

Abstract
Kawasaki disease (KD) is a pediatric self-limited vasculitis characterized by immune-mediated destruction of the arterial wall and myocardium. Neither the trigger that incites the inflammation nor the switch that turns it off is known. To further our understanding of KD pathogenesis and the role of regulatory T-cells in modulating the inflammatory response, we studied circulating effector memory T-cells (CCR7− and IL-15+ Tem) and central memory T-cells (CCR7+ and IL-15+ Tcm) in six KD subjects. In two of the subjects, we cloned the remaining T-cell population by limiting dilution. TaqMan analysis of Tem studied in two KD subjects suggested that Tem are pro-inflammatory CD4+ T-helper 1 cells and CD8+ cytotoxic T-cells. Following memory T-cells over time, we defined that circulating Tem and Tcm are detectable during the acute phase in some KD subjects before treatment with intravenous immunoglobulin. Both Tem and Tcm expand rapidly within 2 weeks of treatment. The circulating Tem pool contracts, while Tcm further proliferate in the convalescent phase. Following depletion of memory T-cells, numerous T-cell clones were derived from two acute KD subjects. The large majority of these T-cells displayed the functional phenotype of peripherally induced regulatory T-cells (Treg). These findings provide insight into the nature and kinetics of the adaptive immune response in KD.
Keywords: Kawasaki disease, T-cell memory, human T-cell clones, immune regulation, Tregs
Introduction
Kawasaki disease (KD) is a pediatric vasculitis that is the most frequent cause of acquired heart disease in children in the United States and Japan [1]. Its etiology is unknown and many details of the immune response have not been characterized, although all arms of the immune system are involved [2]. Genetic association and linkage studies have identified polymorphisms that influence susceptibility to disease and outcome [3–5]. Among these is a functional polymorphism in the inositol 1,4,5-triphosphate 3-kinase (ITPK) C gene that influences T-cell activation and IL-2 production through the calcineurin–NFAT pathway [6]. This and other studies suggest that genetic variation in immune response pathways is important in the pathogenesis of KD.
Few functional studies have addressed the role of different T-cell populations in acute KD. With respect to adaptive immunity, pro-inflammatory T lymphocytes including CD8+ cytotoxic T-cells (CTL) and CD4+ T-helper 1 (Th1) cells clearly participate in the transmural infiltration of the coronary arterial wall based on autopsy studies of children dying during the acute phase [7]. However, functional T-cell studies cannot be performed on these fixed tissues and no previous work has addressed the function of peripheral T-cell clones in these patients.
The self-limited nature of KD suggests that regulatory T-cells (Tregs) play an important role in mitigating the pro-inflammatory effect of pathogenic effector T-cells that participate in the destruction of the media and internal elastic lamina of the coronary artery.
Tregs limit the expansion and mitigate the deleterious effects of pro-inflammatory T-cells by restoring immune tolerance to self-antigens and alloantigens in a variety of immune-mediated conditions, including autoimmune diseases and allograft rejection [8,9]. Two Treg populations have been recently described. One is derived from the thymus during fetal life and is termed natural Tregs [10–13]. A second Treg population, peripherally induced Tregs, arises from naïve T-cells under appropriate conditions (i.e. transforming grow factor (TGF)-β) [14,15] and repeated antigenic stimulation [16]. The relative importance of these two Treg subsets in controlling pediatric inflammatory conditions is unknown and has not been studied in KD.
To address the role and the origin of circulating Tregs in KD, we defined the phenotype and function of peripheral T-cell clones in two acute KD subjects studied before administration of intravenous immunoglobulin (IVIG).
There are several challenges to study the immune response in KD patients. First, because the cause of KD remains unknown [1], there is no antigen-specific system with which to study T-cell function in these patients. Second, tissue-infiltrating T-cells (TIL) in the arterial wall are not accessible for study.
Due to these limitations, we analyzed circulating effector memory T-cells (Tem) and central memory T-cells (Tcm) [17–19] by flow cytometry to capture the pathogenic T-cell population heading toward tissues responding to the pathogenic trigger(s) and then cloned the remaining T-cells by limiting dilution to characterize their phenotype and function. Tem and Tcm were followed over time in acute, subacute, and convalescent KD subjects. These studies have provided new insights into the immunologic response and its downregulation in KD.
