

Abstract
Shown to lower amyloid deposits and improve cognition in APP transgenic mouse models, immunotherapy appears to be a promising approach for the treatment of Alzheimer's disease (AD). Due to limitations in available animal models, however, it has been unclear whether targeting amyloid is sufficient to reduce the other pathological hallmarks of AD—namely, accumulation of pathological, nonmutated tau and neuronal loss. We have now developed two transgenic mouse models (APPSw/NOS2−/− and APPSwDI/NOS2−/−) that more closely model AD. These mice show amyloid pathology, hyperphosphorylated and aggregated normal mouse tau, significant neuron loss, and cognitive deficits. Aβ1–42 or KLH vaccinations were started in these animals at 12 months, when disease progression and cognitive decline are well underway, and continued for 4 months. Vaccinated APPSwDI/NOS2−/− mice, which have predominantly vascular amyloid pathology, showed a 30% decrease in brain Aβ and a 35–45% reduction in hyperphosphorylated tau. Neuron loss and cognitive deficits were partially reduced. In APPSw/NOS2−/− vaccinated mice, brain Aβ was reduced by 65–85% and hyperphosphorylated tau by 50–60%. Furthermore, neurons were completely protected, and memory deficits were fully reversed. Microhemorrhage was observed in all vaccinated APPSw/NOS2−/− mice and remains a significant adverse event associated with immunotherapy. Nevertheless, by providing evidence that reducing amyloid pathology also reduces nonmutant tau pathology and blocks neuron loss, these data support the development of amyloid-lowering therapies for disease-modifying treatment of AD.
Introduction
The amyloid hypothesis of Alzheimer's disease (AD) states that Aβ accumulation and aggregation results in tau hyperphosphorylation and aggregation, which ultimately leads to neuronal death and, finally, dementia (Hardy and Selkoe, 2002). The hypothesis has led to the development of potential therapeutics aimed at either lowering Aβ production or accelerating the clearance of Aβ. One such approach is immunotherapy targeting Aβ using active vaccination or passive immunization (Wilcock and Colton, 2008). Active vaccination uses Aβ combined with an immune adjuvant to stimulate the recipients' immune system to produce anti-Aβ antibodies (Schenk et al., 1999). Passive immunization involves delivery of monoclonal antibodies directed to portions of the Aβ protein (Bard et al., 2000). Both forms of immunotherapy have been shown to lower amyloid levels and improve learning and memory in APP transgenic mice (Janus et al., 2000; Morgan et al., 2000; Wilcock et al., 2004b) and have progressed into clinical trials (Orgogozo et al., 2003; Bayer et al., 2005).
Transgenic mouse models have been useful tools to study AD, but many do not fully recapitulate the cascade of amyloid plaques, neurofibrillary tangles, and neurodegeneration that characterize the human disease (Schwab et al., 2004). We recently developed two transgenic AD mouse models that progress from amyloid deposition to hyperphosphorylated and aggregated native tau pathology with significant neuronal loss. These mice were generated by genetic deletion of nitric oxide synthase 2 (NOS2; iNOS) in the APPSw (Colton et al., 2006) and APPSwDI (Wilcock et al., 2008) mouse strains. The rationale for reducing NOS2 in our mouse models has been detailed in a recent review (Colton et al., 2008) and is based on the immunosuppression of NOS2 in chronic inflammatory diseases (El-Gayar et al., 2003; Gordon, 2003; Minghetti et al., 2005) and on the striking species-specific differences in the NOS2 gene between humans and rodents (Weinberg et al., 1995; Colton et al., 1996; Ganster et al., 2001; Mestas and Hughes, 2004), At 12–13 months of age, APPSw/NOS2−/− mice develop moderate amyloid deposition with moderate vascular involvement, AT8-positive and thioflavin-S-positive tau pathology, 30% hippocampal neuron loss, and significant memory deficits as detected by the radial-arm water maze (Colton et al., 2006). Similarly aged APPSwDI/NOS2−/− animals develop severe amyloid deposition with a high vascular involvement, AT8-positive and thioflavin-S-positive tau pathology, 35% hippocampal neuron loss, and significant memory deficits as detected by the radial-arm water maze (Wilcock et al., 2008).
The impact of amyloid-based therapeutics on the other pathological hallmarks of AD progression has not been adequately examined due to a lack of animal models that sufficiently resemble AD. We show here, for the first time, that lowering amyloid in both APP/NOS2−/− mouse models also significantly reduces tau pathology, protects from neuron loss, and improves learning and memory.
Materials and Methods
Animals.
The APPSwDI/NOS2−/− mice were produced by crossing APPSwDI (Swedish K760N/M671L, Dutch E693Q, and Iowa D694N) (Davis et al., 2004) transgenic mice with NOS2−/− (B6 129P2NOS2tau1Lau/J) (Laubach et al., 1995) mice (The Jackson Laboratory), as described previously (Wilcock et al., 2008). The APPSw/NOS2−/− mice were produced by crossing APPSw (Tg2576, Swedish K760N/M671L) transgenic mice (Hsiao et al., 1996) with NOS2−/− (B6 129P2NOS2tau1Lau/J) mice (The Jackson Laboratory), as described previously (Colton et al., 2006). Mice aged 12 months were assigned to one of two treatment groups. Final sample sizes were six APPSw/NOS2−/− mice receiving control (KLH) vaccination, six APPSw/NOS2−/− mice receiving Aβ vaccination, seven APPSwDI/NOS2−/− mice receiving control vaccination, eight APPSwDI/NOS2−/− mice receiving Aβ vaccination, six NOS2−/− mice receiving control vaccination, and six NOS2−/− mice receiving Aβ vaccination. All mice were killed and brain pathology examined at 16 months of age. The pathologies already present in mice by 12 months of age are summarized in Table 1.
Table 1.
