

Abstract
The skeletons of demosponges, such as Ianthella basta, are known to be a composite material containing organic constituents. Here, we show that a filigree chitin-based scaffold is an integral component of the I. basta skeleton. These chitin-based scaffolds can be isolated from the sponge skeletons using an isolation and purification technique based on treatment with alkaline solutions. Solid-state 13C NMR, Raman, and FT-IR spectroscopies, as well as chitinase digestion, reveal that the isolated material indeed consists of chitin. The morphology of the scaffolds has been determined by light and electron microscopy. It consists of cross-linked chitin fibers approximately 40–100 nm in diameter forming a micro-structured network. The overall shape of this network closely resembles the shape of the integer sponge skeleton. Solid-state 13C NMR spectroscopy was used to characterize the sponge skeleton on a molecular level. The 13C NMR signals of the chitin-based scaffolds are relatively broad, indicating a high amount of disordered chitin, possibly in the form of surface-exposed molecules. X-ray diffraction confirms that the scaffolds isolated from I. basta consist of partially disordered and loosely packed chitin with large surfaces. The spectroscopic signature of these chitin-based scaffolds is closer to that of α-chitin than β-chitin.
Keywords: Sponges, Skeleton, Chitin, Solid-state NMR spectroscopy, Optical spectroscopy
1. Introduction
Chitin has been known to be part of the skeletal structure of various invertebrates for many years. Odier (1823) demonstrated that the exoskeleton of insects is composed of chitin. Arthropods are the most abundant species occurring on earth and chitin provides the chemical basis for their skeletal structures. For a long time, however, chitin has not been recognized to be part of the skeletal formations of sponges (Porifera). Sponges – the first known Metazoans – are the phylogenetically most ancient invertebrates. In sponges, chitin was only found in the inner layer of so-called gemmulae. These microbodies are produced by fresh water sponges prior to their seasonal disappearance under extreme environmental conditions (Simpson, 1984). However, chitin was recently detected within skeletal formations of Verongula gigantea and Aplysina sp. demosponges (Ehrlich et al., 2007a) as well as several glass sponge species (Hexactinellida) (Ehrlich et al., 2007b, 2008; Ehrlich and Worch, 2007). These initial observations indicated an important role of chitin as a skeletal element in sponges and stimulated our present investigations concerning the presence of chitin in other sponge species (especially further members of the order Verongida apart from Aplysina and Verongula species). Chitin as a skeletal material in sponges is of special interest as its presence proves the existence of this important biopolymer several hundred million years before the appearance of chitin in arthropod skeletons.
Verongid sponges form a cohesive and distinct order of marine sponges (Erwin and Thacker, 2007). Although clearly delineated from other orders, the intraordinal relationships among verongid sponges are rather complicated and poorly understood. The skeleton makes up the bulk of the sponge (Bergquist, 1978). A thick skin-like dermis as well as the presence of spherulous cells among the secretory cell content are common to all species. The object of the present study is the marine sponge Ianthella basta Hyatt, 1875 (see Fig. 1A). It is a characteristic representative of the Ianthellidae family, one of the largest families among the verongid sponges. This family consists of 12 species belonging to three genera (Ianthella, Anomoianthella, and Hexadella). It is distinguished from other Verongida sponge families by the presence of eurypylos choanocyte chambers (Erwin and Thacker, 2007). Ianthella sponges are easily identified by their large size (typically 0.5–1 m diameter), their remarkable colour, the fan-shaped morphology of the body, as well as by their characteristic skeleton. This skeleton can be dried and endures in beach-stranded species (Bergquist and Kelly-Borges, 1995). Ianthella sponges are found in the Indian and Pacific oceans. For example, they occur abundantly in South East Asia, Australia, Lord Howe Island, New Caledonia, West Central Pacific, as well as the region between Guam and the West coast of the USA (Hooper and Wiedenmayer, 1994).
Fig. 1.

(A) Underwater photograph of a living marine demosponge Ianthella basta. The diameter of the sponge corresponds to about 1 m. (B) Photographs of the freeze-dried I. basta skeleton (top); a sample after NaOH treatment step 3 (middle) and subsequent H2O2 purification (bottom). (C) SEM images of a freeze-dried I. basta skeleton sample (top) and of an isolated scaffold after treatment step 3 (bottom).
The skeleton of demosponges is known to be a composite material containing various organic constituents. So-called spongin fibers are often found within these skeletal formations. In addition, demosponge skeletons can exhibit mineralized parts containing calcium carbonate or amorphous silica. It was, however, previously stated (Bergquist, 1978) that Verongida sponges do not exhibit a mineralized skeleton. Instead, they were reported to have a collagenous mesohyl stabilized by spongin fibers with a granulated interior region (“pith”) and a laminated exterior “bark”. Apart from spongin, various other organic compounds are found in sponges. The biotechnological and pharmaceutical potential of these compounds is well known: molecules with cytotoxic, antifouling, antitumoral, antibiotic, antiviral, enzyme-inhibitory, and anti-inflammatory activities, as well as favorable properties concerning the treatment of Alzheimer’s disease have been isolated from sponges (Faulkner, 2001). Ianthella basta contains brominated macrocyclic alkaloids, so-called bastadins. Bastadins represent a unique class of natural products based on tyrosine-derived dipeptides (Kazlauskas et al., 1980, 1981; Pordesimo and Schmitz, 1990; Greve et al., 2008) with interesting medicinal properties (Franklin et al., 1996; Aoki et al., 2006; Kotoku et al., 2008). For example, they are cytotoxic against cancer cells. Carotenoid sulfates, bastaxantins, and desulfated bastaxanthiols could also be isolated from I. basta (Hertzberg et al., 1983, 1989). Furthermore, special proteoglycans found in sponges are involved in cell adhesion (Humphreys, 1963; Fernàndez-Busquets and Burger, 2003).
Three different types of chitin are known in nature, namely α-, β-, and γ-chitin (Lotmar and Picken, 1950; Rudall, 1963). Chitin recently isolated from Aplysina sp. as well as Verongula gigantea was found to be α-chitin (Ehrlich et al., 2007a). β-Chitin occurs in the extracellular fibers of diatoms such as Thalassiosira sp. and Cyclotella sp. (McLachlan et al., 1965; Blackwell et al., 1967; Herth and Barthlott, 1979). The crystal structures of α- and β-chitin are well known (Carlström, 1957; Minke and Blackwell, 1978; Gardner and Blackwell, 1975). Chitin has also been studied extensively in the past by solid-state 13C NMR spectroscopy (Peter et al., 1984; Schaefer et al., 1987; Fukamizo et al., 1986; Tanner et al., 1990; Andrade et al., 2002; Schmidt-Rohr and Mao, 2002; Cárdenas et al., 2004; Kono, 2004; Kameda et al., 2005; Webster et al., 2006) as well as by Raman and Infrared (IR) spectroscopy (Pearson et al., 1960; Focher et al., 1992; Cárdenas et al., 2004; Yamaguchi et al., 2005). Therefore, these well-established spectroscopic techniques were used within the present work in order to comprehensively characterize the chitin-based scaffold in I. basta skeletons and to monitor its isolation by the alkali-based treatment proposed by Ehrlich et al. (2007a).
