

Abstract
A major obstacle that hampers the design of drug therapy for traumatic brain injury is the incomplete understanding of the biochemical pathways that lead to secondary cellular injury and contribute to cell death. One such pathway involves reactive species that generate potentially cytotoxic zinc ion fluctuations as a major executor of neuronal, and possibly glial, cell death. Whether zinc ions released during traumatic brain injury are toxic or protective is controversial but can be approached by investigating the exact concentrations of free zinc ions, the thresholds of compromised zinc buffering capacity, and the mechanism of cellular homeostatic control of zinc. Rapidly stretch-injured rat pheochromocytoma (PC12) cells express cellular zinc ion fluctuations that depend on the production of nitric oxide. Chelation of cellular zinc ions after rapid stretch injury, however, increases cellular reactive oxygen species. In a rat model of traumatic brain injury, parasagittal fluid percussion, analysis of the metal load of metallothionein was used as an indicator of changes in cellular zinc ion concentrations. The combined results from the cellular and in vivo investigations caution against interpreting zinc ion fluctuations in the early phase (24 hours) after injury as a primarily cytotoxic event.
Keywords: zinc, pheochromocytoma (PC12) cells, traumatic brain injury, rapid stretch injury, oxidative stress, metallothionein
1. Introduction
Sustained and progressive neuronal degeneration is a characteristic feature of traumatic brain injury (TBI). It is secondary to the primary destruction of tissue caused by the physical impact and involves vascular, inflammatory, cellular, and molecular events (DeWitt and Prough 2003;Enriquez and Bullock 2004;Bramlett and Dietrich 2004;Bramlett and Dietrich 2007;Kochanek et al. 2008). The diffuse nature and the complexity of the secondary injury has thwarted attempts to define effective preventive or therapeutic interventions (Beauchamp et al. 2008). Research on the chemical mechanisms of neurodegeneration focused largely on the toxicity of reactive species, glutamate, and calcium (Hall et al. 1988;Katayama et al. 1990;Bramlett and Dietrich 2007).
More recently, the toxicity of zinc (II) ions in focal and global ischemic brain injury is also receiving experimental scrutiny (Choi and Koh 1998). During transient forebrain ischemia in rats, zinc ions accumulate in degenerating neurons in the hippocampus, cortex, and other brain regions (Koh et al. 1996). Under normal physiological conditions, zinc ions are stored in presynaptic vesicles of some neurons and are believed to be co-released with glutamate, to bind to the N-methyl-D-aspartic acid (NMDA) receptor and other proteins of the postsynaptic membrane or to enter the postsynaptic dendrite and to interact with proteins intracellularly (Frederickson et al. 2004;Sensi et al. 2009). In contrast to ischemic brain disease (Sensi et al. 2009), only a few investigations have addressed the role of zinc in TBI. After TBI, zinc staining with intracellular zinc indicators, such as N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) decreases in the presynaptic neurons, but increases in the postsynaptic hippocampal neurons (Suh et al. 2000). Intracerebroventricular injection of CaEDTA, an extracellular zinc chelator, after transient global cerebral ischemia prevents neuronal death (Koh et al. 1996;Calderone et al. 2004). In TBI of rats, zinc up-regulates neuroprotective genes (Hellmich et al. 2004), but it does not improve learning and memory deficits in behavioral tests following TBI (Hellmich et al. 2008). It has been suggested that release of zinc ions from presynaptic vesicles does not contribute to neuronal damage after TBI (Doering et al. 2007). Thus, intracellular zinc ions mobilized as a consequence of oxidative stress rather than extracellular zinc ions could be the source of potentially cytotoxic zinc ions (Maret 1994;Maret 1995;Frederickson et al. 2004;Dineley et al. 2008). From a large body of data, the sequence of events that links increased cellular zinc ion concentrations and neuronal cell death is thought to begin with glutamate excitotoxicity and to involve calcium influx through calcium channels, calcium-activated nitric monoxide (NO) production by NO synthase (NOS), an increase in zinc ion concentrations, mitochondrial production of reactive oxygen species (superoxide), release of more zinc ions from proteins such as metallothionein (MT), and activation of the mitochondrial pathway of apoptosis (Bossy-Wetzel et al. 2004). Increased concentrations of intracellular zinc ions may promote neuronal death by inhibiting cellular energy production, increasing cellular reactive oxygen species (ROS), changing the mitochondrial membrane potential, and reducing cellular ATP levels (Dineley et al. 2003).