Summarizes the pathologies of the APPSwDI/NOS2−/− and the APPSw/NOS2−/− mice aged 12 months, the age at the beginning of this study
| Mouse | Amyloid deposition (% area) |
Aβ ELISA (pg/mg protein) |
AT8-positive tau | Neuron loss (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| FCX | HPC | SUB | Total soluble | Total insoluble | Total Aβ40 | Total Aβ42 | |||
| APPSw/NOS2−/− | 5.8 ± 0.6 | 4.9 ± 0.3 | 1.6 ± 0.2 | 291 ± 126 | 2881 ± 960 | 2480 ± 800 | 880 ± 250 | Extensive | 30–35 |
| APPSwDI/NOS2−/− | 16.7 ± 1.9 | 18.5 ± 3 | 18.6 ± 4.4 | 1200 ± 61 | 27,544 ± 4500 | 27,000 ± 4400 | 1700 ± 225 | Extensive | 35–40 |
FCX, Frontal cortex; HPC, hippocampus; SUB, subiculum.
Vaccine preparation and administration.
Human Aβ1–42 peptide (rPeptide) or KLH (EMD Biosciences) was suspended in pyrogen-free type I water at 2.2 mg/ml, then mixed with 10× PBS to yield 1× PBS, and incubated overnight at 37°C. For the first vaccination, the antigen suspension was mixed 1:1 with Freund's complete adjuvant (Sigma-Aldrich), and 100 mg of Aβ was injected subcutaneously. For the following three vaccinations, the antigen suspension was mixed 1:1 with Freund's incomplete adjuvant (Sigma-Aldrich). The first two vaccinations were administered biweekly, the next two vaccinations monthly. Four weeks following the final vaccination, at 16 months of age, mice were tested in the radial-arm water maze and killed.
Two-day radial arm water maze.
Mice were initially tested at 12 months of age using the 2 d radial-arm water maze. The 2 d radial-arm water maze has been described in detail previously (Alamed et al., 2006; Wilcock et al., 2008). Briefly, a six-arm maze is submerged in a pool of water, and a platform is placed at the end of one arm. The mouse receives 15 trials per day for 2 d. The mouse begins each trial in a different arm while the arm containing the platform remains the same. The numbers of errors (incorrect arm entries) are counted over a 1 min period. The errors are averaged over three trials, resulting in 10 blocks for the 2 d period (blocks 1–5 are day 1, while blocks 6–10 are day 2). The task was repeated as above immediately before the mice were killed at age 16 months.
Tissue processing and histology.
After injection with a lethal dose of ketamine, blood was collected and the mice were perfused intracardially with 25 ml of normal saline. Brains were rapidly removed and bisected in the midsagittal plane. One half of the brain was immersion fixed in 4% paraformaldehyde, while the other was snap-frozen in liquid nitrogen and stored at −80°C. After being allowed to clot for 2 h at 4°C, the blood samples were centrifuged at 3800 rpm for 15 min, and the serum was collected and stored at −80°C. Tissue was processed, and 25 μm frozen sagittal sections were collected as described previously (Wilcock et al., 2008). Eight 25 μm sections equally spaced 600 mm apart were selected for free-floating immunohistochemistry for Aβ (rabbit polyclonal anti-Aβ N terminal, Biosource, 1:3000), NeuN (mouse monoclonal, Millipore Bioscience Research Reagents, 1:3000) or PHF-tau (AT8, a mouse monoclonal for PHF-tau recognizing phosphorylated Ser202 in tau, Thermo Scientific, 1:250). The method for free-floating immunohistochemistry has been described previously (Wilcock et al., 2004a). Additionally, eight 25 μm sections equally spaced 600 mm apart were selected, mounted on slides, and stained in a 0.5% cresyl violet solution (Sigma-Aldrich) for 5 min at room temperature. The sections were then differentiated in 70 and 95% ethanol solutions before being coverslipped.
Antibody titer measurement.
Serum antibody titers were measured using a method described previously (Morgan et al., 2000). Briefly, 96-well plates were coated with Aβ1–42 for 1 h at 37°C. After blocking, dilutions of serum were prepared in PBS at an initial dilution of 1:4 and subsequent two-fold dilutions were performed in the plate. Using adjacent wells in the 96-well plate, known concentrations of an anti-Aβ monoclonal antibody (6E10; Covance) were added to generate a standard curve. Anti-mouse peroxidase (Sigma-Aldrich) was added, and the ELISA was developed using 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich). The Softmax program (Molecular Devices) was used to calculate antibody concentration using the standard curve generated by the monoclonal antibody.
Aβ load calculation.
Percentage immunoreactive area for Aβ total and vascular Aβ was measured using the Image-Pro Plus software as described previously (Wilcock et al., 2008). Stereological analysis of the NeuN-positive neurons in the CA3 was performed as described previously (Wilcock et al., 2008). The data were exported to an Excel spreadsheet where statistical analysis was performed.
Aβ ELISA.
Soluble and insoluble pools of Aβ1–40 and Aβ1–42 were measured by sandwich ELISA using commercially available kits (Covance) according to manufacturer's instructions. Frozen hemibrains were pulverized using a mortar and pestle on dry ice. Samples were homogenized in TBS containing both protease and phosphatase inhibitor cocktails for the soluble Aβ pool. This sample was centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was removed, and the pelleted material was homogenized with 70% formic acid for the insoluble Aβ pool. Again, this sample was centrifuged at 100,000 × g for 1 h at 4°C and the supernatant removed. The supernatant was neutralized by a 1:20 dilution into 1 m Tris phosphate buffer, pH 11. Protein concentrations were calculated using the BCA protein assay kit (Thermo Scientific) according to manufacturer's instructions. The Softmax program (Molecular Devices) was used to calculate Aβ concentration (in picograms). These values were normalized to protein concentrations (in picograms per milligram). Total Aβ1–40 and Aβ1–42 levels were obtained by adding the values of the soluble and insoluble levels.
Western blotting.
Protein was extracted from pulverized brain powder and quantified using the BCA protein assay kit. Fifteen microgram protein samples from each lysate were run on a denaturing 4–20% SDS-PAGE gel. The gel was transferred onto a nitrocellulose membrane, and Western blots were performed for AT8 (mouse anti-PHF tau antibody 1:200, Thermo Scientific) or AT180 (mouse anti-PHF tau antibody 1:500 Thermo Scientific) as described previously (Wilcock et al., 2008). The blots were stripped using Restore stripping buffer (Thermo Scientific) and reprobed using the above protocol for tau 5 (mouse anti-tau 5, EMD Biosciences, 1:3000). Densitometry was performed as described previously (Wilcock et al., 2008).