2. Experimental
2.1. Sample collection and preparation
Samples were collected at Western Shoals in Apra Harbor (Guam) in fall 2008 and spring 2009. The sponges were collected underwater from depths of 5–12 m by scuba diving. The collected species were visually inspected underwater with respect to their intactness. No signs for fouling or the presence of other invertebrates such as epibionts could be observed for the collected species. Sponges were kept in large Ziploc bags during collection and stored in a cooler during transport to the Guam marine laboratories. The sponges where again carefully inspected with respect to the intactness of their skeletons and the presence of algae or invertebrates using a stereomicroscope. Contaminants or damages were not observed for the collected species. After this inspection, the sponges were frozen at −20 °C and freeze-dried prior to their transport to the bioanalytical laboratories in Dresden (Germany).
2.2. Isolation of chitin-based scaffolds
The isolation of the chitin-based scaffolds was performed according to the following treatment steps:
Step 1: The skeletons were cut into pieces of 10–30 cm2 and treated with 20% acetic acid at 37 °C for 12 h. Afterwards, the samples were rinsed three times with distilled water.
Step 2: The samples were treated with 2.5 M NaOH at 37 °C for 24 h. Afterwards, the samples were rinsed three times with distilled water.
Step 3: Once more, the samples were treated with 2.5 M NaOH at 37 °C – now for 48 h – resulting in total NaOH treatment time of 72 h. Afterwards, the samples were rinsed three times with distilled water.
For final purification, the samples were optionally cleaned in 35% H2O2 at 37 °C.
It should be noted that the NaOH concentration of 2.5 M corresponds to 4% v/v. This is well below the critical concentration of 25–30% v/v where the transformation of β-chitin into α-chitin starts to take place (Noishiki et al., 2003). Therefore, the crystal structure of the chitin-based scaffolds was not influenced by the extraction procedure. A commercially available crab α-chitin sample was purchased from FLUKA. Monomeric N-acetyl-D-glucosamine was purchased from Sigma–Aldrich. β-Chitin was prepared from the diatom species Thalassiosira rotula as follows: The diatom species T. rotula was collected by Prof. M. Sumper (Regensburg, Germany) in the North Sea during an expedition at the German North Sea coast in July 2006. A 20 l culture of T. rotula was grown following the procedures described by Gröger et al. (2009). The culture was harvested and treated with sodium dodecyl sulfate (SDS) and ethylene diamine tetraacetic acid (EDTA) in order to remove loosely attached organic material following the procedures described by Kröger et al. (1999). Finally, the sample was freeze-dried for one day and afterwards dried in a vacuum oven at 40 °C for three days. Solid-state 13C NMR analysis confirmed the high crystallinity and purity of the obtained β-chitin (see below). Note that β-chitin is able to swell in contrast to α-chitin. The solid-state 13C NMR spectrum of β-chitin was measured in the hydrated state and agrees very well with the spectra published previously (Tanner et al., 1990).
2.3. Solid-state 13C NMR spectroscopy
Solid-state 13C NMR experiments were performed on a Bruker Avance 300 spectrometer operating at 75.47 MHz for 13C using a commercial double resonance 4 mm MAS NMR probe. Ramped 1H–13C cross-polarization (CP; Pines et al., 1973; Metz et al., 1994) was used (contact time: 4 ms, number of scans: 40,000, repetition time: 2 s). SPINAL 1H-decoupling (Fung et al., 2000) was applied during signal acquisition. The temperature inside the rotor was calibrated using the 207Pb chemical shift of lead nitrate as a temperature reference (Ferguson and Haw, 1995). The chemical shifts were referenced relative to tetramethylsilane (TMS).
2.4. Raman and FT-IR spectroscopy
Raman spectra were recorded on a Kaiser HoloLab Series 2000 microscope coupled to an f/1.8 Holospec spectrograph (Kaiser Optical Systems, Ann Arbor MI, USA) with a liquid nitrogen cooled CCD camera (Roper Scientific, Trenton NJ, USA). A Toptica XTRA laser (Toptica Photonics AG, Gräfelfing, Germany) with an excitation wavelength of 785 nm was used. The spectra were baseline-corrected using a multi-point linear baseline (at 350, 800, 1700, 1960, 2600, 2750, and 3050 cm−1). Infrared spectra were recorded on a Nicolet 210 FT-IR Spectrometer. About 1–2 mg of the sample was mixed with 400 mg KBr and compressed. 100 scans were recorded at a spectral resolution of 2 cm−1. All spectra were baseline corrected with a two-point linear baseline (at 845 and 1890 cm−1).
3. Results and discussion
Images of the skeleton of the marine demosponge I. basta after different isolation steps are shown in Fig. 1B and C (cf. Experimental section). The alkali-based treatment reveals a scaffold which closely resembles the shape of the I. basta skeleton. It consists of cross-linked fibers forming a two-dimensional network with almost quadratic chambers. After extraction steps 2 and 3, the material still contained a certain amount of pigments giving rise to the red color (Fig. 1B). These pigments were removed by subsequent soaking in H2O2 for 10 min. After this final purification, clean white scaffolds remain (Fig. 1C). SEM investigations show that the scaffolds isolated by NaOH treatment from I. basta skeletons consist of a rather loosely packed and deeply fissured material (see Figs. 2 and 3). The X-ray diffractograms of the scaffolds obtained after NaOH treatment exhibit only a few, broad reflections at positions characteristic for α-chitin (see Supplementary material). In combination with the observations demonstrated in Figs. 2 and 3, this behaviour indicates a nanocrystalline organization of the material. Similar observations were made for the nanocrystalline squid pen β-chitin (Fan et al., 2008) which exhibits similarly broad X-ray reflections. For the I. basta skeleton after the third isolation step, the average particle size is estimated to be of the order of tens of nanometers. It is, therefore, impossible to determine the crystal structure of the isolated material by X-ray diffraction. Therefore, the structural characterization of the isolated I. basta scaffold must rely on spectroscopic techniques such as solid-state 13C NMR or optical spectroscopy. These techniques do not require crystalline samples.
Fig. 2.
