bars: B, 90 μm; D, 60 μm.
To examine whether fibrinogen-bound latent TGF-β was necessary for the induction of scar formation, we injected stereotactically a plasma-derived fibrinogen fraction that does not carry latent TGF-β. Plasma-derived fibrinogen consists primarily of fibrinogen with a molecular weight of 340 kDa (de Maat and Verschuur, 2005). We isolated the low-molecular-weight fibrinogen fraction I-9, which lacks major parts of the Aα chain carbosyl terminal region, and like its intact counterpart is fully coagulable (Mosesson et al., 1972) (Fig. 8 A). Stereotactic injection of fraction I-2 increased GFAP immunoreactivity and neurocan deposition (Fig. 8 C,D) to the levels seen after injection of commercial fibrinogen (Fig. 7 D,E). In contrast to the commercially available fibrinogen and the intact fibrinogen fraction I-2, the fibrinogen fraction I-9 contains dramatically reduced levels of LTBP1 (Fig. 8 B). Stereotactic injection of fraction I-9 resulted in significantly less astrocytosis and neurocan immunoreactivity than intact fibrinogen fraction I-2 (Fig. 8 C,D). As shown by ELISA, fraction I-9 had eightfold less latent TGF-β than fraction I-2 (Fig. 8 E). These results, in combination with the reduction of fibrinogen-induced neurocan deposition by inhibition of the TGF-β receptor (Fig. 7), suggest that the TGF-β receptor pathway is a major contributor to the effects of fibrinogen on scar formation in vivo.
Figure 8.
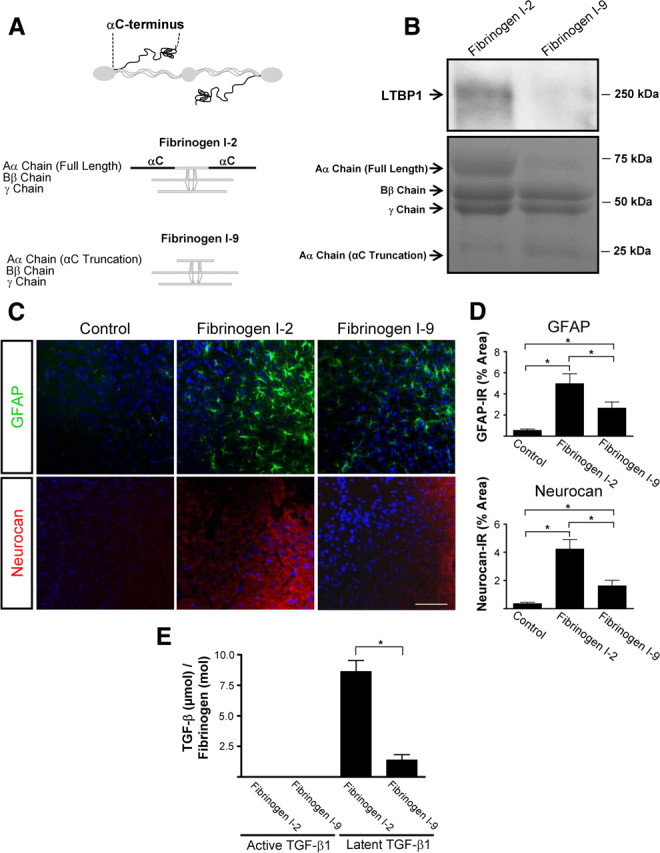
Fibrinogen induces scar formation as a carrier of latent TGF-β. A, Fibrinogen fractions isolated from plasma. Fraction I-2 is intact 340 kDa fibrinogen composed of three pairs of nonidentical polypeptide chains, designated Aα (63.5 kDa), Bβ (56 kDa), and γ (47 kDa). Fraction I-9 lacks residues of the C terminus of the Aα chain. B, Western blotting of plasma fractions I-2 and I-9 shows no LTBP1 in fraction I-9. Coomassie staining revealed the absence of full-length fibrinogen Aα chain in fraction I-9. C, Immunolabeling of astrocytes (GFAP, green) and neurocan (red) shows less astrogliosis and lower neurocan expression after stereotactic injection of fraction I-9 than after injection of fraction I-2. ACSF-injected mice served as controls. D, GFAP and neurocan levels are lower in mice injected with fraction I-9 than in those injected with fraction I-2 (n = 6 per group). E, TGF-β ELISA revealed reduced latent TGF-β1 in plasma-isolated fibrinogen fraction I-9. Values are mean ± SEM of three independent experiments performed in duplicate. *p < 0.001. Scale bar, 90 μm.