Statistics.
The significance of genotype- and treatment-specific behavioral changes were analyzed by the unpaired Student's t test or two-way ANOVA. All immunohistochemical, stereological, ELISA, and Western blot data were analyzed by one-way ANOVA. The statistical analysis software JMP (version 7, SAS) was used for all statistical analyses with p < 0.05 judged as significant. All graphs were made using GraphPad Prism 4.
Results
All mice receiving Aβ vaccination generated significant anti-Aβ antibody titers, with no statistical differences between genotypes. NOS2−/− mice generated titers of 525 ± 170 μg/ml, APPSwDI/NOS2−/− mice generated titers of 314 ± 103 μg/ml, and APPSw/NOS2−/− mice generated titers of 421 ± 94 μg/ml. Also, importantly, Aβ vaccination did not alter either NOS1 or NOS3 mRNA expression levels from control KLH-vaccinated levels. Fold changes in mRNA expression of NOS1 and NOS3 for APPSw/NOS2−/− and APPSwDI/NOS2−/− mice are shown in Table 2. Similar compensatory responses in the expression of NOS1 and NOS3 have been previously observed in NOS2−/− mice (Colton et al., 2006; Wilcock et al., 2008).
Table 2.
Expression of NOS1 and NOS3
| APPSwDI/NOS2−/−
|
APPSw/NOS2−/−
|
|||
|---|---|---|---|---|
| KLH | Aβ VACC | KLH | Aβ VACC | |
| NOS1 mRNA (fold change vs wild type) | 0.70 ± 0.019 | 0.69 ± 0.021 | 0.68 ± 0.014 | 0.66 ± 0.072 |
| NOS3 mRNA (fold change vs wild type) | 1.39 ± 0.031 | 1.41 ± 0.029 | 1.34 ± 0.026 | 1.37 ± 0.035 |
Compensatory changes in mRNA expression for NOS1 and NOS3 are not affected by vaccination (VACC).
Aβ-vaccinated APPSwDI/NOS2−/− mice show deceased Aβ and tau pathology
APPSwDI/NOS2−/− mice receiving 4 months of control (KLH) vaccination showed a typical staining pattern for total Aβ in the brain (Wilcock et al., 2008). Abundant Aβ immunostain was observed in the hippocampus throughout the dentate gyrus and CA3 regions as well as extensive cerebrovascular amyloid deposition in the subiculum (Fig. 1 A). In Aβ-vaccinated APPSwDI/NOS2−/− mice, the extensive, diffuse Aβ staining throughout the dentate gyrus and CA3, as well as the overall staining appearance, was reduced (Fig. 1 B). Immunization with Aβ resulted in a 30% reduction in total Aβ in the hippocampus and frontal cortex, with no significant reduction in the subiculum (Fig. 1 C). The same images used for quantification of total Aβ were also analyzed for vascular amyloid deposition. No significant changes were found when comparing APPSwDI/NOS2−/− mice receiving control or Aβ vaccination. We also performed biochemical measurements of Aβ using ELISA methods on protein extracted from the right hemisphere. We found significant, 40% reductions in both Aβ40 (Fig. 1 E) and Aβ42 (Fig. 1 F). No significant differences were noted when soluble and insoluble Aβ levels were analyzed separately.
Figure 1.
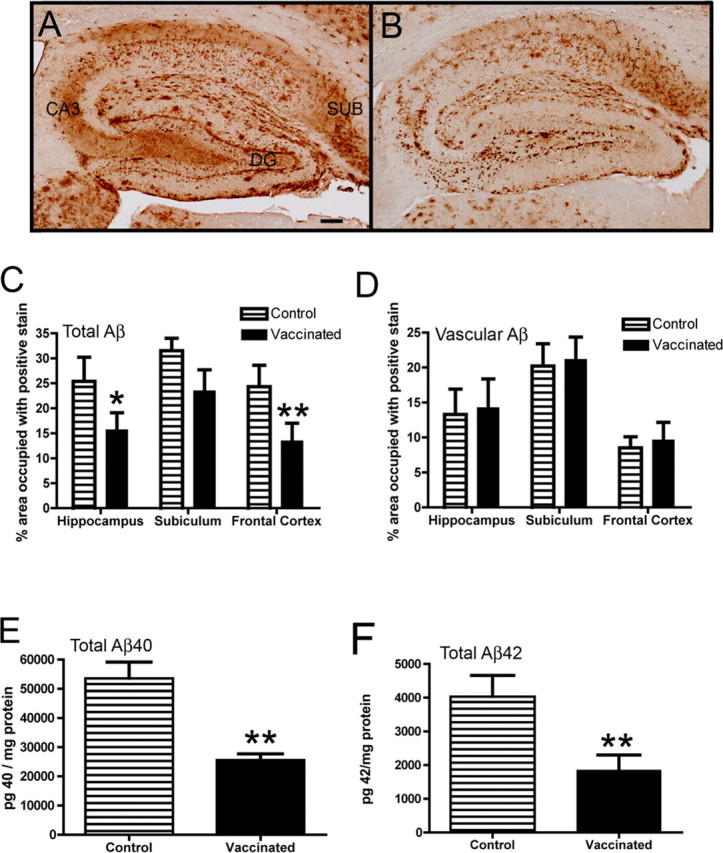
Aβ deposition is reduced in APPSwDI/NOS2−/− mice receiving Aβ vaccination. A and B show Aβ immunohistochemistry in the hippocampus of APPSwDI/NOS2−/− mice receiving either control vaccination (A) or Aβ vaccination (B) for 4 months. DG, Dentate gyrus; CA3, cornu ammonis 3; SUB, subiculum. Magnification, 40×; scale bar, 120 μm. C and D show quantification of percentage area occupied by positive immunostain in the whole field (C) or restricted to the cerebrovasculature (D). E and F show ELISA measurements of Aβ40 (E) or Aβ42 (F). *p < 0.05, **p < 0.01 compared with NOS2−/− mice.
