

Fluorescent imaging is becoming an increasingly important technology for examining the real-time localizations of biologically relevant molecules within model organisms such as the zebrafish. Such imaging often requires the use of 2′,7′-dichlorofluorescein (DCF, Scheme 1), 2′,7′-difluorofluorescein (Oregon Green), or one of their derivatives, since each offers pH insensitivity, compact size, and high quantum yield.[1,2] However, the negatively charged carboxyl groups in these compounds can nonspecifically interact with positively charged biomolecules. Furthermore, the carboxyl group might be responsible when inherently cell-permeable molecules become impermeable following conjugation to fluorescein.
Scheme 1.

Fluorescein derivatives.
Lindqvist and co-workers showed that the carboxyl group of these compounds was not essential for fluorescence emission.[3] This observation was recently corroborated by the Nagano group at the University of Tokyo, who replaced fluorescein’s carboxyl group with a methyl group to form a compound, Tokyo Green, that exhibited no loss of quantum yield.[4] A similar methyl substitution was carried out by the Peterson group at The Pennsylvania State University, this time with Oregon Green,[5] to produce Pennsylvania Green.[6] In live human cells, Pennsylvania Green was more fluorescent than Tokyo Green due to its robust pH insensitivity.[6] While Pennsylvania Green is an excellent fluorescent probe, the synthesis of its conjugation-amenable derivatives is a lengthy process, requiring ten linear steps.[6] In this report, we describe the scalable and concise synthesis of two tissue-permeable DCF derivatives, Pittsburgh Green and Pittsburgh Yellowgreen. Pittsburgh Yellowgreen possesses both the favorable fluorescent and solubility properties of Pennsylvania Green and a “handle” that can be conjugated to target biomolecules.
We envisioned that the replacement of the carboxyl group of DCF with a hydroxymethyl group, rather than a methyl group, would prove to be superior in those instances in which nonspecific hydrophobic interactions and low water solubility are detrimental.[7] Moreover, such compounds could be synthetically more accessible than Tokyo Green or Pennsylvania Green, and conjugation ready via their primary hydroxy groups.
Previously we reported the synthesis of nonfluorescent compounds 1 and 2, and fluorescent compound 3 in a different context, but we now wish to emphasize that these transformations are efficient and produce crystalline solids, thus facilitating large-scale synthesis (Scheme 2).[8] Although the primary hydroxy group of 3 could be used in conjugation chemistry, we wished to introduce a functional group with distinct reactivity compared to the phenolic hydroxy group. Toward this objective, the allyl ether 2 was subjected to a Claisen rearrangement to provide the desired compound 4 as a solid in 80 % yield. Compound 4 is endowed with a terminal olefin, which can react by olefin cross metathesis. For example, this compound was cross-coupled with but-3-en-1-ol in the presence of the nitrated ruthenium catalyst,[9] to generate alkene 5 in 52 % yield. Because olefin cross-metathesis is compatible with many functional groups,[10] various substituents could be introduced into 4. Compound 4 could also be converted to the corresponding aldehyde upon treatment with OsO4 and NaIO4 (unpublished results). These examples demonstrate that compound 4 is conjugation-ready.
Scheme 2.

Synthesis of compounds 4 and 5.
In pH 10 borate buffer, the quantum yields of 3 and 4 were found to be 0.89 and 0.82, respectively. These results demonstrate that these compounds have fluorescent characteristics that are comparable with those of other fluorescein derivatives. The emission maxima of 3 and 4 were determined to be 523 and 535 nm, respectively. Based on these emission spectra, we called 3 and 4 Pittsburgh Green and Pittsburgh Yellowgreen, respectively, to follow the convention in the fluorescein field.