Discussion
A fundamental question in CNS repair has been the identity of the initial molecular inducer that triggers inhibitory scar formation. Our results and previous experiments suggest the following model for the role of fibrinogen as an early inducer of scar formation in the CNS (Fig. 9). (1) In uninjured CNS, the BBB is intact, and fibrinogen bound to latent TGF-β remains sequestered in the blood stream. Indeed, we detected no active TGF-β in plasma-isolated fibrinogen. In accordance, active TGF-β is not detected in normal plasma, whereas it is activated during platelet activation and thrombus formation (Grainger et al., 1995, 2000). (2) CNS injury or disease associated with a compromised BBB allows fibrinogen to leak into the CNS. (3) Fibrinogen carries latent TGF-β, providing a fibrinogen reservoir of TGF-β. (4) Astrocytes express integrins such as αvβ8, which bind LTBP1 and activate TGF-β (Cambier et al., 2005). Fibrinogen-bound latent TGF-β interacts with astrocytes, leading to active TGF-β formation and activation of the TGF-β/Smad signaling pathway. (5) Active TGF-β via TGF-β receptor/Smad signaling in astrocytes induces scar formation and upregulation of neurocan, an inhibitory CSPG (Asher et al., 2000). (6) Fibrinogen triggers the formation of a nonpermissive environment for regeneration via TGF-β-mediated glial scar formation. Because fibrinogen-bound latent TGF-β within the CNS would be expected to become available immediately after injury or BBB disruption, it could serve as the primary astrocyte activation signal initiating astrocyte scar formation. Fibrinogen-carried TGF-β and locally synthesized TGF-β might exert a synergistic effect in a disease or injury setting where active TGF-β is present after transcriptional activation and protein synthesis by CNS or inflammatory infiltrating cells. In this scheme, local provisional fibrin matrices play a critical role in the induction of scar formation in the nervous system by regulating the bioavailability of active TGF-β at sites of vascular damage.
Figure 9.

Proposed model for the role of fibrinogen-bound latent TGF-β activated by reactive astrocytes after injury. In the uninjured CNS, fibrinogen-bound latent TGF-β circulates within the blood stream. BBB disruption or vascular rupture after trauma leads to leakage of fibrinogen-bound latent TGF-β into the CNS. Fibrinogen-bound latent TGF-β interacts with local perivascular astrocytes, leading to active TGF-β formation, which induces reactive astrocytosis by regulating the TGF-β/Smad signaling pathway, resulting in scar formation. Local provisional fibrin matrix thus functions as a primary astrocyte activation signal initiating scar formation in the CNS by regulating the bioavailability of active TGF-β at sites of vascular damage.
Blood proteins leak into the CNS immediately after injury from mechanical damage of CNS vasculature, and also from sustained BBB disruption, which may last for several days after injury and correlate with areas of glial scarring and inflammation (Preston et al., 2001). Thus, fibrinogen leakage might perpetuate as well as trigger glial cell activation. This mechanism might be particularly in play after injury or disease associated with fibrin deposition, such as SCI (Schachtrup et al., 2007), MS (Gay and Esiri, 1991; Marik et al., 2007), ischemia–hypoxia (Adhami et al., 2006), and AD (Paul et al., 2007; Ryu and McLarnon, 2008). Because CSPGs may be upregulated in pathological states within a seemingly intact BBB, additional mechanisms, such as microglial activation and increased inflammation, might contribute to the upregulation of proteoglycans (Fitch et al., 1999). After SCI in vivo, reduction of CSPGs by chondroitinase treatment increases axonal regrowth and functional recovery (Bradbury et al., 2002) and prevents long-distance axonal retraction (Busch et al., 2009). Future studies of fibrin depletion in animal models of SCI will reveal the effects of fibrin in axonal regeneration and dieback.
