

Abstract

Either mono- or di-bromination of benzyl methyl ethers can be achieved by controlling the amount of NBS and the temperature. Elimination of methyl bromide from the monobrominated intermediates produces aromatic aldehydes, whereas hydrolysis of the dibrominated intermediates affords aromatic methyl esters in good yields.
Chemical reactions that result in oxidation of benzyl ether methylene carbons are important chemical transformations because they often convert chemically stable functional groups into reactive groups, including aldehydes1 and esters2 that are widely used in organic synthesis.
Most studies on benzyl ether oxidation with NBS have focused on cleavage to the corresponding aldehyde via formation of N-benzylsuccinimide derivatives followed by acid hydrolysis. Although the first aldehyde formation from benzyl methyl ether was reported by Markees in 1958,3 it has not been widely applied due to its moderate yields and harsh reaction conditions.4,5 Recently, a more efficient method has been reported by Pradhan et al. utilizing an oxoammonium salt.6 On the other hand, the oxidation of benzyl ethers to yield esters was reported using either strong oxidizing agents such as Cr(VI)-periodic acid,7 4-methoxy-TEMPO catalyzed sodium hypochlorite oxidation,8 or heavy metals such as uranium hexafluoride.9 More recently, Strazzolini and Runcio reported a facile method for the oxidation of benzyl ethers to esters by concentrated nitric acid in dichloromethane.10 Such conditions are incompatible with a wide range of functional groups and/or the reagents are expensive.
This article describes a method to selectively convert benzyl methyl ethers to either aromatic aldehydes or aromatic methyl esters by reaction with either one or two equivalents of NBS in carbon tetrachloride. The conversion of benzyl methyl ethers to the corresponding methyl esters by NBS has not been previously reported.
Initially, NBS oxidative cleavage of dichlorobenzyl methyl ether in refluxing CCl4 was studied, utilizing excess NBS. The reaction mixture was illuminated by a normal 60-watt light bulb (Table 1, entry 5). The purification method was optimized by adding dilute NaOH to remove the unreacted NBS and the reaction by-products (succinimide and HBr). As reported, the corresponding aldehyde was obtained in low yield. Interestingly, the major product was the corresponding methyl ester, which may be formed through reaction of the dibromobenzyl intermediate 8 with NaOH (Scheme 1). Next, the conditions for each oxidation type were examined. To determine whether higher temperatures are essential for bromide elimination and methyl ether cleavage to produce the aldehyde, the reaction was conducted at room temperature (entry 3). Interestingly, the only isolable product was the methyl ester, in excellent yield, with no detectable aldehyde.
TABLE 1.
Benzyl Methyl Ether Oxidation Reactions
| entry | sm | conditions |
product | yield (%) | ||
|---|---|---|---|---|---|---|
| NBS (eq) | time (h) | temp. | ||||
| 1 | 1a | 2 | 16 | r.t. |
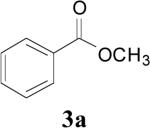
|
82 |
| 2 | 1a | 1 | 1 | Reflux |
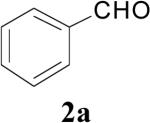
|
65 |
| 3 | 1b | 2 | 16 | r.t. |
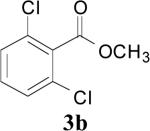
|
92 |
| 4 | 1b | 1 | 1 | Reflux |

|
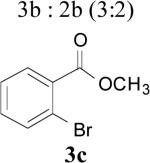
77 |
| 5 | 1b | 3 | 2 | Reflux | 3b : 2b (3:2) | 75 |
| 6 | 1c | 2 | 16 | r.t |
|
81 |
| 7 | 1c | 1 | 1 | Reflux |
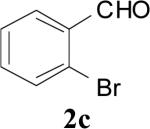
|
60 |
| 8 | 1d | 2 | 12 | r.t |
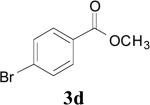
|
91 |
| 9 | 1d | 1 | 1 | Reflux |
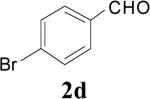
|
63 |
| 10 | 1e | 1 | 12 | r.t. |
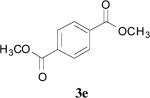
|
20 |
| 11 | 1e | 2 | 12 | r.t. | 38 | |
| 12 | 1e | 2 | 2 | Reflux | 35 | |
| 13 | 1e | 3 | 12 | r.t. | 58 | |
| 14 | 1e | 4a | 12 | r.t. | 73 | |
| 15 | 1e | 4 | 16 | r.t. | 89 | |
| 16 | 1f | 1 | 0.5 | Reflux |
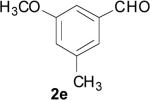
|
45 |
| 17 | 1f | 2 | 12 | r.t. | 63 | |
| 18 | 1g | 1 | 0.5 | Reflux |
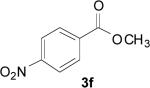
|
45 |
| 19 | 1g | 2 | 2 | r.t. | 91 | |
| 20 | 1h | 1 | 0.5 | Reflux |
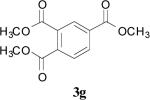
|
30 |
| 21 | 1h | 2 | 2 | r.t. | 81 | |
| 22 | 1i | 2 | 2 | r.t. |
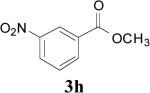
|
85 |
| 23 | 1i | 1 | 1 | Reflux |
|
65 |
Added portionwise.
SCHEME 1.