Tau hyperphosphorylated at Ser202/Thr205 is associated with AD-relevant pathology and is detected by the AT8 antibody (Iqbal and Grundke-Iqbal, 1997). Immunohistochemistry for AT8-positive tau in KLH-immunized 16-month-old APPSwDI/NOS2−/− mice showed a staining pattern previously observed (Wilcock et al., 2008), that is, AT8-immunopositive neuronal cell bodies were found scattered throughout the cortex while strong staining was observed in the subiculum. The cell bodies of cortical neurons appeared densely stained for AT8 in control-treated APPSwDI/NOS2−/− mice (Fig. 2 A). This staining was significantly reduced in APPSwDI/NOS2−/− mice receiving Aβ vaccination (Fig. 2 B).
Figure 2.

Tau pathology and neuron loss are reduced in APPSwDI/NOS2−/− mice receiving Aβ vaccination. A and B show AT8 immunohistochemistry in the cerebral cortex of APPSwDI/NOS2−/− mice receiving either control vaccination (A) or Aβ vaccination (B). Magnification, 200×; scale bar, 25 μm. C shows representative Western blots for AT8, AT180, and tau 5 from APPSwDI/NOS2−/− mice receiving control (control) or Aβ vaccination (Vacc). D shows mean densitometry data of AT8 and AT180 Western blots, normalized to total tau levels. N = 7 mice receiving control vaccination and N = 8 receiving Aβ vaccination. E–H show NeuN immunohistochemistry in the hippocampus (E and F, magnification, 40×; scale bar, 120 μm) and CA3 (G and H, magnification, 200×; scale bar, 25 μm) of APPSwDI/NOS2−/− mice receiving either control (E, G) or Aβ (F, H) vaccination. Dashed boxes in E and F indicate the region shown at higher magnification in G and H. I and J show cresyl violet staining of the CA3 of APPSwDI/NOS2−/− mice receiving either control (I) or Aβ (J) vaccination. Magnification, 200×; scale bar, 25 μm. K shows stereological counts of the CA3 region of NOS2−/−, APPSwDI/NOS2−/− control, and APPSwDI/NOS2−/− mice receiving Aβ vaccination. *p < 0.05, **p < 0.01 compared with NOS2−/− mice.
Another pathologically relevant phosphorylation site on the tau protein is Thr231, which is detected by the AT180 antibody (Iqbal and Grundke-Iqbal, 1997). Protein extracted from the right hemisphere was run on an SDS-PAGE gel and probed for AT8, AT180, and total tau by Western blot. Compared with mice receiving control vaccination, Aβ-immunized APPSwDI/NOS2−/− mice had reduced AT8 and AT180 signal (Fig. 2 C). To control for any changes in total tau due to amyloid changes or vaccination, we performed a Western blot using tau 5, which detects total tau, and found no significant changes in total tau levels in any of the mice examined (Fig. 2 C). Densitometry analysis revealed significant reductions in both AT8 (40%) and AT180 (35%) levels following Aβ vaccination (Fig. 2 D).
Aβ immunization partially relieves neuron loss in APPSwDI/NOS2−/− mice
Neuron loss has previously been shown in the hippocampus and subiculum of the APPSwDI/NOS2−/− mice (Wilcock et al., 2008). The CA3 region appears to be particularly vulnerable and shows thinning, with 35% loss of neurons at 12 months of age. We have previously shown that NOS2−/− mice are indistinguishable from wild-type mice with respect to neuron counts (Wilcock et al., 2008). In the current study, neurons were stained with Nissl and NeuN staining, which are closely correlated with respect to neuron number (Gittins and Harrison, 2004). CA3 thinning was observed using NeuN immunohistochemistry (Fig. 2 E–H) and cresyl violet histology (Fig. 2 I,J) in APPSwDI/NOS2−/− mice receiving control vaccination (Fig. 2 E,G,I). We also observed thinning of the CA3 in Aβ-vaccinated APPSwDI/NOS2−/− mice; however, this appeared less dramatic (Fig. 2 F,H,J). Since NeuN has been shown to be more sensitive in the detection of neuronal lesions than Nissl (Jongen-Rêlo and Feldon, 2002) and since NeuN has the benefit of only labeling neurons with no glial nuclei staining, we counted NeuN-positive cells by stereology. Approximately 50% of NeuN-immunopositive neurons were lost in the CA3 region in KLH-vaccinated APPSwDI/NOS2−/− mice at 16 months of age compared with NOS2−/− mice, while only 40% of the neurons were lost in APPSwDI/NOS2−/− mice immunized with Aβ. These data suggest that Aβ vaccination partially protected against neuron loss in this mouse strain. The decreased loss of neurons in Aβ-vaccinated mice compared with KLH-vaccinated mice was significant at the p < 0.05 level (Fig. 2 K).
Αβ immunization rescues learning defects in APPSwDI/NOS2−/− mice
APPSwDI/NOS2−/− mice and NOS2−/− mice aged 12 months were tested in the 2 d radial arm water maze immediately before initiating vaccination treatments. All mice made the same number of errors at the beginning of the task. By the end of day 2, the NOS−/− mice were making <1 error, indicating full acquisition of the task. In contrast, the APPSwDI/NOS2−/− mice showed no significant learning. The same mice were retested after 4 months of KLH or Αβ vaccination. The KLH-immunized APPSwDI/NOS2−/− animals still made >2 errors by the end of the second day, and were significantly impaired when compared with NOS2−/− mice at the same time point. On the other hand, APPSwDI/NOS2−/− mice receiving Aβ vaccination performed comparably to NOS2−/− mice and were significantly better than the APPSwDI/NOS2−/− mice receiving control vaccination at the end of day 2 (Fig. 3 B).
Figure 3.

APPSwDI/NOS2−/− mice show partial reversal of memory deficits following Aβ vaccination. A and B show radial-arm water maze data for APPSwDI/NOS2−/− mice and NOS2−/− mice before treatment (A) and after 4 months of either control or Aβ vaccination (B). No effect of vaccination was observed in NOS2−/− mice, so they are shown as a single group in B. Blocks 1–5 are day 1 and block 6–10 are day 2. *p < 0.05, **p < 0.01 compared with NOS2−/− mice for the given block number.
