

Abstract
Idd5.1 regulates T1D susceptibility in NOD mice and has two notable candidate genes, Ctla4 and Icos. Reduced expression of one of the four CTLA-4 isoforms, ligand independent CTLA-4 (liCTLA-4), which inhibits in vitro T cell activation and cytokine production similarly to full length CTLA-4 (flCTLA-4), has been hypothesized to increase T1D susceptibility. However, further support of this hypothesis is required since the Idd5.1 haplotypes of the diabetes-susceptible NOD and the resistant B10 strains differ throughout Ctla4 and Icos. Using haplotype analysis and the generation of novel Idd5.1 congenic strains that differ at the disease-associated Ctla4 exon 2 single nucleotide polymorphism (SNP), we demonstrate that increased expression of liCTLA-4 correlates with reduced T1D susceptibility. To directly assess the ability of liCTLA-4 to modulate T1D, we generated liCTLA-4 transgenic NOD mice and compared their diabetes susceptibility to non-transgenic littermates. NOD liCTLA-4 transgenic mice were protected from T1D to the same extent as NOD.B10 Idd5.1 congenic mice, demonstrating that increased liCTLA-4 expression alone can account for disease protection. To further investigate the in vivo function of liCTLA-4, specifically whether liCTLA-4 can functionally replace flCTLA-4 in vivo, we expressed the liCTLA-4 transgene in CTLA-4-/- B6 mice. CTLA-4-/- mice expressing liCTLA-4 accumulated fewer activated effector/memory CD4+ T cells than CTLA-4-/- mice and the transgenic mice were partially rescued from the multiorgan inflammation and early lethality caused by the disruption of Ctla4. These results suggest that liCTLA-4 can partially replace some functions of flCTLA-4 in vivo and that this isoform evolved to reinforce the function of flCTLA-4.
Keywords: Rodent, T Cells, Autoimmunity, Cell Activation, Transgenic / Knockout Mice
Introduction
Genetic studies of type 1 diabetes (T1D) in humans and in the NOD mouse model have identified numerous genes and regions influencing disease susceptibility (1-4). Some of these susceptibility regions have been found to overlap with those influencing other autoimmune diseases (5, 6), raising the possibility that the same genetic element may affect the pathogenesis of multiple autoimmune diseases. Although some T1D genes and their causal variants have strong experimental support (7-19), identification of the causal SNP (or SNPs) within each human or mouse disease gene or gene region remains challenging, especially since regulatory regions harbor many of the suspected polymorphisms (19, 20). One such region is Idd5.1, which is located near the centromere of mouse chromosome 1 and contains the genes encoding the co-stimulatory molecules CTLA-4 and ICOS in addition to two other known genes, Pard3b and Nrp2 (21), also see (http://t1dbase.org/page/Locus/display/?name=Idd5.1&species=Mouse) for a depiction of the Idd5.1 region. NOD congenic mice having the C57BL/10 (B10) diabetes-resistant Idd5.1 haplotype are protected from diabetes when compared to the parental NOD strain (21-23). The orthologous region on human chromosome 2q33 is associated with T1D and other autoimmune diseases such as multiple sclerosis (24), Graves’ disease (25, 26), Hashimoto’s thyroiditis (26, 27), Addison’s disease (27), rheumatoid arthritis (28), and celiac disease (29). Fine-mapping studies in thyroid autoimmunity and type 1 diabetes in humans have identified CTLA4 as the most likely causal gene, although the true causal SNP(s) and the molecular basis of disease have yet to be identified definitively (19). Taken together, these studies suggest that at least some common genetic pathways contribute to both human and mouse autoimmune diseases. This commonality highlights these shared pathways and has focused attention on the definitive identification of the causal variant(s) and understanding their functional consequences. Such goals can be achieved in mice where hypothesized causal genetic variation can be tested by generating appropriate congenic or transgenic mice and examining the effects on autoimmune disease development and other immune-related phenotypes.
Comparative analyses of CTLA-4 mRNA expression by the B10 and NOD alleles revealed that an isoform of CTLA-4, ligand-independent CTLA-4 (liCTLA-4) is differentially expressed by protective and susceptible alleles of Ctla4: NOD mice have the G allele at residue 77 of Ctla4 exon 2 that is hypothesized to be responsible for the reduction in liCTLA-4 mRNA transcript levels due to its position in an exon splicing silencer motif (19, 21). In contrast, autoimmune-resistant B6 and B10 strains carry the A allele, which results in an increased expression of liCTLA-4 transcripts relative to that of the NOD strain (19, 21). Functional analysis of liCTLA-4 revealed that it is constitutively expressed in memory T cells in mice and ectopic expression of liCTLA-4 in T cells limits T cell activation, cytokine production and proximal TcR signaling (30) making liCTLA-4 a logical candidate for causing diabetes susceptibility in NOD mice and that the causal SNP is residue 77 in Ctla4 exon 2.
Previous studies have shown that Icos and Ctla4 have exonic and intronic SNPs in addition to residue 77 of Ctla4 exon 2 that vary between diabetes susceptible NOD and resistant NOD.B10 Idd5.1 congenic mice (21). For example, in addition to the translationally silent SNP in Ctla4 exon 2, a SNP in Icos exon 1 causes a non-conservative amino acid change from arginine to histidine at residue 7 in the putative leader sequence, an alteration that could change the expression of ICOS and the frequency of type 1 diabetes (19, 21). To study the potential role of liCTLA-4 in diabetes susceptibility, we genotyped the Ctla4-Icos region in a panel of inbred mouse strains, and developed new Idd5.1 NOD congenic strains with two of the characterized haplotypes, those from SWR and from CAST. Importantly, we discovered that the alleles of Ctla4 present in the SWR and CAST inbred strains differed at residue 77 in Ctla4 exon 2, with SWR having the NOD SNP and CAST having the B10 SNP. However, the SWR and CAST strains shared many of the other SNPs that differ between the NOD and B10 CTLA-4 and ICOS alleles, including the SNP in Icos exon 1. Therefore, to determine if the Ctla4 SNP or the Icos SNP causes T1D susceptibility, NOD congenic mice having the SWR and CAST haplotypes at the Idd5.1 region (NOD.CAST Idd5.1 and NOD.SWR Idd5.1) were developed and tested for liCTLA-4 expression and their diabetes frequency.
To confirm further the protective role of liCTLA-4 in vivo, we also generated liCTLA-4 transgenic mice on the NOD background to supplement the natural deficiency in liCTLA-4 expression in the NOD strain and compared the diabetes susceptibility between NOD liCLTA-4 transgenic mice, NOD.B10 Idd5.1 congenic mice and wild type NOD mice. To determine further the functional role of liCTLA-4 in vivo, specifically whether liCTLA-4 can functionally replace flCTLA-4, we created transgenic mice expressing liCTLA-4 on the B6 background and crossed these mice to CTLA-4-/- mice in order to study the in vivo function of liCTLA-4 in the absence of flCTLA-4.
Using these new congenic strains of mice and liCLTA-4-tg mice, we clearly show that increased levels of liCTLA-4 correlate with more protection from T1D and can account for the alteration of disease susceptibility mediated by the Idd5.1 region, and in CTLA-4-deficient mice, liCTLA-4 can replace some, but not all, of the functions of flCTLA-4. Expression of liCTLA-4 partially rescued CTLA-4-/- mice from multiorgan inflammatory autoimmune disease and the early lethality normally observed in the CTLA-4-/- mice.
Materials and Methods
Mice
B6, CAST and SWR mice were obtained from The Jackson Laboratory (Bar Harbor, ME). NOD/MrkTac mice were obtained from Taconic (Germantown, NY). NOD.B10 Idd5.1 congenic mice (N15) have been described previously (21) and obtained as line 2193 from the Taconic Emerging Models Program. The 10.37 Mb congenic interval and the Idd5.1 region within its boundaries are derived from the B10 strain and have been detailed previously (31) and is depicted at the following web site: http://t1dbase.org/page/DrawStrains/display (and then select 2193). All mice were maintained in specific pathogen free conditions and the appropriate institutional review committee approved experimental procedures. To develop the NOD strains congenic for the Idd5.1 regions of SWR and CAST, mice were backcrossed to the NOD strain and at the N6 generation were confirmed to be NOD homozygous at Idd1, 3, 4, 6, 9, 10, 17, and 18. The Idd5.2 and Idd5.1 regions on chromosome 1 are in close proximity (31); therefore, incipient congenic mice were screened during the backcrossing phase of strain development for those that had inherited a recombinant chromosome 1 in which the Idd5.1 region remained non-NOD and the closely linked Idd5.2 region was derived from the NOD genome. This was done to prevent the allelic status of the linked Idd5.2 region from influencing the planned frequency study. Subsequent to the development of the NOD.CAST Idd5.1 and NOD.SWR Idd5.1 congenic strains, we discovered a third Idd5 subregion located between Idd5.1 and Idd5.2, called Idd5.3 (31). Idd5.3 had not been previously observed because a protective allele at Idd5.1 masks the effect of a protective allele at Idd5.3; in other words, having a protective allele at both Idd5.1 and Idd5.3 is no more protective than having a protective allele at Idd5.1 only. Therefore, determining if an Idd5.3 allele is T1D protective or susceptible must be ascertained by examining the Idd5.3 congenic segment in the context of NOD alleles at the flanking Idd5.1 and Idd5.2 gene regions. Thus, in the context of the current study, the status of the linked Idd5.3 allele should not affect the interpretation of the T1D frequency.