Our prior studies demonstrated a proinflammatory role for fibrinogen in the nervous system as an activator of microglia via the CD11b/CD18 integrin receptor (Akassoglou et al., 2004; Adams et al., 2007a,b). Depletion of fibrinogen decreases microglial activation in animal models of MS (Akassoglou et al., 2004; Adams et al., 2007b) and AD (Paul et al., 2007). Moreover, specific inhibition of fibrin binding to CD11b suppresses clinical symptoms and demyelination in an animal model of MS without adverse effects in blood coagulation (Adams et al., 2007b). Our current findings suggest that fibrinogen might regulate inflammatory responses not only as a ligand of CD11b/CD18 but also as an inducer of active TGF-β. TGF-β is a pleiotropic cytokine that regulates fibrosis and inflammation. TGF-β functions as a suppressor of inflammation but is also essential for the development of adaptive immune responses (Blobe et al., 2000; Li et al., 2007; Li and Flavell, 2008). TGF-β is also expressed in neurons, in which it exerts neuroprotective functions (Tesseur et al., 2006). Given the pleiotropic functions of TGF-β, fibrinogen as a carrier of TGF-β might exert multiple functions in inflammation and tissue repair. Future studies will shed light in the cellular specificity of fibrinogen-mediated TGF-β activation in the CNS.
The ability of fibrinogen to signal through integrin receptors and to bind precursors of growth factors could underlie its pleiotropic biological functions in CNS disease. Although fibrinogen potently induced the TGF-β receptor (this study) and EGFR (Schachtrup et al., 2007), the mechanisms of activation appear to be quite distinct. Direct binding of ECM ligands to integrins is a well characterized mechanism for EGFR transactivation (Giancotti and Ruoslahti, 1999; Moro et al., 2002). Binding of fibrinogen to αvβ3 integrin transactivates the EGFR in neurons (Schachtrup et al., 2007). However, fibrinogen does not induce EGFR phosphorylation in astrocytes (this study), perhaps because astrocytes lack the molecular machinery (e.g., αvβ3 integrin receptor) (Milner et al., 2001) required for EGFR transactivation (Moro et al., 2002; Schachtrup et al., 2007).
TGF-β receptor signaling in astrocytes is a novel growth factor receptor pathway induced by fibrinogen. TGF-β receptor activation is mediated by the release of active TGF-β from LTBP1, which contains domains that can potentially bind to ECM proteins (Unsöld et al., 2001). Fibrinogen coimmunoprecipitated with LTBP1, suggesting that it binds to latent TGF-β. LTBP1 was not detected in the fibrinogen fraction I-9, which lacks the C terminus of the Aα chain (Spraggon et al., 1998; Burton et al., 2006). The αC domain of fibrinogen affects properties of the fibrin clot (Lord, 2007) and contains binding sites for tissue plasminogen activator (Medved and Nieuwenhuizen, 2003) and α5β1 and αvβ3 integrins (Belkin et al., 2005). Thus, the interaction of fibrinogen with latent TGF-β may occur within the αC domain. It is also possible that loss of interactions within the αC domain other than latent TGF-β may contribute to the reduced scar formation by fibrinogen fraction I-9. Overall, these studies suggest that fibrinogen transactivates the EGFR by binding to αvβ3 integrin (Schachtrup et al., 2007) but activates the TGF-β receptor as a carrier of the latent form of TGF-β.