Postulated Reaction Pathways
The initial step in the proposed mechanism is formation of a monobromo intermediate 4 (Scheme 1) that can either break down, at higher temperature, into an aldehyde, or undergo an immediate second free-radical bromination. The relatively unstable dibromomethoxyl intermediate 8 may react with 0.1 M NaOH to afford the corresponding ester (Scheme 1). The reported3 moderate yields of aldehydes may be due to the use of excess NBS, leading to formation of dibromomethoxymethyl intermediates. These intermediates may decompose at high reaction temperature. Unlike the reported N-benzylsuccinimide intermediates3 obtained by conducting the reaction at high temperature, dibromomethoxymethyl intermediates are hypothesized to be formed under very mild conditions.
With respect to the proposed reaction mechanism, the optimal NBS stoichiometry is informative. In the case of aldehyde formation, using 1.0 equivalent of NBS afforded a slightly better yield than reported.3 In the case of ester formation, 2.0 equivalents of NBS are necessary for optimal yield. Using more than the optimal NBS equivalent(s) resulted in lower yields.
With the optimal conditions identified, the full scope of the methodology was investigated. A range of benzyl methyl ethers were subjected to both reaction conditions (Scheme 2). Unsubstituted phenyl, as well as ortho-, para- and meta-substituted derivatives were utilized (Table 1, entries 1, 2, 6-23). The same trends were observed. The modified reported conditions (1 equiv NBS/reflux) afforded aldehydes in moderate yields (Table 1, entries 2, 4, 7, 9), while our new method (2 equiv NBS/room temperature) afforded esters in high yields (Table 1, entries 1, 3, 6 and 8).
SCHEME 2.

Benzyl Methyl Ether Oxidation Reactions
In the case of electron-deficient aromatic rings, the oxidative ester formation proceeded smoothly in a shorter time (entries 18-21). In contrast, an electron-rich system afforded only the aldehyde (entries 16 and 17).
Based on these findings, the details of reaction mechanism outlined in Scheme 1 may be considered. There are two possible pathways following the first bromination step. First, the aldehyde formation pathway includes elimination of bromide anion to form the resonance-stabilized benzylic carbocation intermediate 5 ↔ 6. The liberated bromide anion may then attack the intermediate 5 ↔ 6 leading to cleavage of the C-O bond to yield the corresponding aldehyde. The other possibility for the monobromo derivative 4, when the reaction is performed at room temperature, is to undergo a second free-radical bromination to form a dibromobenzyl intermediate 8 that decomposes in the presence of hydroxide to afford the esters (Scheme 1).
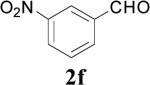
This mechanism is supported by the fact that certain electron-withdrawing groups (e.g. p-NO2) on the aromatic ring completely disfavor aldehyde formation, presumably by increasing the energy of the cationic intermediate 5 ↔ 6 (Scheme 1). On the other hand, a m-NO2 group, which would have less of an effect on the energy of the proposed carbocation intermediate 5 ↔ 6, afforded either aldehyde or ester products as determined by the reaction conditions (Table 1, entries 22-23). Moreover, it was expected that selective aldehyde and/or ester formation could be performed by controlling the reaction conditions involving bis(methoxymethyl)benzene (1e, Scheme 4). It was expected that compounds 16, 17, 18 and 3e might be obtained by treatment of 1e with 1, 2, 3 and 4 equivalent(s) NBS, respectively, and controlling the temperature. Instead, the only isolable product was the diester 3e under all reaction conditions (Table 1, entries 10-15). The only observed difference was the yield. In the first case (Table 1, entry 10), instead of all of the starting material reacting with NBS (1 equiv) to form 1 equivalent monobromo intermediate 11 that could decompose to afford the aldehyde 16 (1 equiv), approximately 20% of the starting material reacted to form the tetrabromo intermediate 13 that decomposed into the diester 3e (0.2 equiv) and 75% of the starting material was recovered. Similarly, approximately 40% of the starting material reacted when 2 equivalents NBS were used (Table 1, entry 11), and 60% in case of using 3 equivalents NBS with 40% of starting material being recovered (Table 1, entry 13). It was assumed that the aldehyde 17 could be obtained by enhancing the cleavage step of the bis(monobromo) intermediate 12 (Scheme 4), and therefore the reaction was conducting using 2 equivalents of NBS at reflux (Table 1, entry 12). Instead, such reaction conditions also furnished the diester 3e in a lower yield (Table 1, entry 12) in comparison with running the reaction at room temperature (Table 1, entry 11). Next, when 4 equivalents NBS were added to the reaction mixture in portions, at a rate of one equivalent every three hours, the diester 3e was obtained in an excellent yield (Table 1, entry 15). Evidently, once bromination starts, all of the brominated intermediates 11, 12, and 15 are rapidly brominated until the tetrabrominated intermediate 13 is formed and all of the available NBS is consumed.
SCHEME 4.

