

Abstract
Calcium-regulated non-receptor Proline-rich Tyrosine Kinase 2 (Pyk2) is a critical mediator of Endothelin-1 (ET-1) signaling in human glomerular mesangial cells (GMC). We aimed to identify which small G-protein is acting downstream of Pyk2. Dominant interfering Pyk2 construct, termed Calcium Regulated Non Kinase (CRNK) or green fluorescent protein (control) were expressed in GMC using adenovirus-mediated gene transfer. ET-1 stimulation resulted in a significant increase of Pyk2 phosphorylation accompanied by GTP-loading of Rap1 and RhoA. CRNK expression inhibited ET-1-induced autophosphorylation of endogenous Pyk2 and diminished Rap1, but not RhoA, activation. The mechanism linking Pyk2 and Rap1 included 1) increased autophosphorylation of Pyk2 associated with p130Cas; 2) augmented p130Cas Y165 and Y249 phosphorylation; 3) enhanced p130Cas-BCAR3 complex formation. CRNK expression prevented p130Cas phosphorylation and attenuated p130Cas association with BCAR3. Downregulation of endogenous BCAR3 protein expression using an siRNA technique led to a significant decrease in Rap1 activation in response to ET-1. We observed that endogenous Pyk2 was important for GMC adhesion and spreading. Our data suggest that ET-1 stimulated the GTPase Rap1 (but neither RhoA nor Ras) by a mechanism involving Pyk2 activation and recruitment of the p130Cas/BCAR3 complex in GMC.
Keywords: Proline-rich Tyrosine Kinase 2, Endothelin-1, glomerular mesangial cells, Calcium Regulated Non Kinase, Rap1, BCAR3
INTRODUCTION
Calcium-regulated non-receptor Proline-rich Tyrosine Kinase 2, or Pyk2 (also known as RAFTK, FAK2, CAKβ and CADTK), is a cytoplasmic protein tyrosine kinase that, together with the focal adhesion kinase (FAK), belongs to a unique focal adhesion family of protein tyrosine kinases (Avraham et al. 2000; Avraham et al. 1995). In different cell types Pyk2 plays a crucial role in the cytoskeleton rearrangements involved in migration, contraction and adhesion (Lev et al. 1995; Martinez et al. 2000; Tolloczko et al. 2000; Watts 2000; Zubkov et al. 2000; Sorokin et al. 2001). Interestingly, Pyk2 shows tissue-specific expression and might exhibit differential roles in a tissue-specific manner (Avraham et al. 1995). Previously we demonstrated that Pyk2 is expressed and can be activated in glomerular mesangial cells (GMC) (Sorokin et al. 2001), which are important regulators of glomerular filtration in normal physiological conditions as well as in renal pathology (diabetic nephropathy, hypertension, and glomerulonephritis) (Sorokin & Kohan 2003; Mene et al. 1989). Nevertheless, the functional role of Pyk2 in GMC remains obscure. In this study we aimed to uncover Pyk2-dependent downstream signaling pathways and the role of Pyk2 in GMC function.
Endothelin-1 (ET-1) is a potent vasoconstrictor peptide involved in the development of glomerulonephritis, diabetic nephropathy, and hypertensive glomerulosclerosis (Levin 1996; Sorokin & Kohan 2003). ET-1 stimulation leads to GMC contraction, proliferation, and induction of pro-fibrotic extracellular matrix synthesis (Gomez-Garre et al. 1996; Simonson et al. 1989; Badr et al. 1989; Herman et al. 1998; Orth et al. 2000; Takeda et al. 1992), directly decreasing glomerular filtration rate and contributing to sclerotic lesion formation (Mene et al., 1989). There are two types of G-protein coupled ET-1 receptors, ETA and ETB, enabling peptide effects in GMC (Simonson & Rooney 1994). ET-1 receptor activation in GMC evokes a wide variety of intracellular signaling events (Sorokin et al. 2002).
One of the typical cellular responses to ligand-dependent G-protein coupled receptor activation shared by ET-1 is mobilization of intracellular calcium (Sorokin & Kohan 2003). The cloning of the calcium-regulated cytoplasmic proline-rich tyrosine kinase Pyk2 suggested the link between G-protein coupled receptors and the inductionof tyrosine phosphorylation via mobilization of intracellularcalcium (Lev et al. 1995; Herzog et al. 1996; Earp et al. 1995; Sasaki et al. 1995; Avraham et al. 1995). It is likely, that the ET-1-induced inflammatory response as well as cytoskeleton-dependent transformation requires engagement of Pyk2, followed by activation of p38 mitogen-activated protein (MAP) kinases (Sorokin et al. 2001; Chen et al. 1997). It should be noted that ET-1 mediated contraction also involves activation of the Rho kinase pathway because Rho kinase inhibition markedly blunts ETB agonist-induced vasoconstriction in different vessels from split hydronephrotic rat kidney (Cavarape et al. 2003). However, details of Pyk2-mediating intracellular cascades of GMC responses to ET-1 remain uncertain. In particular, it is still unknown which particular small G protein is activated downstream of Pyk2. Recently Rho A, Cdc42, and Rac1 had been implicated as effectors of non-receptor tyrosine kinases, participating in the cytoskeleton reorganization in GMC (Murasawa et al. 2000; Ren et al. 2001; Okigaki et al. 2003). Following appropriate stimulation, autophosphorylation of Pyk2 on tyrosine 402 (Y402) recruits and activates Src family kinases resulting in further Pyk2 phosphorylation and enhancement of Pyk2 kinase activity (Park et al. 2004). Interaction of the Pyk2 proline rich site with the SH3 domain of p130Cas (Crk associated substrate) is responsible for Pyk2/p130Cas association (Buensuceso & O’Toole 2000). Tyrosine phosphorylation of p130Cas and its association with Pyk2 results in the recruitment of a multiunit signaling complex to the membrane. Further interaction of Crk SH2 domains with p130Cas substrate domain phosphorylated tyrosines subsequently induces activation of a Crk/C3G/Rap1 signaling pathway. It is intriguing that the p130Cas SH3 domain can directly interact with a C3G amino-terminal proline rich sequence, which is probably different from Crk-binding sites (Kirsch et al. 1998). In addition, another protein with a Ras subfamily GDP exchange-factor-like domain, named BCAR3 (whose murine homologue is known as AND-34), has been implicated in p130Cas signaling (Cai et al. 1999; Gotoh et al. 2000). A p130Cas/BCAR3 complex is regulated by adhesion and inflammatory cytokines and involved in anti-estrogen resistance in breast cancer cell lines (Cai et al. 2003a). Despite the presence of a domain with modest homology to GDP exchange factors, it remains unclear whether AND-34 and two highly related proteins, NSP1 and NSP3/CHAT/SHEP1, activate Ras subfamily members including Rap1 by a direct or indirect mechanism (Gotoh et al., 2000). AND-34 over-expression has also been shown to activate Rac and Cdc42 by an indirect PI3K-mediated mechanism (Cai et al., 2003a) that required both AND-34’s SH2 domain and GEF-like domain to be intact (Felekkis et al. 2005).
Our research was aimed to identify the small GTPase involved in Pyk2-mediated signaling in GMC and to reveal the mechanism responsible for its activation. We find that the small GTPase Rap1 acts downstream of Pyk2 in ET-1-stimulated GMC. The mechanism linking Pyk2 with Rap1 activation includes induction of docking protein p130Cas phosphorylation and recruitment of BCAR3. Using siRNA techniques, we demonstrate for the first time that endogenous BCAR3, but not C3G, is important for regulation of Rap1 GTP-loading in human GMC. Additionally we report that endogenous Pyk2 is involved in GMC adhesion and spreading.
MATERIALS AND METHODS
Materials
Purified human ET-1 and PP2 were from Calbiochem. BQ123, BQ788 were from Alexis Corp. (San Diego, CA). The enhanced chemiluminescence detection system kit was supplied by Amersham Pharmacia Biotech (Baie d’Urfé, QC, Canada). The BCA protein assay kit was from Pierce (Rockford, IL). All other reagents, unless otherwise indicated, were from Sigma (St. Louis, MO).