As depicted in Supplementary Figure 1, the congenic region in the NOD.CAST Idd5.1 strain is defined at the proximal boundary by SNPs rs13475864 and rs13475866, which map to 51.12 and 51.45 Mb on chromosome 1, respectively. The distal boundary is defined by D1Mit77 and D1Mit180 which map to 73.76 and 73.98 Mb on chromosome 1, respectively. The NOD.SWR Idd5.1 strain is defined at the proximal boundary by SNP rs13475866 and D1Mit18, which map to 51.45 Mb and 52.47 Mb on chromosome 1, respectively. The distal boundary is defined by rs3699077 and D1Mit179, which map to 69.31 and 71.72 Mb on chromosome 1, respectively. Thus, the CAST and SWR congenic intervals extend through the Idd5.3 region. The gene encoding long-chain acyl-coenzyme A dehydrogenase (ACADL) is the primary candidate gene in the Idd5.3 region due to its differential expression by the NOD and B10 alleles (31). We have obtained ACADL mRNA expression data showing that CAST has a B6-like ACADL expression pattern, which predicts that CAST has a protective allele at Idd5.3, whereas SWR has a NOD-like ACADL mRNA expression pattern, which predicts a susceptible allele at Idd5.3 (data not shown). To determine the position of the markers used in this study, Mouse Ensembl release 54 was used (www.ensembl.org/Mus_musculus).
Subsequent to the completion of the studies presented in this manuscript, the genetic backgrounds of the NOD.SWR Idd5.1 and NOD.CAST Idd5.1 congenic strains were defined more precisely by genotyping DNA samples using a 5K mouse SNP chip. The assay, performed by ParAllele Biosciences (South San Francisco, CA), demonstrated that the NOD.CAST Idd5.1 strain has no non-NOD SNPs outside of the congenic region and the NOD.SWR Idd5.1 strain has two regions of non-NOD DNA: 8.5 and 0.5 Mb intervals on chromosomes 13 and 14, respectively. The SWR-derived region on chromosome 13 at 38.6-47.1 Mb has the possibility that it includes the Idd14 region, which has been detected in studies using two non-NOD strains, B6 and NON. The B6-derived Idd14 susceptibility allele has a linkage peak at 52-73 Mb (32) and this linkage was confirmed by the development of a NOD.B6 congenic strain (25 Mb to the end of the chromosome at ~116 Mb) (33) that has an increased frequency of T1D. The NON-derived protective Idd14 allele has a linkage peak at 41-81 Mb (34). Based on these localizations, it is likely that the SWR genetic contamination on chromosome 13 is proximal to Idd14. Because the diabetes frequency of both congenic strains was determined in the progeny of F1 parents (see description below), any potential effects from the non-NOD regions on chromosomes 13 and 14 are neutralized due to the fact that they are segregating randomly.
SNP identification for haplotype analysis
NOD and B6 BAC-derived sequences that are available for a portion of the Idd5.1 region (21) were aligned and SNPs and other sequence differences were identified using Sequence Search and Alignment by Hashing Algorithm, SSAHA (35). The sequences were entered into TIDBase (www.T1DBase.org) to generate graphic displays of any polymorphisms in the Idd5.1 region. Novel assays (Supplementary Table I) to detect the allelic status at NOD/B6 SNPs in multiple mouse strains including CAST and SWR were developed by designing primers with Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3). Primers were synthesized by Sigma Genosys (St. Louis, MO), with the forward primer labeled with the fluorescent dye fam. For sequence-based haplotype analysis of selected segments of the CAST and SWR Idd5.1 congenic intervals, nested PCR primers were designed to span the region to be sequenced. Outer primers and inner primers generated 1500 and 500 bp products, respectively, each overlapping by 50 bp to generate a continuous sequence. Big Dye Terminator 3 (Applied Biosystems, Foster City, CA) was used to sequence the products. All sequences generated were entered into T1DBase for SNP identification. Sequences generated for the Ctla4 and Icos genes from the CAST and SWR strains have been submitted to Genbank and have the accession numbers GQ420691 (Icos from SWR), GQ420692 (Ctla4 from CAST), GQ420693 (Ctla4 from SWR) and GQ420694 (Icos from CAST).
liCTLA-4 transgenic mice
cDNA encoding liCTLA-4 was cloned into the EcoR1 site downstream of the CD2 promoter using a pBluescript vector. Plasmids containing the linearized liCTLA-4 were injected directly into the pronuclei of fertilized NOD eggs at the JDRF Center transgenic core facility or C57BL/6 eggs at Brigham and Women’s Hospital transgenic core facility; both at Harvard Medical School. Identification of transgenic mice was accomplished by PCR amplification of DNA obtained from tail biopsies using primers located between the CD2 enhancer and exon 1 of liCTLA-4 and another primer located between exon 1 and exon 3. The liCTLA-4 primer pairs used are listed below:
5′-GCATGGTTCTGGATCTTCAGAGA-3′
5′-TGTGGACTCCACCAGTCTCACTTCAGTTCCTTTTGCA-3′
The samples were amplified using 35 cycles at 94°C for 30 seconds, 60°C for 30 seconds and 72°C for 60 seconds. Amplification was also performed together with the beta-globin housekeeping gene as an internal control for the PCR reaction. NOD liCTLA-4 transgenic mice were bred with wild type NOD mice from Taconic Farms (Germantown, MD). B6 liCTLA-4 transgenic mice were bred with triple knock-out mice lacking B7.1, B7.2 and CTLA-4. Progeny carrying the flCTLA-4 knock-out allele and wild type alleles of B7.1 and B7.2 were identified by PCR as described previously (36).
Analysis of mRNA levels by reverse transcription and quantitative PCR
Total mRNA was isolated from spleen and lymph node cells using the TRIzol method (Invitrogen) followed by an mRNAEasy kit (Qiagen). cDNA was prepared using oligo d(T) 16 primers and random hexamers from Applied Biosystsems. The MMLV reverse transcriptase was used. Quantitative PCR was performed on the ABI Prism 7700 instrument (Applied Biosystems) using ß-actin or ß2 microglobulin as endogenous controls. Primers were used at a final concentration of 300 nM and the probe (FAM) was used at a final concentration of 200 nM. Primer and probe sequences for detecting flCTLA-4 and liCTLA-4 mRNAs (19) and flICOS mRNA (21) have been published previously. Quantitative PCR data were evaluated using an unpaired T test (Prism software, GraphPad, San Diego, CA).
Western blotting
2 × 106 spleen cells from congenic and transgenic mice were lysed using the M2 lysis buffer containing 20 mM Tris pH 7.6, 0.5% Triton 100, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, and one complete mini protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN). Cell extracts were run on an 18% denaturing gel. Fractionated proteins were transferred to a PVDF membrane (BioRad, Hercules, CA) and probed with polyclonal goat anti-CTLA-4 antibodies recognizing the cytoplasmic domain of CTLA-4 (C19, Santa-Cruz Biotechnology, Santa Cruz, CA). The antibodies recognize the cytoplasmic tail shared by the flCTLA-4 and liCTLA-4 isoforms but the two molecules can be differentiated based on size. On a western blot, flCTLA-4 and liCTLA-4 migrate at about 33 and 12 kD, respectively.
Flow cytometric analysis
Spleen, thymus and lymph node cells were prepared and stained for cell surface molecules using the following antibodies: CD3-FITC or allophycocyanin (clone 145-2C11), CD4-PerCP (clone RM4-5), CD8-PerCP (clone 53-6.7), CD25-PE (clone PC61), CD62L-FITC (clone MEL-14), CD69-FITC (clone H1.2F3), CD44-PE (clone IM7), CD28-PE (clone 37.51), and ICOS-PE (clone 7E.17G9). The CD3-FITC and CD69-FITC antibodies were obtained from BD Pharmingen, the remaining antibodies were obtained from BioLegend (San Diego, CA).
T cell proliferation and cytokine assay
5 × 105 lymph node T cells per well were added to 96-well round bottom plates and stimulated with different concentrations of anti-CD3 (clone 145-2C11, BioXCell) antibody and 1 μg /ml of anti-CD28 (clone PVI, BioXCell). Cells were pulsed with 1 μCi of [3H] thymidine after 48 hours and harvested 18 hours after pulsing. The incorporated radioactivity was measured using a Wallac Beta Plate Scintillation Counter (Perkin Elmer). Culture supernatants were collected 40 - 48 hours after activation and analyzed for IL-2, IL-4, IL-10, IFN-γ and IL-17 using standard ELISA methods (BD Biosciences, San Jose, CA).