For biological applications, pH sensitivity under physiological conditions is a major concern. Thus, we measured the fluorescent intensity of 3 at 523 nm from pH 1 to 12. As Figure 1 A shows, the fluorescent signal of compound 3 is constant between pH 5.5 and 9.5, thus indicating the robustness of this reagent. The pKa value of the phenolic hydroxy group was determined to be 4.27, which is nearly identical to that of DCF (4.29) in our experiments. The weaker fluorescence above pH 10 could be explained by invoking a nonfluorescent cyclic ether. The UV absorption spectrum of compound 3 is consistent with a cyclic structure (Figure 1 B); under basic conditions, the peak at 506 nm diminishes and a new peak emerges at 302 nm. Under acidic solutions, compound 3 shows a peak at 457 nm, and that at 506 nm is significantly weaker; this indicates that compound 3 is in the protonated form, but not a cyclic ether form (Figure 1 C).
Figure 1.

A) Fluorescent intensity of 3 at various pH values . Solutions were excited at 497 nm and emissions recorded at 523 nm. B) UV spectra of 3. C) Structures of 3.
The unique advantages of the zebrafish make it an excellent model organism for high-throughput biomolecule screening. These include high fecundity, small embryo size, transparent development ex utero, and demonstrated permeability to certain small molecules.[11] Therefore, we attempted the vital staining of zebrafish embryos with 3 and 4 to assess the permeation and localization of each in vivo. No previous reports describe the successful use of unconjugated fluorescein derivatives for this purpose. Accordingly, embryos exposed to DCF exhibited a fluorescent signal indistinguishable from the faint autofluorescence of controls (Figure 2 A and B), thus suggesting that it did not penetrate the embryos to any observable extent. Remarkably, however, both 3 and 4 effectively stained embryos as evidenced by increased fluorescence at shorter exposure times (Figure 2 C and D).
Figure 2.
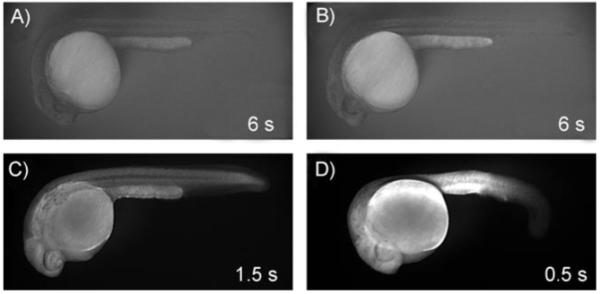
Representative fluorescent images of 24 hpf zebrafish embryos exposed for 1 h to A) a control solution (2 % DMSO in buffered embryo medium) or 25 μm staining solutions of B) DCF, C) 3, or D) 4. All images were captured at 35× magnification with the given exposure times and all other conditions identical.
We next examined whether this staining indicated true tissue penetration or merely a superficial coating effect. To accomplish this, we utilized confocal microscopy to examine compound distribution within the embryo. We chose to use the commercially available BODIPY TR methyl ester as a control stain since it has been successfully used for this purpose.[12]
Preliminary experiments confirmed that incubating zebrafish embryos with 3 or 4 for 1 h yielded relatively consistent vital staining (Figure 3 A and B), comparable to the BODIPY control (Figure 3 C). Confocal projections of the head (Figure 3 D–F) and tail (Figure 3 G, I, K) spanning 80 μm revealed that 3, 4, and BODIPY TR methyl ester permeate live tissue to similar extents. Slices of the dorsal midline (Figure 3 H, J, L) indicate that the vacuoles of the notochord, an early embryonic structure, remain unstained while the surrounding tissue is efficiently labeled. These data support the tissue-specific permeation and localization of 3 and 4 in vivo. It is also noteworthy that 3 and 4 are more water soluble than BODIPY TR methyl ester at the tested concentrations (data not shown).
Figure 3.