Antibodies
To carry out immunoprecipitation, we utilized mouse monoclonal anti-human p130Cas from BD Transduction Laboratories (Lexington, KY), and control mouse IgG antibodies from BD Biosciences Pharmingen. For the Western blotting (WB) analysis, rabbit polyclonal anti-p130Cas, anti-total Pyk2, and mouse monoclonal anti-FLAG antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-actin antibody was from ICN Biochemicals (Costa Mesa, CA). Polyclonal phosphorylation state-specific anti-Pyk2 Y402 antibody was from Biosource International (Camarillo, CA). Rabbit polyclonal phosphorylation state-specific anti-p130Cas Y165 and Y249 were from Cell Signaling Technology (Beverly, MA). Rabbit serum against human BCAR3 has been previously described (Cai et al. 2003b). The horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit immunoglobulins were from BioRad (Hercules, CA).
Cell culture
All materials for cell culturing were purchased from Invitrogen. Previously characterized SV40-transformed human GMC (Sraer et al. 1996) were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 10 mM HEPES, 2mM glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml) in a 37 °C humidified incubator with 5% CO2.
Adenovirus-mediated gene transfer
The recombinant adenoviral vector (Ad) encoding the carboxyl terminus of Pyk2 termed CRNK for calcium-dependent tyrosine kinase-related non-kinase (AdCRNK) was constructed from the replication-deficient adenovirus type 5 with deletions in the E1 and E3 genes as previously described (Sorokin et al., 2001). Mouse C3G was subcloned into the pCMV-Taq3A vector and expression was verified by means of transient transfection of 293 HEK cells. For transfection of GMC, C3G was re-cloned into the pCA3 shuttle vector and an encoding C3G adenovirus (AdC3G) was generated by homologous recombination. Large scale virus preparation and titration was performed as described previously (Foschi et al. 1997). After 24 to 48 hours of serum starvation, GMC were incubated either with adenovirus encoding Green Fluorescent protein (AdGFP), or AdCRNK, or AdC3G at a multiplicity of infection of 40 plaque-forming units/cell as described previously (Rufanova & Sorokin 2006). The efficiency of gene transfer in GMC was about 70–90% as determined by fluorescence microscopy visualization of AdGFP control.
C3G and BCAR3 RNA interference
Transient transfection of cells with C3G- or BCAR3-specific, as well as control scrambled siRNAs labeled with red fluorescent C3Y (C3GsiRNA, BCAR3siRNA, siRNA-C3Y, all from Ambion) siRNA was performed using siPort Amine (Ambion) transfection reagent according to the manufacturer’s instructions. The day before the transfection, cells were typically re-plated in 60-mm Petri dishes at 60–80% confluence. The next day, GMC were transfected over 4h in a serum-free media, and lysed 48 h later. The efficiency of siRNA transfection in GMC was about 100% as determined by fluorescence microscopy visualization of siRNA-C3Y control.
BCAR3 and SH2 mutant of BCAR3 plasmid constructs and transfection
Bicistronic MSCV-IRES-GFP retroviral vector containing HA-AND34/BCAR3 had been described previously (Cai et al., 2003b). HA-tagged AND34/BCAR3 R171K (referred as SH2 mutant of BCAR3) were sub-cloned from corresponding pcDNA1 construct by PCR into XhoI and EcoR1 sites of the retroviral vector. Transfection was done using Lipofectamine2000 (Invitrogen) according to the manufacture’s instructions.
Cell lysis, immunoprecipitation, and immunoblot analysis
After stimulation with ET-1 (100 nM) for the appropriate time, the cells were lysed and immunoprecipitated with antibodies (2 μg) against p130Cas, HA tag or control mouse IgG as described previously (Rufanova & Sorokin 2006; Pratt et al. 2003).
Affinity precipitation assays of RhoA and Rap1 small GTPases
Cells were transfected with AdGFP, AdCRNK, C3GsiRNA, BCAR3siRNA, or control siRNA-C3Y for 48 h. After stimulation with ET-1, cells were washed twice with TBS, and lysed in lysis/wash buffer. To measure the active GTP-bound form of small GTPases in the cell lysates, we performed pulldown assays using recombinant Ral-RBD for Rap1 (Upstate) and Rhotekin-RBD for RhoA (Cytoskeleton) according to manufacture’s protocols. Aliquots (250–500 μg) of the supernatants mixed with glutathione-agarose with 30 μg of Ral GDS-Rap binding domain (Ral-GDS-RBD), or the Rho binding domain of Rhotekin (Rhotekin-RBD) were precipitated by centrifugation. Complexes were boiled in a Laemmli sample buffer and then separated on 4–15% SDS-polyacrylamide gels. The separated proteins were immunoblotted using corresponding antibody against RhoA or Rap1 small GTPases supplied with the each kit.
Adhesion assay
The day before the experiment, wells in 96 wells plate were coated with fibronectin (40 ug/ml), or left untreated at 4 °C overnight. Eighteen to twenty four hours later, coated wells were washed with PBS four times. For negative control we used wells coated with 1% BSA in PBS and placed at 37°C for 1h. AdGFP- and AdCRNK-infected human GMC were detached by trypsinization, re-suspended in serum-free RPMI, and kept on ice. The cell concentration was adjusted to 40,000–60,000 cells per 50 μL for re-plating in 96 wells plate. Serum-free RPMI (50 μL) was added into each well prior to addition of the suspended cells. Each condition within particular experiments was repeated four times. After 30 min or 1 h incubation at 37°C, cell adhesion was stopped by removing the medium, washing twice with PBS, and incubating with 70% ethanol for 20 minutes. Cells were stained with crystal violet, washed four times with PBS, and the dye was extracted with 0.1 M sodium citrate for 30 min. Optical density was measured at 595 nm in an automatic plate reader (Synergy-HT, BioTek Instruments, Inc., Winooski, VT). After background subtraction, data were presented as an average of three experiments.
Spreading assay
Cell spreading of AdGFP- and AdCRNK-infected GMC was analyzed 1h after replating in serum-free RPMI as described previously (Rufanova et al. 2008).
Statistics
Data are reported as means ± SD unless otherwise indicated. For Western blotting data densitometry measurements were normalized to non-simulated control in each experiment. Statistical analysis was performed by one- or two-ways analysis of variance (ANOVA), followed by a Duncan or Newman-Keuls post hoc tests. 2 way ANOVA analysis followed by Tukey test was done to estimate differences between the groups in adhesion experiments. The differences between means were considered significant at p < .05. The digitized images from cell spreading experiments were analyzed in semi-automatic mode by MetaVue 5.0r1 software (Universal Imaging Corp.). Statistical analysis of at least 300 individual cells (one way ANOVA on ranks followed by Dunn’s post hoc comparison) was carried out using SigmaStat 3.5 software (Systat Software, Inc.).
RESULTS
Adenovirus-mediated transfer of Pyk2 CRNK inhibits ET-1-induced activation of small G-protein Rap1, but not RhoA
We first determined which small GTPase is activated by ET-1 in GMC downstream from Pyk2. Previously we have demonstrated that Ras activation in response to ET-1 is independent of Pyk2 phosphorylation in GMC (Sorokin et al., 2001). Also we have obtained evidence that ET-1-induced activation of another GTPase, Cdc42, is Pyk2-independent (A. Chahdi and A. Sorokin, unpublished observation). In the current study, we have examined the activation of two small GTPases, RhoA and Rap1 (Figure 1), using an affinity precipitation assay to separate the activated GTP-bound form of protein. We found that treatment of GMC with ET-1 (100 nM) induced the formation of the GTP-bound form of both small G-proteins (Figure 1). AdCRNK expression, verified by Western blotting with anti-Flag antibody (Figure 1A, lower panel), increased the basal levels of RhoA (Figure 1A, upper panel, Figure 1B) but did not changed the basal level of Rap1 (Figure 1C, upper panel, Figure 1D) activation. The efficiency of endogenous Pyk2 inhibition was confirmed by blotting with phosphospecific anti-Pyk2-Y402 antibody (Figure 1A, third from the top panel). ET-1 stimulates Rap1 in a time-dependent manner, reaching a maximum at 2 min, and returning close to control values by 6 min after stimulation (Figure 1, C and D). Only Rap1 GTP-loading was clearly diminished by inhibition of endogenous Pyk2 phosphorylation using AdCRNK construct (Figure 1D).
Figure 1.
