

Abstract
When combined with Haplotype Fusion PCR (HF-PCR), Ligation Haplotyping is a robust, high-throughput method for empirical determination of haplotypes, which can be applied to assaying both sequence and structural variation over long distances. Unlike alternative approaches to haplotype determination, such as allele-specific PCR and long PCR, HF-PCR and Ligation Haplotyping do not suffer from mispriming or template switching errors. In this method, HF-PCR is used to juxtapose DNA sequences from single molecule templates, that contain single nucleotide polymorphisms (SNPs) or paralogous sequence variants (PSVs) separated by several kilobases. HF-PCR employs an emulsion-based fusion PCR reaction, which can be performed rapidly, and in a 96-well format. Subsequently, a ligation-based assay is performed on the HF-PCR products to determine haplotypes. Products are resolved by capillary electrophoresis. Once optimized, the method is rapid to perform, taking a day and a half to generate phased haplotypes from genomic DNA.
Introduction
Knowledge of the combination of SNPs on the same parental chromosome is extremely valuable to several areas of research: for example, haplotypes can be used i) in evolutionary and population-genetic studies, to detect the influence of selection 1 and population migration 2; ii) for linkage disequilibrium mapping 3; and iii) in association studies, to investigate whether particular combinations of SNPs confer greater susceptibility to diseases (e.g. systemic lupus erythematosus 4). Unlike genotypes, for which several well-established assays are available, there are few reliable empirical methods for the determination of the phase of haplotype for diploid organisms, particularly over long distances, due to the need of a haplotyping assay to function on individual strands of DNA, without giving rise to artefacts.
Commonly used experimental approaches to haplotype determination involve allele-specific PCR 10, long PCR 11 or a combination of both 12. However, optimizing a PCR reaction so that it is truly allele-specific is laborious, as it is difficult to avoid mispriming. Additionally, the risk of ‘jumping PCR’ increases with increasing amplicon length, creating artificial recombinants by template switching 13. The efficiency and robustness of long PCR also decreases with increasing amplicon length 14. Consequently, approaches for haplotype determination that are based on allele-specific PCR or long PCR are unsuitable over distances of several kilobases, and so statistical haplotyping approaches, which reconstruct haplotypes from genotype information, have gained prominence (for a review of statistical haplotyping methods see 6).
Over short distances, statistical haplotyping methods are generally reliable7, 8, but over longer distances, or in parts of the genome with low linkage disequilibrium, statistical approaches suffer from switch errors, rendering them inaccurate. Thus there is a need for a reliable empirical method for the determination of haplotype status, whose accuracy does not decrease with increasing distance 9. Such a method is Ligation Haplotying 15.
The first stage of Ligation Haplotying uses a high-throughput version of Haplotype Fusion PCR (HF-PCR) to generate condensed haplotypes, by juxtaposing DNA sequences from the same single molecule, thus retaining phase information 14 (Fig. 1a). The reaction is carried out in a water-in-oil emulsion – each aqueous compartment is independent from the others, allowing millions of individual reactions to take place in a single PCR tube 17,18. If we know the diameter of these aqueous compartments, we can calculate the total number, and can then perform the PCR reaction with a low compartment : template ratio. In this way, the majority of occupied compartments can be made to contain a single target locus, which ensures that PSVs or SNPs from single molecules become fused together. The HF-PCR reaction generates a condensed haplotype, in which PSVs or SNPs that lie several kb apart in the genomic DNA are brought to within 100-150 bases of one another.
Figure 1. Ligation haplotyping.
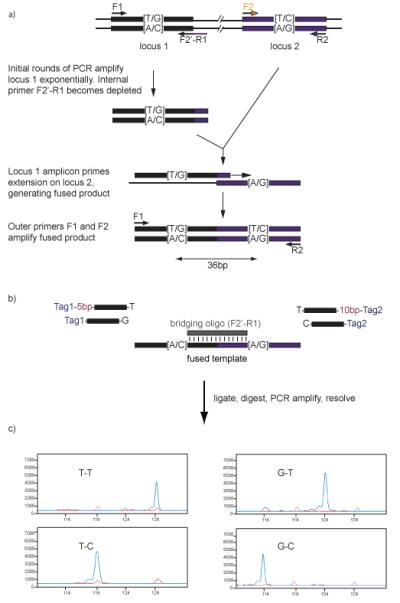
a) Haplotype fusion PCR. The two SNPs to be interrogated can lie several kilobases apart. Primer pairs F1 and R1, and F2 and R2 are designed for the separate loci, as for a typical PCR. The reverse complementary sequence of F2 is appended to the 5′ terminus of primer R1, creating the ‘fusion primer’ F2′-R1. Three of these primers are used in the emulsion PCR reaction: F1, R2, and the fusion primer, F2′-R1, shown as black and black / indigo arrows. The fusion primer is used at a lower concentration than the other primers, so that in the initial rounds of amplification, where locus 1 is amplified exponentially, the fusion primer quickly becomes depleted. At the same time, locus 2 is amplified linearly, by extension of the R2 primer. Because the tail on the fusion primer matches the reverse complement of F2, the amplicon generated by this primer can prime on locus 2. Primers F1 and R2 can then amplify the fused template exponentially for the remainder of the reaction.
b) Ligation Haplotyping. During the ligation reaction, the bridging oligo (in this case, the same oligo as the fusion primer) anneals to the fused template, spanning the junction. Allele-specific oligos, with characteristic sizes, anneal to the positions at either end of the bridging oligo, and when the oligo matches the template, a ligation reaction occurs. For each haplotype, a differently sized product is generated. The allele-specific oligos are tailed with universal primer sequences (Tag 1 and Tag 2), so that all ligation products can be amplified together, with as little bias as possible.
c) Ligated products are amplified by PCR, using a fluorescently labelled primer, which anneals to the Tag 1 sequence, and these fluorescent amplicons are analyzed by capillary electrophoresis.
Figure adapted with permission from Oxford Journals, NAR 2008
Many emulsification methods are not suitable for a high throughput assay because they use large reaction volumes, which makes them expensive, chiefly because of the large quantity of polymerase used 14,17,19, and also because the techniques used to create the emulsions are only suitable for small numbers of simultaneous reactions. This led us to develop a method of emulsification that is effective on low reaction volumes, allowing us to use 2 units of polymerase per reaction instead of more than 35 units for other published methods 17,19. Additionally, 96 emulsions can be generated simultaneously, in 150 seconds, and so this method is both relatively inexpensive and rapid. .