For analysis of expression of activation-induced markers, spleen cells from mice were first stimulated with 1 μg /ml of anti-CD3 (clone 145-2C11, BioXCell) antibody and anti-CD28 (clone PVI, BioXCell) in Dulbecco’s modified Eagles Medium (DMEM)/10% FCS supplemented with 5 × 10-5 M β-mercaptoethanol, 1 mM sodium pyruvate, non-essential amino acids, L-glutamine, and 100 U penicillin/100 μg streptomycin per ml for various periods of time and then analyzed for the expression of cell surface molecules as described above.
Monitoring diabetes
To determine if the SWR Idd5.1 and CAST Idd5.1 congenic regions have a susceptible or resistant allele and differentially affect development of T1D, the diabetes frequency studies were performed using Idd5.1 heterozygous males and females to produce F2 cohorts from each of the two Idd5.1 congenic strains. This method was employed to avoid confounding effects from any residual non-NOD-derived DNA present in the SWR Idd5.1 and CAST Idd5.1 congenic strains since such regions segregate at random in the F2 cohort. All F2 progeny (NOD homozygous at Idd5.1, heterozygous at Idd5.1, and homozygous non-NOD at Idd5.1) were monitored for disease frequency in a blinded fashion and genotyped after developing T1D or at the end of the study (7 months). Idd5.1 congenic mice, liCTLA-4 tg mice, and non-tg littermate controls were screened for diabetes weekly beginning at approximately 3 months of age using Diastix urinalysis strips (Bayer). A mouse was considered to be diabetic when its urine glucose concentration was >500 mg/dL. Diabetic mice also exhibited polydipsia, polyuria, and weight loss. The frequency of diabetes was compared between strains with the Kaplan-Meier log-rank test using Prism software (GraphPad, San Diego, CA).
Histopathology
Heart, lung, and liver were isolated from C57BL/6, CTLA-4-/- and CTLA-4-/- liCTLA-4 tg mice and fixed in 10% formalin. Paraffin embedded sections were stained with hematoxylin and eosin and examined microscopically by an observer blinded to the genotype.
Results
SNP haplotype analysis across the Idd5.1 region in different strains
NOD.B10 Idd5.1 congenic mice are NOD mice in which the Idd5.1 locus has been introgressed from the B10 strain. NOD.B10 Idd5.1 mice differ from the NOD parental strain in their frequency of type 1 diabetes, expression of liCTLA-4 mRNA and protein (19, 21, 30), and cell-surface ICOS expression following activation (37). Due to its importance in regulating T cell activation and because we discovered a putative causal SNP in exon 2 of the gene encoding CTLA-4, we proposed Ctla4 as the most likely gene in Idd5.1 to mediate diabetes susceptibility (21, 37). The Ctla4 SNP is associated with differential expression of the liCTLA-4 isoform, but this SNP is inherited as a haplotype with other SNPs in both Ctla4 and Icos, including a SNP that causes an arginine to histidine substitution in the leader sequence of Icos (21). To determine whether the Ctla4 exon 2 SNP or other SNPs in Ctla4 or Icos alters diabetes susceptibility, we genotyped B6/NOD sequence polymorphisms located in or near Ctla4 and Icos in eleven inbred mouse strains (B10, B6, A/J, CAST, PWK, CZECH, SWR, BALB/c, NOD, NON, and SJL) (Fig. 1a, Supplementary Table1). The A/J haplotype was identical to that of B10 and B6 whereas the BALB/c, NON and SJL had identical SNPs with NOD. Four strains, CAST, SWR, PWK, and CZECH, had haplotypes that appeared to be hybrids of the B10-like and NOD-like SNP patterns. The results from two of the inbred strains, CAST and SWR, were particularly interesting because their SNP haplotypes in the Ctla4-Icos region were more similar to each other than to either NOD or B10. In addition, despite their similarity, the CAST and SWR haplotypes differ at the SNP in exon 2 of Ctla4 (position 77, marker 6 in Fig. 1a, Supplementary Table I). In exon 2 of Ctla4 (position 77), CAST mice harbor the SNP (A) present in strains such as B10, B6 and A/J, whereas SWR mice have the SNP (G) present in NOD, BALB/c, NON and SJL mice.
Figure 1. Multiple Idd5.1 haplotypes in inbred mouse strains influence resistance and susceptibility to T1D.

(A) Haplotype analysis of the Idd5.1 region was performed in DNA from eleven strains of mice. For the SNPs analyzed, those shaded in dark grey have a B10 allele and those shaded in light grey have a NOD allele. Ctla4 and Icos exonic SNPs are shown in the expanded portions of the figure where intermediate shading indicates the CAST-specific SNPs. The frequency of diabetes was monitored in (NOD.CAST Idd5.1 X NOD) F2 (B) and (NOD X NOD.SWR Idd5.1) F2 (C) cohorts as described in the Materials and Methods. As compared to littermates homozygous for the NOD haplotype at Idd5.1, homozygous (P = 0.0002) and heterozygous (P = 0.0017) NOD.CAST Idd5.1 mice are protected from diabetes while NOD.SWR Idd5.1 mice (homozygotes and heterozygotes) have a NOD-like frequency. The frequency of diabetes was compared between strains with the Kaplan-Meier log-rank test
Importantly the CAST and SWR haplotypes both share the B10 haplotype at the SNP in exon 1 of Icos (marker 12 in Fig. 1a, Supplementary Table I) that causes a non-conservative amino acid change from arginine to histidine at residue 7 in the putative leader sequence, as well as the SNP in intron 4 of Icos (marker 13 in Fig. 1, Supplementary Table1). We hypothesized that if the Ctla4 SNP determines diabetes susceptibility in the Idd5.1 region, NOD mice congenic for the CAST Ctla4-Icos region would be resistant to diabetes while NOD mice congenic for the SWR Ctla4-Icos region would be susceptible. However, if, for example, the Icos SNP in the leader sequence (marker 12 in Fig. 1) (21) controls disease susceptibility, both congenic strains would be resistant to diabetes since they both have the B10 SNP. In addition, we predicted that the CAST congenic strain would produce higher levels of the liCTLA-4 isoform than SWR congenic mice. To study the effect of the SWR and CAST Idd5.1 haplotypes on the expression of liCTLA-4 and diabetes susceptibility, we generated congenic mice in which the Idd5.1 interval from CAST and SWR mice was introgressed onto the NOD background.
NOD.CAST Idd5.1 mice are NOD.B10 Idd5.1-like while NOD.SWR Idd5.1 are NOD Idd5.1-like in regard to liCTLA-4 expression and diabetes susceptibility
Although during their development, the NOD.CAST Idd5.1 and NOD.SWR Idd5.1 strains were selected to be NOD homozygous at all known Idd regions differing between the B10 and NOD strains other than Idd5.1, it is still possible that non-NOD alleles at unknown Idd genes not linked to Idd5.1 are still segregating in one or both strains. To control for the potential influence of such unlinked non-NOD alleles, (NOD.CAST Idd5.1 x NOD)F2 and (NOD.SWR Idd5.1 x NOD)F2 littermates were generated and tested for diabetes in a genotype-blinded manner, as they were genotyped only after becoming diabetic or after the end of the 210 day observation period (Fig. 1b and c).
We determined that NOD.CAST Idd5.1 mice were protected from diabetes as compared to mice having two doses of the NOD allele at Idd5.1 (P = 0.0002) (Fig. 1b). Even one diabetes-resistance allele in the heterozygous F2 mice was sufficient to confer significant protection from diabetes (P = 0.0017). On the other hand, two doses of the Idd5.1 segment from the SWR strain did not confer protection from diabetes and the diabetes frequency of these mice was indistinguishable from that of F2 mice having two doses of the NOD allele in the Idd5.1 region (Fig. 1c).