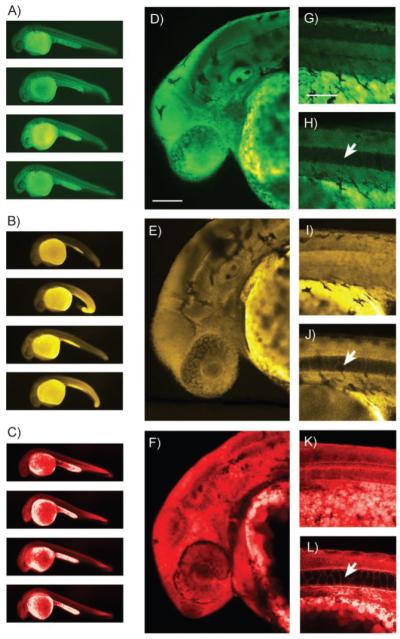
Pseudocolored images of 24 hpf zebrafish embryos stained for 1 h with 25 μm 3 (A, D, G, H), 4 (B, E, I, J), or BODIPY TR methyl ester (C, F, K, L). A)–C) Examples of embryo staining. Representative instances of the slight developmental defects caused by exposure to 4 can be seen in panel (B). D)–F) 80 μm confocal Z projections of the head region. G), I), K) 80 μm Z projections of the dorsal midline (anterior to the left). H), J), L) 1 μm slices from the dorsal midline with notochord vacuoles indicated by arrows. Scale bars = 100 μm.
We also observed that yolk sacs and yolk extensions always appeared brighter than the embryos themselves (Figure 3 A–C). This is likely the result of compound accumulation within the lipidic yolk platelets that comprise almost the entire volume of the yolk.[13] Embryo viability was not significantly decreased by exposure to any of the staining solutions when compared to control solution (n = 60 per compound, p = 0.81). However, we did detect slight developmental abnormalities in the embryos exposed to 25 μm Pittsburgh Yellowgreen, including narrowing of the yolk extension and ventral curling of the tail (Figure 3 B).
Toxicity studies were performed to determine the effects of longer-term exposure to the Pittsburgh compounds on zebrafish development. Embryos were exposed to 3 or 4 standards on a logarithmic scale (25, 8, 2.5, 0.8, 0.25 μm) from 3.5 h post-fertilization (hpf) to 48 hpf. Compound 3 exhibited a significant effect on embryo viability at 25 μm (p < 0.001), but not at 8 μm (Table S1 in the Supporting Information). Compound 4 was slightly more toxic, affecting viability at 25 and 8 μm (p < 0.001), but not at 2.5 μm. These data are consistent with the observations from the staining experiments. The promising vital staining results with these new dyes suggest that these compounds and their derivatives could be useful tools in bioimaging.
The Mannich reactions of DCF have been used to synthesize biologically important fluorescent sensors. However, we find that these compounds are often poorly soluble in commonly used organic solvents, such as CH2Cl2, for further synthetic manipulations. We speculated that the displacement of a carboxy group with a hydroxymethyl group might improve solubility. Of particular interest were previously known compounds 6 and 8, both of which were virtually insoluble in CH2Cl2. To prepare analogues of these compounds, the Mannich reactions of compound 3 and the corresponding amine were employed to generate compounds 7 and 9 in 64 and 45% yield, respectively (Scheme 3). These compounds were found to be sufficiently water soluble for biological experiments (at least up to 10 μm), and yet very soluble in CH2Cl2. Since the carboxy group does not participate in sensing in these sensors, in principle such a replacement would not affect their ability to sense their analytes.
Scheme 3.

Dichlorofluorescein derivatives.