Both long PCR and HF-PCR require the nucleotides of interest to be present on the same template strand, and so for both techniques it is essential to have a good quality, high molecular weight DNA template. If long PCR is attempted on low molecular weight templates, the frequency of template switching will increase 13, leading to false positives. Because single template molecules are amplified independently during Ligation Haplotyping, template switching is prevented: low molecular weight DNA will result in no amplicon being produced. Unlike long PCR, the amplification efficiency of HF-PCR does not decrease over long distances – during the initial stages of the reaction, the two target regions are amplified independently within the aqueous compartment. The fusion reaction then begins once the concentration of these amplicons has increased to a sufficient level 14. The length of haplotype that can be condensed by HF-PCR should be limited only by the size of the template DNA used in the emulsion. We investigated the shearing effect of emulsion preparation on genomic DNA 100-150kb in length by pulsed-field gel electrophoresis, and were unable to detect any reduction in fragment size14, which indicates that haplotype determination in this way should be possible over a longer range than the 25kb offered by long PCR.
The high-throughput Ligation Haplotyping assay itself is performed on the condensed haplotypes generated by HF-PCR (Fig. 1b). There are several assays that exploit the high specificity of ligases at the 3′ side of a ligation junction, such as the ligase detection reaction 20, GoldenGate assay 21 and SNPlex assay 22. In Ligation Haplotyping, we make use of the ligase’s ability to be highly discerning at both the 5′ and 3′ sides of the ligation junction: allele-specific oligos for each SNP anneal to the condensed haplotype at either side of a central bridging oligo, and are ligated (Fig. 1). The assay described here was designed so that this bridging oligo is the same oligo that was used as the fusion primer in the prior reaction. However, in cases where it would be preferable to use PCR primers that do not abut the PSVs or SNPs directly, it is possible to use a bridging oligo that differs from this PCR primer.
The product is linearly amplified by cycles of denaturation and annealing / ligation. Unlike exponential amplification, during this reaction, the ligation product itself does not serve as a template in subsequent cycles. Thus errors caused by misligation are not propagated – each is a de novo event, and this helps to maintain a high level of specificity during the reaction 20. Each allele-specific oligos used in the ligation reaction has a characteristic length so that, once ligated to the central bridging oligo, the size of the products reveals the identity of the haplotypes present. In addition, these oligos have universal sequences at their non-ligating termini, to facilitate simultaneous PCR amplification of all products with fluorescently labelled primers. The fluorescent labels allow the PCR-amplified ligation products to be identified by capillary electrophoresis (Fig. 1c).
The HF-PCR reaction should be amenable to multiplexing. For a given primer pair, there is only one target locus in every ~1000 aqueous compartments, and so additional sets of primers will not compete for resources, because amplification will proceed in separate compartments. Additionally, as with other well-established ligation based assays 20, 21, 22 the Ligation Haplotyping reaction should also be compatible with high levels of multiplexing. In the example presented in this manuscript, oligos were designed so that haplotypes generate peaks differing by 5 bases. This means that a window of 20 nucleotides must be reserved for each set of PSVs / SNPs. To increase the multiplexing capability of peak visualization, it is possible to reduce the difference in size between different haplotypes, to 2 bases, reducing the overall window size to 8 nucleotides. Additionally, it would be possible to use PCR primers with different fluorescent labels. Up to 4 different fluorophores can be used within each dye set, and in this way, overlapping size windows can be used. The assay could easily be adapted for use on addressable arrays, which will increase throughput and decrease cost further.
Experimental design
HF-PCR
Short regions are amplified more efficiently in the HF-PCR, and so it is necessary to select primer pairs that target regions of < 150bp. Primers must be designed for the two SNP- or PSV-containing loci separately, choosing similar annealing / melting temperatures for all primers using an oligonucleotide calculator (such as http://www.cnr.berkeley.edu/~zimmer/oligoTMcalc.html). A melting temperature of approximately 65°C is appropriate. The specificity of primers should be checked by in silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr?command=start).
Once all four primers have been designed, the ‘fusion primer’ is designed by appending the reverse complementary sequence of one primer from one locus onto the 5′ end of one primer from the second locus (Fig. 1a). For locus 1, the forward and reverse PCR primers are F1 and R1 respectively. For locus 2, they are F2 and R2 respectively. A fusion primer that would join the two loci together is F2′-R1 (i.e. the R1 sequence with the reverse complement of F2 attached to its 5′ end). In the PCR reaction, only primers F1, R2 and F2′-R1 are used. In the initial rounds, only locus 1 can amplify, but the strand that is generated by extension of primer F1 acquires the primer F2 sequence at its 3′ end, and so the whole amplicon is able to prime on locus 2. The F2′-R1 fusion primer is present at a lower concentration than primers F1 and R2, and becomes depleted more rapidly. After a few rounds of PCR, amplification of the fused loci proceeds using primers F1 and R2.
There is no requirement for the 3′ termini of primers at either locus to be adjacent to the SNPs of interest, or for the bridging oligo to be the same as the F2′-R1 fusion primer. The main consideration here is that the further the 3′ terminus of F2′ is from the locus 2 SNP, and the further the R1 is from the locus 1 SNP, the further apart the SNPs will be in the condensed haplotype, and hence the longer the bridging oligo will need to be in the ligation reaction, as it is the bridging oligo that must abut both SNPs.
Emulsion formation
When prepared singly by dropwise addition of aqueous phase to oil phase while stirring at 1000rpm, the emulsions produced contain aqueous compartments which, when viewed by light microscopy and compared to fluorescent marker beads, have an average diameter of approximately 15μm 23. If we assume that these compartments are spherical, their volume is 1.77 × 10−15 m3. If we also assume that aqueous compartments are densely packed in the emulsion, then there should be around 5.7 × 105 aqueous compartments per microlitre. If the speed of stirring is increased, the average diameter of aqueous compartments decreases. Increasing the speed of vortexing when preparing emulsions in 96-well format produces the same effect.
For the same mass of DNA in the emulsion, increasing the number of compartments by reducing their volume reduces the frequency of compartments that are occupied by more than one template, and so the background signal is minimised. But because only aqueous compartments that contain both polymerase and template can produce amplification products, increasing the number of compartments reduces the overall efficiency of the reaction. Thus it is necessary to find a compromise between efficient amplification and low background signal. We found that this was achieved by making emulsions with an average aqueous compartment diameter of approximately 5-10μM.
Emulsions made using 200ng genomic DNA contain approximately 30,000 copies of each autosomal target locus. Consequently only 1 in around 900 aqueous compartments contain a suitable template molecule for fusion PCR in the IRF5 assay, and so in most compartments, amplicons are derived from a single template molecule. When made in 96-well format, the volume of aqueous and oil phases are reduced, but the final concentration of reagents is unchanged. Thus, only 50ng of template DNA is required per well, and the ratio of 1 template molecule in 900 aqueous compartments is maintained.