Conversion of 1e to 3e.
Encouraged by the mild conditions and high yields of this reaction, the same reaction conditions were investigated on more complicated structures utilizing several phenylthiazoles (Scheme 3). The corresponding aldehydes and esters were obtained selectively in good yields (Table 2, entries 1-6).
SCHEME 3.

Thiazolyl Methyl Ether Oxidation Reactions
TABLE 2.
Thiazolyl Methyl Ether Oxidation Reactions
| entry | starting material | reaction conditions |
product | yield (%) | ||
|---|---|---|---|---|---|---|
| NBS (eq.) | time (h) | temp. | ||||
| 1 | 1j | 1 | 1 | Reflux | 2g | 74 |
| 2 | 1j | 2 | 24 | r.t. | 3i | 79 |
| 3 | 1k | 1 | 1 | Reflux | 2h | 55 |
| 4 | 1k | 2 | 24 | r.t. | 3j | 71 |
| 5 | 1l | 1 | 1 | Reflux | 2i | 57 |
| 6 | 1l | 2 | 24 | r.t. | 3k | 65 |
In conclusion, a method was developed to generate aldehydes and esters in good yields under mild conditions using an inexpensive and commercially available reagent. Regulating the NBS/ether ratio and temperature controlled formation of aldehydes vs. methyl esters. The advantages of this newly developed methodology include no restrictions on humidity conditions required and no heavy metal used.
Experimental Section
General Procedure for Preparation of Esters
The methyl ether (1 mmol) and NBS (356 mg, 2 mmol) were added to CCl4 (10 mL). The reaction mixture was stirred at room temperature for 3-24 h and illuminated by a 60-watt light bulb. The light source was placed 10 cm away from the flask. The solvent was removed under reduced pressure. An aq NaOH solution (0.1 M, 3 mL) was added to the residue and the mixture was stirred for 15-30 s at room temperature and then EtOAc (10 mL) was added. The organic layer was separated, dried over anhydrous MgSO4, and removed under reduced pressure. The crude products were 92-98 % pure. For further purification, the obtained oil/solid was subjected to chromatography. The reaction worked properly with crystallized NBS, not with the crude form.
General Procedure for Preparation of Aldehydes
The methyl ether (1 mmol) and NBS (178 mg, 1 mmol) were added to CCl4 (15 mL). The reaction mixture was heated at reflux for 1 h and was and illuminated by a 60-watt light bulb. The light source was placed 10 cm away from the flask. The solvent was removed under reduced pressure. The obtained mass was partitioned between EtOAc (10 mL) and NaOH (0.1 M NaOH, 5 mL). The organic layer was separated, dried over anhydrous MgSO4, and removed under reduced pressure. The obtained oil/solid could then be purified through a sodium bisulfite addition compound or by chromatography. The reaction worked properly with crystallized NBS, not with the crude form.
Physical and spectral data of compounds 3i and 2g as representative examples for synthesized esters and aldehydes are shown below.
Dimethyl 2-(4-Chlorophenyl)thiazole-4,5-dicarboxylate (3i)