A major issue in defining the functions of zinc ions in these pathways inducing cellular injury is their cellular concentrations and the capacity of the cell to control them (Maret and Li 2009). Total cellular zinc concentrations are a few hundred micromolar. Most zinc is bound to proteins with high affinity; therefore, the concentrations of cellular free zinc ions are very low. Estimates put them in the picomolar to nanomolar range (Krezel and Maret 2006;Bozym et al. 2006), but higher concentrations can occur when oxidative stress releases zinc from proteins that utilize sulfur ligands for zinc binding (Maret and Vallee 1998;Maret 2006). Thus, exposing cells to oxidizing agents or generating a redox signal, e.g. NO, within the cell, increases cellular free zinc ion concentrations (Turan et al. 1997;Aizenman et al. 2000;St Croix et al. 2002;Spahl et al. 2003;Cima et al. 2006). Such increased free zinc ion concentrations are very potent effectors of proteins (Maret et al. 1999).
The threshold for cellular zinc buffering demarcates cytoprotective (pro-antioxidant) from cytotoxic (pro-oxidant) effects of zinc ions (Hao and Maret 2005). Although these dual activities of zinc ions may appear paradoxical (Cuajungco and Faget 2003), they merely reflect the actions of zinc ions at different concentrations: physiological concentrations confer neuroprotection while pathophysiological concentrations are neurotoxic (Hao and Maret 2005;Maret 2008). For example, sub-lethal ischemia triggers a neuroprotective increase in free zinc ion concentrations in postsynaptic neurons, and this ischemic preconditioning can be blocked by chelating zinc ions with CaEDTA (Lee et al. 2008). Dysregulation of neuronal zinc ion homeostasis, how much cellular free zinc ions increase, and to which targets the released zinc ions bind, are important questions in elucidating the mechanisms of cell death following TBI. In this investigation, changes of zinc ion concentrations during an early phase of sub-lethal injury (within hours) were quantified. Both in vitro (rapid stretch injury, RSI) and in vivo (fluid percussion TBI) models of brain injury were employed. The results demonstrate induced fluctuations of free zinc ions within a physiological range of concentrations and suggest a cytoprotective effect of the released zinc ions in this time period after injury.
2. Results
The free zinc ion concentrations in normal (uninjured) PC12 cells are 0.97 nM in serum-containing media and transiently reach 1.4 nM during proliferation (Li and Maret 2009). Remarkably, during serum withdrawal, PC12 cells mobilize zinc ions from an intracellular source (Li and Maret 2009). With time, free zinc ion concentrations decrease below the normal level and growth arrest ensues (Li and Maret 2009). Thus, measurements of zinc ion concentrations provide critical information on whether or not cells are viable and healthy (Li and Maret 2009).
RSI caused biphasic zinc ion fluctuations in PC12 cells
After subjecting PC12 cells to RSI (up to 60 psi, 50 ms), less than 10% cell death was detected by PI staining (Figure 1). Accordingly, 50 psi (50 ms) was employed as sublethal stretch injury in the following experiments. Within 24 hours, there was a biphasic fluctuation of intracellular zinc ion concentrations (Figure 2). The highest concentration was observed at one hour post-injury (~ 1.4 nM), followed by a sharp decline below the baseline (~ 0.9 nM). The concentrations are then maintained at this low level (~ 0.5 nM).
Figure 1.


PC12 cell viability measured with propidium iodide (PI) staining after rapid stretch injury. (A) Phase contrast and fluorescence microscopic images of PC12 cell cultures at 24 hours after different levels of RSI (0, 20, 30, 40, 50 and 60 psi for a duration of 50 msec). (B) Percentage of dead cells relative to the total number of cells.
Figure 2.

Rapid stretch injury-induced fluctuations of intracellular zinc ion concentrations in PC12 cells. Intracellular zinc ion concentrations were measured fluorimetrically with 0.3 μM FluoZin-3 AM in 1 ml DPBS without Ca2+ and Mg2+ at 37 °C for 30 min at 0, 0.5, 1, 3, 6, 12 and 24 hours after RSI. Data are represented as mean ± SD (n = 3, p < 0.005 by one-way ANOVA).
RSI elevated intracellular reactive species
Sublethally stretched neurons produce high levels of ROS (Arundine et al. 2004). The fluorescent probe CM-H2DCFDA was employed to measure intracellular ROS after RSI of PC12 cells. ROS levels increased immediately after injury and peaked one hour later, coinciding with a time when intracellular zinc ion concentrations were highest (~ 1.4 nM) (Figure 3A). Following the peak at one hour, ROS decreased slightly but increased again and remained high even when intracellular zinc ion concentrations were low. Levels of NO in PC12 cells, measured by DAF-FM over time, displayed a slightly different profile peaking at three hours rather than at one hour after RSI (Figure 3B). Nonetheless, NO levels began to increase immediately after injury. However, they decreased after the peak rather than increased again as observed for ROS.
Figure 3.