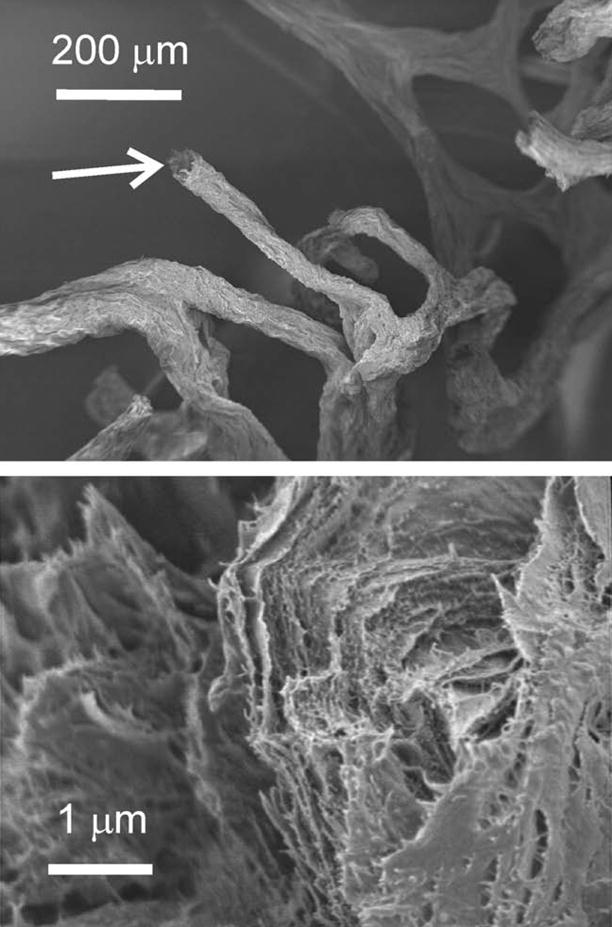
SEM images showing the micro- and nano-organization of a selected chitin fiber from the I. basta skeleton after step 3. The image at the bottom is taken from the disrupted fiber shown in the top image (white arrow).
Fig. 3.

SEM image of an alkali-treated sample showing the nano-organization of the I. basta chitinous fibres (characteristic nanofibrils are marked with white arrows).
The 13C {1H} CP-MAS NMR spectra of the skeleton samples after the different treatment steps are shown in Fig. 4. After step 1, the sample still contains a multitude of overlapping signals indicating the presence of various organic constituents. However, only seven major signals remain after steps 2 and 3 (24 and 72 h NaOH-based treatment, respectively). As can be seen from the pronounced asymmetry, the signal centered around ca. 75 ppm is the superimposition of two signals. The signals remaining after NaOH treatment occur at the characteristic chemical shifts of α-chitin. This observation shows that the isolated scaffolds mainly consist of chitin, and is also confirmed by Raman and FT-IR spectroscopy (see Supplementary material). Chitin consists of poly-N-acetyl-D-glucosamine, and the chemical shifts of the C-1 and C-4 carbon atoms sensitively reflect the polymerization of N-acetyl-D-glucosamine (Webster et al., 2006). 13C chemical shifts of 93.2 ppm (C-1) and 74.6 ppm (C-4) are found for the monomer (see Fig. 5). In contrast, 104.4 ppm (C-1) and 83.3 ppm (C-4) are observed for α-chitin (see Table 1). It should be noted that the latter two signals show up at 104.4 ppm (C-1) and 83.3 ppm (C-4) for I. basta in the freeze-dried samples as well as after the subsequent extraction steps. These chemical shift values are characteristic for α-chitin. In contrast, no significant signal intensity occurs at 93.2 ppm. The presence of larger amounts of monomeric N-acetyl-D-glucosamine can, therefore, be ruled out for all I. basta samples under study.
Fig. 4.

13C {1H} CP-MAS NMR spectra (293 K) of the I. basta skeleton samples measured after the different isolation steps (see Section 2). The spectrum of the α-chitin sample is shown for comparison. Dashed vertical lines indicate the chemical shifts of the signals in α-chitin.
Fig. 5.
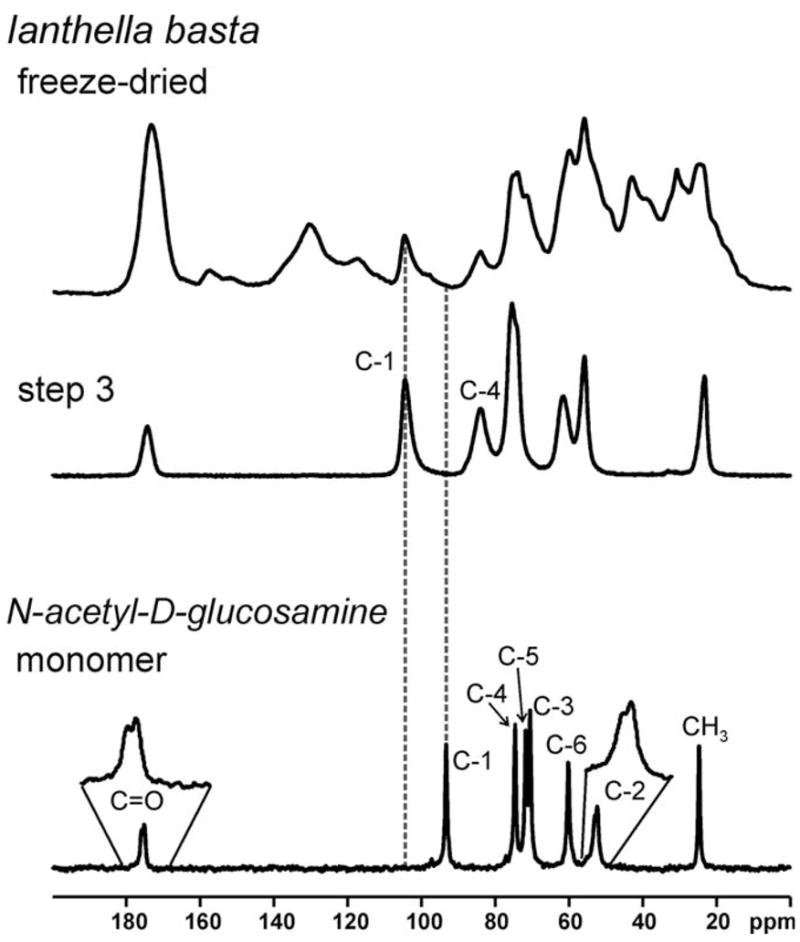
13C {1H} CP-MAS NMR spectra (293 K) of the I. basta skeleton samples measured after the different isolation steps (see Section 2) and monomeric N-acetyl-D-glucosamine. Dashed vertical lines indicate chemical shifts which are particularly indicative. Note that the signals due to C-2 and C=O in the monomer exhibit a splitting due to the same effect as observed for β-chitin (see Fig. 6 and related text).