Methods
Subjects
All subjects met four of the five standard clinical criteria for the diagnosis of KD according to American Heart Association guidelines [20]. The protocol for this study was approved by the institutional review board, and parents of all subjects gave written informed consent. Characteristics of the subjects are shown in Table I.
Table 1.
Characteristics of the study population.
| Subject | Age (years) | Sex | Ethnicity | Illness day | IVIG treatment response | CA status | rs28493229 genotype |
|---|---|---|---|---|---|---|---|
| 1 | 0.67 | M | Hispanic | 4 | Responder | Dilated | GG |
| 2 | 2 | F | Filipino | 8 | Responder | Normal | GG |
| 3 | 4.3 | M | Caucasian | 6 | Responder | Normal | GG |
| 4 | 4.9 | M | Filipino/Japanese | 25 | Late treatment | Dilated | GG |
| 5 | 2.5 | M | Hispanic/Filipino/Caucasian | 5 | Resistant | Dilated | GG |
| 6 | 2.1 | M | Hispanic | 4 | Responder | Normal | GC |
Illness Day 1 was considered to be the first day of fever. Resistance to IVIG was define as persistent of recrudescent fever at least 36 hours after the completion of the initial IVIG infusion. Coronary artery status was determined by transthoracic echocardiography. Patients were considered to have dilated coronary arteries if the CA Z score for either the right or left anterior descending coronary arteries was ≥ 2.0 and returned to normal dimensions within 2 months after illness onset.
Isolation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation (Hystopaque-1119, Sigma Aldrich, St Louis, MO, USA) from 2 ml of blood collected in sterile heparinized tubes.
PBMC were counted and plated in 24-well plates (Falcon, Lincoln Park, NJ, USA) in complete RPMI 1640 (Gibco, Invitrogen Inc., Grand Island, NY, USA) with 5% human AB serum (Mediatech, Manassas, VA, USA) containing 30 U/ml recombinant IL-2 (Peprotech, Rocky Hill, NJ, USA) for 2–3 days. Approximately, 1–2 × 106 PBMC were initially isolated from each subject.
Characterization of Tem and Tcm by flow cytometry
Cultures were harvested at 48–72 h, washed and T-cells separated by density gradient centrifugation (Ficoll) before staining with monoclonal antibodies (MoAbs, used at 100 ng/ml) and analyzed by flow cytometry to characterize the Tem, CCR7−, IL-15 receptor (IL-15r)+, and the Tcm CCR7+ and IL-15+ populations [17]. MoAbs for the studies were as follows: anti-CCR7 (rat Ig2a,k clone 3D12), anti-IL-15r (mouse IgG2b clone eBioJM74A), anti-CD4 (mouse IgG1k clone RPA-T4), and anti-CD8 (mouse IgG1k clone RPA-T8; eBioscience, San Diego, CA, USA).
FACS staining and sorting of the memory T-cell populations were performed with four-color staining (anti-CCR7 PE, anti-CD4 PerCP-Cy5, anti-CD8 APC, and anti-IL-15r FITC) with gating on the CCR7− and CCR7+ T-cell populations. PE rat IG2a,k, FITC mouse IG2b, PerCP-Cy5.5 mouse IG1k, and APC mouse IG1k were used as isotype controls in these experiments.
T-cell cloning
T-cells remaining after the positive selection of Tem and Tem were washed, counted, and cloned by limiting dilution (0.5 cells/w) in 96-well U-bottom plates (Falcon) in the presence of 0.1 μg/ml phytohemagglutinin (PHA; Sigma L-4144) and allogeneic, irradiated feeder cells (1 × 105/well).
PCR analysis
Tem cells and PHA-expanded T-cell clones were washed, counted, and plated for 6 h (Tem) or 24 h (T-cell clones) with an agonist anti-CD3 MoAb (clone UCHT1, mouse IGg1k, BD Bioscience, Franklin Lakes, NJ, USA) before harvest of the cells for PCR analysis. RNA was extracted using Trizol (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized using oligo (dT) 20 (Invitrogen) and superscript III (Invitrogen). mRNA levels of interferon (IFN)-γ, IL-17, TGF-β, IL-10, IL-4, and the transcription factor forkhead box P3 (FoxP3) were measured using TaqMan 5′-nuclease gene expression assays (Applied Biosystems, Foster City, CA, USA; Hs 00174143-ml IFNγ, Hs 00174383-ml IL-17, Hs 171257-ml TGF-β, Hs 00961622 IL-10, Hs 00929862 IL-4, and Hs 00203958-ml FoxP3) and cDNA derived from 25 ng of total RNA. Results were normalized to the housekeeping gene TAF1B (Applied Biosystems; Hs 01057259) and expressed as relative expression units.