Rapid stretch injury-induced levels of reactive oxygen and nitrogen species in PC12 cells. (A) ROS and (B) NO were measured fluorimetrically at 0, 0.5, 1, 3, 6, 12 and 24 hours after RSI. Cells (1 × 106/ml) were incubated with 5 μM CM-H2DCFDA or 3 μM DAF-FM in 1 ml DPBS without Ca2+ and Mg2+ at 37 °C for 30 min for ROS or NO measurement, respectively. The level of ROS was calibrated by Fmax, which is the fluorescence with the presence of 300 μM tert-butyl hydroperoxide, and was normalized to the fluorescence of PC12 cells prior to serum starvation as the control. The level of NO was normalized to total cell numbers. Data are represented as mean ± SD (n = 3, p < 0.0005 for ROS, and p < 0.01 for NO by one-way ANOVA).
The NOS inhibitor, L-NAME, abolished the increase in intracellular zinc ion concentrations caused by RSI
To determine whether zinc ion fluctuations depend on the production of NO, the generation of NO was inhibited by adding 500 μM L-NAME to the PC12 cell culture immediately after RSI. Intracellular zinc ion concentrations decreased significantly one hour after L-NAME treatment following RSI, indicating that NO was involved in the increase of zinc ion concentrations (Figure 4).
Figure 4.
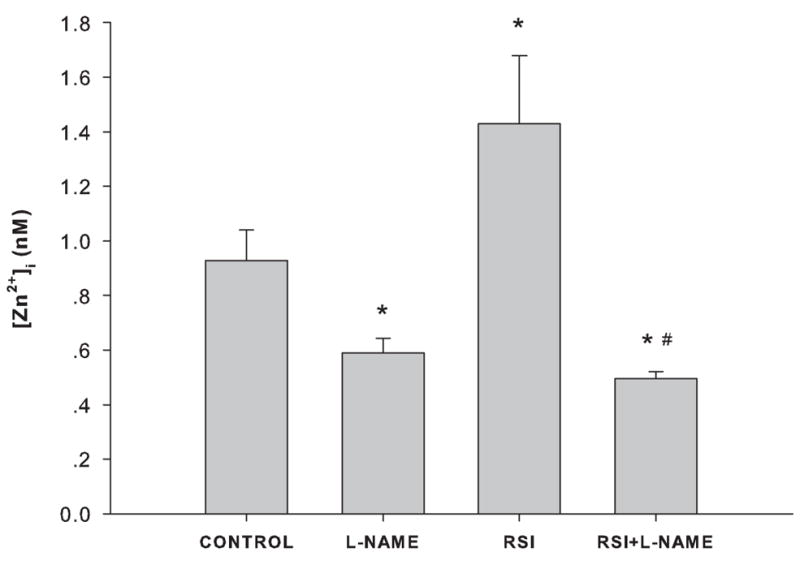
Effect of NOS inhibition on intracellular zinc ion concentrations in PC12 cells. CONTROL: no RSI, no L-NAME treatment; L-NAME: treated with 500 μM L-NAME in complete culture medium for 1 hour; RSI: 1 hour after RSI; and RSI+L-NAME: treated with 500 μM L-NAME immediately after RSI for 1 hour. Data are represented as mean ± SD (n = 3, *p < 0.001 compared to CONTROL, and #p < 0.001 compared to RSI by one-way ANOVA with Bonferroni adjustment for multiple comparisons).
Zinc chelation potentiated the generation of ROS
To examine the relationship between the increased zinc ion concentrations and the levels of intracellular ROS, an intracellular zinc chelator, TPEN (50 nM), was added immediately after RSI, and intracellular ROS was measured after one hour. Instead of suppressing the production of ROS, zinc chelation potentiated it (Figure 5).
Figure 5.

Effect of zinc chelation on ROS generation in PC12 cells. CONTROL: no RSI, no TPEN treatment; TPEN: treated with 50 nM TPEN in complete culture medium for 1 hour; RSI: 1 hour after RSI; and RSI+TPEN: treated with 50 nM TPEN immediately after RSI for 1 hour before measurement. Data are represented as mean ± SD (n = 3, *p < 0.001 compared to CONTROL, and #p < 0.001 compared to TPEN and RSI by one-way ANOVA with Bonferroni adjustment for multiple comparisons).