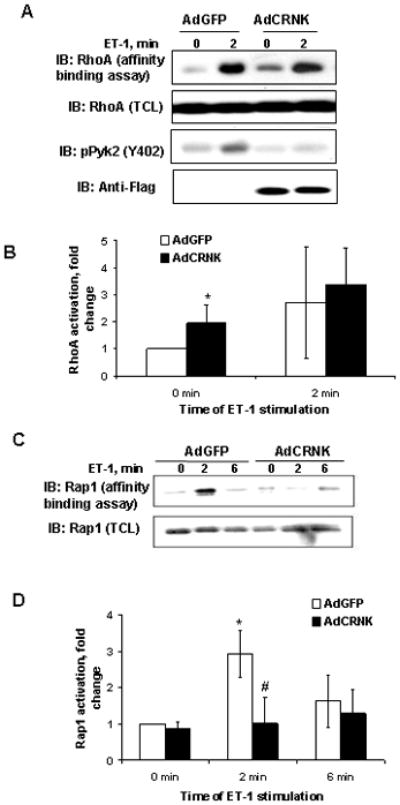
RhoA and Rap1 activation after changes in Pyk2 phosphorylation. A. Lysates from AdGFP- and AdCRNK-infected human GMC, quiescent or stimulated with endothelin-1 (100 nM) for indicated periods of time (min), were either resolved by SDS-PAGE and immunoblotted with anti-RhoA (panel second from the top), anti-phospho-Pyk2 (panel third from the top), and anti-FLAG (lower panel) antibodies or subjected to RhoA affinity binding assay.. AdCRNK overexpression was confirmed by anti-Flag antibody (lower panel). In response to ET-1 Pyk2 autophosphorylation on Y402 was increased in control GMC (third panel from the top). B. Summary of four separate experiments analyzing RhoA activation is shown. Basal RhoA activity was significantly increased after overexpression of CRNK construct compared to GFP control and in AdGFP group after ET-1 stimulation. C. Adenovirus-mediated transfer of Pyk2 CRNK inhibits ET-1-induced activation of small G-protein Rap1. Similarly to RhoA activation analysis obtained and treated lysates from AdGFP- and AdCRNK-infected human GMC were immunoblotted with anti-Rap1 (lower panel), or subjected to Rap1 (upper panel) affinity binding assay. AdCRNK overexpression resulted in inhibition of Rap1 activation. Results of Rap1-GTP affinity precipitation from three independent experiments are summarized in D. ET-1 stimulatesRap1 in a time-dependent manner, reaching the maximum at 2 min, and this activation decreases 6 min after stimulation. * - statistically significant difference (p<0.05) compare to 0 min. # - statistically significant difference (p<0.05) compare to AdGFP at the same time-point.
We used a pharmacological approach to determine what type of ET-1 receptors is responsible for Pyk2 and Rap1 activation. Pre-incubation (30 min, 100 nM) with the ETA receptor antagonists BQ123 or with the ETB receptor antagonist BQ788 prevented ET-1 induced phosphorylation of Pyk2 and Rap1 activation as assessed by affinity binding assay for Rap1-GTP and Western blotting (Figure 2). Our results suggest that in this GMC cell line, the both ETA and ETB receptors appear to play a role in ET-mediated activation of Pyk2 and Rap1.
Figure 2.

ET A and B type receptors involved in Pyk2-Rap1 activation in human mesangial cells. A. Untreated GMC (lanes 1 and 2) and GMC pretreated for 30 min with BQ123 (100 nM; lane 3), or with BQ788 (100 nM; lane 4) were left unstimulated (lane 1) or were incubated with ET-1 (100 nM, 2 min, lanes 2–4) prior to affinity precipitation of GTP-bound form of Rap1. Equal amounts of total cell lysates (upper and lower panels) and precipitates (middle panel) were analyzed by SDS-PAGE, and immunoblotted with phospho-specific anti-Pyk2 anti-Tyr-402 antibody (pPyk2Y401; upper panel) and with anti-Rap1 antibody (middle and lower panels). B. Summary of six separate experiments analyzing Pyk2 activation and four experiments for Rap1 activation are shown. For Rap1 activation data presented as median + 75% and −25%. * - statistically significant difference (p<0.05) compare to unstimulated control.
We next sought to determine what molecular mechanism linked Pyk2 with Rap1. Numerous GDP-exchanging factors (GEF) have been described for Rap1 (Stork 2003; Bos 2005; Kooistra et al. 2007). However, only few of them, including C3G and BCAR3, are implicated in the signal transduction cascade initiated by phosphorylated Pyk2. Both Rap1 GEF candidates C3G and BCAR3 have been shown to physically associate with the docking protein p130Cas. p130Cas is a large (calculated molecular weight 93 kDa, PAGE migration of 130 kD) scaffolding protein located at the cytoplasmic membrane that contains an SH3 domain, a substrate binding domain (SD) and 31 potential tyrosine phosphorylation sites, 15 of which are in the SD. The Rap1 GEF C3G directly binds to the p130Cas SH3 domain (Kirsch et al. 1998), but can also associate with the scaffolding protein indirectly via CrkII, an adaptor protein whose SH2 domain interacts with phospho-tyrosines in the p130Cas SD. BCAR3 also forms a complex with p130Cas (Goto et al., 2000) and might associate via its proline sequence with SH3 domain of CrkII (Rufanova & Sorokin 2006). Thus, the proteins described above are found in several multiunit signaling complexes whose formation may be regulated by p130Cas modification. For this reason, we sought to determine whether C3G or BCAR3 might be responsible for Rap1 activation in GMC.
Changes in C3G protein level does not influence Rap1 activation in response to ET-1
The level of C3G protein in cells infected with AdC3G was significantly increased when compared to GFP expressing control cells infected with AdGFP (Figure 3A, third panel from top). Correspondently, endogenous C3G protein level was noticeably reduced after transfection with siRNA construct targeting C3G when compared to siRNA control (Figure 3B, third panel from top). In agreement with our previous findings, ET-1 stimulation caused significant increase in Rap1-GTP amount at first 2 minutes of treatment in cells infected with control adenoviral construct encoding GFP protein (Figure 3A, upper panel and graphical insert on the right side) or transfected with control siRNA (Figure 3B, upper panel and graphical insert on the right side). Neither C3G protein overexpression (with AdC3G), nor C3G protein siRNA inhibition (with C3GsiRNA) changed the ET-1-dependent increase in Rap1 activation (Figure 3A and 3B). Equal protein loading was confirmed by immunoblotting of cell lysates with anti-Rap1 and anti-α-actin antibodies (Figure 3, A and B, second and fourth panels from the top).
Figure 3.
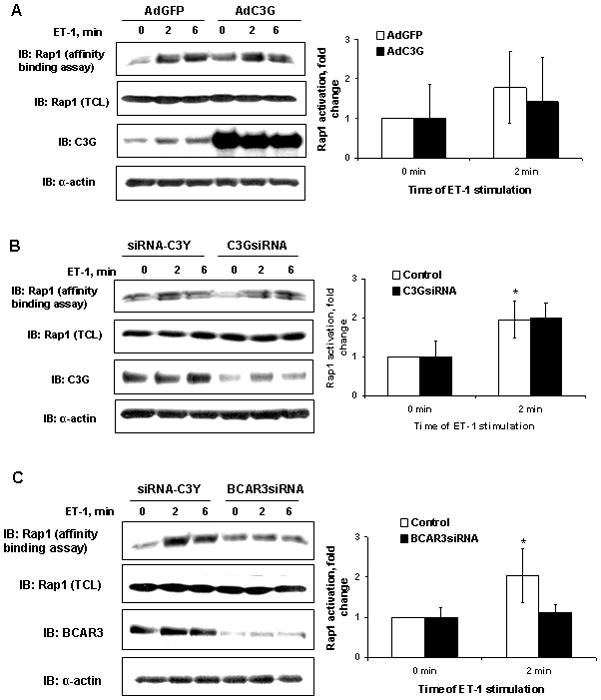
Guanine exchange factor C3G is not important for Rap1 activation in human mesangial cells. Lysates from control AdGFP (A) or siRNA-C3Y (B) and AdC3G (A) or C3GsiRNA (B) -transfected human GMC stimulated with 100 nM ET-1 for the indicated periods of time (in minutes) were subjected to Rap1 affinity precipitation assay as described previously (A and B, upper panel). Overexpression C3G protein and inhibition of endogenous C3G expression using siRNA construct was verified by immunoblotting with anti-C3G antibodies (A and B, third panel from the top). Rap1 activation was not changed by AdC3G or C3GsiRNA (A and B, top panel and graphic summary of three independent experiments for each C3G protein level). C. RNA inhibition of BCAR3 expressions decreases Rap1 GTP-loading in response to ET-1. Lysates from control siRNA-C3Y and BCAR3siRNA -transfected human GMC stimulated with 100 nM ET-1 for the indicated periods of time (in minutes) were subjected to Rap1 affinity precipitation assay (upper panel).. Inhibition of endogenous BCAR3 expression using siRNA construct was verified by immunoblotting with anti-BCAR3 antibodies (third panel from the top). Rap1 activation in response to ET-1 stimulation was decreased by BCAR3siRNA (top panel). Results of Rap1-GTP affinity precipitation assay from five independent experiments are summarized in graph on the right. * - statistically significant difference (p<0.05) compare to unstimulated control. In each experiment the equal amount of Rap1 in total cell lysates was confirmed by immunoblot analysis with anti-Rap1 and anti-α-actin antibodies.