For genotype, DNA was extracted as previously described [4] and PCR was performed for polymorphism (rs28493229; Applied Biosystems, C_25932098_10) with 20 ng of DNA.
Cytokine quantitation
T-cell clones were harvested at days 10–12 after PHA stimulation of the cultures, washed and plated at a concentration of 1 × 105 cells/well (200 μl total volume/well) in 96-well, U-bottom plates (Falcon) in the presence of 0.1 μg/ml anti-CD3 MoAb as an agonist (clone UCHT1 mouse IGg1k BD Bioscience). T-cells were cultured for 24 h at 37°C with 5% CO2. Cytokine levels in culture supernatants were determined using ELISA assays according to the manufacturer’s instructions (BD Bioscience for IL4, IL10, and IFN-γ and eBioscience for TGF-β).
Results
Isolation and characterization of circulating Tem in acute KD
To determine if there were circulating Tem during the acute phase of KD, we sorted PBMC that had been cultured 48–72 h in IL-2 using anti-CCR7 and anti-IL-15r antibodies. We obtained CCR7− CD4+ IL-15r+ and CCR7− CD8+ IL-15r+ T-cells from both KD subjects studied (Figure 1(A), upper and lower panels).
Figure 1.

Tem isolation and characterization in two acute KD subjects. (A) CCR7-negative cells were FACS sorted to obtain CD4+ and CD8+ IL-15r+ T-cells. (B) mRNA levels of TGF-β, IL-4, IL-10, FoxP3, IFN-γ, and IL-17 measured after 6 h stimulation with anti-CD3 using TaqMan 5′-nuclease gene expression assays.
To functionally characterize these circulating T-cells and to determine if Tem represented the T-cell population heading to tissues, we measured mRNA expression of cytokines and FoxP3 that define their functional phenotype.
Expression levels of TGF-β, IL-4, IL-10, and FoxP3 were low or undetectable in the CD4+ and CD8+ Tem derived from both KD subjects (Figure 1(B)), indicating the absence of regulatory T-cells in the Tem compartment. In contrast, IFN-γ mRNA transcript levels were high in both CD4+ and CD8+ Tem derived from both KD subjects (Figure 1(B)), indicating that the CD4+ T-cells were Th-1 cells and that the CD8+ cells were conventional CTL (Figure 1(B)).
No IL-17 transcript was detectable in the Tem population, suggesting that Th-17+ cells, described in the initial stage of many inflammatory conditions [21,22], are not circulating at this time point in acute KD patients (Figure 1(B)).
Analysis of the memory T-cell compartments in acute and convalescent KD subjects
To further define the role of T-cell memory and its evolution over time in KD, we studied four KD subjects during the acute phase before IVIG treatment, subacute phase (2 weeks later), and convalescent phase (1–3 months).
The results indicate that Tem (CCR7− and IL-15+) peak in the circulation during the subacute phase, but taper in number in the convalescent stage (Figure 2(A)). However, the Tcm (CCR7+ and IL-15+) compartment expands with the Tem population in the subacute phase and continues to expand in the convalescent phase (Figure 2, panel B).
Figure 2.

Tem and Tcm in four acute, subacute, and convalescent KD subjects. (A) Analysis of the IL-15+ T-cells within the CCR7− population. (B) Analysis of IL-15+ T-cells within the CCR7+ population.
Subject 6, who was heterozygous for a functional polymorphism in the ITPKC gene that influences T-cell activation and IL-2 production through the calcineurin–NFAT pathway [6], differed from the other subjects studied by having a larger number of memory T-cells, numerous population of circulating IL-15+ T-cells, and some double positive CD4+ CD8+ T-cells (Figure 2(A),(B), lower panels).