Parasagittal fluid percussion TBI induced changes in MT/T ratios
To date, there is no method to monitor real-time changes of cellular zinc ion concentrations directly in animal tissues. The metal load and the redox state of MT depend on the cellular environment (Krezel and Maret 2007;Li and Maret 2008). Therefore, it is possible to use analyses of the MT protein as a specific redox indicator and indicator of zinc ion availability (Yang et al. 2001;Haase and Maret 2004;Krezel and Maret 2007). Under normal conditions, the brain level of T is approximately half of the total MT level, namely, the MT/T ratio is about one (Yang et al. 2001;Krezel and Maret 2007). The basal concentrations of MTtot (MT + T) and T in the hippocampus and cortex of male Sprague-Dawley rats were 0.2 and 0.1 μmol/g, respectively (Figure 6). Since there were no significant differences between the injured and uninjured sides of the hippocampus or the cortex, data were pooled, and a comparison was made between sham-treated rats and rats that received TBI. Both sham and moderate parasagittal fluid percussion caused an overall decrease in total MT + T levels (Figure 7). At 30 minutes, total MT + T concentrations became significantly higher (p < 0.05) in TBI groups in both hippocampal and cortical regions. A significant effect of time (p < 0.001) for the MT/T ratio in both sham- and TBI-operated animals was observed when the data were analyzed by two-way ANOVA (Figure 8). During the acute phase after sham or TBI (< 4 hours), the MT/T ratio increased significantly in both hippocampal and cortical regions, indicating a transient increase in intracellular zinc ion bioavailability and subsequent zinc binding to metallothionein. After this peak at 4 hours, the ratio decreased. At 24 hours after TBI, the ratio of MT/T was significantly lower than at the beginning of the experiment. While analysis using two-way ANOVA revealed no significant overall effect of injury (sham vs TBI) (p > 0.05), individual time points (hippocampus: 0.5 hours; cortex: 2 and 4 hours) differ significantly in sham- and TBI-injured animals when analyzed by Student’s t-test (p < 0.05). TBI tended to increase the MT/T ratio at the acute phase (< 4 hours), suggesting that intracellular zinc ion concentrations are elevated after TBI, followed by a tendency for decreased zinc ion availability as indicated by smaller MT/T ratios at later times. Sham operation resulted in qualitatively similar changes of the MT/T ratio but there was a smaller MT/T peak at the acute phase (< 4 hours).
Figure 6.

Basal concentrations of MTtot (MT + T) and T in rat hippocampus and cortex. (n =6)
Figure 7.


Total levels of MT plus T in rat (A) hippocampus and (B) cortex after sham and TBI treatments (n = 6, *p < 0.05 compared to sham by Student’s t-test).
Figure 8.


The ratio of MT to T in rat (A) hippocampus and (B) cortex after sham and TBI treatments (n = 6, *p < 0.05 compared to sham by Student’s t-test; p < 0.0001 for time effect by two-way ANOVA).
3. Discussion
This study demonstrates changes in intracellular free zinc ion concentrations during the early (0–24 hours) time period after injury. The RSI cell culture model (Ellis et al. 1995) was among the first used to investigate how mechanical force causes cellular injury. It provides the opportunity to study the effects of pure mechanical stretch, which is the major mechanical force when the head is impacted, thus eliminating other factors such as the production of reactive species due to a lack of oxygen during hypoxia/reperfusion or the need to apply excitatory agents, such as NMDA and glutamate extracellularly. RSI caused mitochondrial dysfunction and decreased ATP in mixed cultures of astrocytes and cortical neurons (Ahmed et al. 2000). It elicited a rapid, yet transient, elevation in intracellular Ca2+ concentration in astrocytes, with persistent alterations in calcium-mediated signal transduction (Rzigalinski et al. 1998). It enhanced both NMDA- and AMPA-mediated ion currents, but the mechanisms for the enhancement differed: RSI reduced the Mg2+ blockade of the NMDA receptor (Zhang et al. 1996), but it desensitized the AMPA receptor (Goforth et al. 1999;Goforth et al. 2004). Oxidative stress is one of the major consequences of RSI (Arundine et al. 2004). To date, the effects of RSI on cellular zinc ions have not been reported.
Our results demonstrate intracellular zinc ion fluctuations within the physiological range (0.4 – 1.4 nM, Figure 2) of zinc ion concentrations in undifferentiated PC12 cells (Li and Maret 2009) as a consequence of sub-lethal RSI, and an NO-mediated increase of zinc ions. Instead of inducing the production of ROS, i.e. exerting a pro-oxidant effect, the increased zinc ion concentrations had a pro-antioxidant effect because levels of ROS were higher when cellular zinc ion concentrations were lowered with a cell-permeable chelating agent (Figure 5). Zinc ions may protect cells from oxidative damage by binding to thiols and preventing their oxidation, and/or by activating metal response element (MRE)-binding transcription factor-1 (MTF-1) and antioxidant response element (ARE)-binding transcription factors, such as Nrf2 (Maret 2006;Cortese et al. 2008). In zinc ion-mediated ischemic preconditioning, sub-lethal increases in zinc ion concentrations induced activated caspase 3 to levels that do not induce apoptosis but are sufficient to promote the cleavage of poly(ADP-ribose) polymerase 1 (PARP1), thereby blocking the downstream damaging effects of this enzyme (Lee et al. 2008).