Table 1.
Chemical shifts and line widths of the various solid-state 13C NMR signals observed for the chitin samples under study. Chemical shifts of asymmetric or split signals (C=O, C-2 in β-chitin) determine the center of gravity of the corresponding signals. Line widths are the full widths measured at half maximum height of the signals. Experimental errors are ±0.2 ppm for the chemical shifts and ±5 Hz for the line widths.
| Sample | C=O |
C-1 |
C-4 |
C-5 |
C-3 |
C-6 |
C-2 |
CH3 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
δ (ppm) |
Δν1/2 (Hz) |
|
| I. basta | 174.2 | 270 | 104.4 | 170 | 83.3 | 215 | 76.0 | 195 | 73.6 | 115 | 61.3 | 345 | 55.3 | 180 | 23.2 | 110 |
| α-chitin (reference) | 174.2 | 270 | 104.4 | 125 | 83.3 | 185 | 76.0 | 145 | 73.6 | 100 | 61.2 | 260 | 55.3 | 125 | 23.1 | 75 |
| β-chitin (T. rotula) | 175.8 | 115 | 105.3 | 30 | 84.4 | 25 | 75.4 | 40 | 73.0 | 40 | 59.8 | 70 | 55.6 | 125 | 22.7 | 40 |
Residual impurities such as pigments were still present after the third isolation step (see Fig. 1B). To remove these impurities, the sample was further cleaned using 35% H2O2. Fig. 6 exhibits the solid-state 13C NMR spectrum of α-chitin, β-chitin, and the chitin-based scaffold isolated from I. basta after H2O2 purification. It should be noted that the spectra measured for the α- and β-chitin reference samples agree very well with previously published spectra (see for example Tanner et al., 1990; Cárdenas et al., 2004; Kono, 2004; Kameda et al., 2005). After H2O2 treatment, the solid-state 13C NMR spectrum of the I. basta skeleton agrees even more with that of α-chitin than the spectrum after step 3 of the NaOH-based isolation procedure (cf. Fig. 4). Within the experimental error, the signals of the chitin-based I. basta scaffold are found at the characteristic positions of α-chitin (cf. also Table 1). In contrast, several carbon atoms in I. basta chitin exhibit significantly different chemical shifts than observed for β-chitin. This is particularly true for C=O, C-1, C-4, and C-6 (see Fig. 6 and Table 1). It is therefore concluded that the structure of the chitin-based scaffold isolated from I. basta is closer to α-chitin than β-chitin. The native I. basta skeleton contains about 7 wt.% of chitin as could be determined by measuring the amount of chitin isolated from a freeze-dried skeleton sample of known weight.
Fig. 6.
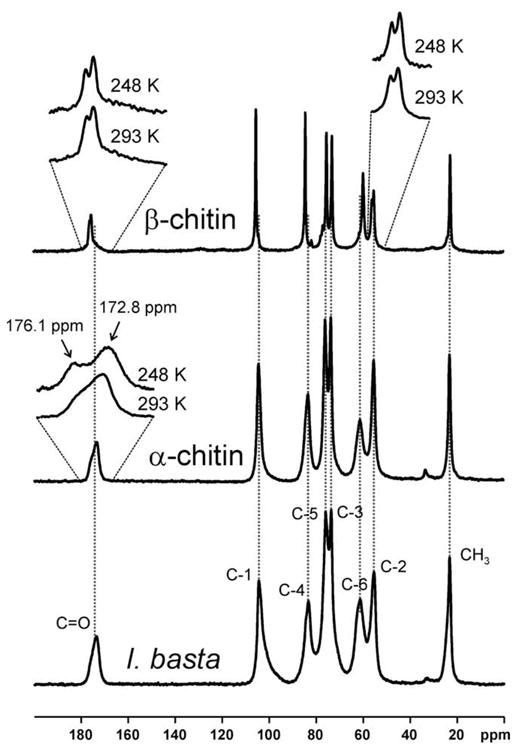
13C {1H} CP-MAS NMR spectra of β-chitin from T. rotula, α-chitin from crabs, and an I. basta scaffold after NaOH treatment and subsequent H2O2 purification measured at 293 K. Inserts: Selected signals measured at room temperature and at 248 K. Dashed vertical lines indicate the chemical shifts of the signals in α-chitin.
An interesting effect is observed for the carbonyl 13C NMR signal in the α-chitin spectrum. The signal is broad and asymmetric which has previously been explained by the existence of two unresolved, overlapping signals (Kameda et al., 2005). These two signals are caused by single and double hydrogen bonded carbonyl groups within the hydrogen-bond network of α-chitin. At elevated temperatures, the two signals merge into a single symmetric line due to rapid conformational exchange processes (Kameda et al., 2005). In order to slow down this exchange process, the samples were cooled to 249 K. Cooling resulted in the observation of two separate signals (see Fig. 6). The intensity ratio of the two lines at 172.8 and 176.1 ppm was approximately 3:2. The C=O and C-2 signals of β-chitin, as well as N-acetyl-D-glucosamine exhibits a temperature-independent splitting of ca. 50–60 Hz due to the residual dipolar coupling1 with the neighboring quadrupolar 14N nucleus (Tanner et al., 1990) as can be seen in Figs. 5 and 6. However, this splitting is much smaller than the temperature-dependent splitting of ca. 250 Hz observed at 248 K for C=O in α-chitin (see Fig. 6), which is caused by the aforementioned hydrogen-bond influence (Kameda et al., 2005). The characteristic asymmetry is not observed for I. basta chitin after extraction step 3. The C=O signal in I. basta chitin occurs at an isotropic chemical shift of 174.2 ppm. The latter value agrees well with the weighted average of 174.1 ppm for the two signals observed in α-chitin at 248 K. This weighted average corresponds to the center of gravity of the asymmetric C=O signal for α-chitin at 293 K. In contrast, a significantly different value of 175.5 ppm is observed for the center of gravity of the C=O signal in β-chitin. These observations indicate that the hydrogen-bond network characteristic for highly crystalline α-chitin is perturbed in I. basta chitin. This may be due to the presence of minor amounts of impurities such as pigments. Nevertheless, the average C=O chemical shift observed for I. basta agrees well with α-chitin as do the other signals.