Yellow solid (79%): mp 72-73 °C. 1H NMR (CDCl3) δ 7.90 (d, J = 8.4 Hz, 2 H), 7.43 (d, J = 8.4 Hz, 2 H), 4.00 (s, 3 H), 3.94 (s, 3 H); 13C NMR (CDCl3) δ 169.8, 160.3, 151.6, 137.7, 130.4, 129.3, 128.1, 53.1, 53.0; MS (m/z, rel intensity) 336 (MNa+, 36.7), 334 (MNa+, 100); HRMS (ESI), m/z MNa+ 333.9921 calcd for C13H10ClNO4SNa 333.9917.
Methyl 2-(4-Chlorophenyl)-4-formylthiazole-5-carboxylate (2g)
White solid (74%): mp 139-140 °C. 1H NMR (CDCl3) δ 10.61 (s, 1 H), 7.96 (d, J = 8.7 Hz, 2 H), 7.45 (d, J = 8.7 Hz, 2 H), 3.99 (s, 3 H); 13C NMR (CDCl3) δ 184.8, 169.8, 160.4, 155.7, 138.1, 135.3, 130.1, 129.3, 128.4, 53.3; MS (m/z, rel intensity) 284 (MH+, 36.7), 282 (MH+, 100); HRMS (ESI), m/z MH+ 281.9987 calcd for C12H9ClNO3S 281.9992.
Supplementary Material
Acknowledgment
This work was sponsored by the NIH/NIAID Regional Center of Excellence for Bio-defense and Emerging Infectious Diseases Research (RCE) Program. Support is gratefully acknowledged from the Region V ‘Great Lakes’ RCE (NIH award 1-U54-AI-057153). This research was also supported by a fellowship to A. S. M. from the Egyptian government.
Footnotes
Supporting Information Available. Preparation of methyl ethers, characterization data, copies of NMR spectra for all new and known compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.a Feltenberger JB, Hayashi R, Tang Y, Babiash ESC, Hsung RP. Org. Lett. 2009;11:3666. doi: 10.1021/ol901434g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rayabarapu DK, Zhou A, Jeon KO, Samarakoon T, Rolfe A, Siddiqui H, Hanson PR. Tetrahedron. 2009;65:3180. doi: 10.1016/j.tet.2008.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Batsomboon P, Phakhodee W, Ruchirawat S, Ploypradith P. J. Org. Chem. 2009;74:4009. doi: 10.1021/jo900504y. [DOI] [PubMed] [Google Scholar]
- 2.a Chen Y, Cho C, Larock RC. Org. Lett. 2009;11:173. doi: 10.1021/ol8021287. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martinez C, Alvarez R, Aurrecoechea JM. Org. Lett. 2009;11:1083. doi: 10.1021/ol8028687. [DOI] [PubMed] [Google Scholar]; c Iwai T, Fujihara T, Terao J, Tsuji Y. J. Am. Chem. Soc. 2009;131:6668. doi: 10.1021/ja901778y. [DOI] [PubMed] [Google Scholar]; d Mahboobi S, Dove S, Sellmer A, Winkler M, Eichhorn E, Pongratz H, Ciossek T, Baer T, Maier T, Beckers T. J. Med. Chem. 2009;52:2265. doi: 10.1021/jm800988r. [DOI] [PubMed] [Google Scholar]
- 3.Markees DG. J. Org. Chem. 1958;23:1490. [Google Scholar]
- 4.Lovins RE, Andrews LJ, Keef RM. J. Org. Chem. 1963;28:2847. [Google Scholar]
- 5.Micheal EM, Hangenah JA. Heterocycles. 1987;25:117. [Google Scholar]
- 6.Pradhan PP, Bobbitt JM, Bailey WF. J. Org. Chem. 2009;74:9524. doi: 10.1021/jo902144b. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Xu L, Trudell ML. Synthesis. 2005;11:1757. [Google Scholar]
- 8.a Cho SN, Park CH. Bull. Korean Chem. Soc. 1994;15:924. [Google Scholar]; b Cho SN, Park CH. J. Korean Chem. Soc. 1995;39:657. [Google Scholar]
- 9.Goosen A, McCleland CW, Johnannes P, Vender MW. S. Africa J. Chem. 1987;40:30. [Google Scholar]
- 10.Strazzolini P, Runcio A. Eur. J. Org. Chem. 2003;2:526. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.