Inhibition of BCAR3 expression decreases Rap1 activation in response to ET-1
The level of endogenous BCAR3 protein, as evaluated by Western blot analysis of cell lysates, was significantly (p<0.0001, n=5) decreased in cells treated with BCAR3 siRNA, when compared to cells treated with control siRNA (Figure 3C, third panel from the top). At 48 h after introducing BCAR3 siRNA, the cellular content of BCAR3 was decreased by an average of 3.9±0.8-fold. A representative experiment is shown in Figure 3C. In accordance with our previous results, ET-1 stimulation caused a significant increase in Rap1-GTP amount after 2 minutes of treatment in cells transfected with control siRNA (Figure 3C). In contrast, BCAR3 siRNA inhibition markedly diminished the ET-1-dependent increase in Rap1 activation (Figure 3C, upper panel). Equal protein loading was confirmed by immunoblotting of cell lysates with anti-Rap1 and anti-α-actin antibodies (Figure 3C, second and fourth panels from the top).
p130Cas association with phosphorylated Pyk2 and p130Cas SD phosphorylation are increased in response to ET-1
Since, the most likely connection between Pyk2 and BCAR3 is mediated by the scaffolding protein p130Cas, we investigated ET-1 regulation of Pyk2-p130Cas association. It had been demonstrated previously that Pyk2 can be co-precipitated with p130Cas from cell lysates. Here we confirmed the same observation in human GMC (Figure 4A). Moreover, in our experiments we observed constitutive association of Pyk2 with p130Cas (Figure 4A, second panel from the top). ET-1 stimulation leads to enhanced association of p130Cas with phosphorylated Pyk2, and this effect was diminished by expression of the dominant negative Pyk2 CRNK construct (Figure 4A, upper panel). The specificity of immunoprecipitation with anti-p130Cas antibody was confirmed using immunoprecipitation with control mouse immunoglobulins (Fig. 4A set of right panels).
Figure 4.

Pyk2-p130Cas-BCAR3 interaction in response to ET-1 stimulation. Increase of p130 Cas phosphorylation (B) and association with phosphorylated Pyk2 (A) in ET-1-treated cells are inhibited by AdCRNK. Lysates from AdGFP- and AdCRNK-infected human GMC stimulated with 100 nM ET-1 for the indicated periods of time (in minutes) were either immunoprecipitated with anti-Cas antibodies (A, left panels) or control mouse immunoglobulins (IgG) (A, right panels) prior to SDS-PAGE and immunoblotting with phosphospecific anti-Pyk2-Y402, anti-Pyk2, anti-p130Cas antibodies (A) or were directly subjected to SDS-PAGE and immunoblotting with phosphospecific anti-Pyk2-Y402, phosphospecific anti-p130Cas Y165 and Y249 antibodies (B). Equal loading was verified by immunoblotting with anti-α-actin antibodies. Shown is representative of three experiments. C. Src kinase inhibition by PP2 decreases p130Cas activation in response to ET-1. Quiescent GMC were preincubated with PP2 (10 μM, 10 min) and stimulated with ET-1 (100 nM) for the times indicated (in minutes). Cell lysates were analyzed using phospho-specific anti-p130Cas anti-Tyr-165 antibody (p130CasY165; upper panel) and with anti-actin antibody (lower panel). Shown is representative of four experiments. D. Adenovirus-mediated transfer of Pyk2 CRNK decreases p130 Cas association with BCAR3 in response to ET-1. Lysates from AdGFP- and AdCRNK-infected human mesangial cells stimulated with ET-1 for the indicated periods of time (in minutes) were immunoprecipitated with anti-p130Cas antibodies and immunoblotted either with anti-BCAR3 antibodies (upper panel), or anti-p130Cas antibodies (middle panel). Equal expression of BCAR3 was verified using immunoblotting of cell lysates with anti BCAR3 antibodies (lower panel). Shown is representative of four experiments.
Pyk2 activation regulates p130Cas tyrosine phosphorylation primarily by recruitment of Src family kinases that phosphorylate the SD, suggesting that endothelin might also regulate p130Cas SD phosphorylation (Ruest et al. 2001). In support of this hypothesis, we found that p130Cas SD phosphorylation, as evaluated by antibodies specific to phosphorylated p130Cas tyrosines Y165 and Y249, is increased in response to ET-1 (Figure 4B). Furthermore, inhibition of endogenous Pyk2 activation by transient transfection with the Pyk2 CRNK construct resulted in a significant reduction in p130Cas Y165 and Y249 phosphorylation (Figure 4B, second and third panels from the top). Significance of Src kinases activity for p130Cas SD phosphorylation was confirmed using Src inhibitor PP2 (Figure 4C).
Adenovirus-mediated transfer of Pyk2 CRNK decreases p130Cas association with BCAR3 in response to ET-1
p130Cas SD tyrosine phosphorylation enables p130Cas interaction with a number of signaling proteins. Given the siRNA experiments shown above that implicated BCAR3 in endothelin-associated Rap1 activation, we next examined the effect of endothelin on BCAR3/p130Cas association. ET-1 stimulation led to significant increase in p130Cas-BCAR3 complex formation as early as 1 min of treatment. Adenovirus-mediated transfer of CRNK prevented ET-1 induced p130Cas-BCAR3 association as analyzed by immunoprecipitation and Western blotting technique (Figure 4D, upper panel). Equal protein loading was confirmed by immunoblotting of cell lysates with anti-BCAR3 antibody (Figure 4D, third panel from top). Equal amount of p130Cas protein analyzed during immunoprecipitation was confirmed by blotting the precipitates with anti-p130Cas antibody (Figure 4D, second panel from top).
SH2 domain of BCAR3 is not involved in association with p130Cas in response to ET-1
To analyze how endothelin regulated BCAR3/p130Cas complex formation, we expressed HA-tagged forms of mouse wild type BCAR3 and an SH2 mutant of BCAR3 in GMC. The SH2 BCAR3 mutant contains an R171K mutation in the SH2 domain that renders SH2 domains nonfunctional and correspondingly all interactions mediated by the BCAR3 SH2 domain should be repressed. Thus, if the BCAR3-p130Cas interaction depends on an interaction of the SH2 domain of BCAR3 with phosphotyrosines in p130Cas, a decrease in the interaction of the SH2 BCAR3 mutant should be detected in response to ET-1 stimulation when compared to wild type BCAR3. Human GMC were transfected with either vector alone, wild type BCAR3, or with the SH2 mutant of BCAR3. Since the vector also encoded GFP, we were able to verify by Western blotting that transfection has been equally efficient in all variants (Figure 5, bottom panel). p130Cas-BCAR3 association as analyzed by immunoprecipitation of HA-tagged proteins followed by Western blotting with anti-p130Cas and anti-HA antibody were unchanged by either mutation of the BCAR3 SH2 domain or ET-1 stimulation (Figure 5, first and second panels from the top). The equal expression and immunoprecipitation of HA-tagged constructs was confirmed by blotting the precipitates with anti-HA antibody (Figure 5, second panel from the top). Equal protein loading was confirmed by immunoblotting of cell lysates with anti-p130Cas antibody (Figure 5, third panel from the top). In contrast to our experiments that examined endogenous BCAR3 and p130Cas, we failed to detect an increase in interaction between p130Cas and HA-tagged BCAR3 in ET-1-treated cells. In these experiments, both the wild-type HA-BCAR3 and HA-BCAR3 SH2 mutants were constitutively associated with endogenous p130Cas regardless of ET-1 stimulation. These data argue that the physiologic ET-1-mediated regulation of interaction between these two signaling proteins we previously observed is disrupted by BCAR3 over-expression.