Crystalline β-chitin exhibits extremely narrow solid-state 13C NMR signals (see Tanner et al., 1990; cf. also Fig. 6 and Table 1). In contrast, the signals of the α-chitin sample are considerably broader. The signals in the I. basta chitin spectrum are even broader than in the crab α-chitin sample (see Fig. 6 and Table 1). Moreover, the C-1 signal is highly asymmetric. It should be noted that squid pen β-chitin also exhibits a solid-state 13C NMR spectrum with broadened signals compared with highly crystalline β-chitin samples (see, e.g., Tanner et al., 1990 and references therein). Tanner and coworkers explained this effect as arising from the poorly crystalline state of squid pen chitin which was found to be caused by a significant amount of surface-exposed polysaccharide chains. This interpretation was supported by recent X-ray diffraction analyses (Fan et al., 2008). The diameter of squid pen β-chitin fibrils is only 2–3 nm. The chitin-based I. basta scaffolds obviously exhibit a similar nanoscale organization. The surface of the material is rough and deeply fissured, as can be seen in the corresponding SEM images, and the X-ray diffraction patterns are broadened (see above). It is likely that this organization of the chitin results in a large amount of surface-exposed polysaccharide chains. These observations also explain the broadened solid-state 13C NMR signals and may be a further reason for the aforementioned perturbation of the hydrogen-bond network. In addition, the native sponge skeleton contains other organic material such as proteins and bromotyrosine-related compounds which were removed from the skeleton and probably hydrolysed by the NaOH treatment (see Fig. 1). In the native sponge skeleton, these constituents are intercalated into the chitin-based scaffold or may even be cross-linked with the chitin, thus preventing it from forming unperturbed crystalline chitin. Similar results were reported for chitin isolated from beetle larva. Intercalated catechol compounds lead to an amorphous structure of the chitin-based cuticle (Zhang et al., 2000).
Chitinase digestion experiments (see Supplementary material) again confirmed the chitinuous nature of the isolated I. basta scaffolds. Furthermore, these experiments revealed another interesting phenomenon. In contrast to the freeze-dried skeletons, purified samples were readily digested in chitinase solution. Previously, it was suggested that the skeletal fibers of sponges may be involved in the sequestration and accumulation of brominated compounds (Turon et al., 2000; Teeyapant et al., 1993). Tabudravu et al., (2002) reported that brominated tyrosine-derived compounds isolated from the verongid sponge Aplysinella rhax inhibit bacterial chitinase. This behaviour may explain why the freeze-dried skeleton samples withstand the chitinase test. Possibly, bromotyrosine-related compounds present in the skeleton inhibit the chitinase thus protecting the chitin-based scaffolds of Verongida sponges. Therefore, these compounds may be crucial for the survival of Verongida sponges in the marine environment.
The results of Raman and infrared spectroscopic studies are shown in Figs. 7 and 8, respectively. The band assignment is based on previous publications (Pearson et al., 1960; Focher et al., 1992; Cárdenas et al., 2004; Yamaguchi et al., 2005). For comparison, spectra of α- and β-chitin are shown in addition to the spectra of the I. basta scaffold after NaOH treatment and H2O2 purification. The spectra of I. basta agree very well with the spectra of α-chitin (see also Tables 2 and 3), while characteristic differences to the β-chitin spectra were observed. In both, the Raman and IR spectra, β-chitin exhibits narrower bands than I. basta chitin. Furthermore, the relative intensities of several bands are different. For example, β-chitin exhibits a narrow Raman band at 995 cm−1 which is almost absent and strongly broadened in I. basta chitin. A significant shift is also observed for the Raman band at 1413 cm−1 in I. basta chitin. This band occurs at 1420 cm−1 in β-chitin. Infrared spectra of β-chitin reveal two additional bands for CHx deformations at about 1455 and 1374 cm−1; these bands were not observed in I. basta. Additionally, bands in the region of 1300–1200 cm−1 further confirmed the similarity of I. basta chitin to α-chitin: The C–O–C and C–O stretching vibration region (1200–950 cm−1) of β-chitin exhibits a greater number of narrower bands with different intensities than I. basta chitin. Another characteristic marker is the CH deformation of the β-glycosidic bond. This band shifts from 890 cm−1 in β-chitin to 895 cm−1 in α-chitin and I. basta chitin. Therefore, optical spectroscopy confirms the solid-state 13C NMR spectroscopic finding that the chitin-based scaffolds isolated from I. basta are closer to α-chitin than β-chitin.
Fig. 7.

Raman spectra of β-chitin from T. rotula, α-chitin from crabs, and an I. basta scaffold after NaOH and H2O2 treatment.
Fig. 8.
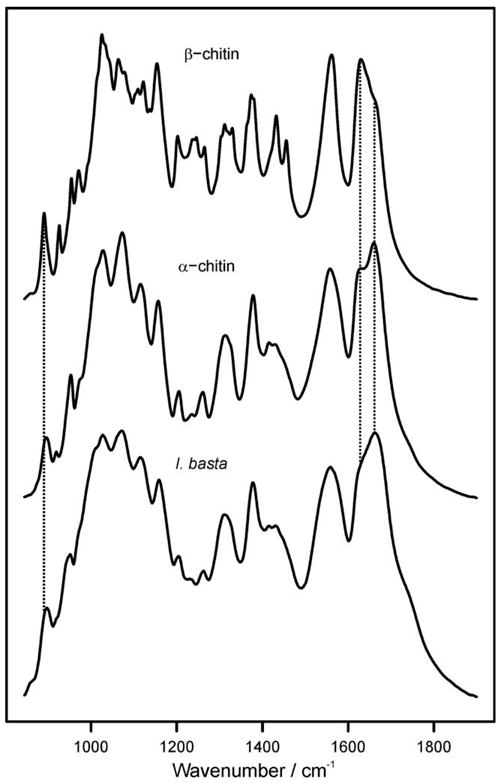
Infrared absorption spectra of β-chitin from T. rotula, α-chitin from crabs, and I. basta chitin after NaOH and H2O2 treatment. Dashed vertical lines are drawn to highlight characteristic differences between α- and β-chitin. The band due to the β-glycosidic bond occurs at 895 cm−1 in α-chitin and at 890 cm−1 for β-chitin. The amide I band splits into two components at ca. 1630 and 1660 cm−1 due to the influence of hydrogen bonding. Note the intensity differences observed between α-and β-chitin.
Table 2.