At twenty-four hours after mechanical injury, the cellular zinc ion concentrations were below normal baseline values (0.9 nM), suggesting that PC12 cells develop a cellular “zinc ion deficiency”. We observed recently that serum starvation leads to a low threshold of 0.4 nM cellular zinc ion in PC12 cells (Li and Maret 2009). Prolonged incubation in serum-starved medium will induce apoptosis, which can be prevented by addition of zinc ions (Adamo et al. 2009). The deficit of zinc ions after sub-lethal RSI therefore can be detrimental for cellular functions because free zinc ions are a metabolically active pool of zinc in cellular regulation and signaling (Maret and Li 2009). Such a “zinc ion deficiency” refers to the inability of the cell to control this pool of zinc and is different from a deficiency of total cellular zinc. The intracellular zinc ion concentration measured by FluoZin-3 AM is the total zinc ion concentration in the cytoplasm. This method does not provide any information about the source of the zinc ions released after injury.
Although cell and tissue culture models permit mechanistic studies of pathways contributing to cell death and tissue degeneration, validation of the in vitro findings in animal models is required to formulate interventions for preventing and treating brain injuries.
In the CA3 and the dentate gyrus regions of the hippocampus, TBI causes selective neuronal loss that may be associated with learning and memory deficits in both experimental animals (Hamm et al. 1996;Floyd et al. 2002;Grady et al. 2003;Dietrich et al. 1994;Dietrich and Allen 1998) and humans (McAllister 1992;Kesner and Hopkins 2006). The hippocampus and the cerebral cortex also are regions that stain most intensely for histochemically reactive zinc ions. Staining of rat brain sections with TSQ (Suh et al. 2000) or Newport Green (Hellmich et al. 2004) 24 hours after brain injury demonstrated that zinc ions accumulate in degenerating hippocampal and cortical neurons. Although the presence of free zinc ions can be demonstrated histochemically, an effective method to quantify them in animal tissues does not exist. However, MT levels and MT metal loads can be employed as indicators of free zinc ion concentrations (Yang et al. 2001;Krezel and Maret 2007).
Using metallothionein as a reporter molecule, our results demonstrated transient increases in cellular free zinc ion concentrations monitored as increased MT/T ratios in the hippocampus and cortex of adult male sham or moderately injured Sprague-Dawley rats four hours after TBI. Increases in intracellular zinc ions were also observed in sham-operated rats, suggesting that anesthesia and/or surgical stress alone increased zinc ion concentrations in the same brain regions. Compared to sham operation, a difference was not detected in TBI at a significance level of 0.05 when the data were analyzed by two-way ANOVA.
The observed initial increase in intracellular zinc ion concentrations followed by a decrease several hours after TBI is consistent with the results from rapid stretch-injured PC12 cells. The biphasic changes in zinc ion concentrations demarcate two phases with potentially different and opposite functions of zinc ions during progression of the injury. Thus, time is a critical parameter when investigating the functions of zinc ions in brain injuries. In addition, similar patterns of changing MT/T ratios after sham and TBI where observed in the hippocampus and the cortex. However, the individual time points at the acute phase of injury when TBI showed a significantly higher MT/T ratio compared to sham are different in the hippocampus and the cortex. The overall decrease in the total concentrations of MT and T and in the MT/T ratio suggest that a zinc ion deficiency develops over a longer period after TBI, which is also apparent for the rapidly stretch-injured PC12 cells.
MT and T levels and their ratios provide a surrogate of direct measurements of zinc ion concentrations, which at present cannot be determined directly in animal tissues. It is possible that during tissue homogenization, with the breakdown of the barrier between intracellular and extracellular compartments, extracellular zinc ions contribute to a change in the MT/T ratio. Therefore, observed changes in MT/T ratios may be due to fluctuations of zinc ions intracellularly and/or extracellularly. However, with the possible exception of zinc ions released from presynaptic zinc-rich vesicles, the contribution from extracellular zinc ions on the MT/T ratios must be small given the very low extracellularly available zinc ion concentration and the low ratio of interstitial/cytoplasmic fluid. Nonetheless, if extracellular zinc were depleted and not repleted, a resulting systemic zinc deficiency in the brain would compromise repair and recovery processes. Our method determines the metal load of metallothioneins but does not differentiate among the different MT isoforms. The cell-specific expression of MTs and their variable metal load and redox states under different experimental conditions continues to be a major challenge in their analysis (Li and Maret 2008).
Such a cellular zinc ion deficiency might precede a systemic zinc deficiency that develops in brain trauma patients (McClain et al. 1986). Protective effects of zinc supplementation have been observed in both long-term ischemia-reperfusion (Atahan et al. 2009) and in brain trauma (Young et al. 1996). Our findings indicate that a loss of cellular zinc ions is equally, if not more, detrimental as the pathological increase of intracellular zinc ions, especially when sustained for a longer period of time after mild or moderate TBI. As a matter of fact, our in vitro results support a pro-antioxidant effect of the rise in zinc ion concentrations within the physiological range. The function of such an increase as a physiological response counteracting injury and protecting the cell against further injury may explain the failure of zinc chelation at certain time points as a therapeutic strategy to brain injury.