The expression of liCTLA-4 and ICOS mRNA and protein by splenocytes from NOD, NOD.CAST Idd5.1, NOD.SWR Idd5.1 and NOD.B10 Idd5.1 congenic mice was also examined (Fig. 2). Cells from NOD.B10 Idd5.1 congenic mice, with a diabetes-resistant Idd5.1 allele, had 4.2 fold higher liCTLA-4 mRNA compared with cells from NOD mice (Fig. 2a) (P = 1.9 × 10-7), which is consistent with our previous data (19, 21). As expected, cells from diabetes resistant NOD.CAST Idd5.1 congenic mice expressed more (13.7 fold) liCTLA-4 mRNA than NOD mice as well (Fig. 2a) (P = 1.1 × 10-9). Unexpectedly spleen cells from NOD.CAST Idd5.1 congenic mice had a 3.2-fold higher expression of liCTLA-4 mRNA than cells from NOD.B10 Idd5.1 mice (P = 5.3 × 10-6). In contrast to mice with the B10 and CAST haplotyopes, diabetes-susceptible NOD and NOD.SWR Idd5.1 mice produced equivalent amounts of liCTLA-4 mRNA (1.2 fold, P = 0.28). The increased expression of liCTLA-4 in cells from NOD.CAST Idd5.1 mice as compared with NOD.B10 Idd5.1 mice was not due to an overall increase in flCTLA-4 mRNA in NOD.CAST Idd5.1 spleen cells (Fig. 2b). The small expression differences in flCTLA-4 mRNA noted in Figure 2b were not observed in all experiments, whereas the liCTLA-4 mRNA expression differences were consistently observed in all experiments (compiled in Figure 2c after normalizing to flCTLA-4 mRNA). We also did not observe expression differences amongst the strains for the mRNA species encoding the soluble CTLA-4 isoform (mRNA formed by splicing exons 1, 2, and 4) or the exon 1/4 CTLA-4 mRNA formed by splicing exon 1 to exon 4 (data not shown). The increased expression of liCTLA-4 in cells from NOD.CAST Idd5.1 and NOD.B10 Idd5.1 congenic mice, compared with NOD and NOD.SWR Idd5.1 mice, was further confirmed at the protein level by Western blotting (Fig. 2e). In contrast to the liCTLA-4 mRNA and protein expression patterns, ICOS expression on CD4+ and CD8+ T cells as measured by cell surface staining following various stimulation conditions was not altered by the SNP in Icos exon 1 (data not shown). However, a small but consistent increase in ICOS mRNA was observed in cells from the B10 and CAST haplotypes (Fig. 2d). The ICOS mRNA difference did not correlate with the SNP in Icos exon 1, instead it correlated with the Ctla4 exon 2 SNP since the NOD and SWR ICOS mRNA levels were both slightly lower than those of B10 and CAST. These results confirm that a NOD haplotype at Ctla4, even in the context of a B10-like SNP in Icos exon 1 (the hybrid haplotype present in the SWR strain), is sufficient to cause decreased levels of liCTLA-4 mRNA and protein as compared to strains having a B10 haplotype at Ctla4. Thus, a B10-like SNP in Ctla4 exon 2 (position 77) is sufficient to increase the expression of liCTLA-4 and supports the hypothesis that this SNP, and not a SNP in Icos, causes an expression difference in liCTLA-4 and influences T1D susceptibility.
Figure 2. Differential expression of liCTLA-4 mRNA and protein in Idd5.1 congenic strains.

mRNA expression levels in NOD, NOD.SWR Idd5.1, NOD.CAST Idd5.1, and NOD.B10 Idd5.1 splenocytes were compared for (A) liCTLA-4, (B) flCTLA-4, (C) liCTLA-4 from multiple experiments normalized to flCTLA-4 mRNA for each individual mouse, and (D) flICOS. Spleen cells were activated in vivo for 90 min using 1 μg of anti-CD3 (clone 145-2C11) (A, B and D) or with one of two activation protocols (90 min/1 μg or 6 h/5 μg in vivo) (C) prior to mRNA isolation. The ΔCt for each sample was determined using the following formula: Cttest gene-CtB2M. Data are presented as the mean +/- SE. In C, ΔΔCt values were determined following the normalization of average Ct values using the following formula: CtflCTLA-4-CtliCTLA-4. Quantitative PCR data were evaluated using an unpaired T test. (E) Western blot analysis of liCTLA-4 and flCTLA-4 protein expression in spleen cells from NOD, NOD.SWR Idd5.1, NOD.CAST Idd5.1, NOD.B10 Idd5.1, and B6 mice. Lysates from cells were immunoblotted with anti-CTLA-4 antibody C-19 (Santa Cruz Biotech).
Because we observed a higher expression of liCTLA-4 mRNA in NOD.CAST Idd5.1 splenocytes compared to NOD.B10 Idd5.1 splenocytes, all exons, intronic regions encompassing donor and acceptor splice sites and branch points of the CAST and SWR Ctla4 and Icos alleles were sequenced in order to define the haplotypes in more detail. Whereas further sequencing reinforced the hypothesis that SWR is NOD-like at Ctla4 and B10-like at Icos, two and five CAST-specific exonic SNPs were found in exon 2 of Ctla4 and Icos, respectively (Fig. 1). It is notable that both CAST-specific Ctla4 SNPs (263 and 332 bp in exon 2) alter exon splicing enhancer motifs in regions that are conserved in human CTLA4 (Supplementary Figure 2) thereby providing a likely molecular basis for the enhanced production of liCTLA-4 by cells having the CAST haplotype. All of the CAST-specific Ctla4 and Icos SNPs are synonymous except for the Icos SNP at position 122 that causes an amino acid change from glutamic acid to aspartic acid. As a potential caveat to our interpretation of the sequence and genotyping data we have obtained and its correlation with gene expression and T1D frequency, it should be emphasized that we did not sequence the entirety of the Idd5.1 region present in the CAST, B10, SWR or NOD strains. Other SNPs in addition to those described here are certainly present and it is possible that they contribute to the T1D frequency and gene expression results obtained in this study. In addition, although it is clear from the NOD-like T1D frequency shown by the NOD.SWR Idd5.1 cogenic strain that the B6 allele at Icos is not sufficient to mediate T1D protection, it is possible that the B6 Icos allele could be required in conjunction with the B6 allele at Ctla4 for full Idd5.1-associated protection. Finally, an alternate explanation for the increased T1D protection present in the NOD.CAST Idd5.1 strain as compared to the NOD.B10 Idd5.1 strain is that the CAST Idd5.3 allele contributes a portion of the protection, although we believe this scenario to be unlikely (see Materials and Methods). Because of these caveats, we sought an additional method to support our hypothesis that increased expression of liCTLA-4 reduces T1D susceptibility.
Generation of liCTLA-4 transgenic mice on NOD and B6 CTLA-4-/- background
Based on the haplotype analysis and mRNA expression levels, we could clearly deduce that the A/G SNP at position 77 in Ctla4 exon 2 correlated with different levels of liCTLA-4 expression and diabetes susceptibility whereas the amino acid-changing SNP in the leader sequence of Icos did not correlate with flCTLA-4, liCTLA-4, or ICOS mRNA expression or disease. To better understand the function of liCTLA-4 in vivo, we generated two transgenic mouse models. In the first model, we created liCTLA-4 transgenic (tg) mice on the NOD background to directly test whether expression of liCTLA-4 can supplement the natural liCTLA-4 deficiency of the NOD strain and confer protection from T1D. In the second model, we generated liCTLA-4 tg x CTLA-4-/- mice to test the hypothesis that expression of liCTLA-4 in the absence of flCTLA-4 can rescue the CTLA-4-/- mice from fatal autoimmunity.
To generate these transgenic mice, cDNA encoding liCTLA-4 was cloned downstream of the CD2 promoter and 3′ UTR CD2 enhancer using the pBluescript vector (Fig. 3a), as described in the Materials and Methods section, and injected directly into NOD or B6 eggs. The presence of the liCTLA-4 transgene was detected by PCR using genomic DNA purified from tail biopsies. liCTLA-4 mRNA and protein expression was further confirmed by reverse transcription followed by quantitative PCR (Fig. 3b) and Western blotting (Fig. 3c), respectively. We did not detect any differences in the expression of flCTLA-4 mRNA or protein.
Figure 3. liCTLA-4 mRNA and protein expression in liCTLA-4 transgenic mice.

(A) Schematic structure of the liCTLA-4 vector construct. liCTLA-4 was subcoloned into the EcoR1 site of pBluescript vector encoding 5′ CD2 promoter and 3′ CD2 enhancer. (B) liCTLA-4 and flCTLA-4 mRNA expression was compared between non-transgenic and liCTLA-4 transgenic mice. Spleen cells were activated in vivo for 90 minutes using 1 μg of anti-CD3 (clone 145-2C11) prior to mRNA isolation. The ΔCt for each sample of spleen cells was determined using the following formula: CtliCTLA-4-CtB2M. Data are presented as the mean ΔCt +/- SE. Quantitative PCR data were evaluated using an unpaired T test. (C) For Western analysis, 2 × 106 spleen cells were immunoblotted with anti-CTLA-4 antibody (C-19, Santa Cruz Biotech).
NOD liCTLA-4 tg mice are protected from autoimmune diabetes
To determine if liCTLA-4 transgene expression alters the cellular phenotype of NOD mice, we examined the expression of CD25, CD62L, CD44, CD69, and CD28, ICOS and flCTLA-4 in naïve and activated T cells by flow cytometry. We were particularly interested to know whether liCTLA-4 transgenic expression would alter the expression of flCTLA-4 and ICOS. No differences in cell subsets and expression of activation markers were observed in the thymus, spleen and lymph nodes of transgenic mice and their non-transgenic littermate controls (Supplemental Figure 3). The lack of any differences is notable since overexpression of the negative-signaling CTLA-4 cytoplasmic domain in T cells might have led to a substantial reduction in T cell signaling thereby causing an alteration in T cell development. However, these results, together with those from studies in which flCTLA-4 and various CTLA-4 mutants were expressed in CTLA-4 sufficient and CTLA-4 KO mice (38-40), suggest that engagement and phosphorylation of transgenic CTLA-4 isoforms is relatively physiologic, possibly due to a limiting amount of one or more signaling molecules required to recruit CTLA-4.