Figure 5.

SH2 domain of BCAR3 is not important for ET-1 stimulated association with p130Cas. Lysates from BCAR3- and SH2mutant of BCAR3-infected human mesangial cells stimulated with ET-1 for the indicated periods of time (in minutes) were immunoprecipitated with anti-HA antibodies and immunoblotted either with anti-HA antibodies (upper panel), or anti-p130Cas antibodies (middle panel). Equal expression of plasmids was verified using immunoblotting of cell lysates with anti-GFP antibodies (lower panel). Equal expression of p130Cas was verified using immunoblotting of cell lysates with anti-p130Cas antibodies (second from the bottom panel). Shown is representative of four experiments.
Adenovirus-mediated transfer of Pyk2 CRNK decreases GMC adhesion and spreading
We studied GMC adhesion and spreading as the most likely cell functions which can be linked to Pyk2 in numerous cell types (Ostergaard & Lysechko 2005). First, we determined the phosphorylation status of endogenous Pyk2 in suspension and 1h after replating on regular cell culture treated plastic (RCCTP) (Figure 6A). Pyk2 activation, as measured by Western blotting with anti-phosphoY402 Pyk2 antibody, was markedly increased in plated cells compare to cells in suspension. CRNK overexpression caused a decrease of endogenous Pyk2 activation when compared to GFP control (Figure 6A, upper panels). Equal protein loading was confirmed by immunoblotting of cell lysates with anti-α-actin antibodies (Figure 6A, lower panels).
Figure 6.

Glomerular mesangial cell adhesion depends on endogenous Pyk2 activity. A. Representative example of three independent experiments to analyze endogenous Pyk2 phosphorylation either during GMC spreading or in suspension. Endogenous Pyk2 phosphorylation, as assessed by Western blotting with anti-phospho-Tyr402 antibody, was increased during GMC spreading on plate. Introduction of CRNK construct into GMC reduced Pyk2 phosphorylation. B. Illustration of decreased adhesion of AdGFP and AdCRNK-infected human glomerular mesangial cells (GMC) 1h after plating on regular cell culture treated plastic plates before and after washing unbound cells. Pictures were taken either under fluorescent (left panels) or bright field conditions. C. Summary of three independent experiments with GMC adhesion to Fibronectin or BSA (negative control) 30 min and 1h after plating. P<0.05 was considered significant and marked as * on the graph.
The majority of control GMC-expressing GFP was attached to RCCTP 1 h after replating (Figures 6B and 6C). Expression of the CRNK construct in GMC caused a noticeable decrease in the number of cells attached to substrate (Figures 6B and 6C). For adhesion experiments, we used fibronectin (FN) as this extracellular matrix protein has been shown 1) to stimulate Pyk2 activation via β1 integrins (Loeser et al. 2003); 2) to be secreted by and to be involved into regulation of GMC function (Gupta et al. 2000); 3) to be an important pathological factor in several renal diseases (Ruiz-Torres et al. 2005). AdGFP infected GMC demonstrated a significant increase in the number of cells bound to FN between 30 min and 1 h after plating (Figure 6C, white and grey-on-white bars). AdCRNK infected GMC had no such obvious time-dependent changes in attachment to FN over the same time period (Figure 6C, dark grey and dark-grey-on-light-grey bars). As shown in Figure 9B, we found that GMC transiently transfected with CRNK had reduced attachment to FN when compared with those transfected with GFP control vector after 1 h incubation (Figure 6C, grey-on-white and dark-grey-on-light-grey bars).
Cell spreading is a unique process actively engaging not only integrins, but also the actin cytoskeleton (Defilippi et al. 1999). Since Pyk2 is involved into both aspects of this cellular event (Ostergaard & Lysechko 2005), we subsequently analyzed GMC spreading. One hour after replating, GFP expressing control cells had a significantly larger planar cell area compared to CRNK expressing GMC (Figure 7A, compare AdGFP with AdCRNK, Phalloidine staining). Another noticeable consequence of endogenous Pyk2 inhibition by CRNK was enriched peripheral cortical F-actin staining compared to GFP control which demonstrated a diffuse F-actin distribution pattern. As such an effect could be associated with altered ROCK signaling, we used a ROCK inhibitor to assess Pyk2’s potential role in a RhoA-ROCK cascade role during GMC spreading. Spreading data from AdGFP and AdCRNK GMC populations with and without ROCK inhibition are presented in Figure 7B. Expression of CRNK dominant-negative construct in GMC resulted in significantly decreased planar cell area compare to GFP expressing control cells after 1 h spreading. ROCK inhibition abolished this difference between GFP and CRNK groups, leading to a marked increase in cell size in both populations when compared to unstimulated cells.
Figure 7.

Glomerular mesangial cell spreading depends on endogenous Pyk2 activity. (A) Confocal microscopy images of AdGFP- (green) and AdCRNK-infected human glomerular mesangial cells (GMC) stained with Palloidine-AlexaFluor568 (red) after 1h spreading. AdCRNK expression led to decreased cell planar area, and increased cortical F-actin ring thickness compare to GFP-expressing control. Scale bars - 30 μm. (B) Histograms represent the cell distribution (Y axes) depending on cell size (X axes). Cell spreading assay was done exactly as described in Methods. Number at the top portion of each histogram presents the medain value of that particular GMC distribution. One way ANOVA on ranks analysis was done to estimate differences between the groups. P<0.05 was considered significant and marked as * on the graph. Double head arrows point to comparable groups with significant differences in cell distribution. AdCRNK expression led to decreased cell size at basal spreading. Application of ROCK kinase inhibitor Y27632 (10 μM) promoted spreading in both GMC populations to similar extends. The results shown are representative of three independent GMC preparations and experiments.
DISCUSSION
The distinctive finding of the current study is that small GTPase Rap1 is activated downstream from the non-receptor tyrosine kinase Pyk2 in response to ET-1 stimulation in human GMC. The mechanism linking Pyk2 and Rap1 activation may include induction of docking protein p130Cas tyrosine phosphorylation as well as increased levels of association of p130Cas with BCAR3, a protein whose over-expression has previously been linked to Rap1 activation. Rap1 is a principal player in force initiated signal transduction and couples p130Cas to p38 MAP kinase (Sawada et al. 2001; Tamada et al. 2004; Sawada et al. 2006). Our data clearly indicate that in GMC, Rap1 is a downstream mediator of Pyk2 activation. It is of note that in B lymphocytes (McLeod et al. 2004) and vascular endothelial cells (Satoh et al. 2001), Rap1 was shown to act upstream of Pyk2. A possible explanation for such a discrepancy may lie in the nature of the cell types examined, as all three cell types are characterized by very unique properties and signaling specificity. Taken together these data from GMC, lymphocytes and endothelial cells suggest the existence of multiple interaction pathways between Pyk2 and Rap1. Indeed, it has been shown that Rap1 can activate itself via a positive feedback loop involving the phospholipase C (PLC) γ-isoform (Stork 2003). PLCγ contains a CDC25 homology domain that can activate Rap1. In parallel with PLCγ/Rap1 interaction, the Pyk2/Rap1 pathway proposed here might be part of another positive feedback loop extending Rap1 activation in pathological conditions such as tumorogenesis, inflammation or proliferative disease (Stork 2003).