Wavenumbers and assignment of the bands observed in the Raman spectra of the chitin samples shown in Fig. 7.
| I. basta Wavenumber/cm−1 | α-Chitin Wavenumber/cm−1 | β-Chitin (T. rotula) Wavenumber/cm−1 | Assignment |
|---|---|---|---|
| 895 | 895 | 890 | CHx deformation (o. o. p.) |
| 918 weak | 919 | ||
| 950 | 952 | 952 | |
| 1000 | C–O–C/C–O stretching | ||
| 1057 | 1058 | 1048 | |
| 1108 | 1108 | 1102 | |
| 1132 | |||
| 1146 | 1147 | 1147 | |
| 1202 | 1203 | 1201 | Amide III |
| 1243 | |||
| 1264 | 1264 | 1268 | |
| 1311 | |||
| 1326 | 1326 | 1326 | |
| 1360 | |||
| 1375 | 1375 | 1376 | CHx deformation |
| 1411 | 1413 | 1417 | |
| 1449 | 1448 | 1447 |
Table 3.
Wavenumbers and assignment of the bands observed in the FT-IR spectra of the chitin samples shown in Fig. 8.
| I. basta Wavenumber/cm−1 | α-Chitin Wavenumber/cm−1 | β-Chitin (T. rotula) Wavenumber/cm−1 | Assignment |
|---|---|---|---|
| 897 | 895 | 890 | CH deformation (o.o.p.), C1 axial (β bond) |
| 920* | 919 | CHX deformation (o.o.p.) | |
| 926 | |||
| 951 | 953 | 954 | |
| 973* | 976* | 971 | |
| 993* | C–O–C/C–O stretching | ||
| 1013* | 1012* | 1014* | |
| 1027 | 1028 | 1025 | |
| 1032* | |||
| 1041* | |||
| 1064 | |||
| 1071 | 1073 | 1078 | |
| 1103* | |||
| 1109 | |||
| 1115 | 1115 | ||
| 1122 | |||
| 1136 | |||
| 1159 | 1157 | 1154 | |
| 1204 | 1205 | 1202 | Amide III |
| 1230* | 1235 | 1237 | |
| 1246 | |||
| 1263 | 1261 | 1265 | |
| 1305* | |||
| 1310 | 1312 | 1312 | |
| 1330 | |||
| 1364* | CHX deformation | ||
| 1374 | |||
| 1378 | 1378 | 1379 | |
| 1415 | 1415 | 1420* | |
| 1430 | 1429 | 1432 | |
| 1456 | |||
| 1559 | 1558 | 1562 | Amide II |
| 1633* | 1630 | 1630 | Amide I |
| 1662 | 1660 | 1660* |
= shoulder, o.o.p. = out of plane.
The observation of chitin forming an integral part of sponge skeletons is an intriguing finding. Is this chitin of exogenous or endogenous nature? Previously, it was reported that chitin in several sponges may be caused by a variety of micro-invertebrates harboured by the sponges (Dauby and Jeuniaux, 1986) such as polychaete worms (Haplosyllis basticola) that colonize the fan-like sponge I. basta. Indeed, H. basticola species were observed (Sarda et al., 2002) inside the characteristic small inhalant pores (ostia), oscules, and internal channels of I. basta (Hooper and Wiedenmayer, 1994). These worms do not cause wounding or other damage to the sponges (Sarda et al., 2002). However, our field and laboratory observations on fresh I. basta material as well as light and electron microscopic studies did not yield any hints as to the presence of foreign invertebrates nor were remnants of their skeletons/cuticles found in the I. basta skeleton samples under study. Additionally, no evidence for bacterial or fungal activities could be observed. Instead, our studies prove the presence of a filigree chitin-based scaffold as an integral constituent of the sponge skeleton. This scaffold exactly resembles the shape of the sponge skeleton (see Fig. 1A and B).
In summary, it can be concluded that our solid-state 13C NMR, Raman, and FT-IR spectroscopic as well as electron microscopic studies unequivocally prove that the scaffolds isolated from the marine demosponge I. basta are chitin-based networks made up of cross-linked fibers of ca. 40–100 nm diameter (see Fig. 1B and C). These fibers consist of loosely packed chitin with rough and deeply fissured surfaces. In agreement with this observation, the X-ray diffractogram of I. basta chitin is strongly broadened. The solid-state 13C NMR signals of the isolated chitin-based scaffolds were also broader than in the crystalline α-chitin sample indicating a high amount of disordered polysaccharide chains, possibly in the form of surface-exposed molecules. Taking all these results together, our investigations show that the uniquely structured, two-dimensional chitin-based scaffolds found in I. basta skeletons consist of a special type of partially disordered chitin. The structure of this material is closer to α-chitin than to β-chitin. Since the processing of chitin is technologically difficult, such chitin-based sponge scaffolds may be of interest for various applications, in particular because it is possible to generate the required amount of material from natural sources, e.g. marine ranching or primmorph cultivation of sponges.
Supplementary Material
Acknowledgments
This research work is supported by a joint grant of the German Academic Exchange Service (DAAD: Ref-325; A/06/91419) and the Ministry of Education and Science of the Russian Federation (RNP.2.2.2.3.10063) – “Mikhail Lomonosov” 2007/2008 as well as by the Erasmus Mundus External Co-operation Programme of the European Union 2009. The authors wish to thank Professor M. Sumper (Regensburg) for providing the T. rotula culture. Thanks are further due to Dr. A.L. Stelling, Ms. Annett Bachmann, and Ms. Renate Schulze for carefully proofreading the manuscript.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jsb.2009.06.018.
Footnotes
The magnetic dipole–dipole interaction between a spin-1/2 nucleus and a nucleus bearing an electric quadrupole moment (i.e., a spin with S > 1/2) can result in a characteristic residual dipolar broadening and splitting of the MAS NMR signal of the spin-1/2 nucleus (see for example Böhm et al. 1983; Olivieri 1989).