Aβ vaccine markedly reduces amyloid and tau pathology in APPSw/NOS2−/−mice
Compared with the APPSwDI/NOS2−/− strain, APPSw/NOS2−/− mice have significantly less total Aβ deposition in the brain (Colton et al., 2006). In the hippocampus, the primary regions of Aβ deposition are the dentate gyrus, the hippocampal fissure separating the dentate gyrus from the CA1, CA2 regions, and the subiculum. The APPSw/NOS2−/− mice receiving control vaccination showed this typical staining pattern (Fig. 4 A). In contrast, Aβ-immunized APPSw/NOS2−/− mice had weaker total Aβ immunohistochemical staining, and amyloid deposits were more sparsely distributed, despite similar localization (Fig. 4 B). Quantification of total Aβ immunohistochemistry showed an 85% reduction in the hippocampus and frontal cortex, while a more subtle 20% reduction was observed in the subiculum (Fig. 4 C). Analyzing the same images for Aβ staining in the cerebrovasculature, we found no significant difference between APPSw/NOS2−/− mice receiving either control or Aβ vaccination (Fig. 4 D). Using ELISA to measure levels of Aβ protein extracted from the right hemisphere, we found a 50% reduction in Aβ40 (Fig. 4 E) and a >65% reduction in Aβ42 (Fig. 4 F). No significant differences were noted when soluble and insoluble levels were analyzed separately.
Figure 4.
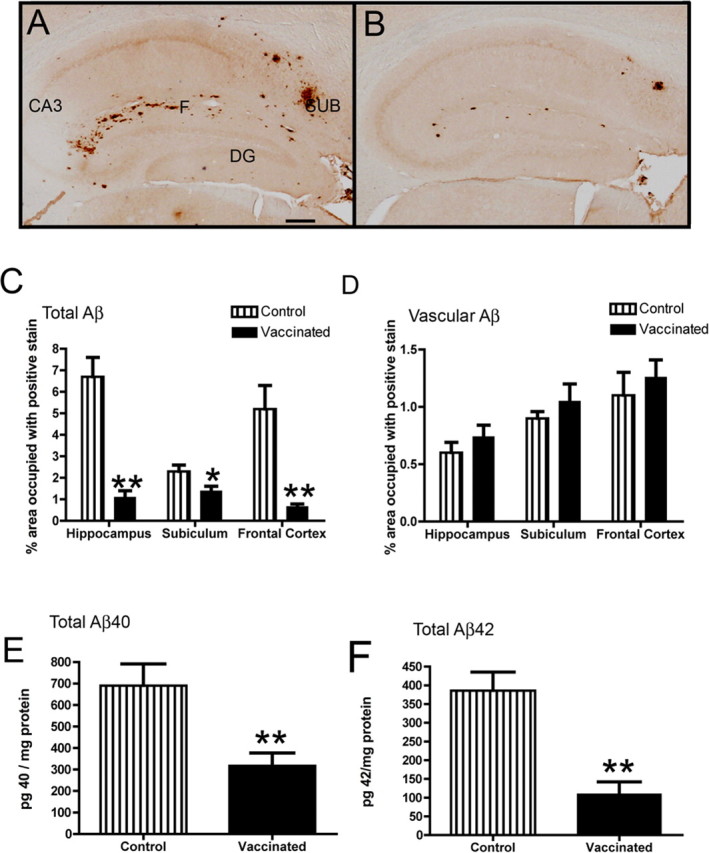
Aβ deposition is reduced in APPSw/NOS2−/− mice receiving Aβ vaccination. A and B show Aβ immunohistochemistry in the hippocampus of APPSw/NOS2−/− mice receiving either control vaccination (A) or Aβ vaccination (B) for 4 months. DG, Dentate gyrus; CA3, cornu ammonis 3; SUB, subiculum; F, hippocampal fissure. Magnification, 40×; scale bar, 120 μm. C and D show quantification of percentage area occupied by positive immunostain in the whole field (C) or restricted to the cerebrovasculature (D). E and F show ELISA measurements of Aβ40 (E) or Aβ42 (F). *p < 0.05, **p < 0.01 compared with NOS2−/− mice.
We have previously shown that AT8-positive staining is detected in the cell bodies of neurons throughout the cerebral cortex and hippocampus of 12-month-old APPSw/NOS2−/− mice (Colton et al., 2006). In the current study, we observe the same staining pattern in APPSw/NOS2−/− mice receiving control vaccination, with numerous cell bodies positive for hyperphosphorylated tau (Fig. 5 A). Following Aβ vaccination, there are fewer AT8-positive neurons, and the AT8 signal appears less intense (Fig. 5 B). Protein extracted from the right hemisphere was run on an SDS-PAGE gel and probed for AT8, AT180, and total tau by Western blot. Both AT8 and AT180 in APPSw/NOS2−/− mice receiving Aβ vaccination showed significant reductions in signal compared with KLH-treated control mice (Fig. 5 C). Densitometry analysis showed significant reductions in both AT8 (60%) and AT180 (50%) levels, when normalized to total tau levels, following Aβ vaccination (Fig. 5 D).
Figure 5.

Tau pathology and neuron loss are reduced in APPSw/NOS2−/− mice receiving Aβ vaccination. A and B show AT8 immunohistochemistry in the cerebral cortex of APPSw/NOS2−/− mice receiving either control vaccination (A) or Aβ vaccination (B). Magnification, 100×; scale bar, 50 μm. C shows representative Western blots for AT8, AT180, and tau 5 from APPSw/NOS2−/− mice receiving control (control) or Aβ vaccination (Vacc). D shows mean densitometry data of AT8 and AT180 Western blots, normalized to total tau levels. N = 5 mice receiving control vaccination and N = 5 receiving Aβ vaccination. E–H show NeuN immunohistochemistry in the hippocampus (E and F, magnification, 40×; scale bar, 120 μm) and CA3 (G and H, magnification, 200×; scale bar, 25 μm) of APPSwDI/NOS2−/− mice receiving either control (E, G) or Aβ (F, H) vaccination. Dashed boxes in E and F indicate the region shown at higher magnification in G and H. I and J show cresyl violet staining of the CA3 of APPSw/NOS2−/− mice receiving either control (I) or Aβ (J) vaccination. Magnification, 200×; scale bar, 25 μm. K shows stereological counts of the CA3 region of NOS2−/−, APPSw/NOS2−/− control, and APPSw/NOS2−/− mice receiving Aβ vaccination. *p < 0.05, **p < 0.01 compared with NOS2−/− mice.