In conclusion, zinc ion fluctuations were detected in sub-lethally injured cells. These fluctuations are within a physiological range of cellular concentrations and may protect the cell against secondary injury. A protective effect of mobilized zinc ions has been observed in ischemic pre-conditioning (Lee et al. 2008;Aras et al. 2009). However, our results do not rule out that zinc homeostasis and zinc status are compromised over 24 hours after injury when zinc ion concentrations are below baseline and contribute to cell death directly or indirectly. In future studies, the thresholds of cellular zinc buffering, the role of different pathways and compartments in the induction of zinc ion fluctuations at different time points, and the different proteins that become targets at different zinc ions concentrations all need to be considered.
4. Methods and Materials
Cell culture
Pheochromocytoma (PC12) cells [(Greene and Tischler 1976) ATCC, CRL-1721] were maintained in complete growth medium [RPMI 1640 (GIBCO) supplemented with 10% (v/v) heat-inactivated horse serum (GIBCO), 5% (v/v) fetal bovine serum (FBS) (HyClone) and 1% (w/v) penicillin/streptomycin (GIBCO)] at 37 °C in a humidified, 5% CO2 incubator.
Fluorescence assay for intracellular zinc ion concentrations
Intracellular zinc ion concentrations were determined with FluoZin-3 (Krezel and Maret 2006). Briefly, cells were detached from culture dishes by gentle scraping. Following two washes with DPBS without Ca2+ and Mg2+ at 37 °C, cell suspensions (1 × 106 cells/ml) were aliquoted into Eppendorf tubes and incubated at 37 °C for 30 min. For all measurements of intracellular zinc ion concentrations, 0.3 μM FluoZin-3 acetoxymethyl ester (FluoZin-3 AM) (Invitrogen, Molecular Probes), was used. The concentration of FluoZin-3 AM (0.3 μM) was chosen from a quantitative assay that employs a series of different concentrations of FluoZin-3 AM and an extrapolation to a zero probe concentration to determine the absolute intracellular zinc ion concentration (Krezel and Maret 2006). It is the lowest concentration of the probe that allows accurate measurements and exerts a minimal effect on cellular zinc buffering of PC12 cells (Li and Maret 2009). Cells were then washed three times with DPBS without Ca2+ and Mg2+ at 37 °C to remove any residual fluorescence probe, and incubated another 30 min at 37 °C. Fluorescence was measured at 25 °C with 492 nm excitation and 517 nm emission in a spectrofluorimeter. Six Eppendorf tubes of cells were prepared for each measurement, and they were randomly assigned into two groups afterwards with three tubes in each group to receive two different treatments for 10 min to measure Fmin and Fmax. Fmin is the background fluorescence of the dye measured in the presence of 50 μM N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN) (Sigma-Aldrich), and Fmax is the maximum of fluorescence in the presence of 100 μM pyrithione (Sigma-Aldrich), a zinc ion ionophore that facilitates zinc entry into cells, and 250 μM ZnSO4 to saturate the probe. The concentrations of zinc ions were calculated by using the following equation. [Zn2+] = Kd (F-Fmin)/(Fmax-F) with Kd = 8.9 nM.
The calibration employing Fmin and Fmax makes the determination independent of cell number.
Fluorescence assays for reactive species
Intracellular reactive oxygen species (ROS) was measured with 5′-(and-6′)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester, mixed isomers (CM-H2DCFDA) (Invitrogen, Molecular Probes), a probe that detects superoxide (O2•−), hydrogen peroxide (H2O2), hydroxyl free radical (HO•), and peroxynitrite (ONOO−) (Fekete et al. 2008). Cells were detached from culture dishes by gentle scraping. After washing twice with DPBS without Ca2+ and Mg2+ at 37 °C, the cell suspensions (1 × 106 cells/ml) were aliquoted into Eppendorf tubes and incubated for 30 min with 5 μM CM-H2DCFDA with (Fmax) or without (F) 300 μM tert-butyl hydroperoxide (Sigma-Aldrich) at 37 °C. Cells were washed three times with DPBS without Ca2+ and Mg2+ at 37 °C to remove any residual fluorescence probe, and incubated for another 30 min at 37 °C. Fluorescence was measured at 25 °C with 492 nm excitation and 517 nm emission in a spectrofluorimeter. The level of ROS was calibrated by determination of Fmin, which is the basal level of ROS in control cells, and Fmax, which is the maximum fluorescence in the presence of tert-butyl hydroperoxide that is used to oxidize all the probe molecules. The relative normalized fluorescence was calculated by using the equation:
The concentration of NO was measured with 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate (Invitrogen, Molecular Probes). Cells were detached from culture dishes by gentle scraping. After washing twice with DPBS without Ca2+ and Mg2+ at 37 °C, the cell suspensions (1 × 106 cells/ml) were aliquoted into Eppendorf tubes and incubated with 3 μM DAF-FM diacetate at 37 °C for 30 min. Cells were washed three times with DPBS without Ca2+ and Mg2+ at 37 °C to remove any residual fluorescence probe, and incubated for another 30 min at 37 °C. Fluorescence was measured at 25 °C with 492 nm excitation and 517 nm emission in a spectrofluorimeter. The level of NO was normalized to total cell numbers, which were determined using a Coulter Counter, Model ZF (Coulter Electronics, Inc., Hialeah, FL).