Ligations
The allele-specific oligos used in the ligation reaction are designed to be complementary to the HF-PCR product, and to have an annealing temperature of approximately 65°C. This is achieved simply by obtaining the sequence of the strand of the fused template that is complementary to the bridging oligo (see Fig 1b.), measuring the annealing temperature of the strand between the SNP and the end of the strand in an oligonucleotide calculator (e.g.http://www.cnr.berkeley.edu/~zimmer/oligoTMcalc.html) and reducing the length from the non-ligating end until the desired Tm is achieved. This is performed for both SNPs at both sides of the bridging oligo. Allele-specific oligos designed in this way may differ in length by one or two nucleotides, because the SNPs themselves will influence the Tm of the oligo. This is not problematic, since it is necessary to make the allele specific oligos from each locus differ from each other in length anyway. In the example given here, we added non-complementary ‘spacer nucleotides’ to one oligo from each pair, so that the oligos at the 5′ end of the bridging oligo differed in length by 5 nucleotides and those at the 3′ end of the bridging oligo differed by 10 nucleotides. The universal annealing sequence is then added to the non-ligating end of the allele-specific oligos.
Thus the overall length of the allele-specific oligos are governed by the universal sequence, the spacer region and their Tm, so will differ from assay to assay. The length of the bridging oligo is governed by the distance between the SNPs. When allele-specific oligos ligate to the central bridging oligo, four different combinations are possible, and the combinations differ in length by 5 nucleotides.
Prior to PCR amplification, it is necessary to digest unligated oligonucleotides using exonucleases, and so it is necessary to add blocking groups to the non-ligating end of each allele-specific oligo. In this way, after successful ligation, products are protected from digestion at both ends, whereas unligated oligos are not. The exact nature of the blocking groups is not critical, but we have found suitable modifications to be four 2′ O-methyl uracil bases added to the 3′ end 5′-specific oligos, and a single amino-C6 modification added to the 5′ end of 3′-specific oligos.
Template DNA
For a successful HF-PCR reaction, it is essential that the strands of template DNA are longer than the distance between the SNPs that are being assayed. Commercial DNA preparations typically have a fragment size in excess of 100kb, whereas biological specimens can be shorter, depending upon the extraction method used. If in doubt, fragment size and DNA integrity can be assayed by pulsed-field gel electrophoresis 14. Buffers that contain EDTA should be avoided, because this can inhibit PCR reactions.
Controls
To monitor reagent contamination, it is necessary to prepare reaction blanks for all enzymatic reactions. Here, an equivalent volume of ultrapure water is used instead of the DNA sample. The formation of reaction products indicates contamination.
Materials
Reagents
Oil phase for preparation of emulsions singly
Span 80 (Sigma cat. no. S6760)
Tween 80 (Sigma cat. no. P8074)
Triton X-100 (Sigma cat. no. T9284)
Light mineral oil (Sigma cat. no. M5904)
Oil phase for preparation of emulsions in 96-well format
Silicone polyether / cyclopentasiloxane (Dow Corning cat. no. DC5225C)
Cyclopentasiloxane / trimethylsiloxysilicate (Dow Corning cat. no. DC749)
AR20 silicone oil (Aldrich cat. no. 10838)
10μM fluorescent beads (Beckman Coulter cat. no. 6601329)
Aqueous phase for emulsions
Phusion DNA polymerase (Finnzymes; NEB cat. no. F-530L) CRITICAL – avoid the use of polymerases that do not generate blunt ended products.
5X Phusion HF buffer (Finnzymes; NEB cat. no. F-518S)
50mM MgCl2 (Finnzymes; supplied with NEB cat. no. F-518S)
dNTPs (NEB cat. no. N0447L)
Genomic DNA
HF-PCR primers for IRF5 SNPs (Sigma)
IRF5_F1 5′ CGGGATGAAGACTGGAGTAGG
IRF5_R2 5′ GACAAGGAGGAGTAAGCAAGGAAC
IRF5_F2′-R1 5′ GGGTGCCTACAGCAGGGTTCTGACCCTGGCAGGTCC
Disruption of emulsions
diethyl ether (Sigma cat. no. 309958) or hexane (Sigma cat. no. 34859) – CAUTION: highly flammable
Proteinase K (Sigma cat. no. P4850)
Ligation Haplotyping reaction
10X T4 ligase buffer (supplied with NEB cat. no. M0202S)
T4 polynucleotide kinase (NEB cat. no. M0201L)
Thermus thermophilus (Tth) ligase (ABGene cat. no. AB-0325)
10x Tth ligase buffer (supplied with ABGene cat. no AB-0325)
Ligation oligos for IRF5 SNPs
IRF5_F2′-R1 (Sigma, as above)
IRF5_5T (IDT; TGAGCATCACCAATGTGACCTATCCTATCCGTGCCAGCAAGATCCAATCTAG A-2′UOMe-2′UOMe-2′UOMe-2′UOMe
IRF5_5C (IDT; CGAGCATCACCAATGTGACCGTGCCAGCAAGATCCAATCTAGA-2′UOMe-2′UOMe-2′UOMe-2′UOMe)
IRF5_3G (IDT; AmC6-GGGTTCCCTAAGGGTTGGAGAAAGCGAGCTCGGGG)
IRF5_3T (IDT; AmC6-GGGTTCCCTAAGGGTTGGATTTCGGAAAGCGAGCTCGGGT)
Ligation cleanup
Lambda exonuclease (NEB cat. no. M0262L)
10x Lambda exonuclease buffer (supplied with NEB cat. no. M0262L)
E. coli exonuclease I (USB cat. no. 70073Z)
Reamplification PCR
Phusion DNA polymerase (Finnzymes; NEB cat. no. F-530L) CRITICAL – avoid the use of polymerases that do not generate blunt ended products.
5X Phusion HF buffer, containing 7.5mM MgCl2 (Finnzymes; NEB cat. no. F-518S)
50mM MgCl2 (Finnzymes; supplied with NEB cat. no. F-518S)
dNTPs (NEB cat. no. N0447L)
MLPAF (Sigma; 5′ 6FAM GGGTTCCCTAAGGGTTGGA 3′)
MLPAR (Sigma; 5′ GTGCCAGCAAGATCCAATCTAGA 3′)
Visualizing reaction products
Agarose (Invitrogen cat. no. 16500100 )
10x TBE solution (Invitrogen cat. no. 15581028)
10mg / ml ethidium bromide (Sigma cat. no. E1510) – CAUTION: toxic – wear nitrile gloves when handling, and avoid formation of aerosols.