Aβ vaccine reduces neuron loss in APPSw/NOS2−/− mice
Approximately 30% of the neurons in the hippocampus and subiculum were lost at 12 months of age in the APPSw/NOS2−/− mice (Colton et al., 2008). To detect further neuron loss in KLH- or Aβ-vaccinated APPSw/NOS2−/− mice at 16 months of age, NeuN immunohistochemistry (Fig. 5 E–H) and cresyl violet histology (Fig. 5 I,J) were again used to detect the density of neurons in the CA3 region. Visible thinning of the CA3 was observed using both stains in the APPSw/NOS2−/− mice receiving control vaccination (Fig. 5 E,G,I). While some thinning of the CA3 was seen in APPSw/NOS2−/− mice receiving Aβ vaccination, the overall density of neurons appeared greater than control vaccinated mice (Fig. 5 F,H,J). These data were confirmed using unbiased stereology. Stereological counts of NeuN-positive CA3 neurons revealed a 45% loss of neurons in KLH-immunized APPSw/NOS2−/− mice compared with NOS2−/− mice. In contrast, Aβ-vaccinated APPSw/NOS2−/− animals lost only 30% of their neurons, suggesting that the vaccine protected against neuron loss in APPSw/NOS2−/− animals (Fig. 5 K).
Aβ immunization reverses cognitive decline in APPSw/NOS2−/− mice
APPSw/NOS2−/− mice and NOS2−/− mice (12 months old) were tested in the 2 d radial arm maze before vaccination. APPSw/NOS2−/− mice were significantly impaired in this task compared with the NOS2−/− mice (Fig. 6 A). While the NOS2−/− mice appeared to fully acquire the task by the end of the second day, the APPSw/NOS2−/− mice demonstrated slowed learning, making ∼3 errors at this time point. The same mice were retested after 4 months of KLH control or Aβ vaccination. The KLH-immunized APPSw/NOS2−/− mice made >3 errors by the end of the second day, and were significantly impaired when compared with NOS2−/− mice. In contrast, APPSw/NOS2−/− mice receiving Aβ vaccination were indistinguishable from NOS2−/− mice, making <1 error at the end of the second day (Fig. 6 B). This suggests that deficits that were present before treatment were no longer detectable following Aβ vaccination.
Figure 6.

APPSw/NOS2−/− mice show complete reversal of memory deficits following Aβ vaccination. A and B show radial-arm water maze data for APPSw/NOS2−/− mice and NOS2−/− mice before treatment (A) and after 4 months of either control or Aβ vaccination (B). No effect of vaccination was observed in NOS2−/− mice, so they are shown as a single group in B. Blocks 1–5 are day 1 and block 6–10 are day 2. *p < 0.05, **p < 0.01 compared with NOS2−/− mice for the given block number.
Microhemorrhage in Aβ-vaccinated APPSw/NOS2−/− but not APPSwDI/NOS2−/− mice
We used the Prussian blue histological stain to label hemosiderin, a ferric oxide material produced in the breakdown of hemoglobin. Extravenous blood in the brain leads to microglial phagocytosis of the erythrocytes and breakdown of the hemoglobin within them. Hence, ferric oxide-containing microglia serve as markers of past hemorrhage (Koeppen, 1995). Microhemorrhages are typically seen in very small numbers in APPSw mice (Wilcock et al., 2004a), and this was also the case in the APPSw/NOS2−/− mice, where we observed microhemorrhages in 3 of 6 mice. In these 3 mice, the microhemorrhages were small and only detectable on 1–2 sections. In contrast, APPSw/NOS2−/− mice receiving Aβ vaccination all showed microhemorrhages, with bleeds ranging in size from large, diffuse areas (Fig. 7 A) to small, single-cell-sized blue profiles (Fig. 7 B). The location of these bleeds was not predictable. They appeared in leptomeningeal vessels (Fig. 7 C) and in intraparenchymal vessels (Fig. 7 D) of the cerebral cortex, thalamus, hippocampus, and striatum. Quantification of microhemorrhages was performed by counting the number of microhemorrhage profiles per brain section as shown previously (Wilcock et al., 2004a). The average was 3 and showed an almost 10-fold increase over mice receiving control vaccination (Fig. 7 E). Importantly, no microhemorrhages were detected in the APPSwDI/NOS2−/− mice (data not shown).
Figure 7.

Microhemorrhages are significantly increased in APPSw/NOS2−/− receiving Aβ vaccination. A–D show examples of the microhemorrhages observed in Aβ-vaccinated APPSw/NOS2−/− mice. The images show Prussian blue-positive microhemorrhages (blue) counterstained with neutral red (red) to provide background. Magnification, 200×;Y scale bar, 25 μm. E shows quantification of microhemorrhages in APPSw/NOS2−/− mice receiving either control or Aβ vaccination and is expressed as mean number of Prussian blue profiles per section. **p < 0.01 compared with control vaccination.
Discussion
Amyloid-lowering therapies, such as Aβ immunotherapy, represent one of the most promising approaches for the treatment of AD. Though a number of studies have documented benefits in neuronal function (Klyubin et al., 2005) and in learning and memory (Morgan et al., 2000; Dodart et al., 2002), the impact of amyloid lowering on normal, nonmutant tau pathology and neuron loss has not yet been described. By using two mouse models that develop amyloid deposits, native mouse tau hyperphosphorylation and neuron loss, we show here, for the first time, that lowering amyloid levels by active Aβ vaccination in fact reduces native mouse tau pathology and protects from neuron loss. The APPSwDI/NOS2−/− mouse showed moderate amyloid reductions associated with significant reductions in tau pathology, partial protection from neuron loss, and partial reversal of memory deficits. The APPSw/NOS2−/− mouse showed >85% reduction in amyloid deposition, significantly reduced tau pathology, complete protection from neuron loss, and complete reversal of memory deficits. A previous study by Vasilevko et al. (2007) found no significant reductions in Aβ levels in the APPSwDI transgenic mouse following Aβ immunotherapy. There are several distinct differences between the current study and that of Vasilevko et al. (2007). First, the model used in the current study includes deletion of the NOS2 gene, which may modify the response to Aβ immunotherapy. Vasilevko et al. (2007) showed that centrally administered antibodies did, indeed, clear Aβ deposits. Therefore, it is possible that more antibodies entered the CNS in our current study. Second, we used Freund's adjuvant for our vaccine formulations, while Vasilevko et al. (2007) used Quil-A. Adjuvants can significantly modify the response to the Aβ immunization, as has been previously shown (Cribbs et al., 2003).