In conclusion, we have developed new fluorescein derivatives. The synthesis of 3 and 4 only requires three steps for each and is scalable, thus indicating manufacturing potential. Furthermore, compounds 3 and 4 exhibited desirable staining and permeation in zebrafish embryos. Since these new fluorescein derivatives are soluble in both water and organic solvents, they should facilitate chemical syntheses and biological studies of related compounds. We also improved the physical properties of previously known fluorescein-based sensors. We are currently synthesizing 4-based probes as part of our efforts toward sensor development.[2,14]
Experimental Section
Preparation of 4
A solution of compound 2 (1.43 g, 3.35 mmol) in Ph2O (5 mL) was stirred at 150°C for 12 h. After being cooled to 24°C, the reaction mixture was transferred directly to a silica gel column. The column chromatography was performed with 10 % isopropyl alcohol in hexanes to afford compound 4 as a redorange solid (1.15 g, 80 %). M.p. 186–188°C; Rf = 0.59 (50 % EtOAc in hexanes); 1H NMR (300 MHz, CDCl3, 293 K): δ = 7.42–7.35 (m, 2 H), 7.30–7.28 (m, 1 H), 6.89–6.87 (m, 3 H), 6.77 (s, 1 H), 6.01 (ddt, J = 17.4, 9.9, 6.3 Hz, 1 H), 5.29 (br s, 2 H), 5.14 (dd, J = 17.4, 1.7 Hz, 1 H), 5.05 (dd, J = 9.9, 1.5 Hz, 1 H), 3.63 (br d, J = 6.3 Hz, 2 H); 13C NMR (75 MHz, CD3OD, 293 K): δ = 155.3, 152.5, 151.2, 149.2, 145.3, 140.1, 138.7, 130.4, 129.8 (2 C), 127.6, 124.6, 122.2, 118.6, 118.1, 117.5, 117.3, 117.1, 115.6, 104.4, 85.0, 73.0, 29.0; IR (in CH2Cl2): 3305 (br, O-H), 3074, 2923, 2855, 1635, 1598, 1482, 1446, 1356, 1278, 1213, 737 cm−1; HRMS (EI+) calcd for C23H16Cl2O4: 426.0426 [M]+, found: 426.0419.
Preparation of 5
A solution of compound 4 (34 mg, 0.075 mmol) in 1,2-dichloroethene (DCE; 0.8 mL) was added to a solution of the ruthenium catalyst (7.5 mg, 0.011 mmol) in DCE (0.5 mL) under a nitrogen atmosphere at 24°C. The resulting mixture was stirred for 5 min at the same temperature, and but-3-en-1-ol (20 μL, 0.225 mmol) was added dropwise at 24°C. The resulting mixture was heated at 45°C for 3 h. Subsequently, the reaction mixture was cooled to 24°C, and the solvent was removed in vacuo. The residue was purified by flash column chromatography (5→50 % EtOAc in hexanes containing 1 % HOAc) to afford compound 5 as a red solid (15 mg, 42 %; 52 % based on 9 mg of recovered 4). For characterization, trans-5 (major product) and cis-5 (minor product) were separated by HPLC (20→100 % CH3CN in H2O; C18 column). The major product was fully characterized. Data for trans-5: Rf = 0.15 (60 % EtOAc in hexanes containing 1 % HOAc); 1H NMR (300 MHz, CDCl3, 293 K): δ = 7.40–7.35 (m, 2 H), 7.31–7.26 (m, 1 H), 6.89 (s, 1 H), 6.88 (br d, J = 7.5 Hz, 1 H), 6.86 (s, 1 H), 6.76 (s, 1 H), 5.79–5.68 (m, 1 H), 5.62–5.51 (m, 1 H), 5.28 (br s, 2 H), 3.65–3.59 (m, 2 H), 2.27 (appt q, J = 12.3, 6.0 Hz, 2 H), 2.11 (s, 1 H, OH); 13C NMR (75 MHz, CDCl3, 293 K): δ = 151.9, 150.3, 149.7, 147.9, 143.8, 138.7, 130.2, 128.7, 128.6, 128.4, 127.5, 125.8, 123.7, 121.0, 118.3, 118.0, 115.4, 115.3, 115.2, 103.8, 83.4, 72.3, 61.9, 35.9, 27.2; IR (CH2Cl2): 3300 (br, O-H), 3070, 2926, 2854, 1632, 1582, 1482, 1443, 1354, 1283, 1212, 1014, 736 cm−1; LRMS (ESI+)C25H20Cl2O5: 493 (18 %) [M+H]+, 493 (35 %) [M+Na]+, 493 (35 %) [M+ K]+; (EI+) 469 (30 %) [M−H]+, 470 (22 %) [M]+, 471 (25 %) [M+H]+.