Loading dye (Qiagen cat. no. 239901)
Low molecular weight DNA ladder (NEB cat. no. N3233L)
8μl formamide (Sigma cat. no. F5786) – CAUTION: toxic – handle with care and avoid breathing vapour.
1μl GeneScan ROX 350 size standard (Applied Biosystems cat. no. 401735).
Equipment
Parafilm M (Sigma cat. no. P7793)
50ml Falcon Tubes (VWR cat. no. 734-0448)
Thermal cycler with 96-well heating blocks (e.g. MJ Research product no. PTC-225)
100ml and 1l measuring cylinders (e.g. Sigma cat. nos. Z324183 and Z650641 respectively)
Thin-walled 200μl PCR tubes (VWR cat. no. 732-0548)
3mm tungsten carbide beads (Qiagen cat. no. 69997).
96-well poly propylene PCR plates (Greiner cat. no. 652290)
Microseal ‘A’ plate seal (Bio-Rad cat. no. MSA-5001)
Microseal ‘F’ plate seal (Bio-Rad cat. no. MSA-1001)
Vortex mixer, fitted with a microtitre plate adapter (e.g. VWR cat. no. 444-0486 and VWR cat. no. 444-5919).
Plate centrifuge (e.g. Eppendorf product no. 5804, fitted with A-2-DWP rotor)
2ml polypropylene round-bottomed Cryogenic Vial (Corning cat. no. 430289) CRITICAL: the shape of vial influences the stability of the resulting emulsion.
8mm × 3mm magnetic bar with pivot ring (VWR International cat. no. 442-4500) CRITICAL: the size of magnetic stirrer and the presence / absence of a pivot ring influences the stability of the resulting emulsion.
Stirring plate, capable of 1,000 rpm (e.g. IKAMAG Midi MR1; Sigma cat. no. Z403733)
250ml conical flask (e.g. Sigma cat. no. 308811)
Horizontal electrophoresis gel tank (e.g. Sigma cat. no. Z338818)
Electrophoresis power supply (e.g. Sigma cat. no. Z654361)
UV transilluminator (e.g. Sigma cat. no. Z363839)
Fluorescence microscope (e.g. Olympus IX71)
Glass microscope slides (e.g. Cole-Parmer cat. no. WZ-48500-00)
ABI 3100 capillary electrophoresis sequencer with 36cm capillary array
Procedure
Optimization of PCRs (total time: 7 hours)
- 1. Optimize PCRs in solution before attempting them in emulsion. For locus 1, this is achieved using primer F1 and F2′-R1 and for locus 2 using F2 and R2 as forward and reverse primers respectively. For each locus, set up 12 identical PCR reactions as follows, adding the polymerase last. Each reaction will be run with a different annealing temperature, in order to find the most suitable one for the fusion reaction. Also include a reaction in which water is used instead of the DNA template, to monitor reagent contamination. Keep reagents on ice, apart from the polymerase, which should be kept at −20°C until it is needed:
5× Phusion HF buffer 10μl (→ 1× final conc.) 50mM MgCl2 1μl (→2.5mM TOTAL final conc.) 10mM dNTP mix 1.25μl (→ 250μM each final conc.) 10μM forward primer 1.5μl (→ 300nM final conc.) 10μM reverse primer 1.5μl (→ 300nM final conc.) 50ng / μl genomic DNA (or water) 2μl (→ 2ng / μl final conc.) 2 U / μl Phusion polymerase 0.5μl (→ 0.02 U / μl final conc.) water 32.25μl Total volume 50μl per reaction 2. Mix well by vortexing and spin down. Transfer 50μl of each reaction to a 200μl thin walled PCR tube and spin down.
- 3. Transfer tubes to a thermal cycler and perform standard PCR conditions with a gradient for the annealing temperature. The upper and lower range should be set at 5°C higher and lower than the mean Tm for both primers. For primers designed as described in the Experimental Design section, suitable cycling conditions are:98°C for 30 seconds33 cycles of 98°C for 10 seconds60-70°C for 30 seconds72°C for 15 secondsthen 72°C for 5 minutes4°C indefinitely
4. Prepare 1l of 1x TBE buffer by adding 900ml deionized water to 100ml 10x TBE in a 1l measuring cylinder, and mixing well.
5. Add 150ml of 1x TBE to 3g of agarose in a 250ml conical flask, and heat in a microwave oven on full power until the liquid just begins to boil.
6. Remove the conical flask from the microwave and allow to cool, mixing frequently, until the flask can be held comfortably in a gloved hand.
7. Add 6μl of 10mg / ml ethidium bromide to the gel and mix well. Pour into the gel cassette, with a suitable comb, and allow to solidify. Submerge the gel in 1x TBE, also containing 0.4μg/ml ethidium bromide.
8. After PCR, take 5μl of each reaction from Step 3, mix with 2μl of loading dye in a 200μl PCR tube and load each reaction into a separate well of the gel from Step 7. Also include a well containing low molecular weight DNA ladder, for size estimation.
9. Run the gel at 6V / cm, until the yellow dye is close to the bottom of the gel (takes approximately 45 minutes for a 14cm gel).
10. Visualise the gel by UV light on a transilluminator.
11. Identify the highest annealing temperature at which the amplification of both loci produces a strong band, representing a single product of the intended size. Use this temperature as the optimal annealing temperature in subsequent reactions. TROUBLESHOOTING
12. Take the PCR reactions for each locus that were performed at the optimal annealing temperature, and prepare a 10x dilution with water.
- 13. Perform a PCR using 1μl of both diluted templates from Step 12 to confirm that the fusion product is formed successfully. Prepare the following, in a 200μl thin-walled PCR tube:
5× Phusion HF buffer 10μl (→ 1× final conc.) 50mM MgCl2 1μl (→2.5mM TOTAL final conc.) 10mM dNTP mix 1.25μl (→ 250μM each final conc.) 10μM F1 primer 1.5μl (→ 300nM final conc.) 10μM R2 primer 1.5μl (→ 300nM final conc.) diluted template 1 1μl diluted template 2 1μl 2 U / μl Phusion polymerase 0.5μl (→ 0.02 U / μl final conc.) water 32.25μl Total volume 50μl per reaction Mix well and spin down 14. Transfer to a thermal cycler and begin the PCR cycle described in Step 3, but using the optimal annealing temperature selected in Step 11. Increase the extension time, if necessary, so that it is suitable for amplification of the full length fusion product. As an approximate guide, 20 seconds extension time is sufficient for a 300bp amplicon.