Rapid stretch injury (RSI)
Intracellular zinc ions and generation of reactive species were monitored fluorimetrically over a time period of up to 24 hours after injuring PC12 cells with the rapid stretch injury model described by Ellis et al., 1995. To perform RSI, cells were plated in 6-well Flex I® culture plates with a silastic membrane bottom coated with collagen I (FlexCell International Corporation) at a density of 1 × 105/ml in 1 ml complete culture medium. After 24 hours of incubation, the culture plates were connected to a 94A Cell Injury Controller (Biomedical Engineering Facility, Medical College of Virginia), which employs a nitrogen gas pulse to deform the silastic membrane and achieve a predetermined degree of stretch for a predetermined duration (Ellis et al. 1995). Viability of PC12 cells 24 hours after different levels of stretch injury (0, 20, 30, 40, 50 and 60 psi for a duration of 50 msec) were determined by propidium iodide (PI, Calbiochem, San Diego, CA) staining. Cells were stained with 5 μM PI solution in medium for 30 min and counted under fluorescent microscope. For all of the RSI experimental procedures, the pulse pressure/duration was 50 psi/50 msec, which generated sub-lethal mechanical stretch to PC12 cells. After RSI, cells were kept in complete culture medium at 37 °C in a humidified, 5% CO2 incubator for 0.5, 1, 3, 6, 12 and 24 hours before measuring intracellular zinc ion concentrations or reactive species.
For the NOS inhibition experiment, 500 μM Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME) (Sigma-Aldrich) was added immediately after RSI and the cells were kept at 37 °C in a humidified, 5% CO2 incubator before measuring intracellular zinc ion concentrations. PC12 cell cultures were divided randomly into four groups: Group 1 served as a control without either RSI or L-NAME treatment; Group 2 was treated only with L-NAME; Group 3 received only RSI; and Group 4 received both RSI and treatment with L-NAME. Since the highest intracellular zinc ion concentrations were observed one hour after RSI, L-NAME was added to cell cultures with or without RSI one hour before zinc ions were measured. At high concentrations, L-NAME is non-selective and inhibits all three types of nitric oxide synthases: eNOS, nNOS, and iNOS (Liaudet et al. 1998). For the zinc chelation experiment, 50 nM TPEN was added immediately after RSI and the cells were maintained at 37 °C in a humidified, 5% CO2 incubator for one hour, since the highest ROS levels were observed one hour after RSI. PC12 cell cultures were divided randomly into four groups: Group 1 served as a control without either RSI or TPEN treatment; Group 2 was treated only with TPEN; Group 3 received only RSI; and Group 4 received both RSI and treatment with TPEN.
Parasagittal fluid percussion injury
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch, Galveston, Texas. Male Sprague-Dawley rats weighing 400–500 g were anesthetized, intubated and mechanically ventilated with 1.5–2.0% isoflurane in air:oxygen (70:30) and prepared for parasagittal fluid percussion TBI (Mathew et al. 1999). Briefly, the rats were placed in a stereotaxic frame and a craniotomy was performed 1 mm lateral right to the sagittal suture, midway between the bregma and lambda sutures. A modified LuerLok syringe hub was placed in the craniotomy site and anchored in place with dental hygienic acrylic resin. Rats were then connected to the trauma device and subjected to either a sham injury or moderate (2.0 atm) TBI. Rats were randomly assigned into the sham injury (n = 6 per time point) or moderate TBI (n = 6 per time point) group. The acrylic cap was removed, wounds were sutured with 4-0 prolene, the isoflurane was discontinued, and the rats were extubated and permitted to awaken from anesthesia. After survival for 0.5, 2, 4, 16 or 24 hours, rats were re-anesthetized with 4% isoflurane, decapitated and the brains were rapidly removed for dissection of the cortex and hippocampus from the ipsilateral (injured) and contralateral sides of the brain to measure MT. The basal concentration of total MT and the MT to thionein (T) ratio were also obtained by collecting brain tissues from six rats receiving no surgery and sacrificed under anesthesia with 4% isoflurane.