T-cell clonal repertoire in acute KD
To study circulating T-cells in acute KD, we cloned the remaining cells by limiting dilution. A total of 7 T-cell clones were obtained from subject 1 and 15 from subject 2. FACS staining for the expression of CD4 and CD8 co-receptors with specific MoAbs demonstrated that all the 7 T-cell clones from subject 1 were CD4+, while 13 of the 15 T-cell clones from subject 2 were CD4+ and 2 were CD8+.
The functional phenotype of these T-cells was characterized by measuring FoxP3 transcript levels and levels of TGF-β, IL-10, IL-4, and IFN-γ in culture supernatants by ELISA. Overall, 3 of the 7 CD4+ T-cell clones from subject 1, and 5 of the 13 CD4+ T-cell clones from subject 2, produced high levels of suppressive lymphokines TGF-β and IL-10 and variable amounts of IL-4 (Figure 3).
Figure 3.

Characterization of Treg clones in acute KD. Lymphokine profile and FoxP3 mRNA transcript levels of three CD4+ T-cell clones derived from subject 1 and six CD4+ T-cell clones derived from subject 2 demonstrating a Treg phenotype. Cytokine levels (TGF-β, IL-10, and IL-4) in culture supernatants were determined using ELISA and are shown in (A), (B), and (C). FoxP3 mRNA transcript levels are shown in (D).
One clone (L3) from subject 2 produced IL-10 and IL-4 and lower levels of TGF-β. All of the T-cell clones tested expressed detectable FoxP3 (Figure 3(D)). In summary, CD4+ T-cell clones with a regulatory phenotype comprised 42 and 40% of the clones for subjects 1 and 2, respectively. One CD4+ T-cell clone derived from subject 1 co-secreted high levels of TGF-β and IFN-γ in the presence of abundant expression of FoxP3 (Figure 4).
Figure 4.

TGF-β secretion by a Th-1 clone derived from subject 1. Lymphokine profiles measured by ELISA and FoxP3 RNA transcript levels of a CD4+ T-cell clone derived from subject 1 on day 4 of illness which produced both TGF-β and IFN-γ.
Conventional Th-1 T-cell clones (IFN-γ > 5 ng/ml) and Th-2 T-cell clones (production of IL-4 and IL-10 in the absence of IFN-γ) were identified from subject 2 and represented 23% (3 of 13) and 8% (1 of 13) of the CD4+ T-cell clonal repertoire (data not shown). Th-0 clones were defined as CD4+ T-cells secreting measurable amounts of IFN-γ and IL-4. Th-0 clones represented 43% (3 of 7) and 31% (3 of 13) of the T-cell clonal repertoire in subjects 1 and 2, respectively (data not shown).
CD8+ Treg clones in the acute phase of KD
Two CD8+ T-cell clones derived from subject 2 secreted low levels of IFN-γ in conjunction with high levels of TGF-β and did not secrete detectable amounts of IL-10 or IL-4 (Figure 5(A)). FoxP3 expression was also detected in these T-cell clones (Figure 5, panel B), suggesting a peripherally induced regulatory CD8+ phenotype [15,16].
Figure 5.