15. Run the PCR product on a 2% agarose gel, as described in Steps 4-10, alongside the locus 1 and 2 amplicons from Step 3, to confirm that a single fusion product of the expected size is produced. TROUBLESHOOTING
HF-PCR
16. Emulsions can be prepared and HF-PCR carried out either singly or in 96-well format. Once the vortexing speed has been optimized, the 96-well format is simpler and costs less per reaction, but this format uses a different oil phase, which makes the post-emulsion cleanup slightly lengthier.
A. Preparation of emulsions and HF-PCR singly (Timing: 1 hour to make emulsions, 1.5 hours to perform PCR)
- i. In a 100ml measuring cylinder, add:
- 4.5ml Span 80
- 400μl Tween 80
- 50μl Triton X-100
ii. Make the volume up to 100ml with light mineral oil.
iii. Cover the top of the measuring cylinder with Parafilm and mix thoroughly by vortexing / shaking.
iv. Transfer to 2x 50ml Falcon Tubes
PAUSE POINT
This oil phase can be stored indefinitely at room temperature (20°C).
CRITICAL STEP
the components of this oil phase are very viscous, so are difficult to pipette accurately. For the Triton X-100, it is helpful to use a larger volume pipette (e.g. P1000) than would normally be used, as smaller volume pipettes can struggle to draw the desired volume of liquid into the pipette tip. When drawing liquids up, it is also essential to leave the pipette tip in the liquids for several seconds, to ensure that the intended volume has been taken, and to dispense slowly, so that no liquid is left behind. It is also possible to rinse tips out with light mineral oil, collecting all liquid in the 100ml measuring cylinder.
- v. Prepare 100μM aqueous phase for each reaction, adding the polymerase last. Keep reagents on ice, apart from the polymerase, which should be kept at −20°C until it is needed:
5× Phusion HF buffer 20μl (→ 1× final conc.) 50mM MgCl2 2μl (→2.5mM TOTAL final conc.) 10mM dNTP mix 2.5μl (→ 250μM each final conc.) 10μM F1 primer 10μl (→ 1μM final conc.) 10μM R2 primer 10μl (→ 1μM final conc.) 1μM F2′-R1 primer 1μl (→ 10nM final conc.) 50ng / μl genomic DNA 4μl (→ 2ng / μl final conc.) 2 U / μl Phusion polymerase 8μl (→ 0.16 U / μl final conc.) water 42.5μl Total volume 100μl Mix well by vortexing and spin down. vi. Add 200μl oil phase to a round bottomed Cryogenic Vial containing an 8mm × 3mm magnetic bar, and position in the centre of a stirring plate set to 1,000 rpm. This is most easily done by holding the tube in a foam rack, making sure that the bottom of the tube is in contact with the stirrer. Move the tube until the sound of the spinning magnetic bar is quietest; this indicates that the magnetic bar is in the centre of the tube.
vii. Add the 100μl of aqueous phase from step v. dropwise, one drop every five seconds, to the centre of the cryogenic vial, starting a timer as the first drop is added.
viii. Continue stirring for a total of five minutes since the first drop of aqueous phase was added and stop stirring.
ix. This emulsification method creates 300μl of emulsion, which is more than can fit into a single 200μl PCR tube, so either it can be divided between two tubes, by pipetting 125μl into each, creating duplicate reactions, or the surplus can be discarded. Overlay reactions with one drop / 30μl of light mineral oil.
- x. Transfer to a thermal cycler and begin the following PCR program, using the optimal annealing temperature established in Step 11 (if using an MJ cycler, enter 100μl as the reaction volume).98°C for 30 seconds33 cycles of 98°C for 10 secondse.g. 65°C for 30 seconds72°C for 30 secondsthen 72°C for 5 minutes4°C indefinitely
B. Preparation of emulsions and HF-PCR in 96-well format (Timing: 4 hours for initial optimization, 1 hour subsequently, and 1.5 hours to perform PCR)
- i. In a 100ml measuring cylinder, add:
- 40ml Silicone polyether / cyclopentasiloxane
- 30ml Cyclopentasiloxane / trimethylsiloxysilicate
- 30ml AR20 silicone oil
ii. Cover the top of the measuring cylinder with Parafilm and mix thoroughly by vortexing / shaking.
iii. Transfer to 2x 50ml Falcon Tubes
CRITICAL STEP
This oil phase may have a cloudy appearance, which will clear over time. Mix thoroughly before use.
PAUSE POINT
This oil phase can be stored for six months at room temperature.
- iv. To allow optimisation of the emulsion preparation, prepare mock aqueous phase:
5× HF buffer 5μl 15μm fluorescent beads 1μl water 19μl Total volume 25μl Mix thoroughly and spin down v. Transfer the mock aqueous phase from Step 16Biv to a well in the centre of a 96-well polypropylene PCR plate, along with a 3mm tungsten carbide bead, and add 50μl of oil phase from Step 16Biii.
vi. Apply a Microseal ‘A’ plate seal, pressing firmly, and centrifuge briefly in a plate centrifuge with the plate seal downmost. Holding the ‘short spin’ button for 10 seconds is sufficient. The purpose is to get the bead and both oil and aqueous phases to the correct part of the well.
vii. Vortex the plate for 150 seconds, inverted, at speed 5 on a Vortex Genie 2, fitted with a microtitre plate adapter.
At the same setting, different vortexers will operate at slightly different speeds, and so it is necessary to optimize the vortexing step to determine the settings required to obtain aqueous compartments that are 5-10μM in diameter.
viii. Revert the plate and centrifuge extremely briefly. Again, holding the ‘short spin’ button for 10 seconds is sufficient. The purpose is to get the bead and emulsions to the bottom of the wells before removing the plate seal. CRITICAL STEP: Excessive centrifugation may cause separation of the emulsions.
ix. Using a pipette tip, spread 10μl of the emulsion thinly on a clean microscope slide and inspect the emulsion by fluorescence microscopy. Estimate the average diameter of aqueous compartments by comparison with the fluorescent beads. This should be 5-10μm (Fig. 2 a-d); if it is not, repeat Steps 16Bv-ix using different vortexing settings: a lower vortexing speed will increase the diameter of aqueous compartments, whereas a faster speed will reduce the diameter. Note the setting on the vortexer at Step 16Bvii necessary to obtain 5-10μm average diameter of aqueous compartments, as this setting will be used for all subsequent emulsification. TROUBLESHOOTING
- x. For HF-PCR, prepare aqueous phase as follows, adding the polymerase last. Keep reagents on ice, apart from the polymerase, which should be kept at −20°C until it is needed:
5× Phusion HF buffer 5μl (→ 1× final conc.) 50mM MgCl2 0.5μl (→2.5mM TOTAL final conc.) 2.5mM dNTP mix 2.5μl (→ 250μM each final conc.) 10μM F1 primer 2.5μl (→ 1μM final conc.) 10μM R2 primer 2.5μl (→ 1μM final conc.) 0.5μM F2′-R1 primer 0.5μl (→ 10nM final conc.) 50ng / μl genomic DNA 1μl (→ 2ng / μl final conc.) 2 U / μl Phusion polymerase 2μl (→ 0.16 U / μl final conc.) water 9.5μl Total volume 25μl Mix well by vortexing and spin down. xi. Add one 3mm tungsten carbide bead to each well of a 96-well polypropylene PCR plate, and then add 50μl of oil phase from Step 16Biii and 25μl of aqueous phase from Step 16Bx to each well.