References
- Andrade CT, Silva KMP, Tavares MI, Simão RA, Achete C, Pérez CA. Comparative study on structural features of a chitin from Xiphopenaeus kroyeri and its precipitated product from phosphoric acid solution. J Appl Polym Sci. 2002;83:151–159. [Google Scholar]
- Aoki S, Cho SH, Hiramatsu A, Kotoku N, Kobayashi M. Bastadins, cyclic tetramers of brominated-tyrosine derivatives, selectively inhibit the proliferation of endothelial cells. J Nat Med. 2006;60:231–235. doi: 10.1007/s11418-006-0045-3. [DOI] [PubMed] [Google Scholar]
- Bergquist PR. Sponges. Hutchinson and Company; London: 1978. [Google Scholar]
- Bergquist PR, Kelly-Borges M. Systematics and biogeography of the genus Ianthella (Demospongiae: Verongida: Ianthellida) in the South-West Pacific. The Beagle, Rec Northern Terr Mus Arts Sci. 1995;12:151–176. [Google Scholar]
- Blackwell J, Parker KD, Rudall KM. Chitin fibers of the diatoms Thalassiosira fluviatilis and Cyclotella cryptica. J Mol Biol. 1967;28:383–385. doi: 10.1016/s0022-2836(67)80018-4. [DOI] [PubMed] [Google Scholar]
- Böhm J, Fenzke D, Pfeifer H. Effects of quadrupolar nuclei on NMR spectra of I = 1/2 nuclei in magic angle spinning experiments. J Magn Reson. 1983;55:197–204. [Google Scholar]
- Cárdenas G, Cabrera G, Taboada E, Miranda SP. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J Appl Polym Sci. 2004;93:1876–1885. [Google Scholar]
- Carlström D. The crystal structure of α-chitin (poly-N-acetyl-D-glucosamine) J Biophysic Biochem Cytol. 1957;3:669–683. doi: 10.1083/jcb.3.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauby P, Jeuniaux C. Origine exogene de la chitine d’cel’e chez les Spongiares. Cah Biol Mar. 1986;28:121–129. [Google Scholar]
- Ehrlich H, Maldonado M, Spindler KD, Eckert C, Hanke T, Born R, Goebel C, Simon P, Heinemann S, Worch H. First evidence of chitin as a component of the skeletal fibers of marine sponges. Part I Verongidae (Demospongia: Porifera) J Exp Zool (Mol Dev Evol) 2007a;308B:347–356. doi: 10.1002/jez.b.21156. [DOI] [PubMed] [Google Scholar]
- Ehrlich H, Krautter M, Hanke T, Simon P, Knieb C, Heinemann S, Worch H. First evidence of the presence of chitin in skeletons of marine sponges. Part II Glass sponges (Hexactinellida: Porifera) J Exp Zool (Mol Dev Evol) 2007b;308B:473–483. doi: 10.1002/jez.b.21174. [DOI] [PubMed] [Google Scholar]
- Ehrlich H, Worch H. Sponges as natural composites: from biomimetic potential to development of new biomaterials. In: Hajdu E, editor. Porifera Research: Biodiversity, Innovation & Sustainability, Museu Nacional, Rio de Janeiro. 2007. pp. 217–223. [Google Scholar]
- Ehrlich H, Janussen D, Simon P, Heinemann S, Bazhenov VV, Shapkin NP, Mertig M, Erler C, Born R, Worch H, Hanke T. Nanostructural organisation of naturally occuring composites: Part II. Silica-chitin-based biocomposites. J Nanomater. 2008 (published online, Article ID 670235, 8 pages) [Google Scholar]
- Erwin PM, Thacker RW. Phylogenetic analyses of marine sponges within the order Verongida: a comparison of morphological and molecular data. Invert Biol. 2007;126:220–234. [Google Scholar]
- Fan Y, Saito T, Isogai A. Preparation of chitin nanofibers from squid pen β-chitin by simple mechanical treatment under acidic conditions. Biomacromolecules. 2008;9:1919–1923. doi: 10.1021/bm800178b. [DOI] [PubMed] [Google Scholar]
- Faulkner J. Marine natural products. Nat Prod Rep. 2001;19:1–48. doi: 10.1039/b009029h. [DOI] [PubMed] [Google Scholar]
- Ferguson DB, Haw JF. Transient methods for in situ NMR of reactions on solid catalysts using temperature jumps. Anal Chem. 1995;67:3342–3348. [Google Scholar]
- Fernàndez-Busquets X, Burger MM. Circular proteoplycans from sponges: first members of the spongican family. Cell Mol Life Sci. 2003;60:88–112. doi: 10.1007/s000180300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focher B, Naggi A, Torri G, Cosani A, Terbojevich M. Structural differences between chitin polymorphs and their precipitates from solutions – evidence from CP-MAS 13C-NMR, FT-IR and FT-Raman spectroscopy. Carbohydr Polym. 1992;17:97–102. [Google Scholar]
- Franklin MA, Penn SG, Lebrilla CB, Lam TH, Pessah IN, Molinski TF. Bastadin 20 and bastadin O-sulfate esters from Ianthella basta: novel modulators of the Ry1R FKBP12 receptor complex. J Nat Prod. 1996;59:1121–1127. doi: 10.1021/np960507g. [DOI] [PubMed] [Google Scholar]
- Fukamizo T, Kramer KJ, Mueller DD, Schaefer J, Garbow J, Jacob GS. Analysis of chitin structure by nuclear magnetic resonance spectroscopy and chitinolytic enzyme digestion. Arch Biochem Biophys. 1986;249:15–26. doi: 10.1016/0003-9861(86)90555-2. [DOI] [PubMed] [Google Scholar]
- Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Gardner KH, Blackwell J. Refinement of the structure of β-chitin. Biopolymers. 1975;14:1581–1595. doi: 10.1002/bip.1975.360140804. [DOI] [PubMed] [Google Scholar]
- Greve H, Kehraus S, Krick A, Kelter G, Maier A, Fiebig HH, Wright AD, König GM. Cytotoxic bastadin 24 from the Australian sponge Ianthella quadrangulata. J Nat Prod. 2008;71:309–312. doi: 10.1021/np070373e. [DOI] [PubMed] [Google Scholar]
- Gröger C, Lutz K, Brunner E. NMR studies of biomineralisation. Prog Nucl Magn Reson Spectrosc. 2009;54:54–68. [Google Scholar]
- Herth W, Barthlott W. The site of β-chitin fibril formation in centric diatoms. J Ultrastruct Res. 1979;68:6–15. doi: 10.1016/s0022-5320(79)90137-0. [DOI] [PubMed] [Google Scholar]
- Hertzberg S, Ramdahl T, Johansen JF, Liaaen-Jensen S. Carotenoid sulphates II. Structural elucidation of bastaxanthin. Acta Chem Scand B Org Chem Biochem. 1983;37:267–279. [Google Scholar]
- Hertzberg S, Bergquist P, Liaaen-Jensen S. Further occurrence of sulfated carotenoids in Ianthella sp (Demospongiae) Biochem Syst Ecol. 1989;17:51–53. [Google Scholar]
- Hooper JNA, Wiedenmayer F. Porifera. In: Wells A, editor. Zoological Catalogue of Australia. Vol. 12. CSIRO Australia xiii; Melbourne: 1994. p. 624. [Google Scholar]