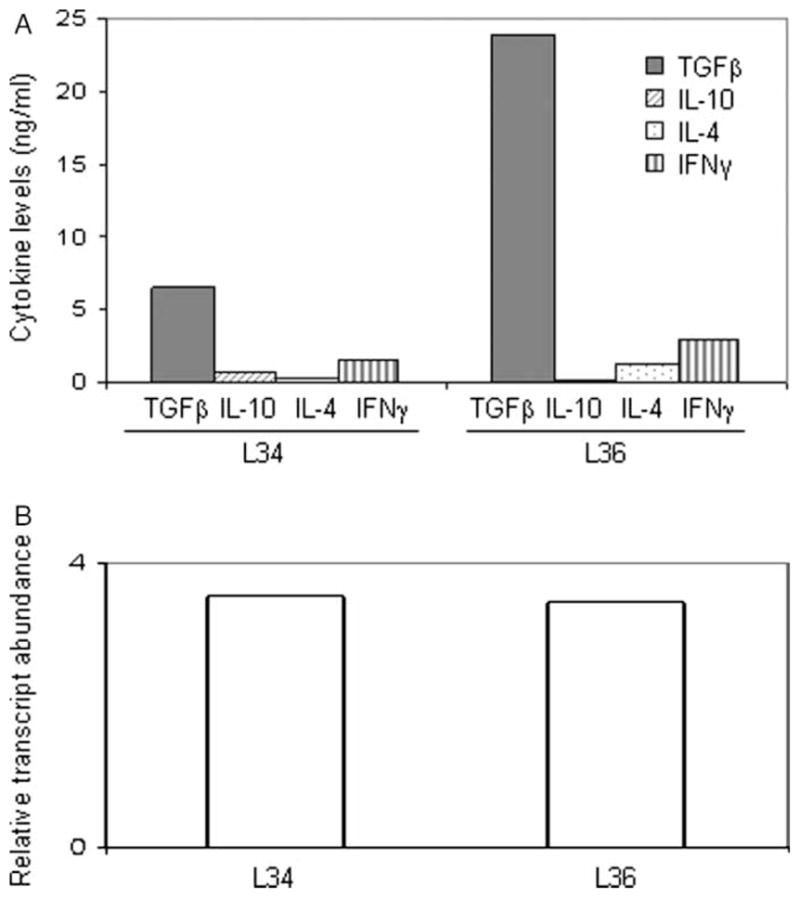
Functional characterization of two CD8+ Treg clones derived from subject 2. Lymphokine profiles measured by ELISA (panel A) and FoxP3 RNA transcript levels (B) of two CD8+ T-cell clones, derived from subject 2 on day 8 of illness which produced IFN-γ and TGF-β.
Frequency of Tregs in PBMC from acute KD subjects
The high frequency of Treg clones isolated from the two acute KD subjects is in agreement with the elevated FoxP3, IL-10, and TGF-β transcript levels measured in unsorted PBMC ex vivo from the same subjects during the acute phase (Figure 6). FoxP3 and IL-10 transcript levels were lower in PBMC from four age-matched control subjects (data not shown).
Figure 6.

Ex vivo Treg frequency in bulk PBMC. TGF-β, IL-10, and FoxP3 RNA transcript levels have been measured in bulk PBMC derived from subject 1 and subject 1 before T-cell cloning.
Discussion
Characterization of peripheral T-cells from subjects with acute KD demonstrated that Tregs are present in the circulating T-cell pool during the acute phase of the disease. Our studies on the two memory T-cell compartments in KD suggest that the antigenic exposure that triggers the immune response may have occurred days to weeks before the onset of fever and the clinical syndrome. This conclusion is supported by the detection of circulating Tem cells with a pro-inflammatory functional phenotype in the acute phase (Figures 1 and 2(A)) that rapidly expand within 2 weeks (Figure 2(A)) [17,23]. The circulating Tcm population also expanded during the subacute phase of the illness (Figure 2(B)), further supporting the idea of an earlier antigenic exposure. In the convalescent phase (1–3 months from disease onset), the circulating effector memory T-cell population contracted (Figure 2(A)), while the central memory T-cell population expanded (Figure 2(B)).
It is intriguing that subject 6 who was heterozygous for a functional polymorphism in the ITPKC gene [6] differed from the other subjects by having numerous circulating IL-15r+ T-cells in the acute phase and a broader expansion of both Tem and Tcm memory T-cell compartments in the subacute phase (Figure 2(A),(B), lower panels). It has been demonstrated in vitro that the C allele at this locus leads to reduced transcript abundance for ITPKC, which catalyzes the conversion of IP3 to IP4. This, in turn, leads to higher levels of IL-2 secretion in Jurkat cells studied in vitro [6]. Additional subjects of known genotype at this locus should be studied to further characterize the impact of this polymorphism on the peripheral T-cell repertoire.
The characterization of the T-cell clonal repertoire revealed that many of the CD4+ and CD8+ circulating T-cells were functionally Tregs. The Treg clones studied in our two KD subjects have the lymphokine profile and the ability to proliferate in culture consistent with peripherally induced Tregs (TR1 and CTR1). The presence of these T-cells in the circulation suggests antecedent repeated antigenic stimulation [24].