xii. Seal the plate firmly with a Microseal ‘A’ plate seal, and centrifuge briefly, as described in Step 16Bvi.
xiii. Vortex the plate for 150 seconds, inverted, at the setting determined by the optimization (Steps 16Bv-ix).
xiv. Revert the plate and centrifuge extremely briefly as described in Step 16Bviii.
- xv. Transfer to a thermal cycler and begin the following PCR program, using the optimal annealing temperature established in Step 11 (if using an MJ cycler, enter 100μl as the reaction volume).98°C for 30 seconds33 cycles of 98°C for 10 secondse.g. 65°C for 30 seconds72°C for 30 secondsthen 72°C for 5 minutes4°C indefinitely
xvi. Proceed to Step 17 as soon as possible, ideally within an hour of the PCR reaching 4°C, as emulsions may begin to separate if left for longer.
Figure 2. Preparation of emulsions.

We prepared emulsions by vortexing and stirring as described, but we also included 10μm fluorescent microspheres in the aqueous phase, to allow us to estimate the diameter of aqueous compartments (green). Aqueous compartments prepared by vortexing were typically 5-10μm (panel a), whereas those present in stirred emulsions were larger, approximately 15μm in diameter (panel b). Regardless of the method of emulsion preparation, the diameter of aqueous compartments tends to decrease slightly during the PCR reaction (see panel a compared to c, and b compared to d).
Figure adapted with permission from Oxford Journals, NAR 2008
Disruption of emulsions (timing: 1 hour)
Transfer emulsions to a clean plate / tube using a multichannel pipette, taking care to leave any separated aqueous phase behind.
CRITICAL STEP
For emulsions in plate format, it is helpful to add 50μl of 1x Phusion HF PCR buffer to each well before transfer, to increase the volume.
CRITICAL STEP
Some separation of emulsions can occur during PCR. This will be evident as clear liquid at the bottom of wells / tubes. Fusion PCR that occurred in this aqueous phase will not necessarily result in the joining of loci from the same template molecule, and so it is essential to transfer intact emulsion to a clean plate / tube and to discard the separated aqueous phase. It is not necessary to recover the entire emulsion, and it is preferable to discard some than to retain this aqueous phase.
17. In a fume cupboard, add an equal volume of diethyl ether or hexane to emulsions, seal plates firmly with a foil seal and vortex, until a homogenous mixture is achieved.
18. For tubes, centrifuge at 13,000g, and for plates at 3000g, both for 3 minutes at 20°C.
19. In a fume cupboard, remove and discard the upper (solvent) layer using a multichannel pipette.
20. Repeat steps 18. to 20 until the solvent and aqueous phases separate cleanly.
21. Leave the plate / tubes uncovered for 15 minutes in a fume cupboard, to allow remaining ether / hexane to evaporate.
22. Add 0.8 units of Proteinase K to the recovered aqueous phase from Step 22 and incubate at 56°C for 1h to digest the polymerase.
23. Incubate at 95°C for 10 minutes to denature the Proteinase K.
24. Make the volume up to 200μl with water.
PAUSE POINT
reactions can be stored at −20°C for several weeks.
Preparation of ligation oligonucleotides (timing: 1 hour)
- 26.Phosphorylate unblocked 5′ termini in a 200μl thin-walled PCR tube. Add:
10μM IRF5_5T 5μl (→ 1μM final conc.) 10μM IRF5_5C 5μl (→ 1μM final conc.) 10μM IRF5_FusF4 10μl (→ 2μM final conc.) 10× T4 ligase buffer 5μl (→ 1× final conc.) 10U / μl T4 polynucleotide kinase 1μl (→ 0.2U / μl final conc.) water 24μl Total volume 50μl Mix thoroughly and spin down 27.Incubate for 30 minutes at 37°C
-
28.Denature the kinase by heating to 65°C for 20 minutes
PAUSE POINT: phosphorylated oligos can be stored in 10μl aliquots at −20°C for several weeks.
29.Add 5μl of 10μM primers IRF5_3G and IRF5_3T, along with water to a total volume of 100μl.
Ligation Haplotying Reaction (total time 5 hours: ligation haplotying reaction - 2 hours; digestion of surplus oligos - 1.5 hours; amplification of ligated products - 1.5 hours)
- 30.Set ligation reactions up in 200μl thin-walled PCR tubes as follows:
2μl primer mix 0.5μl 50 U / μl Tth ligase 0.4μl (→ 0.02 U / μl final conc.) water 15.1μl Total volume 20μl Mix by vortexing and spin down - 31.Transfer tubes to a thermal cycler and begin following ligation program: 95°C for 120 seconds20 cycles of 95°C for 30 seconds64°C for 240 secondsthen 4°C indefinitely
32.Add 2μl of 0.8 U / μl Proteinase K to each reaction, mix and spin down, and incubate as described in steps 23 and 24.
- 33.To digest surplus oligos, add:
10× Lambda exonuclease buffer 5μl (→ 1× final conc.) 5 U / μl Lambda exonuclease 0.2μl (→ 0.02 U / μl final conc.) 10 U / μl E. coli exo I 0.05μl (→ 0.001 U / μl final conc.) water 22.75μl Total volume 50μl Mix thoroughly and spin down 34.Incubate tubes at 37°C for an hour, followed by 65°C for 20 minutes to denature the exonucleases.
- 35.To amplify ligated oligos and to incorporate a fluorescent label, perform a final PCR reaction, in a 200μl thin-walled PCR tube. Use a proofreading DNA polymerase to avoid the addition of adenosine to the 3′ end of PCR products, to give single peaks after capillary electrophoresis:
5× Phusion HF 10μl (→ 1× final conc.) 2.5mM dNTP mix 4μl (→ 200μM each final conc.) 10μM primer MLPAF 1.5μl (→ 300μM final conc.) 10μM primer MLPAR 1.5μl (→ 300μM final conc.) Ligation product 2.5μl 2 U / μl Phusion polymerase 0.5μl (→ 0.02 U / μl final conc.) water 20μl Total volume 50μl Mix thoroughly and spin down - 36.Transfer tubes to a thermal cycler and begin the PCR program:98°C for 30 seconds33 cycles of 98°C for 10 seconds63°C for 30 seconds72°C for 30 secondsthen 72°C for 5 minutes4°C indefinitely
PAUSE POINT
Reactions can be stored at −20°C for several weeks.