The present study is the first demonstration that ET-1 activates the entire Pyk2/p130Cas/BCAR3/Rap1 cascade in human GMC and that association of p130Cas with BCAR3 correlates with ET-1-mediated p130Cas tyrosine phosphorylation. A schematics illustrating the overall model is shown in Figure 8. Report by Kodama et al. demonstrates that the focal adhesion-dependent p130Cas/Crk/Pyk2/c-Src-mediated pathway is selectively involved in ET-induced JNK MAP kinase activation in cardiomyocytes (Kodama et al. 2003). However, in the cited study, the authors did not link a signaling cascade to Rap1 or any other small G-protein. Our findings regarding Pyk2-dependent p130Cas tyrosine phosphorylation, p130Cas/BCAR3 association and BCAR3 involvement into Rap1 activation are in good correspondence with data obtained in other cell systems (Cai et al. 1999; Gotoh et al. 2000; Kodama et al. 2003). ET-1-induced p130Cas tyrosine phosphorylation can also involve Src family tyrosine kinases acting upstream of Pyk2. Recruitment of Src to a molecular complex with ET receptor may be mediated via β-arrestin-1 (Imamura et al. 2001), whereas adhesion-dependent activation of Src could be achieved via interaction with FAK (Cazaubon et al. 1997). ET-1 may also activate Src, at least partially, via activation of Ca2+/calmodulin-dependent protein kinase II in GMC (Wang et al. 2003). These alternative pathways leading to p130Cas phosphorylation could be responsible for the incomplete inhibition of p130Cas phosphorylation we observe in ET-1-treated cells expressing the dominant negative Pyk2 construct (Figure 4).
Figure 8.

Schematic representation of proposed signaling via Pyk2/p130Cas/BCAR3/Rap1 cascade. Pyk2 is constitutively bound to p130Cas through Cas amino-terminal SH3 domain. Carboxy-terminal GEF-like domain (GLD) of BCAR3 interacts with carboxy-terminal p130Cas. ET-1 triggers recruitment of Src to Pyk2 resulting in activation of Src and Pyk2 tyrosine kinase activities. Consequently, p130Cas becomes Tyr phosphorylated and progressively more associates with BCAR3, which causes GTP-loading of Rap1 followed by changes in actin cytoskeleton, cell adhesion and spreading. Whether SH2 domain of BCAR3 plays role in increase of endogenous BCAR3 binding to p130Cas in ET-1 treated cells remains to be determined. It also remains to be established whether BCAR3 is acting itself as a GDP exchange factor for Rap1, or operates via another GEF.
Although BCAR3 contains a carboxy terminal domain with modest homology to cdc25-like Ras subfamily GDP exchange factors, it remains to be established whether BCAR3 is itself a bonafide GDP exchange factor. Several studies have reported that over-expression of BCAR3 can activate both Ras subfamily (Ral, Rap1 and R-Ras) and Rho subfamily (Rac, Cdc42) GTPases but have not determined whether the activation of the GTPases is by a direct or indirect mechanism (Cai et al. 2003a; Gotoh et al. 2000). In order to avoid non-specific effects resulting from over-expression of BCAR3, we have used an siRNA approach to evaluate the role of endogenous BCAR3. Our data indicate that endogenous BCAR3 is at least partially responsible for ET1-induced Rap1 activation in human GMC since siRNA-mediated silencing of BCAR3 gene resulted in a significant decrease of ET-1-dependent GTP loading of Rap1 (Figure 3). It is highly unlikely that BCAR3 is solely responsible for Rap1 activation in ET-1-treated GMC. On the contrary, we hypothesize that a complex interplay of several proteins influencing Rap1 activity may be involved. The overexpression of highly related protein NSP3/CHAT/SHEP1 activates Rap1 via C3G pathway in 293T cells (Sakakibara et al. 2002). Among three members of the NSP family of proteins (NSP1, AND-34/BCAR3/NSP2 and NSP3/CHAT/SHEP1), BCAR3’s capacity to confer anti-estrogen resistance in MCF-7 cells is the greatest and is accompanied by disruption of cell-cell junctions (Near et al. 2007). According to our data, the canonical p130Cas-associated Rap1 activation pathway via the Rap1 GEF C3G is not important for Rap1 activation by ET-1 in human GMC since C3G expression level changes did not influence ET-1 induced Rap1 activation. Previously, we demonstrated that BCAR3 can be found in complexes with CrkII (Rufanova & Sorokin 2006). Therefore, BCAR3 could modulate other CrkII-Rap1GEFs pathways. The CrkII adaptor protein interacts with phosphorylated tyrosines of p130Cas SD through Crk SH2 domain and then can recruit the recently described guanine nucleotide exchange factor DOCK4, which acts as a direct activator of Rap1 (Yajnik et al. 2003; Ichiba et al. 1999; Buensuceso & O’Toole 2000). The C-terminus of BCAR3 including its GEF-like domain is responsible for interaction with unique C-terminal sequence of p130Cas (Gotoh et al. 2000) suggesting that p130Cas could be a negative regulator of BCAR3 signaling. The presence of multiple targets of protein-protein interaction domains in the signaling molecules involved in Rap1 activation could serve as a basis for the assembly of protein-protein conglomerates, resulting in complex regulation of Rap1 activity, which could differ significantly depending on cell type and agonist stimulation.
Here we report for the first time that Pyk2 is important for cultured human GMC adhesion and spreading. GMC provide structural support for and regulate blood flow of the glomerular capillaries by the contractile activity toward fenestrated endothelium and glomerular basement membrane, decreasing the surface area of the basement membrane therefore directly affecting glomerular filtration rate. Molecular mechanisms of GMC action include anchoring to substrates such as endothelial cells and basement membrane consisting of extracellular matrix proteins (one of which is fibronectin), and managing the intracellular acto-myosin dynamics, accessible by cell spreading experiments. The multiple GMC adhesions have extended to neighbouring substrates resulting in blood flow regulation of the involved capillaries. Since Pyk2 autophosphorylation was found to be involved in focal adhesion formations (Du et al. 2001), it is reasonable that decreased Pyk2 activation correlated with diminished human GMC attach in our experiments.
Currently, it is believed that GTP binding of Rho leads to acto-myosin contraction through activation of ROCKs, one of which phosphorylates and inactivates a myosin light-chain phosphatase (Kimura et al. 1996). Increased phosphorylation of myosin light chain correlates with increased acto-myosin contraction and precedes the formation of stress fibers and focal adhesions (Wakatsuki et al. 2003; Ren et al. 2000). During cell adhesion and spreading, maturation of these cellular structures usually requires engagement of several kinases, including Pyk2, independent of paracrine or autocrine factors that initiate receptor-mediated signaling. Therefore, our observation of RhoA activation in response to ET-1 stimulation in CRNK expressing GMC is in accordance with prior studies. Interestingly, in several different cell types, both Pyk2 autophosphorylation and the kinase domain were important for peripheral actin ring formation (Rodriguez-Fernandez et al. 2001; Du et al. 2001; Chellaiah et al. 2007). However, here we used a CRNK construct, which consists of the proline rich region and the focal adhesion targeting (FAT) domain. In contrast to findings in other cell types, GMC CRNK overexpression led to visible cortical actin ring formation. This observation allows us to speculate that the Pyk2 FAT domain is important for dynamic actin regulation in GMC. Increased actin assembly recently have been described for Pyk2-null osteoclasts with enhanced Rho activity (Gil-Henn et al. 2007). Thus, it is plausible that the observed enrichment in peripheral actin filaments and decreased basal spreading in CRNK-expressing GMC occurs as a result of an increase in basal Rho activation. Indeed, the difference in spreading between cell populations was completely abolished by application of a ROCK inhibitor. Pyk2 may be important for ROCK activity as a result of basal RhoA activation during spreading of cells on substrate, but not for ET-1-mediated stimulation when all focal adhesions are formed.
In summary, we have demonstrated that in human GMC, ET-1 induces activation of a Pyk2/p130Cas/BCAR3/Rap1 signaling cascade and that Pyk2 activation is involved in GMC adhesion and spreading regulation. Based on previously published data and the data obtained in our study, we propose that the described pathway may be of pathophysiological significance in renal disease.
Supplementary Material
Acknowledgments
DK 41684 and HL 22563 from the National Institutes of Health to A. Sorokin, AT 003984 from the National Institutes of Health to T. Wakatsuki and by Postdoctoral Fellowship from AHA 0525801Z to V. Rufanova (Petrukhina).
This study was supported by NIH research grants DK 41684 and HL 22563 to A. Sorokin, AT 003984 to T. Wakatsuki and by Postdoctoral Fellowship from AHA 0525801Z to V. Rufanova (Petrukhina).