Other threads of evidence, such as the unexplained normocytic, normochromic, and non-regenerative anemia in these patients, are also consistent with an inflammatory process that predates the onset of fever and clinical signs [25]. Failure to find the etiologic agent of KD despite intense efforts further suggests that the agent(s) that triggers the immunologic response may have come and gone by the time the patient is diagnosed with clinical signs and symptoms of KD [26,27].
Our findings, if confirmed through study of additional subjects, would suggest that the self-limited nature of KD results from the development of T-cell regulation. Current paradigms divide Tregs into naturally occurring Tregs (CD25high) that do not expand in vitro upon antigenic stimulation [10–13] and peripherally induced Tregs (CD25low) that are defined by their lymphokine profile and the ability to expand in vitro under appropriate stimulation conditions [14,15,24]. Based on these characteristics, all the Treg clones found in our subjects, including CD8+ T-cell clones, were peripherally induced.
A previous study measured thymic-derived natural Tregs (CD4+ CD25high+ PBMC) in 54 acute and convalescent KD patients before and after therapy with IVIG [28]. The authors concluded that the thymic-derived natural CD4+ CD25high+ Treg population was low in acute KD compared to healthy control children and children with different viral infections, contributing to the persistence of inflammation in these patients. These low levels of natural Tregs may contribute to KD susceptibility. In our T-cell clonal study, we found peripherally induced Tregs as the largest circulating T-cell population in acute KD, suggesting that this population may be responsible for the self-limited nature of KD.
Furthermore, evidence to support that the Treg clones in KD were peripherally differentiated is the finding of a CD4+ T-cell clone that produced IFN-γ (Th-1) and also IL-10 and TGF-β. Such ‘self-controlled’ Th-1 cells suggest peripheral differentiation of effector T-cells into Tregs [29,30]. Th-1 T-cells that acquire a Treg phenotype have been described in murine experimental infections [31–33] but have not been previously described as part of the in vivo human T-cell repertoire.
We recognize several limitations to our study. First, the results are drawn from only six subjects and the generalizability of the findings must be approached with caution. Additional subjects should be studied and the addition of age-matched controls suffering from viral illnesses would provide an informative comparator group. Another limitation is that, in the absence of being able to study TIL, studies on the T-cell repertoire in KD patients must be confined to the circulating compartment. Without an antigen-specific system, further mechanistic studies with the T-cell clones are not possible.
In summary, these preliminary studies on the T-cell repertoire in acute KD subjects raise questions about the nature and timing of the antigenic stimulus that sets the inflammatory process in motion. Our findings, suggest that an antigenic stimulus may occur days or weeks before the onset of fever and the self-limited nature of the vasculitis is due to the expansion of peripherally derived Tregs.
The study of T-cell memory over time is novel in a clinical setting. Our results support the idea that KD is pathogen induced: the rapid expansion of circulating Tem and Tcm in subacute subjects and the contraction of the Tem population in convalescent subjects may correlate with the elimination of the pathogen itself. As a future extension of this work, the ability to establish T-cell clones from acute KD patients is a prerequisite for T-cell recognition studies, which provides another tool for the investigation of candidate antigens that may trigger the disease.