Latent TGF-β is converted into its biologically active form by various mechanisms, including matrix metalloproteinases, thrombospondin 1, and integrins (Saharinen et al., 1999; Wipff and Hinz, 2008). Integrins αvβ6 and αvβ8 are major activators of latent TGF-β (Annes et al., 2003). In mice, genetic depletion of β6 integrin produced a phenotype similar to that of targeted knock-out of TGF-β (Huang et al., 1996). Similar phenotypes are produced by knock-out of the integrin subunits αv (Annes et al., 2004) and β8 (Cambier et al., 2005; Araya et al., 2006). Integrin αvβ6 binding to latent TGF-β induces mechanical conformational changes that render TGF-β accessible for binding to TGF-β receptor (Munger et al., 1999; Wipff et al., 2007). On the other hand, αvβ8-induced TGF-β1 activation is dependent on proteolytic degradation of LAP, which results in the release of active TGF-β1 into the extracellular environment (Mu et al., 2002). Indeed, astrocytes express αvβ6 and αvβ8 integrins (Milner et al., 2001; Cambier et al., 2005), and αvβ8 binding to latent TGF-β is a major mechanism of TGF-β activation in astrocytes (Cambier et al., 2005). Interestingly, αvβ8 is a known receptor for fibrinogen (Chernousov and Carey, 2003). It is therefore possible that fibrinogen binding to αvβ8 might contribute to the liberation of active TGF-β. Fibrinogen might exert a synergistic effect when active TGF-β is present through other mechanisms, such as inflammation attributable to vascular damage or infiltrating cells at sites of injury. In the absence of endogenous active TGF-β, fibrinogen appears to be a prime carrier of latent TGF-β to sites of injury. Although TGF-β-deficient mice would be ideal to dissect the contribution of direct and synergistic effects of fibrinogen and TGF-β in scar formation, embryonic (Shull et al., 1992) or early postnatal (Sanford et al., 1997) lethality may limit such studies after injury in the adult CNS.
Our study investigated an important aspect of vascular damage by exploring the molecular link between blood leakage in the CNS and scar formation. Pharmacologic depletion of fibrinogen with ancrod reduced active TGF-β levels in the CNS after injury and decreased neurocan deposition. We propose that fibrin matrices play a key role in establishing a nonpermissive environment for tissue repair in the CNS by activating TGF-β signaling in astrocytes. Fibrinogen also inhibits neurite outgrowth (Schachtrup et al., 2007) and activates microglia/macrophages (Adams et al., 2007b). Thus, fibrinogen might contribute to the inhibitory environment after traumatic injury in the CNS by inducing deposition of inhibitory proteoglycans and by directly inhibiting axonal regeneration and activating the inflammatory response. Given the multifaceted functions of fibrinogen as a proinflammatory and profibrotic blood protein at sites of vascular damage, anticoagulant therapies that inhibit fibrin formation or inhibition of fibrinogen binding to integrin receptors (Adams et al., 2007a) or growth factors might be beneficial for tissue repair.
Footnotes
This work was supported by a German Research Foundation (Deutsche Forschungsgemeinschaft) postdoctoral fellowship (to C.S.), an American Heart Association postdoctoral fellowship (to J.K.R.), and National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants R01NS051470 and R01NS052189 (to K.A.). We thank Aristoteles A. Alexandrou, Caroline Miller, and Jo Dee Fish for technical assistance and Stephen Ordway for editorial assistance.