Visualizing reaction products (timing: 1 hour)
37.Mix 1μl of PCR product from Step 36 with 8μl formamide and 1μl GeneScan ROX 350 size standard in a 96-well polypropylene PCR plate and spin down.
38.Heat to 96°C for 3 minutes and transfer immediately to ice
- 39.Spin plate down again, load into capillary sequencer and run, following the manufacturer’s instructions.
Timing 1 Optimization of PCRs 7 hours 16 HF-PCR A. Preparation of emulsions singly 1 hour to make emulsions, 1.5
hours to perform PCRB. Preparation of emulsions in 96-well format 4 hours for initial optimization
1 hour subsequently, and 1.5
hours to perform PCR17 Disruption of emulsions 1 hour 26 Preparation of ligation oligonucleotides 1 hour 30 Ligation Haplotying reaction 2 hours 33 digestion of surplus oligos 1.5 hours 35 amplification of ligated products 1.5 hours 37 Visualizing reaction products 1 hour Step
numberProblem Possible reason Solution 11 More than one
amplicon per PCRPrimers
insufficiently specificRedesign primers and
confirm specificity as
described in steps 1-1011 No amplicons
produced by PCRPCR master mix
prepared incorrectly,
or reagents have
deterioratedRepeat PCR with fresh
reagents step 111 Temperature
gradient range is too
highRepeat gradient PCRs
using a lower range of
annealing temperatures –
step 311 No annealing
temperature that
works for both lociAnnealing
temperature of
primer pairs does
not matchAdjust length of primers to
give more similar melting
temperatures step 115 No fusion PCR
productIncorrectly designed
fusion primerCheck sequence of fusion
primer16 B ix Aqueous
compartments too
largeVortexing speed too
lowRepeat optimization with
higher vortexing speed16 B ix Aqueous
compartments too
smallVortexing speed too
highRepeat optimization with
lower vortexing speed16 B x Emulsion is not stable
before PCRIncorrect volumes of
reagents used in oil
phaseThe components of the oil
phase are very viscous.
Make fresh oil phase,
taking care to pipette the
components slowly and
accurately16 B xii Leakage of emulsion
from platePlate sealed with
insufficient pressureMake sure that plate seal is
Microfilm ‘A’ and that it is
pressed into wells firmly16 B xvi Emulsion separates
during PCRIncorrect volumes of
aqueous and oil
phases usedThe oil phase is very
viscous. Repeat plate,
taking care to pipette the oil
phase slowly and
accurately16 B xvi Incorrect volumes of
reagents used in oil
phaseSee above 21 Some traces of
emulsion remain after
solvent stepsInadequate mixing
of solvent and
emulsionRepeat solvent washes,
taking care to mix very
thoroughly. However, it is
not essential to obtain a
completely clean aqueous
phase39 More than 2 peaks
observed per
individualAqueous phase
recovered along
with emulsionRepeat HF-PCR, taking
care only to recover intact
emulsion39 No peaks visible Sequencing run
failedRepeat sequencing run 39 Only size standard
peaks visibleNo ligation in
haplotyping reaction
due to failure of
phosphorylationRepeat oligonucleotide
preparation using fresh T4
ligase buffer. Make sure
that all solids have
dissolved before use.39 No ligation in
haplotyping reaction
due to failure of
ligaseRepeat oligonucleotide
preparation using fresh T4
ligase buffer. Make sure
that all solids have
dissolved before use.
Warm buffer to 37°C and
vortex thoroughly. Inspect
visually39 Peaks too large (i.e.
go off the Y axis
scale on visualisation
software)Too much DNA
used in sequencing
reactionRepeat sequencing with
less DNA39 Peaks differ in size by
> 10xSuboptimal
concentration of
oligos used in
ligation reactionDouble concentration of
oligos that generate small
peaks, halve concentration
of oligos that generate
large peaks
Anticipated results
The allele-specific oligos for each SNP at each position differ in length, as described in the ‘Experimental Design - Ligations’ section, differ in length. In the example given here, the two oligonucleotides at SNP 1 were designed to differ by 5 bases, and the two oligonucleotides at SNP 2 were designed to differ by 10 bases. After the fusion ligation reaction, products have a different size, depending upon the two SNPs present in the condensed haplotype template.
Ligation products are amplified by PCR, using one fluorescently labelled primer, and are resolved by capillary electrophoresis. This generates a peak, the size of which indicates the SNPs present. For autosomal haplotypes, an individual who is homozygous at the interrogated positions generates a single peak, whereas heterozygous individuals generate two peaks, representing the haplotypes on each parental chromosome. (Fig. 1).
Different allele-specific oligos will ligate with different efficiencies during the ligation haplotyping reaction, and this will give rise to different (Fig. 3). The concentration of allele-specific oligos given in steps 28 and 29 can be optimised to produce more uniform peak heights: the concentration of bridging oligo is kept constant, and for low peak heights the concentration of the relevant allele-specific oligo is doubled, whereas for large peak heights, the concentration is halved. This is usually sufficient, though the concentration can be halved or doubles once more, if necessary.
Figure 3. Optimization of Ligation Haplotyping reactions.
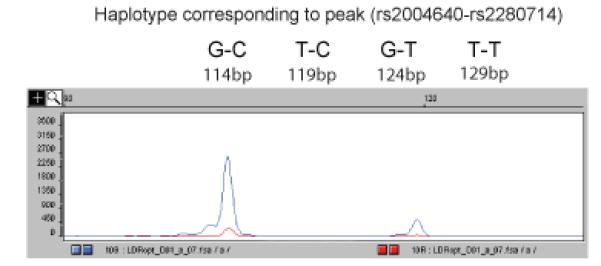
Peak heights may differ considerably after the Ligation Haplotyping reaction. To generate peaks with a more even size, the oligo concentration in the ligation reaction should be optimized. The 114bp GC peak is much larger than the 124bp GT peak – the 3G oligo is common to both peaks, indicating that the concentration of the 5T oligo should be increased.