UV-visible spectroscopy
Absorption spectra were acquired on a Cary 50 UV/Vis spectrometer, under the control of Windows-based PCs running the manufacturer’s supplied software.
Fluorescence spectroscopy
All fluorescence measurements were acquired in a 1 cm × 1 cm quartz cell by using a Spex Fluorolog fluorometer with 0.7 nm bandwidth slits, λex = 490 nm, collecting λem = 496–620 nm. All spectra were corrected for emission intensity by using the manufacturer’s supplied photomultiplier curves.
Relative quantum yields
To determine quantum yields relative to fluorescein, stock solutions of 3 and 4 were prepared in DMSO (1 mm) and diluted in borate buffer (pH 10) to OD490 = 0.12. The samples were excited at λ = 490 nm, and the integrated emission spectra were compared. The quantum yields of all compounds were referenced to fluorescein in 0.1 n NaOH (Φ = 0.95).
Vital staining of zebrafish embryos
Embryos were stained as previously described,[15] but with some modifications. Stock solutions (10 mm) of Pittsburgh Green (3), Pittsburgh Yellowgreen (4), and DCF were each prepared in DMSO. BODIPY TR methyl ester (Invitrogen) was supplied as a 5 mm stock solution in DMSO. All stock solutions were protected from light and stored in aliquots at −20°C. Stock solutions were diluted in HEPES-buffered embryo medium (5 mm HEPES, pH 7.2, 5 mm NaCl, 0.33 mm CaCl2, 0.33 mm MgSO4, 0.17 mm KCl) to yield 25 μm staining solutions with normalized DMSO contents of 2 % (v/v).
At 24 h post-fertilization (hpf), embryos were manually dechorionated with fine forceps and added in groups of 20 to agarose-coated wells of a 12-well plate containing HEPES-buffered embryo medium. Excess medium was carefully removed from each well, avoiding exposure of the embryos to air, and replaced with staining solution or control solution (1 mL; 2 % DMSO in HEPES-buffered E3). The plates were then covered in foil and incubated at room temperature with gentle shaking for 1 h. After being stained, the embryos were transferred to agarose-coated 60 mm dishes and washed in HEPES-buffered embryo medium (3 × 10 min each) with gentle shaking.
Imaging of zebrafish embryos
Embryos used for imaging were anesthetized in HEPES-buffered embryo medium containing Tricaine (ethyl 3-aminobenzoate methanesulfonate salt; 0.64 mm; Sigma–Aldrich). Digital photographs were taken by using a stereomicroscope (Leica Microsystems, Wetzlar, Germany) and CCD camera (QImaging, Burnaby, Canada). For fluorescent imaging, filter sets (Chroma Technology, Rockingham, VT) were used to visualize Pittsburgh Green, Pittsburgh Yellowgreen, DCF (all at λex = 480/40, λem = 535/50) or BODIPY TR methyl ester (λex = 575/50, λem = 640/50).
Confocal optical sections were collected at 1 mm intervals with a 20× objective by using a laser scanning confocal head (Leica Microsystems) mounted on an inverted microscope (Leica Microsystems). Excitation was provided by an argon laser at 488 nm (Pittsburgh Green, Pittsburgh Yellowgreen, and DCF) and a HeNe laser at 543 or 594 nm (BODIPY TR methyl ester). Image stacks were rendered and analyzed with image analysis software (ImageJ, version1.37v; Wayne Rasband, NIH/NIMH, http://rsb.info.nih.gov/ij/).