NOD and NOD liCTLA-4 lymph node T cells were tested for proliferation and cytokine production following stimulation with anti-CD28 and different concentrations of anti-CD3. NOD liCTLA-4 tg mice showed significantly lower proliferation and lower production of the pro-inflammatory cytokines IL-17 and IFN-γ compared to their non-transgenic littermates (Fig. 4a). We did not detect any IL-10 or IL-4 production from the activated lymph node cells (data not shown). These data are reminiscent of the results obtained when liCTLA-4 was ectopically expressed in T cells resulting in reduced proliferation and IFN-γ production following stimulation (30). Also similar are the results from the PYAA mutant CTLA-4 transgene, which encodes a CTLA-4 variant that fails to bind B7 molecules and is likely to be functionally equivalent to the liCTLA-4 isoform, where a reduced proliferative response was observed when it was expressed in the BALB/c CTLA-4-sufficient background (38).
Figure 4. T cell proliferation, IL-17 and IFN-γ production, and diabetes frequency are reduced in NOD liCTLA-4 transgenic mice.

(A) Lymph node T cells from NOD liCTLA-4 tg and non-tg littermates were stimulated with the indicated concentrations of anti-CD3 antibody and 1 μg /ml of anti-CD28. T cell proliferation was measured with a [3H] thymidine incorporation assay and the data are presented as the mean cpm of triplicate wells. Culture supernatants were assayed by ELISA in triplicate for detection of IFN-γ and IL-17. IL-10 and IL-4 were not detected in the supernatants (data not shown). Data represent the average of three independent experiments with at least two mice in each group for each experiment. A 95% level of confidence was used to calculate the error bars. (B) NOD liCTLA-4 tg mice are protected from diabetes when compared to their non-transgenic littermate controls (P = 0.0115) The frequency of diabetes in NOD liCTLA-4 tg mice is similar to that in NOD.B10 Idd5.1 congenic mice (C), which are protected as compared to NOD mice (P = 0.0095). The frequency of diabetes was compared between strains with the Kaplan-Meier log-rank test.
The frequency of spontaneous diabetes in NOD liCTLA-4-tg mice was compared to that of non-transgenic littermates; significant protection from disease was conferred by the transgene (Fig. 4b, P = 0.01). This level of protection was not complete and was nearly identical to the disease protection observed in the NOD.B10 Idd5.1 congenic strain compared to a NOD cohort (Fig. 4c, P = 0.01). These results suggest that increased expression of liCTLA-4 in NOD mice can compensate for the effects caused by the inherent genetic deficiency of liCTLA-4 in this strain. It also strengthens our hypothesis that the Ctla4 exon 2 variant at residue 77 is sufficient to explain the disease susceptibility mediated by Idd5.1.
Expression of liCTLA-4 inhibits early activation of T cells in CTLA-4-/- mice
The NOD liCTLA-4 tg mice allowed us to explore the function of liCTLA-4 in the presence of flCTLA-4. However, we also wanted to determine whether liCTLA-4 could confer disease protection in the absence of flCTLA-4. Mice deficient in CTLA-4 develop massive lymphoproliferation and fatal multiorgan tissue damage. The lymphocytes in these mice spontaneously acquire an activated phenotype with increased expression of CD25, CD69, CD44, ICOS and CD28 and an increase in the number of CD62L low cells compared to WT mice (39, 41, 42). Therefore, we compared B6 CTLA-4-/- mice with and without transgenic expression of liCTLA-4; for this purpose we generated liCTLA-4 transgenic mice directly on the B6 background and crossed these mice onto the CTLA-4-/- background. CTLA-4-/- mice generally were smaller in size than their littermates. The frequencies of thymic CD4 and CD8 single positive cells were not different among CTLA-4-/-, CTLA-4-/- liCTLA-4 tg and WT mice (data not shown), suggesting that thymocyte development is not altered in CTLA-4-/- and CTLA-4-/- liCTLA-4 tg mice. Figure 5 shows a summary of the mean fluorescence intensities (MFI) of CD25, CD69, CD62L, CD44, ICOS and CD28 on CD4+ T cells from CTLA-4-/-, CTLA-4-/- liCTLA-4 tg and WT mice at 3-7 weeks of age. Representative FACS profiles of these data are in Supplemental Figure 4. Activation marker expression was higher on T cells from CTLA-4-/- mice than from WT mice (Fig. 5). In contrast, T cells from CTLA-4-/- liCTLA-4 tg mice appeared more like WT T cells with respect to expression of T cell activation markers, although spontaneous activation and expression of cell surface activation markers on T cells in the CTLA-4-/- liCTLA-4 tg mice varied as the mice aged. The MFIs of CD25, CD69, CD44 and CD28 on CD4+ T cells from CTLA-4-/- liCTLA-4 tg mice were not significantly different from the WT mice (Fig. 5). However, expression of CD62L and ICOS on CD4+ T cells from CTLA-4-/- liCTLA-4 tg mice was significantly different compared to the expression in WT mice (P = 0.0012 and 0.0031, respectively), which likely represents an underlying partial activation of the transgenic T cells. These data indicate that liCTLA-4 expression reduces the upregulation of positive costimulatory molecules such as CD28 and ICOS, thereby reducing the early T cell activation that occurs in CTLA-4-/- mice. Importantly, CD28 and ICOS levels on T cells from wild-type B6 mice were not altered by expression of the liCTLA-4 tg (data not shown).
Figure 5. CTLA-4-/- liCTLA-4 tg T cells share most cellular phenotypes with WT T cells.

Average MFI of the activation markers CD25, CD69, CD62L, CD44, ICOS and CD28, in CTLA-4-/-, CTLA-4-/- liCTLA-4 tg, wild type mice (3-7 weeks of age, both sexes). Note that group sizes are not equal for each activation marker since not all antibody reagents were available each day that cells from individual mice were evaluated. P values shown in the figure were determined using the unpaired T test.
To compare the function of WT, CTLA-4-/-, and CTLA-4-/- liCTLA-4 tg T cells, proliferation and cytokine secretion assays were performed (Fig. 6). Since CTLA-4 deficient mice become moribund by 6 weeks of age, responses to anti-CD3 and anti-CD28 stimulation were examined using mice from each strain at three weeks of age. Although the proliferation of CTLA-4-/- liCTLA-4 tg T cells was comparable to that of CTLA-4-/- and WT T cells, there were dramatic alterations in the cytokine profiles of the activated T cells. As described previously by others (38), we confirmed that WT T cells from B6 mice produced both IFN-γ and IL-4 whereas B6 CTLA-4-/- T cells produced copious amounts of IFN-γ and low levels of IL-4 (Fig. 6). In contrast, T cells from B6 CTLA-4-/- liCTLA-4 transgenic mice displayed a dramatic reversal of the CTLA-4-/- T cell cytokine pattern; IFN-γ was essentially undetectable whereas IL-4 was secreted without stimulation and increased levels were produced in response to as little as 0.1 μg/ml of anti-CD3 (Fig 6). Masteller et al. (40) observed a similar IL-4 skewing when the tailless CTLA-4 variant was used as a transgene in CTLA-4-/- mice on the B6 background. IL-2 and IL-10 production differed from IFN-γ and IL-4 in that similar responses were made by CTLA-4-/- and CTLA-4-/- liCTLA-4 tg T cells which differed from wild type T cells. Of particular note however was the increased production of IL-17 from CTLA-4-/- liCTLA-4 tg T cells at the highest level of anti-CD3 and anti-CD28 activation, which was reproducibly higher than the production by CTLA-4-/- T cells.
Figure 6. Decreased IFN-γ and IL-2 with increased IL-17, IL-4, and IL-10 production by CTLA-4-/- liCTLA-4 tg cells.

Lymph node T cells from male and female 3-week-old CTLA-4-/-, CTLA-4-/- liCTLA-4 tg and WT mice were stimulated with the indicated concentrations of anti-CD3 and 1 μg /ml of anti-CD28 antibody. T cell proliferation was measured by [3H]thymidine incorporation and represented as mean CPM of triplicates (upper left). Culture supernatants were assayed by ELISA in triplicate for detection of IFN-γ, IL-4, IL-10, IL-2, and IL-17 after 48 hours. Data are representative of 3 independent experiments with 4 mice in each group. A 95% level of confidence was used to calculate the error bars.
liCTLA-4 rescues CTLA-4-deficient mice from multiorgan autoimmunity and early death
CTLA-4-/- mice develop lymphoproliferative disease with massive mononuclear cell infiltration and tissue destruction of multiple organs, e.g. heart, lung, pancreas and liver resulting in death between 3 and 6 weeks of age (39, 41-44). In our colony, all of the CTLA-4-/- mice died by 50 days of age (Fig. 7) whereas CTLA-4-/- liCTLA-4 tg mice were partially rescued from this early death, i.e., 26.7% of CTLA-4-/- liCTLA-4 tg mice survived more than 50 days (p=2.2 × 10-5, compared to CTLA-4-/- mice, Fig. 7). Heart, lung, and liver samples were harvested from 4-6 weeks old WT (n=5), CTLA-4-/- (n=3) and CTLA-4-/- liCTLA-4 tg mice (n=6) and examined histologically (Fig. 8). No mononuclear cell infiltrates were observed in the organs from WT mice (Fig. 8a, d and g), while CTLA-4-/- mice had severe myocarditis (Fig. 8c), increased bronchus-associated lymphoid tissue (BALT) (Fig. 8f), and moderate to severe hepatitis (Fig. 8i). In contrast, tissues from CTLA-4-/- liCTLA-4 tg mice showed milder inflammation and BALT (Fig. 8b, e and h). Interestingly, older CTLA-4-/- liCTLA-4 tg mice (15 - 29 week-old) that survived early lethality had no, or very mild, inflammation in the heart, lung and liver when they were euthanized (data not shown).