References
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Adams RA, Passino M, Sachs BD, Nuriel T, Akassoglou K. Fibrin mechanisms and functions in nervous system pathology. Mol Interv. 2004;4:163–176. doi: 10.1124/mi.4.3.6. [DOI] [PubMed] [Google Scholar]
- Adams RA, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 2007a;14:2925–2936. doi: 10.2174/092986707782360015. [DOI] [PubMed] [Google Scholar]
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007b;204:571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Strickland S. Nervous system pathologies: the fibrin perspective. Biol Chem. 2002;383:37–45. doi: 10.1515/BC.2002.004. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Yu WM, Akpinar P, Strickland S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron. 2002;33:861–875. doi: 10.1016/s0896-6273(02)00617-7. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, Probert L, Strickland S. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:6698–6703. doi: 10.1073/pnas.0303859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Cambier S, Morris A, Finkbeiner W, Nishimura SL. Integrin-mediated transforming growth factor-beta activation regulates homeostasis of the pulmonary epithelial-mesenchymal trophic unit. Am J Pathol. 2006;169:405–415. doi: 10.2353/ajpath.2006.060049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L, Wrana JL, López-Casillas F, Massagué J. TGF-beta receptors and actions. Biochim Biophys Acta. 1994;1222:71–80. doi: 10.1016/0167-4889(94)90026-4. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Transglutaminase-mediated oligomerization of the fibrin(ogen) alphaC domains promotes integrin-dependent cell adhesion and signaling. Blood. 2005;105:3561–3568. doi: 10.1182/blood-2004-10-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RA, Tsurupa G, Medved L, Tjandra N. Identification of an ordered compact structure within the recombinant bovine fibrinogen alphaC-domain fragment by NMR. Biochemistry. 2006;45:2257–2266. doi: 10.1021/bi052380c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Silver DJ, Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009;29:9967–9976. doi: 10.1523/JNEUROSCI.1151-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, Einheber S, Boudreau N, Nishimura SL. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernousov MA, Carey DJ. alphaVbeta8 integrin is a Schwann cell receptor for fibrin. Exp Cell Res. 2003;291:514–524. doi: 10.1016/s0014-4827(03)00409-9. [DOI] [PubMed] [Google Scholar]
- de Maat MP, Verschuur M. Fibrinogen heterogeneity: inherited and noninherited. Curr Opin Hematol. 2005;12:377–383. doi: 10.1097/01.moh.0000169287.51594.3b. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanakis DK, Neerman-Arbez M, Scheiner T, Henschen A, Hubbs D, Nagaswami C, Weisel JW. Homophenotypic Aalpha R16H fibrinogen (Kingsport): uniquely altered polymerization associated with slower fibrinopeptide A than fibrinopeptide B release. Blood Coagul Fibrinolysis. 2007;18:731–737. doi: 10.1097/MBC.0b013e3282f10157. [DOI] [PubMed] [Google Scholar]
- Gay D, Esiri M. Blood-brain barrier damage in acute multiple sclerosis plaques. An immunocytological study. Brain. 1991;114:557–572. doi: 10.1093/brain/114.1.557. [DOI] [PubMed] [Google Scholar]
- Gentry LE, Nash BW. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry. 1990;29:6851–6857. doi: 10.1021/bi00481a014. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent TGF-beta in blood clots during dissolution with plasmin. Nat Med. 1995;1:932–937. doi: 10.1038/nm0995-932. [DOI] [PubMed] [Google Scholar]
- Grainger DJ, Mosedale DE, Metcalfe JC. TGF-beta in blood: a complex problem. Cytokine Growth Factor Rev. 2000;11:133–145. doi: 10.1016/s1359-6101(99)00037-4. [DOI] [PubMed] [Google Scholar]
- Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV, Jr, Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399–411. doi: 10.1016/s0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- Lagord C, Berry M, Logan A. Expression of TGFbeta2 but not TGFbeta1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol Cell Neurosci. 2002;20:69–92. doi: 10.1006/mcne.2002.1121. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Lin AH, Luo J, Mondshein LH, ten Dijke P, Vivien D, Contag CH, Wyss-Coray T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J Immunol. 2005;175:547–554. doi: 10.4049/jimmunol.175.1.547. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol. 2007;14:236–241. doi: 10.1097/MOH.0b013e3280dce58c. [DOI] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost. 2003;89:409–419. [PubMed] [Google Scholar]