Our data show that native mouse tau hyperphosphorylation is significantly reduced following removal of amyloid by vaccination. In APPSwDI/NOS2−/− mice, where Aβ levels were reduced by 40%, AT8-positive tau levels were significantly lower (∼40%) than in KLH-immunized control littermates, In APPSw/NOS2−/− mice, where Aβ levels were reduced by 50–65%, AT8-positive tau was reduced by 60% compared with KLH-vaccinated control mice. AT180 levels showed reductions similar to AT8, with a 35% reduction in the APPSwDI/NOS2−/− mice and a 50% reduction in the APPSw/NOS2−/− mice. While both APP/NOS2−/− transgenic mice were the same age at the start of treatment, the APPSwDI/NOS2−/− mice have significantly greater Aβ levels compared with the APPSw/NOS2−/− mice (Table 1).
Both AT8 and AT180 detect specific phosphorylation sites in the tau protein, and are associated with pathological changes in AD (Iqbal et al., 1994). AT8 detects phosphorylation at residues Ser202 and Thr205 (Goedert et al., 1995) and has been shown to correlate well with the staging of AD (Braak et al., 2006). It has been suggested that tau phosphorylation at Thr231, detected by AT180, is a necessary step for the conversion of normal tau to pathological tau in human AD (Wang et al., 2007). Reduction in both AT8 and AT180 by Aβ vaccination suggests that removal of Aβ significantly affects disease-relevant tau pathology. A study by Oddo et al. (2004), using their triple transgenic mouse model (Oddo et al., 2003b), did not find reductions in AT8 or AT180 following administration of anti-Aβ antibodies intracranially, though they did observe significant reductions in early, nonphosphorylation dependent, tau. It is possible that the mutated human tau overexpressed in this triple transgenic mouse model is more resistant to modification following removal of amyloid. Importantly, since this mouse carries the P301L human tau transgene, which is associated with frontotemporal dementia (Lewis et al., 2000), this mouse would develop tau pathology in the absence of amyloid, despite the fact that amyloid influences the rate of tau pathology in this mouse (Oddo et al., 2003a).
In human AD, tau pathology and neuron loss are closely related (Fukutani et al., 1995). The 12-month-old APPSwDI/NOS2−/− mice demonstrate visible thinning of the CA3 region of the hippocampus and stereological counting of neurons shows a 35% loss in the CA3 (Wilcock et al., 2008). At 16 months of age, following 4 months of vaccination, KLH-immunized APPSwDI/NOS2−/− mice show a 50% loss of CA3 neurons, while those receiving Aβ vaccination showed only a 40% loss of CA3 neurons. While both lost significantly more neurons than seen in 12-month-old mice before immunization, the mice receiving Aβ vaccination did show less severe degeneration. This indicates that Aβ vaccination partially protected the APPSwDI/NOS2−/− mice from further neuron loss. Aβ vaccination provided complete protection from neuron loss in the APPSw/NOS2−/− mice. At 12 months of age, APPSw/NOS2−/− mice show a 30% loss of neurons in the CA3 region (Colton et al., 2008). After 4 months of KLH control vaccinations, APPSw/NOS2−/− mice showed a 45% loss of neurons in the CA3 region. Aβ-immunized APPSw/NOS2−/− mice, however, showed only a 30% loss of CA3 neurons, indicating that little to no further neuron loss occurred after the initiation of Aβ vaccination. This is the first report suggesting that Aβ immunotherapy provides protection from neuron loss.
In the radial-arm water maze task, Aβ vaccination resulted in partial reversal of learning deficits in APPSwDI/NOS2−/− mice and complete reversal of these losses in APPSw/NOS2−/− mice. The behavior data itself is not necessarily surprising since Aβ immunotherapy has been shown in multiple studies to improve learning and memory in APP transgenic mouse models where memory deficits are observed in the absence of significant neuron loss (Janus et al., 2000; Morgan et al., 2000; Wilcock et al., 2004a). In the current study, we were able to show that the mice were originally impaired immediately before the initiation of vaccination, an age where we have shown significant neuron loss. Despite the protection from neuron loss, be it partial or complete, the neuron loss that is present at 12 months remains. Despite this, we observe significant improvements in learning and memory, to the extent that the Aβ-vaccinated APPSw/NOS2−/− mice are indistinguishable from normal NOS2−/− mice. These data suggest that the pathways of learning and memory tested in the radial-arm water maze (and likely other similar tasks) are more sensitive to levels of amyloid, and possibly to levels of abnormal tau protein, than they are to the actual number of neurons. This is not necessarily unexpected, as it has previously been shown that pilocarpine-induced hippocampal lesions result in normal spatial memory (Mohajeri et al., 2003).
One adverse effect of Aβ immunotherapy is the increased occurrence of microhemorrhages. This has been reported to occur in aged APP transgenic mice following passive immunization (Pfeifer et al., 2002; Wilcock et al., 2004a; Racke et al., 2005), active Aβ vaccination (Wilcock et al., 2007), and passive immunization in younger APP transgenic mice (Schroeter et al., 2008). Sometimes this increased incidence is associated with increased cerebral amyloid angiopathy (CAA) levels (Wilcock et al., 2004a, 2007), while at other times it is not (Pfeifer et al., 2002; Schroeter et al., 2008). Most importantly, increased incidence of microhemorrhage was observed in the AN1792 active Aβ vaccination clinical trial (Boche et al., 2008). In the current study, we found significantly increased incidence of microhemorrhage in APPSw/NOS2−/− mice despite the absence of significant CAA changes. The microhemorrhages were frequent, occurring multiple times in every mouse receiving Aβ vaccination. The location of the microhemorrhages varied; however, the bleeds were always located in brain regions with other pathologies, including CAA. While endothelial NOS (NOS3) is increased in NOS2−/− mice, NOS3 mRNA levels were not changed following vaccination; however, we do not rule out this increased eNOS expression changing the susceptibility of the mice to microhemorrhage following Aβ vaccination. It is possible that the high antibody titers observed in our study contribute to the high incidence of microhemorrhage.