Reference List
- Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–133. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- Avraham S, London R, Fu Y, Ota S, Hiregowdara D, Li J, Jiang S, Pasztor LM, White RA, Groopman JE. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- Badr KF, Murray JJ, Breyer MD, Takahashi K, Inagami T, Harris RC. Mesangial cell, glomerular and renal vascular responses to endothelin in the rat kidney. Elucidation of signal transduction pathways. J Clin Invest. 1989;83:336–342. doi: 10.1172/JCI113880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Buensuceso CS, O’Toole TE. The association of CRKII with C3G can be regulated by integrins and defines a novel means to regulate the mitogen-activated protein kinases. J Biol Chem. 2000;275:13118–13125. doi: 10.1074/jbc.275.17.13118. [DOI] [PubMed] [Google Scholar]
- Cai D, Clayton LK, Smolyar A, Lerner A. AND-34, a novel p130Cas-binding thymic stromal cell protein regulated by adhesion and inflammatory cytokines. J Immunol. 1999;163:2104–2112. [PubMed] [Google Scholar]
- Cai D, Felekkis KN, Near RI, O’Neill GM, van Seventer JM, Golemis EA, Lerner A. The GDP exchange factor AND-34 is expressed in B cells, associates with HEF1, and activates Cdc42. J Immunol. 2003a;170:969–978. doi: 10.4049/jimmunol.170.2.969. [DOI] [PubMed] [Google Scholar]
- Cai D, Iyer A, Felekkis KN, Near RI, Luo Z, Chernoff J, Albanese C, Pestell RG, Lerner A. AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates Rac, PAK1, and the cyclin D1 promoter. Cancer Res. 2003b;63:6802–6808. [PubMed] [Google Scholar]
- Cavarape A, Endlich N, Assaloni R, Bartoli E, Steinhausen M, Parekh N, Endlich K. Rho-kinase inhibition blunts renal vasoconstriction induced by distinct signaling pathways in vivo. J Am Soc Nephrol. 2003;14:37–45. doi: 10.1097/01.asn.0000039568.93355.85. [DOI] [PubMed] [Google Scholar]
- Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and -independent pathways. J Neurosci. 1997;17:6203–6212. doi: 10.1523/JNEUROSCI.17-16-06203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellaiah MA, Kuppuswamy D, Lasky L, Linder S. Phosphorylation of a Wiscott-Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem. 2007;282:10104–10116. doi: 10.1074/jbc.M608957200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sun Y, Lin J, Zhou A, Wang H. Research on the mechanism of endothelin inflammatory effects on human mesangial cells. Chin Med J(Engl) 1997;110:530–534. [PubMed] [Google Scholar]
- Defilippi P, Olivo C, Venturino M, Dolce L, Silengo L, Tarone G. Actin cytoskeleton organization in response to integrin-mediated adhesion. Microsc Res Tech. 1999;47:67–78. doi: 10.1002/(SICI)1097-0029(19991001)47:1<67::AID-JEMT7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Du QS, Ren XR, Xie Y, Wang Q, Mei L, Xiong WC. Inhibition of PYK2-induced actin cytoskeleton reorganization, PYK2 autophosphorylation and focal adhesion targeting by FAK. J Cell Sci. 2001;114:2977–2987. doi: 10.1242/jcs.114.16.2977. [DOI] [PubMed] [Google Scholar]
- Earp HS, Huckle WR, Dawson TL, Li X, Graves LM, Dy R. Angiotensin II activates at least two tyrosine kinases in rat liver epithelial cells. Separation of the major calcium-regulated tyrosine kinase from p125FAK. J Biol Chem. 1995;270:28440–28447. doi: 10.1074/jbc.270.47.28440. [DOI] [PubMed] [Google Scholar]
- Felekkis KN, Narsimhan RP, Near R, Castro AF, Zheng Y, Quilliam LA, Lerner A. AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. Mol Cancer Res. 2005;3:32–41. [PubMed] [Google Scholar]
- Foschi M, Chari S, Dunn MJ, Sorokin A. Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK cascade and phosphatidylinositol 3-kinase. EMBO J. 1997;16:6439–6451. doi: 10.1093/emboj/16.21.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, Schlessinger J. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(−/−) mice. J Cell Biol. 2007;178:1053–1064. doi: 10.1083/jcb.200701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Garre D, Ruiz-Ortega M, Ortego M, Largo R, Lopez-Armada MJ, Plaza JJ, Gonzalez E, Egido J. Effects and interactions of endothelin-1 and angiotensin II on matrix protein expression and synthesis and mesangial cell growth. Hypertension. 1996;27:885–892. doi: 10.1161/01.hyp.27.4.885. [DOI] [PubMed] [Google Scholar]
- Gotoh T, Cai D, Tian X, Feig LA, Lerner A. p130Cas regulates the activity of AND-34, a novel Ral, Rap1, and R-Ras guanine nucleotide exchange factor. J Biol Chem. 2000;275:30118–30123. doi: 10.1074/jbc.M003074200. [DOI] [PubMed] [Google Scholar]
- Gupta S, Clarkson MR, Duggan J, Brady HR. Connective tissue growth factor: potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000;58:1389–1399. doi: 10.1046/j.1523-1755.2000.00301.x. [DOI] [PubMed] [Google Scholar]
- Herman WH, Emancipator SN, Rhoten RL, Simonson MS. Vascular and glomerular expression of endothelin-1 in normal human kidney. Am J Physiol. 1998;275:F8–17. doi: 10.1152/ajprenal.1998.275.1.F8. [DOI] [PubMed] [Google Scholar]
- Herzog H, Nicholl J, Hort YJ, Sutherland GR, Shine J. Molecular cloning and assignment of FAK2, a novel human focal adhesion kinase, to 8p11.2-p22 by nonisotopic in situ hybridization. Genomics. 1996;32:484–486. doi: 10.1006/geno.1996.0149. [DOI] [PubMed] [Google Scholar]
- Ichiba T, Hashimoto Y, Nakaya M, Kuraishi Y, Tanaka S, Kurata T, Mochizuki N, Matsuda M. Activation of C3G guanine nucleotide exchange factor for Rap1 by phosphorylation of tyrosine 504. J Biol Chem. 1999;274:14376–14381. doi: 10.1074/jbc.274.20.14376. [DOI] [PubMed] [Google Scholar]
- Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, Miller WE, Lefkowitz RJ, Olefsky JM. beta -Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem. 2001;276:43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kirsch KH, Georgescu MM, Hanafusa H. Direct binding of p130(Cas) to the guanine nucleotide exchange factor C3G. J Biol Chem. 1998;273:25673–25679. doi: 10.1074/jbc.273.40.25673. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fukuda K, Takahashi E, Tahara S, Tomita Y, Ieda M, Kimura K, Owada KM, Vuori K, Ogawa S. Selective involvement of p130Cas/Crk/Pyk2/c-Src in endothelin-1-induced JNK activation. Hypertension. 2003;41:1372–1379. doi: 10.1161/01.HYP.0000069698.11814.F4. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Levin ER. Endothelins as cardiovascular peptides. Am J Nephrol. 1996;16:246–251. doi: 10.1159/000169004. [DOI] [PubMed] [Google Scholar]
- Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003;278:24577–24585. doi: 10.1074/jbc.M304530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MC, Randriamboavonjy V, Ohlmann P, Komas N, Duarte J, Schneider F, Stoclet JC, Andriantsitohaina R. Involvement of protein kinase C, tyrosine kinases, and Rho kinase in Ca(2+) handling of human small arteries. Am J Physiol Heart Circ Physiol. 2000;279:H1228–H1238. doi: 10.1152/ajpheart.2000.279.3.H1228. [DOI] [PubMed] [Google Scholar]
- McLeod SJ, Shum AJ, Lee RL, Takei F, Gold MR. The Rap GTPases regulate integrin-mediated adhesion, cell spreading, actin polymerization, and Pyk2 tyrosine phosphorylation in B lymphocytes. J Biol Chem. 2004;279:12009–12019. doi: 10.1074/jbc.M313098200. [DOI] [PubMed] [Google Scholar]
- Mene P, Simonson MS, Dunn MJ. Physiology of the mesangial cell. Physiol Rev. 1989;69:1347–1424. doi: 10.1152/physrev.1989.69.4.1347. [DOI] [PubMed] [Google Scholar]
- Murasawa S, Matsubara H, Mori Y, Masaki H, Tsutsumi Y, Shibasaki Y, Kitabayashi I, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Iba S, Iwasaka T. Angiotensin II initiates tyrosine kinase Pyk2-dependent signalings leading to activation of Rac1-mediated c-Jun NH2-terminal kinase. J Biol Chem. 2000;275:26856–26863. doi: 10.1074/jbc.M909999199. [DOI] [PubMed] [Google Scholar]