- Milev P, Maurel P, Häring M, Margolis RK, Margolis RU. TAG-1/axonin-1 is a high-affinity ligand of neurocan, phosphacan/protein-tyrosine phosphatase-zeta/beta, and N-CAM. J Biol Chem. 1996;271:15716–15723. doi: 10.1074/jbc.271.26.15716. [DOI] [PubMed] [Google Scholar]
- Milner R, Relvas JB, Fawcett J, ffrench-Constant C. Developmental regulation of alphav integrins produces functional changes in astrocyte behavior. Mol Cell Neurosci. 2001;18:108–118. doi: 10.1006/mcne.2001.1003. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Hellman U, Wernstedt C, Heldin CH. Latent high molecular weight complex of transforming growth factor beta 1. Purification from human platelets and structural characterization. J Biol Chem. 1988;263:6407–6415. [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Boeri Erba E, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- Mosesson MW, Sherry S. The preparation and properties of human fibrinogen of relatively high solubility. Biochemistry. 1966;5:2829–2835. doi: 10.1021/bi00873a008. [DOI] [PubMed] [Google Scholar]
- Mosesson MW, Finlayson JS, Umfleet RA, Galanakis D. Human fibrinogen heterogeneities. I. Structural and related studies of plasma fibrinogens which are high solubility catabolic intermediates. J Biol Chem. 1972;247:5210–5219. [PubMed] [Google Scholar]
- Mosesson MW, Siebenlist KR, Meh DA. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci. 2001;936:11–30. doi: 10.1111/j.1749-6632.2001.tb03491.x. [DOI] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Niederöst BP, Zimmermann DR, Schwab ME, Bandtlow CE. Bovine CNS myelin contains neurite growth-inhibitory activity associated with chondroitin sulfate proteoglycans. J Neurosci. 1999;19:8979–8989. doi: 10.1523/JNEUROSCI.19-20-08979.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passino MA, Adams RA, Sikorski SL, Akassoglou K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science. 2007;315:1853–1856. doi: 10.1126/science.1137603. [DOI] [PubMed] [Google Scholar]
- Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med. 2007;204:1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston E, Webster J, Small D. Characteristics of sustained blood-brain barrier opening and tissue injury in a model for focal trauma in the rat. J Neurotrauma. 2001;18:83–92. doi: 10.1089/089771501750055794. [DOI] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med. 2008;13:2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen J, Hyytiäinen M, Taipale J, Keski-Oja J. Latent transforming growth factor-beta binding proteins (LTBPs)–structural extracellular matrix proteins for targeting TGF-beta action. Cytokine Growth Factor Rev. 1999;10:99–117. doi: 10.1016/s1359-6101(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Sahni A, Francis CW. Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood. 2000;96:3772–3778. [PubMed] [Google Scholar]
- Sahni A, Altland OD, Francis CW. FGF-2 but not FGF-1 binds fibrin and supports prolonged endothelial cell growth. J Thromb Haemost. 2003;1:1304–1310. doi: 10.1046/j.1538-7836.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- Sahni A, Guo M, Sahni SK, Francis CW. Interleukin-1beta but not IL-1alpha binds to fibrinogen and fibrin and has enhanced activity in the bound form. Blood. 2004;104:409–414. doi: 10.1182/blood-2004-01-0126. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Lu P, Jones LL, Lee JK, Lu J, Sachs BD, Zheng B, Akassoglou K. Fibrinogen inhibits neurite outgrowth via beta3 integrin-mediated phosphorylation of the EGF receptor. Proc Natl Acad Sci U S A. 2007;104:11814–11819. doi: 10.1073/pnas.0704045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci. 1999;11:3648–3658. doi: 10.1046/j.1460-9568.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Spraggon G, Applegate D, Everse SJ, Zhang JZ, Veerapandian L, Redman C, Doolittle RF, Grieninger G. Crystal structure of a recombinant alphaEC domain from human fibrinogen-420. Proc Natl Acad Sci U S A. 1998;95:9099–9104. doi: 10.1073/pnas.95.16.9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsöld C, Hyytiäinen M, Bruckner-Tuderman L, Keski-Oja J. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J Cell Sci. 2001;114:187–197. doi: 10.1242/jcs.114.1.187. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. 2008;87:601–615. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]