Figure 7. liCTLA-4 prevents early lethality from CTLA-4 deficient mice.
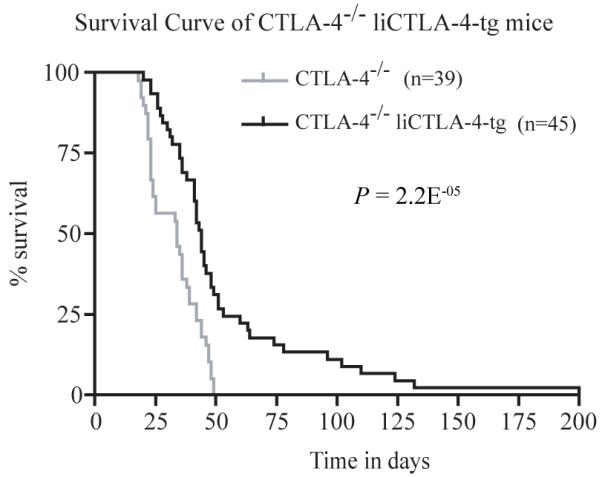
Survival curves for CTLA-4-/- mice (grey line) and CTLA-4-/- liCTLA-4 tg mice (black line). Survival was compared with the Kaplan-Meier log rank test.
Figure 8. Histopathological findings in WT, CTLA-4-/- liCTLA-4 tg and CTLA-4-/- mice reveals that CTLA-4-/- liCTLA-4-tg mice have an intermediate phenotype.

Representative findings in heart (A, B and C), lung (D, E and F), and liver (G, H and I) tissues are shown. WT mice show no inflammation; CTLA-4-/- liCTLA-4-tg mice have mild myocarditis, hepatitis and increased BALT. CTLA-4-/- mice have severe myocarditis, hepatitis and large BALT. Hematoxylin and eosin, original magnifications: A-C, G-I, 160X; D-F, 80X.
Discussion
In this study, we have performed haplotype mapping of Idd5.1 and have generated new Idd5.1 congenic and NOD liCTLA-4 tg transgenic mice to address the genetic and functional role of Ctla4 as the causative gene in Idd5.1 that regulates susceptibility to T1D. Through haplotype analysis, we discovered that the SWR haplotype is NOD-like at most of the sequence polymorphisms in and near Ctla4, including the exon 2 SNP at residue 77 that is associated with changes in liCTLA-4 expression, but is B10-like at Icos, including a SNP in Icos exon 1 that causes a non-conservative amino acid change in the putative leader sequence. Since alterations in the expression of either liCTLA-4 or ICOS could conceivably alter the T1D frequency, the hybrid SWR haplotype allowed us to test whether the T1D protection conferred by the B10 Idd5.1 haplotype would be lost or retained in the SWR Idd5.1 haplotype. In a frequency study, the SWR Idd5.1 haplotype was shown to be unable to protect NOD mice from T1D, clearly in support of the hypothesis that increased liCTLA-4 expression in the B10 haplotype is required for T1D protection.
In contrast to the SWR haplotype, the CAST Idd5.1 haplotype was shown to be protective, which is consistent with its high liCTLA-4 expression (Fig. 2) and again supports the hypothesis that increased liCTLA-4 protects from T1D. However, since with further sequencing, we discovered that the CAST haplotype had additional CAST-specific SNPs in Ctla4 and Icos, our study took on an added dimension. We propose that the additional 2 CAST-specific SNPs in exon 2 of Ctla4 are responsible for even higher expression of liCTLA-4 as compared to the protected B10 haplotype (Fig. 2a and 2c) and that the molecular basis for this increase is probably due to alteration of exon splicing enhancer motifs (Supplemental Figure 2). The selection and retention of a second, distinct haplotype that produces high levels of liCTLA-4 suggests that an important function for this isoform has evolved in mice. On a practical level, the CAST allele at Ctla4 is a tool, that in conjunction with the B10 and NOD alleles, creates a hierarchy of liCTLA-4 expression levels with over a 10-fold difference at the mRNA level between the highest and lowest producing alleles. Although the CAST and B10 Idd5.1 haplotypes have yet to be compared directly in a T1D frequency experiment, the increased expression of liCTLA-4 in activated splenocytes from NOD.CAST Idd5.1 congenic mice, as compared with splenocytes from NOD.B10 Idd5.1 mice, is correlated with an increased level of protection from T1D by the CAST versus B10 Idd5.1 alleles, since (NOD.CAST Idd5.1 x NOD) F1 mice remain protected from T1D (Fig. 1b) whereas (NOD.B10 Idd5.1 x NOD) F1 are not (31). Future experiments addressing the downstream molecular and cellular events determined by differing levels of liCTLA-4 should benefit from a comparison of Idd5.1 congenic strains. The apparent ability of exonic sequences to alter splicing preferences in the case of liCTLA-4 can also be made use of to model the expression changes by developing exon 2 knock-in mice having the CAST, B10, or NOD exon 2 SNPs.
Since in addition to Ctla4 and Icos, Pard3b and Nrp2 are within the Idd5.1 interval, we generated liCTLA-4 transgenic mice on the NOD background to test the hypothesis that increased liCTLA-4 expression is sufficient to explain the protective effect of the B10 Idd5.1 allele on the frequency of type 1 diabetes. Indeed, when liCTLA-4 is expressed transgenically, it confers protection from diabetes as compared to non-transgenic littermates and with an incidence remarkably similar to NOD.B10 Idd5.1 congenic mice (Fig. 4). Together with the haplotype analyses in the congenic strains discussed above, these positive results from mice with transgenic overexpression of liCTLA-4 make it unlikely that non-NOD alleles at any of the other Idd5.1 region genes are required to regulate the diabetes phenotype.
liCTLA-4 has been hypothesized to have a major role in the downregulation of T-cell responses (19, 30). Our present data demonstrate that NOD T-cells secrete high levels of IL-17 upon anti-CD3 stimulation, which is reduced by expression of the liCTLA-4 transgene (Fig. 4). IFN-γ levels are also decreased in the liCTLA-4 transgenic NOD mice. In addition to increased IL-17 production by NOD T cells, it has also been hypothesized that NOD mice have a defect in the regulatory T cell repertoire resulting from abnormalities in thymic selection (45, 46). Since liCTLA-4 has been shown to be highly expressed in the activated/memory T cells of diabetes resistant strains (21, 30), higher levels of liCTLA-4 may function to reduce effector and memory cell function presumably by raising the activation threshold of such cells. The naturally high expression of liCTLA-4 on activated/memory T cells of mice having a resistance allele at Ctla4 may serve to keep the immune system from responding following exposure to weak antigens or low affinity self-antigens, thus providing an important checkpoint for inhibiting T cell activation and maintaining self-tolerance.
We have also demonstrated that transgenic expression of liCTLA-4 even in the absence of flCTLA-4 is capable of negatively regulating T cell activation in vivo. However, a functional signaling domain alone is not sufficient to completely reverse the autoimmunity and inflammation present in CTLA4-/- mice (Figs. 7 and 8). Chikuma et al (38) reached a similar conclusion when they studied CTLA4-/- mice receiving a CTLA-4 transgene mutated in the B7 binding domain. CTLA-4-/- T cells are spontaneously activated and readily secrete massive amounts of cytokines such as IFN-γ□ and IL-10 upon further activation with anti-CD3 and anti-CD28. Expression of liCTLA-4 inhibited the production of IFN-γ but not IL-10 or IL-17 from the CTLA-4-/- T cells. Interestingly, we observed a reproducible, increased production of IL-17 by CTLA-4-/- liCTLA-4-tg T cells as compared to CTLA-4-/- T cells. Recently, McGeachy et al (47) demonstrated that cells co-producing IL-17 and IL-10 acquire a non-pathogenic phenotype that could potentially confer these cells with regulatory function. This raises the possibility that the appearance of IL-17 producing T cells that co-produce IL-10 in the CTLA-4-/- liCTLA-4 tg mice may be regulatory and not pathogenic T cells, thereby increasing the number of cells capable of restraining pathogenic effectors in the CTLA-4-/- liCTLA-4 tg mice. Alternatively, there is some support for the hypothesis that there is antagonism between IFN-γ (Th1) and IL-17 (Th17) producing cells (48), suggesting that the inhibition of IFN-γ by liCTLA-4 expression in CTLA4-/- T cells could lead to the expansion of Th17 cells. Since the decrease in IFN-γ production by cells from CTLA-4-/- liCTLA-4 tg is enough to reduce inflammation and prolong the survival of these mice, this hypothesis suggests that the inflammatory phenotype of CTLA-4-/- mice is partly mediated by Th1 cells.