No microhemorrhages were observed in APPSwDI/NOS2−/− mice with or without Aβ vaccination. This mouse model develops extensive vascular amyloid pathology, most of which is localized to the microvasculature (Davis et al., 2004). It is likely that these vessels become occluded before the development of vascular leakage. Though microhemorrhages did not appear to significantly affect learning and memory as measured by the radial-arm water maze, it is unlikely the microhemorrhages have no functional effect. In humans, the clinical effect of the microhemorrhages depends on the localization in the brain. Thalamic microhemorrhages cause a decline in memory performance (Vermeer et al., 2003), while microhemorrhages in the mammillothalamic tract have been shown to impair episodic memory and executive function (Kramer et al., 2002). High variability in the location of microhemorrhages following immunotherapy likely makes it difficult to detect a distinct functional outcome. For these reasons, we believe that microhemorrhage remains a significant obstacle for the long-term success of this approach.
Together, these data support amyloid as a therapeutic target for the disease-modifying treatment. In two novel mouse models of AD demonstrating amyloid, tau pathology, and neuron loss, we show that lowering amyloid by immunotherapy reduces tau pathology, protects neurons from degeneration, and reverses memory deficits. One major issue with this particular therapeutic approach remains, which is increased incidence of microhemorrhage. This effect must be addressed for the long-term success of Aβ-targeted immunotherapy.
Footnotes
This work was supported by National Institutes of Health Grants AG030942 (D.M.W.), AG19780 (M.P.V.), AG19740 (C.A.C.), and NS55118 (W.V.N.) and Alzheimer's Association Grant IIRG-07-59802 (C.A.C.).
References
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc. 2006;1:1671–1679. doi: 10.1038/nprot.2006.275. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA. Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–3310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, Dubois-Dalcq M. Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol. 1996;28:15–20. doi: 10.1007/BF02815200. [DOI] [PubMed] [Google Scholar]
- Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:12867–12872. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008;15:571–587. doi: 10.3233/jad-2008-15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- El-Gayar S, Thüring-Nahler H, Pfeilschifter J, Röllinghoff M, Bogdan C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J Immunol. 2003;171:4561–4568. doi: 10.4049/jimmunol.171.9.4561. [DOI] [PubMed] [Google Scholar]
- Fukutani Y, Kobayashi K, Nakamura I, Watanabe K, Isaki K, Cairns NJ. Neurons, intracellular and extracellular neurofibrillary tangles in subdivisions of the hippocampal cortex in normal ageing and Alzheimer's disease. Neurosci Lett. 1995;200:57–60. doi: 10.1016/0304-3940(95)12083-g. [DOI] [PubMed] [Google Scholar]
- Ganster RW, Taylor BS, Shao L, Geller DA. Complex regulation of human inducible nitric oxide synthase gene transcription by Stat 1 and NF-kappa B. Proc Natl Acad Sci U S A. 2001;98:8638–8643. doi: 10.1073/pnas.151239498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittins R, Harrison PJ. Neuronal density, size and shape in the human anterior cingulate cortex: a comparison of Nissl and NeuN staining. Brain Res Bull. 2004;63:155–160. doi: 10.1016/j.brainresbull.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Mechanism of Alzheimer neurofibrillary degeneration and the formation of tangles. Mol Psychiatry. 1997;2:178–180. doi: 10.1038/sj.mp.4000269. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso AC, Gong CX, Khatoon S, Singh TJ, Grundke-Iqbal I. Mechanism of neurofibrillary degeneration in Alzheimer's disease. Mol Neurobiol. 1994;9:119–123. doi: 10.1007/BF02816111. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Jongen-Rêlo AL, Feldon J. Specific neuronal protein: a new tool for histological evaluation of excitotoxic lesions. Physiol Behav. 2002;76:449–456. doi: 10.1016/s0031-9384(02)00732-1. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Koeppen AH. The history of iron in the brain. J Neurol Sci. 1995;134(Suppl):1–9. doi: 10.1016/0022-510x(95)00202-d. [DOI] [PubMed] [Google Scholar]
- Kramer JH, Reed BR, Mungas D, Weiner MW, Chui HC. Executive dysfunction in subcortical ischaemic vascular disease. J Neurol Neurosurg Psychiatry. 2002;72:217–220. doi: 10.1136/jnnp.72.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Ajmone-Cat MA, De Berardinis MA, De Simone R. Microglial activation in chronic neurodegenerative diseases: roles of apoptotic neurons and chronic stimulation. Brain Res Brain Res Rev. 2005;48:251–256. doi: 10.1016/j.brainresrev.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Saini K, Li H, Crameri A, Lipp HP, Wolfer DP, Nitsch RM. Intact spatial memory in mice with seizure-induced partial loss of hippocampal pyramidal neurons. Neurobiol Dis. 2003;12:174–181. doi: 10.1016/s0969-9961(02)00031-1. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Khan K, Barbour R, Doan M, Chen M, Guido T, Gill D, Basi G, Schenk D, Seubert P, Games D. Immunotherapy reduces vascular amyloid-β in PDAPP mice. J Neurosci. 2008;28:6787–6793. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Hosokawa M, McGeer PL. Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol. 2004;188:52–64. doi: 10.1016/j.expneurol.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Vasilevko V, Xu F, Previti ML, Van Nostrand WE, Cribbs DH. Experimental investigation of antibody-mediated clearance mechanisms of amyloid-β in CNS of Tg-SwDI transgenic mice. J Neurosci. 2007;27:13376–13383. doi: 10.1523/JNEUROSCI.2788-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, Wood ER, Smith GK, McDonald B, Bachus KE, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood. 1995;86:1184–1195. [PubMed] [Google Scholar]
- Wilcock DM, Colton CA. Anti-amyloid-beta immunotherapy in Alzheimer's disease: relevance of transgenic mouse studies to clinical trials. J Alzheimers Dis. 2008;15:555–569. doi: 10.3233/jad-2008-15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004a;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]