Acknowledgments
This work has been supported by NIH grant R21 HL91494-01 to J. C. Burns and A. Franco. We thank Joan Pancheri for blood sample collection, DeeAnna Scherrer for RNA purification, Cheryl Kim for Tem FACS sorting, and Judy Norberg for FACS analysis.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Burns JC, Glodé MP. Kawasaki syndrome. Lancet. 2004;364:533–544. doi: 10.1016/S0140-6736(04)16814-1. [DOI] [PubMed] [Google Scholar]
- 2.Burns JC. The riddle of Kawasaki disease. N Engl J Med. 2007;356:659–661. doi: 10.1056/NEJMp068268. [DOI] [PubMed] [Google Scholar]
- 3.Brugner D, Davila S, Breunis WB, Ng SB, Bonnard C, Ling L, Wright VJ, Thalamuthu A, Odam M, Shimizu C, Burns JC, Levin M, Kuijpers TW, Hibberd ML. A genomic wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 2009;5:e1000319. doi: 10.1371/journal.pgen.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns JC, Shimizu C, Shike H, Newburger JW, Sundel RP, Baker AL, Matsubara T, Ishikawa Y, Brophy VA, Cheng S, Grow MA, Steiner LL, Kono N, Cantor RM. Family-based association analysis implicates IL-4 in susceptibility of Kawasaki disease. Genes Immun. 2005;6:438–444. doi: 10.1038/sj.gene.6364225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burns JC, Shimizu C, Gonzalez E, Kulkarni H, Patel S, Shike H, Sundel RS, Newburger JW, Ahuja SK. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis. 2005;192:344–349. doi: 10.1086/430953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, Nakamura Y, Yanagawa H, Wakui K, Fukushima Y, Kishi F, Hamamoto K, Terai M, Sato Y, Ouchi K, Saji T, Nariai A, Kaburagi Y, Yoshikawa T, Suzuki K, Tanaka T, Nagai T, Cho H, Fujino A, Sekine A, Nakamichi R, Tsunoda T, Kawasaki T, Nakamura Y, Hata A. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40:35–42. doi: 10.1038/ng.2007.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. 2001;184:940–943. doi: 10.1086/323155. [DOI] [PubMed] [Google Scholar]
- 8.Franco A, Albani S. Translating the concept of suppressor/regulatory T cells to clinical applications. Inter Rev Immunol. 2006;25:27–47. doi: 10.1080/08830180500544506. [DOI] [PubMed] [Google Scholar]
- 9.Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and to alloantigens in humans. Nat Rev. 2007;7:585–598. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- 10.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 11.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigens. Nat Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 12.Walker LSK, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2008;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen WJ. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 14.Chen WJ, Jin W, Hardegen N, Lei K, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig M, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1–9. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 16.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2003;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 17.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subset of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 18.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 19.Sallusto F, Baggiolini M. Chemokines and leukocyte traffic. Nat Immunol. 2008;9:949–952. doi: 10.1038/ni.f.214. [DOI] [PubMed] [Google Scholar]
- 20.Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, Shulman ST, Bolger AF, Ferrieri P, Baltimore RS, Wilson WR, Baddour LM, Levison ME, Pallasch TJ, Falace DA, Taubert KA. Diagnosis, treatment and long term management of Kawaski disease: A statement for helath professionals from the Committee for Rheumatic Fever, Endocarditis and Kawaski Disease, Council on Cardiovascular Diseases in the Young, American Heart Association. Circulation. 2004;110:2747–2771. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 21.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:549–551. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 22.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 23.Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol. 2005;16:82–89. doi: 10.1016/j.coi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Apostolou I, von Boemer H. In vivo instruction od suppressor committment in naive T cells. J Exp Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burns JC, Mason WH, Glode MP, Shulman ST, Melish ME, Meissner C, Bastian J, Beiser AS, Meyerson HM, Newburger JW. Clinical and epidemiologic characteristics of patients referred for evaluation of possible Kawasaki disease. United States Multicenter Kawasaki Disease Study Group. J Pediatr. 1991;118:680–686. doi: 10.1016/s0022-3476(05)80026-5. [DOI] [PubMed] [Google Scholar]
- 26.Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease—cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. 2008;6:394–401. doi: 10.1038/nrmicro1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, Crawford SE, Russo PA, Hammadeh R, Takahashi K, Orenstein JM. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLoS ONE. 2008;2:e1582. doi: 10.1371/journal.pone.0001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faruno K, Yuge T, Kasuhara K, Takada H, Nishio H, Khajoee V, Ohno T, Hara T. CD25+CD4+ regulatory T cells in patients with Kawaski disease. J Pediat. 2004;145:385. doi: 10.1016/j.jpeds.2004.05.048. [DOI] [PubMed] [Google Scholar]
- 29.Trinchieri G. Interleukin-10 production by effector cells: Th1 cells show self control. J Exper Med. 2007;204:239–243. doi: 10.1084/jem.20070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Garra A, Vieira P. Th1 cells control themselves by producing IL-10. Nat Rev. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 31.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4+ CD25-Foxp3-Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nylen S, Radheshyam M, Eidsmo L, Das Manandhar K, Sundar S, Sacks D. Splenic accumulation of Il-10 mRNA in T cells disinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med. 2007;204:805–817. doi: 10.1084/jem.20061141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, Wynn TA, Kamanaka M, Flavell FA, Sher A. Conventional T-bet+ Foxp3- Th1 cells are the major source of host protective regulatory IL10-during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]