As detailed above, in the NOD liCTLA-4 transgenic T cells where liCTLA-4 is expressed in the presence of flCTLA-4 as well as the other isoforms of CTLA-4, IFN-γ and IL-17 are both inhibited. Considering these data, one might speculate that the inhibition of IFN-γ and increased production IL-17 in CTLA4-/- liCTLA-4 tg mice is due to the non-physiologic situation in which liCTLA-4 is present in the absence of flCTLA-4. Thus, the lack of complete protection and the presence of residual inflammation observed in the CTLA-4-/- liCTLA-4 tg mice may be due to the increased number of IL-17-producing Th17 cells even though disease in CTLA-4-/- mice is possibly Th1-driven.
liCTLA-4 has been reported to heterodimerize with flCTLA-4 and recruit SHP-2 to dephosphorylate the TcRς chain (30). However, when liCTLA-4 is expressed in the absence of flCTLA-4, it interacts with the TcRς chain independently of flCTLA-4 and the recruitment of SHP-2 (30). The inability to recruit SHP-2 when liCTLA-4 and flCTLA-4 are expressed separately could pose an impediment to the complete negative signaling of T cell responses by flCTLA-4 when liCTLA-4 levels are low because of genetically-determined expression levels. The formation of liCTLA-4 and flCTLA-4 heterodimers could stabilize the lattice resulting in an increased affinity in binding of SHP-2 to the TcRς complex. In the present study undertaking a detailed clinical and histopathological analysis on a large cohort of animals, we observed that CTLA4-/- liCTLA-4 tg mice were only partially protected from the lethal autoimmune lymphoproliferative disease observed in CTLA-4-/- mice. This partial protection from disease is consistent with the results of Chikuma et al (38), who generated mice transgenic for a point-mutated CTLA-4 lacking B7 binding capacity and demonstrated that this ligand-nonbinding mutant CTLA-4 delayed lethal lymphoproliferative disorder of CTLA-4-/- mice (38). Overall, our combined results indicate that CTLA-4 needs to bind to B7 molecules to completely abrogate the abnormal T cell activation present in CTLA-4-/- mice, however, the signaling domain alone is sufficient to provide some protection from the activated phenotype present in CTLA-4-/- mice. These results have an interesting parallel with the results from Masteller et al (40) where only partial protection from disease in CTLA-4-/- mice was provided by a transgene encoding a CTLA-4 molecule lacking the signaling domain. Thus, perhaps not surprisingly, both the B7 binding IgV domain and the signaling domain in the cytoplasmic tail are critical for the full functioning of the CTLA-4 molecule.
Overall, our present data suggest that both the ligand-independent and the ligand binding, B7-dependent, CTLA-4 isoforms are required for delivering maximal negative signal into T cells. These previous findings (38-40) support our current data that when liCTLA-4 is expressed in wild type NOD mice where flCTLA-4 is also present, liCTLA-4 is able to inhibit T-cell expansion and production of both inflammatory cytokines, IFN-γ and IL-17. In contrast, expression of liCTLA-4 in CTLA-4-/- mice only inhibited production of IFN-γ.
In this paper, using multiple approaches, we have determined that the SNP in exon 2 (position 77) of Ctla4 is the most likely SNP causing the differential expression of the liCTLA-4 isoform. Formal proof that the exon 2 SNP of Ctla4 alone causes the full protective effect of Idd5.1, rather than a model in which variation in ICOS or another gene within the Idd5.1 interval contributes a portion of the protective effect from T1D, will require the knock-in of the NOD allele at residue 77 of exon 2 into the B6 Ctla4 allele with subsequent backcrossing of the Idd5.1 region to the NOD background for the analysis of T1D. For example, until such an experiment is done it remains a possibility that the B6 Icos allele could be required in conjunction with the B6 allele at Ctla4 for full Idd5.1-associated protection to be observed, even though we have demonstrated that the B6 Icos allele in the absence of the B6 Ctla4 allele is not sufficient on its own to mediate T1D protection. If the residue 77 SNP is the sole cause of protection for the B6 allele of Idd5.1, this NOD.B6 Idd5.1 residue 77 knockin congenic mouse strain will have a T1D frequency equal to that of the NOD parental strain. If the knockin strain still has protection from T1D as compared to the NOD strain, then another B6-derived gene within the interval contributes to the Idd5.1-mediated T1D effect. Finally, we have shown by congenic and transgenic approaches that higher expression of liCTLA-4 in NOD mice correlates with protection from T1D and that overexpression of liCTLA-4 in CTLA-4-/- mice can partially rescue them from early lethality. Overall, our data suggest liCTLA-4 contributes to preventing activation and maintaining tolerance to self-antigens thus, preventing autoimmunity.
Supplementary Material
Acknowledgments
We thank David Lee for technical assistance.
Abbreviations used in this paper
- T1D
type 1 diabetes
- Idd
insulin-dependent diabetes
- T1D
type 1 diabetes
- flCTLA-4
full length CTLA-4
- liCTLA-4
ligand independent CTLA-4
- tg
transgenic
- WT
wild type
- MFI
mean fluorescence intensity
- SHP-2
Src homology region 2 domain-containing phosphatase 1
- Ct
threshold cycle
- SNP
single nucleotide polymorphism
- ACADL
long-chain acyl-coenzyme A dehydrogenase
Footnotes
LSW is supported by grants from the Juvenile Diabetes Research Foundation, the Wellcome Trust and the National Institutes of Health (P01 AI39671). LSW is a JDRF/WT Principal Research Fellow. VKK is supported by grants from the NIH RO1 AI044880, PO1 AI 56299, PO1 AI 39671, NS046414 and JDRF center for Immunological tolerance at Harvard. M. A. is supported by a postdoctoral fellowship from the Juvenile Diabetes Research Foundation. Cambridge Institute for Medical Research (CIMR) is in receipt of a Wellcome Trust Strategic Award (079895). The availability of NOD congenic mice through the Taconic Farms Emerging Models Program has been supported by grants from the Merck Genome Research Institute, NIAID, and the JDRF.
Disclosures
The authors have no conflict of interest.
hese two authors contributed equally to this work.