- Near RI, Zhang Y, Makkinje A, Vanden Borre P, Lerner A. AND-34/BCAR3 differs from other NSP homologs in induction of anti-estrogen resistance, cyclin D1 promoter activation and altered breast cancer cell morphology. J Cell Physiol. 2007;212:655–665. doi: 10.1002/jcp.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld DP, Sheetz MP, Schlessinger J. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci USA. 2003;100:10740–10745. doi: 10.1073/pnas.1834348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth SR, Amann K, Gehlen F, Unger L, Wagner J, Raschack M, Ritz E. Adult human mesangial cells (HMCs) express endothelin-B-receptors which mediate endothelin-1-induced cell growth. J Cardiovasc Pharmacol. 2000;36:S232–S237. doi: 10.1097/00005344-200036051-00069. [DOI] [PubMed] [Google Scholar]
- Ostergaard HL, Lysechko TL. Focal adhesion kinase-related protein tyrosine kinase Pyk2 in T-cell activation and function. Immunol Res. 2005;31:267–282. doi: 10.1385/IR:31:3:267. [DOI] [PubMed] [Google Scholar]
- Park SY, Avraham HK, Avraham S. RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J Biol Chem. 2004;279:33315–33322. doi: 10.1074/jbc.M313527200. [DOI] [PubMed] [Google Scholar]
- Pratt PF, Bokemeyer D, Foschi M, Sorokin A, Dunn MJ. Alterations in subcellular localization of p38 MAPK potentiates endothelin-stimulated COX-2 expression in glomerular mesangial cells. J Biol Chem. 2003;278:51928–51936. doi: 10.1074/jbc.M309256200. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000;113(Pt 20):3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- Ren XR, Du QS, Huang YZ, Ao SZ, Mei L, Xiong WC. Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J Cell Biol. 2001;152:971–984. doi: 10.1083/jcb.152.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fernandez JL, Sanchez-Martin L, Rey M, Vicente-Manzanares M, Narumiya S, Teixido J, Sanchez-Madrid F, Cabanas C. Rho and Rho-associated kinase modulate the tyrosine kinase PYK2 in T-cells through regulation of the activity of the integrin LFA-1. J Biol Chem. 2001;276:40518–40527. doi: 10.1074/jbc.M102896200. [DOI] [PubMed] [Google Scholar]
- Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Mol Cell Biol. 2001;21:7641–7652. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufanova VA, Lianos E, Alexanian A, Sorokina E, Sharma M, McGinty A, Sorokin A. C3G overexpression in glomerular epithelial cells during anti-GBM-induced glomerulonephritis. Kidney Int. 2008 doi: 10.1038/ki.2008.448. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufanova VA, Sorokin A. CrkII associates with BCAR3 in response to endothelin-1 in human glomerular mesangial cells. Exp Biol Med(Maywood) 2006;231:752–756. [PubMed] [Google Scholar]
- Ruiz-Torres MP, Lopez-Ongil S, Griera M, Diez-Marques ML, Rodriguez-Puyol M, Rodriguez-Puyol D. The accumulation of extracellular matrix in the kidney: consequences on cellular function. J Nephrol. 2005;18:334–340. [PubMed] [Google Scholar]
- Sakakibara A, Ohba Y, Kurokawa K, Matsuda M, Hattori S. Novel function of Chat in controlling cell adhesion via Cas-Crk-C3G-pathway-mediated Rap1 activation. J Cell Sci. 2002;115:4915–4924. doi: 10.1242/jcs.00207. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- Satoh K, Ichihara K, Landon EJ, Inagami T, Tang H. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors block calcium-dependent tyrosine kinase Pyk2 activation by angiotensin II in vascular endothelial cells. involvement of geranylgeranylation of small G protein Rap1. J Biol Chem. 2001;276:15761–15767. doi: 10.1074/jbc.M009165200. [DOI] [PubMed] [Google Scholar]
- Sawada Y, Nakamura K, Doi K, Takeda K, Tobiume K, Saitoh M, Morita K, Komuro I, De Vos K, Sheetz M, Ichijo H. Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase. J Cell Sci. 2001;114:1221–1227. doi: 10.1242/jcs.114.6.1221. [DOI] [PubMed] [Google Scholar]
- Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson MS, Rooney A. Characterization of endothelin receptors in mesangial cells: evidence for two functionally distinct endothelin binding sites. Mol Pharmacol. 1994;46:41–50. [PubMed] [Google Scholar]
- Simonson MS, Wann S, Mene P, Dubyak GR, Kester M, Nakazato Y, Sedor JR, Dunn MJ. Endothelin stimulates phospholipase C, Na+/H+ exchange, c-fos expression, and mitogenesis in rat mesangial cells. J Clin Invest. 1989;83:708–712. doi: 10.1172/JCI113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin A, Foschi M, Dunn MJ. Endothelin signalling and regulation of protein kinases in glomerular mesangial cells. Clin Sci(Lond) 2002;103(Suppl 1):132S–136S. doi: 10.1042/CS103S132S. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Kohan DE. Physiology and pathology of endothelin-1 in renal mesangium. Am J Physiol Renal Physiol. 2003;285:F579–F589. doi: 10.1152/ajprenal.00019.2003. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Kozlowski P, Graves L, Philip A. Protein-tyrosine kinase Pyk2 mediates endothelin-induced p38 MAPK activation in glomerular mesangial cells. J Biol Chem. 2001;276:21521–21528. doi: 10.1074/jbc.M008869200. [DOI] [PubMed] [Google Scholar]
- Sraer JD, Delarue F, Hagege J, Feunteun J, Pinet F, Nguyen G, Rondeau E. Stable cell lines of T-SV40 immortalized human glomerular mesangial cells. Kidney Int. 1996;49:267–270. doi: 10.1038/ki.1996.38. [DOI] [PubMed] [Google Scholar]
- Stork PJ. Does Rap1 deserve a bad Rap? Trends Biochem Sci. 2003;28:267–275. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- Takeda M, Breyer MD, Noland TD, Homma T, Hoover RL, Inagami T, Kon V. Endothelin-1 receptor antagonist: effects on endothelin- and cyclosporine-treated mesangial cells. Kidney Int. 1992;41:1713–1719. doi: 10.1038/ki.1992.245. [DOI] [PubMed] [Google Scholar]
- Tamada M, Sheetz MP, Sawada Y. Activation of a signaling cascade by cytoskeleton stretch. Dev Cell. 2004;7:709–718. doi: 10.1016/j.devcel.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Tolloczko B, Tao FC, Zacour ME, Martin JG. Tyrosine kinase-dependent calcium signaling in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1138–L1145. doi: 10.1152/ajplung.2000.278.6.L1138. [DOI] [PubMed] [Google Scholar]
- Wakatsuki T, Wysolmerski RB, Elson EL. Mechanics of cell spreading: role of myosin II. J Cell Sci. 2003;116:1617–1625. doi: 10.1242/jcs.00340. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mishra R, Simonson MS. Ca(2+)/Calmodulin-Dependent Protein Kinase II Stimulates c-fos Transcription and DNA Synthesis by a Src-Based Mechanism in Glomerular Mesangial Cells. J Am Soc Nephrol. 2003;14:28–36. doi: 10.1097/01.asn.0000043180.18456.47. [DOI] [PubMed] [Google Scholar]
- Watts SW. 5-Hydroxytryptamine-induced potentiation of endothelin-1- and norepinephrine-induced contraction is mitogen-activated protein kinase pathway dependent. Hypertension. 2000;35:244–248. doi: 10.1161/01.hyp.35.1.244. [DOI] [PubMed] [Google Scholar]
- Yajnik V, Paulding C, Sordella R, McClatchey AI, Saito M, Wahrer DC, Reynolds P, Bell DW, Lake R, van den HS, Settleman J, Haber DA. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell. 2003;112:673–684. doi: 10.1016/s0092-8674(03)00155-7. [DOI] [PubMed] [Google Scholar]
- Zubkov AY, Rollins KS, Parent AD, Zhang J, Bryan RM., Jr Mechanism of endothelin-1-induced contraction in rabbit basilar artery. Stroke. 2000;31:526–533. doi: 10.1161/01.str.31.2.526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.