- Humphreys T. Chemical dissolution and in vitro reconstruction of sponge cell adhesions I. Isolation and functional demonstration of the components involved. Develop Biol. 1963;8:27–47. doi: 10.1016/0012-1606(63)90024-1. [DOI] [PubMed] [Google Scholar]
- Kameda T, Miyazawa M, Ono H, Yoshida M. Hydrogen bonding structure and stability of α-chitin studied by 13C solid-state NMR. Macromol Biosci. 2005;5:103–106. doi: 10.1002/mabi.200400142. [DOI] [PubMed] [Google Scholar]
- Kazlauskas R, Lidgard RO, Murphy PT, Wells RJ. Brominated tyrosine-derived metabolites from the sponge Ianthella basta. Tetrahedron Lett. 1980;21:2277–2280. [Google Scholar]
- Kazlauskas R, Lidgard RO, Murphy PT, Wells RJ, Blount JF. Brominated tyrosine-derived metabolites from the sponge Ianthella basta. Austr J Chem. 1981;34:765–786. [Google Scholar]
- Kono H. Two-dimensional magic angle spinning NMR investigation of naturally occurring chitins: precise 1H and 13C resonance assignment of α- and β-chitin. Biopolymers. 2004;75:255–263. doi: 10.1002/bip.20124. [DOI] [PubMed] [Google Scholar]
- Kotoku N, Hiramatsu A, Tsujita H, Hirakawa Y, Sanagawa M, Aoki S, Kobayashi M. Structure-activity relationships study of bastadin 6, an anti-angiogenic brominated tyrosine derived metabolite from marine sponge. Arch Pharm Chem Life Sci. 2008;341:568–577. doi: 10.1002/ardp.200700231. [DOI] [PubMed] [Google Scholar]
- Kröger N, Deutzmann R, Sumper M. Polycationic peptides from diatom biosilica that direct silica nanosphere formation. Science. 1999;286:1129–1132. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- Lotmar W, Picken LER. A new crystallographic modification of chitin and its distribution. Experientia. 1950;6:58–59. [Google Scholar]
- McLachlan J, McInnes AG, Falk M. Studies on the chitan (chitin: poly-N-acetylglucosamine) fibers of the diatom Thalassiosira fluviatilis Hustedt. Can J Bot. 1965;43:707–713. [Google Scholar]
- Metz G, Wu X, Smith SO. Ramped-amplitude cross polarization in magic-angle-spinning NMR. J Magn Reson A. 1994;110:219–227. [Google Scholar]
- Minke R, Blackwell J. The structure of α-chitin. J Mol Biol. 1978;120:167–181. doi: 10.1016/0022-2836(78)90063-3. [DOI] [PubMed] [Google Scholar]
- Noishiki Y, Takami H, Nishiyama Y, Wada M, Okada S, Kuga S. Alkali-induced conversion of β-chitin to α-chitin. Biomacromolecules. 2003;4:869–899. doi: 10.1021/bm0257513. [DOI] [PubMed] [Google Scholar]
- Odier A. Memoire sur la composition chimique des parties cornees des insects. Mem Soc Hist Nat Paris. 1823;1:29–42. [Google Scholar]
- Olivieri AC. Quadrupolar effects in the CPMAS NMR spectra of spin-1/2 nuclei. J Magn Reson. 1989;81:201–205. [Google Scholar]
- Pearson FG, Marchessault RH, Liang CY. Infrared spectra of crystalline polysaccharides. V. Chitin. Journal of Polymer Science. 1960;43:101–116. doi: 10.1016/0006-3002(60)91486-4. [DOI] [PubMed] [Google Scholar]
- Peter MG, Grün L, Förster H. CP/MAS-13C-NMR spectra of sclerotized insect cuticle and of chitin. Angew Chem Int Ed. 1984;23:638–639. [Google Scholar]
- Pines A, Gibby MG, Waugh JS. Proton-enhanced NMR of dilute spins in solids. J Chem Phys. 1973;59:569–590. [Google Scholar]
- Pordesimo EO, Schmitz FJ. New bastadins from the sponge Ianthella basta. J O Chem. 1990;55:4704–4709. [Google Scholar]
- Rudall KM. The chitin/protein complexes of insect cuticles. Adv Insect Physiol. 1963;1:257–313. [Google Scholar]
- Sarda R, Avila C, Paul J. An association between a syllid polychaete, Haplosyllis basticola n. sp., and the sponge Ianthella basta. Micronesica. 2002;34:165–175. [Google Scholar]
- Schaefer J, Kramer KJ, Garbow JR. Aromatic cross-links in insect cuticle: detection by solid-state 13C and 15N NMR. Science. 1987;235:1200–1204. doi: 10.1126/science.3823880. [DOI] [PubMed] [Google Scholar]
- Schmidt-Rohr K, Mao JD. Selective dephasing of OH and NH proton magnetization based on 1H chemical-shift anisotropy recoupling. J Magn Reson. 2002;157:210–217. doi: 10.1006/jmre.2002.2589. [DOI] [PubMed] [Google Scholar]
- Simpson TL. The Cell Biology of Sponges. Springer Verlag; New York: 1984. [Google Scholar]
- Tabudravu JN, Eijsink VGH, Gooday GW, Jaspars M, Komander D, Legg M, Synstad B, van Aalten DMF. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg Med Chem. 2002;10:1123–1128. doi: 10.1016/s0968-0896(01)00372-8. [DOI] [PubMed] [Google Scholar]
- Tanner SF, Chanzy H, Vincendon M, Roux JC, Gaill F. High-resolution solid-state carbon-13 nuclear magnetic resonance study of chitin. Macromolecules. 1990;23:3576–3583. [Google Scholar]
- Teeyapant R, Woerdenbag HJ, Kreis P, Hacker J, Wray V, Witte L, Proksch P. Antibiotic and cytotoxic activity of brominated compounds from the marine sponge Verongia aerophoba. Z Naturforsch. 1993;48:939–945. doi: 10.1515/znc-1993-11-1218. [DOI] [PubMed] [Google Scholar]
- Turon X, Becerro MA, Uriz MJ. Distribution of brominated compounds within the sponge Aplysina aerophoba: coupling of X-ray microanalysis with cryofixation techniques. Cell Tissue Res. 2000;301:311–322. doi: 10.1007/s004410000233. [DOI] [PubMed] [Google Scholar]
- Webster A, Osifo PO, Neomagus HWJP, Grant DM. A comparison of glycans and polyglycans using solid-state NMR and X-ray powder diffraction. Solid-state Nucl Magn Reson. 2006;30:150–161. doi: 10.1016/j.ssnmr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Nge TT, Takemura A, Hori N, Ono H. Characterization of uniaxially aligned chitin film by 2D FT-IR spectroscopy. Biomacromolecules. 2005;6:1941–1947. doi: 10.1021/bm0492172. [DOI] [PubMed] [Google Scholar]
- Zhang M, Haga A, Sekiguchi H, Hirano S. Structure of insect chitin isolated from beetle larva cuticle and silkworm (Bombyse mori) Pupa exuvia. Int J Biol Macromol. 2000;27:99–105. doi: 10.1016/s0141-8130(99)00123-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.