Figure adapted with permission from Oxford Journals, NAR 2008
For the example given (Fig. 3), the 114bp GC peak is much larger than the 124bp GT peak, indicating that the concentration of the 5T oligo should be increased. In practice, it is not necessary to have identical peak heights to determine haplotypes unambiguously. It should be noted that the fragment size estimated by the sequencing software may not correspond exactly to the true size. It is helpful to perform the Ligation Haplotyping reaction on fusion PCR products that were generated in solution rather than emulsion, and to run the Ligation Haplotyping reaction products on the capillary sequencer. This will generate peaks of all four sizes, and will reveal any inaccuracy in the size calls.
IRF5 SNPs
The expression levels of several unique IRF5 isoforms is controlled by two SNPs: presence of the T allele of SNP rs2004640, in exon 1 of IRF5, permits expression of these isoforms by creating a splice site that is absent from the G allele. The T allele of the second SNP, rs2280714 is associated with higher expression levels. The effect is acts in cis, increasing the likelihood that individuals with the T SNP at both positions on the same parental chromosome will develop systemic lupus erythematosus (SLE) (Fig. 4a) 4,24.
Figure 4. Ligation haplotyping on genomic DNA.
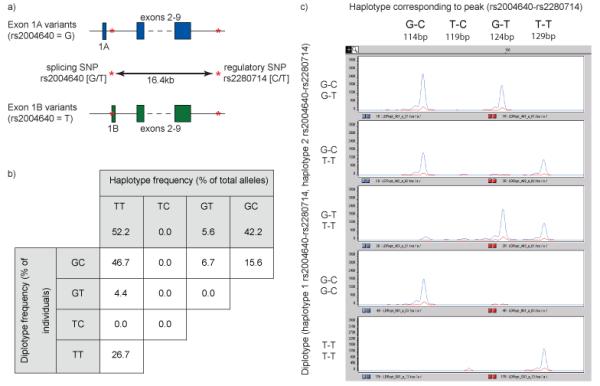
a) The G allele of rs2004640 creates IRF5 splice variants that are initiated at exon 1A. The T allele causes expression of isoforms from exon 1B. At the second locus, rs2280714, the T allele is associated with elevated gene expression compared to the C allele, and the effect acts in cis. Consequently, the T-T haplotype is associated with a heightened risk of developing systemic lupus erythematosus.
b) Haplotype inheritance was observed to be Mendelian, and the frequencies of diplotypes obeyed the Hardy-Weinberg equilibrium. None of the individuals had the TC haplotype. The GT haplotype was present at a low frequency, and no GTGT homozygotes were observed.
c) Five diplotypes were observed in the CEPH individuals used in this study, and examples of each are shown. IRF5 is on Chromosome 7, so individuals have two haplotypes, and heterozygotes will generate two peaks.
Figure adapted with permission from Oxford Journals, NAR 2008
To test the method, we obtained statistically derived haplotypes from the 30 Centre d’Etude du Polymorphisme Human (CEPH) HapMap trios 4,24, and compared these to the results obtained from Ligation Haplotyping. Both sets of results were in agreement in every case 15. Additionally, our results showed haplotype inheritance to be Mendelian, and the frequencies of diplotypes obeyed the Hardy-Weinberg equilibrium, as expected (Fig. 4b). Not all of the possible SNP combinations were observed in these samples: none of the individuals had the TC haplotype, and because of the low frequency (5.6%) of the GT haplotype, there were no GTGT homozygotes (Figs. 4b and 4c).
Acknowledgements
This work was funded by the Wellcome Trust [grant number 077014/Z/05/Z]. The authors would like to thank Chris Tyler-Smith for his work on the primary Ligation Haplotyping paper, and for his invaluable comments on this manuscript. We would also like to thank Robert Graham for statistically derived IRF5 haplotype data and Oxford Journals for permission to reuse figures from the primary research paper.
Footnotes
Competing Financial Interests The authors declare that they have no competing financial interests
Contributor Information
Daniel J. Turner, Email: djt@sanger.ac.uk.
Matthew E. Hurles, Email: meh@sanger.ac.uk.
References
- 1.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 2.Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci U S A. 2001;98:4563–4568. doi: 10.1073/pnas.081068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 4.Sigurdsson S, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salem RM, Wessel J, Schork NJ. A comprehensive literature review of haplotyping software and methods for use with unrelated individuals. Hum Genomics. 2005;2:39–66. doi: 10.1186/1479-7364-2-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchini J, et al. A comparison of phasing algorithms for trios and unrelated individuals. Am J Hum Genet. 2006;78:437–450. doi: 10.1086/500808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium TIH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S, Cutler DJ, Zwick ME, Chakravarti A. Haplotype inference in random population samples. Am J Hum Genet. 2002;71:1129–1137. doi: 10.1086/344347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo YM, et al. Direct haplotype determination by double ARMS: specificity, sensitivity and genetic applications. Nucleic Acids Res. 1991;19:3561–3567. doi: 10.1093/nar/19.13.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald OG, Krynetski EY, Evans WE. Molecular haplotyping of genomic DNA for multiple single-nucleotide polymorphisms located kilobases apart using long-range polymerase chain reaction and intramolecular ligation. Pharmacogenetics. 2002;12:93–99. doi: 10.1097/00008571-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Michalatos-Beloin S, Tishkoff SA, Bentley KL, Kidd KK, Ruano G. Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res. 1996;24:4841–4843. doi: 10.1093/nar/24.23.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paabo S, Irwin DM, Wilson AC. DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem. 1990;265:4718–4721. [PubMed] [Google Scholar]
- 14.Turner DJ, et al. Assaying chromosomal inversions by single-molecule haplotyping. Nat Methods. 2006;3:439–445. doi: 10.1038/nmeth881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner DJ, Tyler-Smith C, Hurles ME. Long-range, high-throughput haplotype determination via haplotype-fusion PCR and ligation haplotyping. Nucleic Acids Res. 2008;36:e82. doi: 10.1093/nar/gkn373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yon J, Fried M. Precise gene fusion by PCR. Nucleic Acids Res. 1989;17:4895. doi: 10.1093/nar/17.12.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 19.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barany F. Genetic disease detection and DNA amplification using cloned thermostable ligase. Proc Natl Acad Sci U S A. 1991;88:189–193. doi: 10.1073/pnas.88.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan JB, et al. Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol. 2003;68:69–78. doi: 10.1101/sqb.2003.68.69. [DOI] [PubMed] [Google Scholar]
- 22.Tobler AR, et al. The SNPlex genotyping system: a flexible and scalable platform for SNP genotyping. J Biomol Tech. 2005;16:398–406. [PMC free article] [PubMed] [Google Scholar]
- 23.Ghadessy FJ, Ong JL, Holliger P. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci U S A. 2001;98:4552–4557. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham RR, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]