References
- 1.Leiter EH. Nonobese diabetic mice and the genetics of diabetes susceptibility. Curr Diab Rep. 2005;5:141–148. doi: 10.1007/s11892-005-0042-z. [DOI] [PubMed] [Google Scholar]
- 2.Maier LM, Wicker LS. Genetic susceptibility to type 1 diabetes. Curr Opin Immunol. 2005;17:601–608. doi: 10.1016/j.coi.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AA, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JM, Guja C, Ionescu-Tirgoviste C, Simmonds MJ, Heward JM, Gough SC, Dunger DB, Wicker LS, Clayton DG. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, Jones R, Ring SM, McArdle W, Pembrey ME, Strachan DP, Dunger DB, Twells RC, Clayton DG, Todd JA. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet. 2005;76:773–779. doi: 10.1086/429843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, Barratt BJ, Guja C, Ionescu-Tirgoviste C, Savage DA, Dunger DB, Widmer B, Strachan DP, Ring SM, Walker N, Clayton DG, Twells RC, Gough SC, Todd JA. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes. 2004;53:3020–3023. doi: 10.2337/diabetes.53.11.3020. [DOI] [PubMed] [Google Scholar]
- 6.Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, Gwilliam R, Takeuchi F, McLaren WM, Holmes GK, Howdle PD, Walters JR, Sanders DS, Playford RJ, Trynka G, Mulder CJ, Mearin ML, Verbeek WH, Trimble V, Stevens FM, O’Morain C, Kennedy NP, Kelleher D, Pennington DJ, Strachan DP, McArdle WL, Mein CA, Wapenaar MC, Deloukas P, McGinnis R, McManus R, Wijmenga C, van Heel DA. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert AP, Gillespie KM, Thomson G, Cordell HJ, Todd JA, Gale EA, Bingley PJ. Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J Clin Endocrinol Metab. 2004;89:4037–4043. doi: 10.1210/jc.2003-032084. [DOI] [PubMed] [Google Scholar]
- 8.Cucca F, Lampis R, Congia M, Angius E, Nutland S, Bain SC, Barnett AH, Todd JA. A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Hum Mol Genet. 2001;10:2025–2037. doi: 10.1093/hmg/10.19.2025. [DOI] [PubMed] [Google Scholar]
- 9.Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet. 1995;9:284–292. doi: 10.1038/ng0395-284. [DOI] [PubMed] [Google Scholar]
- 10.Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, Concannon P, Gharani N, McCarthy MI, Olavesen MG, McCormack R, Guja C, Ionescu-Tirgoviste C, Undlien DE, Ronningen KS, Gillespie KM, Tuomilehto-Wolf E, Tuomilehto J, Bennett ST, Clayton DG, Cordell HJ, Todd JA. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes. 2004;53:1884–1889. doi: 10.2337/diabetes.53.7.1884. [DOI] [PubMed] [Google Scholar]
- 11.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, Chen SL, Rosa R, Cumiskey AM, Serreze DV, Gregory S, Rogers J, Lyons PA, Healy B, Smink LJ, Todd JA, Peterson LB, Wicker LS, Santamaria P. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet. 2006;38:479–483. doi: 10.1038/ng1766. [DOI] [PubMed] [Google Scholar]
- 13.Kahles H, Ramos-Lopez E, Lange B, Zwermann O, Reincke M, Badenhoop K. Sex-specific association of PTPN22 1858T with type 1 diabetes but not with Hashimoto’s thyroiditis or Addison’s disease in the German population. Eur J Endocrinol. 2005;153:895–899. doi: 10.1530/eje.1.02035. [DOI] [PubMed] [Google Scholar]
- 14.Qu H, Tessier MC, Hudson TJ, Polychronakos C. Confirmation of the association of the R620W polymorphism in the protein tyrosine phosphatase PTPN22 with type 1 diabetes in a family based study. J Med Genet. 2005;42:266–270. doi: 10.1136/jmg.2004.026971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng W, She JX. Genetic association between a lymphoid tyrosine phosphatase (PTPN22) and type 1 diabetes. Diabetes. 2005;54:906–908. doi: 10.2337/diabetes.54.3.906. [DOI] [PubMed] [Google Scholar]
- 16.Ladner MB, Bottini N, Valdes AM, Noble JA. Association of the single nucleotide polymorphism C1858T of the PTPN22 gene with type 1 diabetes. Hum Immunol. 2005;66:60–64. doi: 10.1016/j.humimm.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton-Williams EE, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, Slattery RM. Transgenic rescue implicates beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (NOD) mice. Proc Natl Acad Sci U S A. 2001;98:11533–11538. doi: 10.1073/pnas.191383798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Razavi R, Chan Y, Afifiyan FN, Liu XJ, Wan X, Yantha J, Tsui H, Tang L, Tsai S, Santamaria P, Driver JP, Serreze D, Salter MW, Dosch HM. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell. 2006;127:1123–1135. doi: 10.1016/j.cell.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 20.Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, Clayton DG, Wicker LS, Todd JA. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 21.Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL, Howson JM, Smink LJ, Kingsnorth A, Lyons PA, Gregory S, Rogers J, Todd JA, Peterson LB. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol. 2004;173:164–173. doi: 10.4049/jimmunol.173.1.164. [DOI] [PubMed] [Google Scholar]
- 22.Lamhamedi-Cherradi SE, Boulard O, Gonzalez C, Kassis N, Damotte D, Eloy L, Fluteau G, Levi-Strauss M, Garchon HJ. Further mapping of the Idd5.1 locus for autoimmune diabetes in NOD mice. Diabetes. 2001;50:2874–2878. doi: 10.2337/diabetes.50.12.2874. [DOI] [PubMed] [Google Scholar]
- 23.Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes. 2000;49:1744–1747. doi: 10.2337/diabetes.49.10.1744. [DOI] [PubMed] [Google Scholar]
- 24.Ligers A, Xu C, Saarinen S, Hillert J, Olerup O. The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol. 1999;97:182–190. doi: 10.1016/s0165-5728(99)00072-7. [DOI] [PubMed] [Google Scholar]
- 25.Heward JM, Allahabadia A, Armitage M, Hattersley A, Dodson PM, Macleod K, Carr-Smith J, Daykin J, Daly A, Sheppard MC, Holder RL, Barnett AH, Franklyn JA, Gough SC. The development of Graves’ disease and the CTLA-4 gene on chromosome 2q33. J Clin Endocrinol Metab. 1999;84:2398–2401. doi: 10.1210/jcem.84.7.5820. [DOI] [PubMed] [Google Scholar]
- 26.Braun J, Donner H, Siegmund T, Walfish PG, Usadel KH, Badenhoop K. CTLA-4 promoter variants in patients with Graves’ disease and Hashimoto’s thyroiditis. Tissue Antigens. 1998;51:563–566. doi: 10.1111/j.1399-0039.1998.tb02993.x. [DOI] [PubMed] [Google Scholar]
- 27.Donner H, Braun J, Seidl C, Rau H, Finke R, Ventz M, Walfish PG, Usadel KH, Badenhoop K. Codon 17 polymorphism of the cytotoxic T lymphocyte antigen 4 gene in Hashimoto’s thyroiditis and Addison’s disease. J Clin Endocrinol Metab. 1997;82:4130–4132. doi: 10.1210/jcem.82.12.4406. [DOI] [PubMed] [Google Scholar]
- 28.Seidl C, Donner H, Fischer B, Usadel KH, Seifried E, Kaltwasser JP, Badenhoop K. CTLA4 codon 17 dimorphism in patients with rheumatoid arthritis. Tissue Antigens. 1998;51:62–66. doi: 10.1111/j.1399-0039.1998.tb02947.x. [DOI] [PubMed] [Google Scholar]
- 29.Holopainen P, Arvas M, Sistonen P, Mustalahti K, Collin P, Maki M, Partanen J. CD28/CTLA4 gene region on chromosome 2q33 confers genetic susceptibility to celiac disease. A linkage and family-based association study. Tissue Antigens. 1999;53:470–475. doi: 10.1034/j.1399-0039.1999.530503.x. [DOI] [PubMed] [Google Scholar]
- 30.Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, Peterson LB, Hafler DA, Freeman GJ, Sharpe AH, Wicker LS, Kuchroo VK. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity. 2004;20:563–575. doi: 10.1016/s1074-7613(04)00110-4. [DOI] [PubMed] [Google Scholar]
- 31.Hunter K, Rainbow D, Plagnol V, Todd JA, Peterson LB, Wicker LS. Interactions between Idd5.1/Ctla4 and other type 1 diabetes genes. J Immunol. 2007;179:8341–8349. doi: 10.4049/jimmunol.179.12.8341. [DOI] [PubMed] [Google Scholar]
- 32.Morahan G, McClive P, Huang D, Little P, Baxter A. Genetic and physiological association of diabetes susceptibility with raised Na+/H+ exchange activity. Proc Natl Acad Sci U S A. 1994;91:5898–5902. doi: 10.1073/pnas.91.13.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brodnicki TC, Quirk F, Morahan G. A susceptibility allele from a non-diabetes-prone mouse strain accelerates diabetes in NOD congenic mice. Diabetes. 2003;52:218–222. doi: 10.2337/diabetes.52.1.218. [DOI] [PubMed] [Google Scholar]
- 34.McAleer MA, Reifsnyder P, Palmer SM, Prochazka M, Love JM, Copeman JB, Powell EE, Rodrigues NR, Prins JB, Serreze DV, Delarato NH, Wicker LS, Peterson LB, J SN, Todd JA, Leiter EH. Crosses of NOD mice with the related NON strain. A polygenic model for IDDM. Diabetes. 1995;44:1186–1195. doi: 10.2337/diab.44.10.1186. [DOI] [PubMed] [Google Scholar]
- 35.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandelbrot DA, McAdam AJ, Sharpe AH. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) J Exp Med. 1999;189:435–440. doi: 10.1084/jem.189.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greve B, Vijayakrishnan L, Kubal A, Sobel RA, Peterson LB, Wicker LS, Kuchroo VK. The diabetes susceptibility locus Idd5.1 on mouse chromosome 1 regulates ICOS expression and modulates murine experimental autoimmune encephalomyelitis. J Immunol. 2004;173:157–163. doi: 10.4049/jimmunol.173.1.157. [DOI] [PubMed] [Google Scholar]
- 38.Chikuma S, Abbas AK, Bluestone JA. B7-independent inhibition of T cells by CTLA-4. J Immunol. 2005;175:177–181. doi: 10.4049/jimmunol.175.1.177. [DOI] [PubMed] [Google Scholar]
- 39.Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol. 1999;162:5784–5791. [PubMed] [Google Scholar]
- 40.Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural analysis of CTLA-4 function in vivo. J Immunol. 2000;164:5319–5327. doi: 10.4049/jimmunol.164.10.5319. [DOI] [PubMed] [Google Scholar]
- 41.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 42.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 43.Chambers CA, Cado D, Truong T, Allison JP. Thymocyte development is normal in CTLA-4-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9296–9301. doi: 10.1073/pnas.94.17.9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–5820. [PubMed] [Google Scholar]
- 45.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 46.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 47.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 48.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.