Supplementary Material
Acknowledgements
This work was supported by the University of Pittsburgh and the National Science Foundation (CHE-0616577). E.D.D. and N.A.H. are supported by R01HD053287 from NIDDK. A.L.G. and V.D.M are recipients of the Novartis Graduate Fellowship and The Organic Division Undergraduate Research Fellowship, respectively. We wish to thank Anastasiya Kastrama for preliminary experiments. The ruthenium catalyst was a gift from Boehringer Ingelheim.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- [1].a) Walkup GK, Burdette SC, Lippard SJ, Tsien RY. J. Am. Chem. Soc. 2000;122:5644. doi: 10.1021/ja010059l. [DOI] [PubMed] [Google Scholar]; b) Kenmoku S, Urano Y, Kanda K, Kojima H, Kikuchi K, Nagano T. Tetrahedron. 2004;60:11067. [Google Scholar]
- [2].Sparano BA, Koide K. J. Am. Chem. Soc. 2005;127:14954. doi: 10.1021/ja0530319. [DOI] [PubMed] [Google Scholar]
- [3].Martin MM, Lindqvist L. J. Lumin. 1975;10:381. [Google Scholar]
- [4].Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J. Am. Chem. Soc. 2005;127:4888. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- [5].Sun WC, Gee KR, Klaubert DH, Haugland RP. J. Org. Chem. 1997;62:6469. [Google Scholar]
- [6].a) Mottram LF, Boonyarattanakalin S, Kovel RE, Peterson BR. Org. Lett. 2006;8:581. doi: 10.1021/ol052655g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mottram LF, Maddox E, Schwab M, Beaufils F, Peterson BR. Org. Lett. 2007;9:3741. doi: 10.1021/ol7015093. During the preparation of this manuscript, the same group reported an improved version, in which the four-step synthesis gave 28 % overall yield; see. [DOI] [PubMed] [Google Scholar]
- [7].Dowlut M, Hall DG, Hindsgaul O. J. Org. Chem. 2005;70:9809. doi: 10.1021/jo051503w. [DOI] [PubMed] [Google Scholar]
- [8].a) Sparano BA, Shahi SP, Koide K. Org. Lett. 2004;6:1947. doi: 10.1021/ol049537y. [DOI] [PubMed] [Google Scholar]; b) Song F, Garner AL, Koide K. J. Am. Chem. Soc. 2007;129:12354. doi: 10.1021/ja073910q. [DOI] [PubMed] [Google Scholar]
- [9].Grela K, Harutyunyan S, Michrowska A. Angew. Chem. 2002;114:4210. doi: 10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:4038. [Google Scholar]
- [10].a) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. 2005;117:4564. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; Chem. Int. Ed. 2005;44:4490. Examples from our laboratory: [Google Scholar]; b) Albert BJ, Sivaramakrishnan A, Naka T, Koide K. J. Am. Chem. Soc. 2006;128:2792. doi: 10.1021/ja058216u. [DOI] [PubMed] [Google Scholar]; c) Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. J. Am. Chem. Soc. 2007;129:2648. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Peterson RT, Link BA, Dowling JE, Schreiber SL. Proc. Natl. Acad. Sci. USA. 2000;97:12965. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cooper MS, Szeto DP, Sommers-Herivel G, Topezewski J, Solnica-Krezel L, Kang HC, Johnson I, Kimelman D. Dev. Dyn. 2005;232:359. doi: 10.1002/dvdy.20252. [DOI] [PubMed] [Google Scholar]
- [13].Cooper MS, D’Amico LA, Henry CA. Methods Cell Biol. 1999;59:179. doi: 10.1016/s0091-679x(08)61826-9. [DOI] [PubMed] [Google Scholar]
- [14].Sparano BA, Koide K. J. Am. Chem. Soc. 2007;129:4785. doi: 10.1021/ja070111z. [DOI] [PubMed] [Google Scholar]
- [15].Cooper MS, Szeto DP, Sommers-Herivel G, Topezewski J, Solnica-Krezel L, Kang HC, Johnson I, Kimelman D. Dev. Dyn. 2005;232:359. doi: 10.1002/dvdy.20252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.