Fluorescence assay for MT
Fluorimetric assays allow determination of the metal load of MT (Yang et al. 2001;Krezel and Maret 2007). The fraction of the protein that is not saturated with metal ions is referred to as T. Hippocampi and cortices of Sprague-Dawley rats were dissected immediately after sacrifice for MT measurements (Yang et al. 2001;Krezel and Maret 2007). The tissues were homogenized by a Kontes electric pellet pestle (Fisher Scientific) in homogenizing buffer (0.2 M mannitol, 0.05 M sucrose, 0.01 M KCl, 0.01 M HEPES, pH 7.4) in a microliter volume that corresponds to 4 times the weight of the tissue (in mg). After centrifugation at 10,000 × g for 5 min, 10 μl supernatant was diluted in 20 mM Tris-HCl, pH 7.4 to determine total protein concentrations using the Pierce Micro BCA™ protein assay kit. The remaining supernatant was collected and treated with 40% (v/v) acetonitrile (EM Science) for 15 min to precipitate large and hydrophobic proteins. The suspension was spun at 10,000 × g for 5 min and the supernatant was labeled immediately with 1 mM ammonium 7-fluorobenz-2-oxa-1,3-diazole-4-sulfonamide (ABD-F) (Invitrogen, Molecular Probes) in 35 mM borate buffer, pH 7.4. Tris-(2-carboxyethyl)phosphine, hydrochloride (TCEP) (Invitrogen, Molecular Probes) was added to a final concentration of 15 mM because ABD-F can label only reduced thiols. Twenty-five mM Na2EDTA (Sigma-Aldrich) was added for total metallothionein measurement, but omitted for T measurement. After incubation at 60 °C for 10 min, samples were separated on a reversed-phase C4 Phenomenex Jupiter 5μ column (250 mm × 4.60 mm2) with a Phenomenex pre-column at a flow rate of 1 ml/min, using a Beckman Coulter System Gold HPLC system with eluent A (5 mM Tris-HCl, pH 7.4) and eluent B (50% 2-propanol in A). The condition was 90% eluent A and 10% eluent B with a double step gradient to 40% eluent B at 5 min and to 100% eluent B at 12 min. The fluorescence signal was detected with a JASCO Intelligent Fluorescence Detector Model 2020 with excitation of 384 nm and emission of 510 nm and a detector gain at ×100. All aspects of the experiment were performed at 25 °C. During analysis, however, samples were cooled at 4 °C in the auto-sampler.
Statistical analysis
Results were analyzed by using one-way ANOVA to determine significance at the level of 0.05 between different time points or treatments for in vitro experiments. When ANOVA detected significance, the significance of differences was determined by the Student’s t-test with Bonferroni adjustment for multiple comparisons. Two-way ANOVA was used to analyze the effect of in vivo treatment (sham vs TBI) and time for MT/T measurements. The Student’s t-test was used to compare the difference between sham and TBI at individual time points.
Acknowledgments
This work was supported by National Institutes of Health grant NS042849 (to DSP), and in part by a predoctoral fellowship (to BEH) from the National Institutes of Environmental Health Sciences training grant ES007254. We thank Margaret A. Parsley for her technical assistance and Dr. Alai Tan for help with the statistical analyses.
Abbreviations used
- ABD-F
7-fluorobenz-2-oxa-1,3-diazole-4-sulfonamide
- CM-H2DCFDA
5′-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester, mixed isomers
- DAF-FM diacetate
4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate
- FluoZin-3 AM
FluoZin-3 acetoxymethyl ester
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- MT
metallothionein
- NO
nitric monoxide
- NOS
nitric oxide synthase
- PI
propidium iodide
- ROS
reactive oxygen species
- RSI
rapid stretch injury
- T
thionein
- TBI
traumatic brain injury
- TPEN
N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- TSQ
N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamo AM, Zago MP, Mackenzie GG, Aimo L, Keen CL, Keenan A, Oteiza PI. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox Res. 2009 doi: 10.1007/s12640-009-9067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SM, Rzigalinski BA, Willoughby KA, Sitterding HA, Ellis EF. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem. 2000;74:1951–1960. [PubMed] [Google Scholar]
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Aras MA, Hara H, Hartnett KA, Kandler K, Aizenman E. Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning. J Neurochem. 2009;110:106–117. doi: 10.1111/j.1471-4159.2009.06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Aarts M, Lau A, Tymianski M. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci. 2004;24:8106–8123. doi: 10.1523/JNEUROSCI.1362-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atahan E, Ergun Y, Kurutas EB, Alici T. Protective effect of zinc aspartate on long-term ischemia-reperfusion injury in rat skeletal muscle. Biol Trace Elem Res. 2009 doi: 10.1007/s12011-009-8568-6. [DOI] [PubMed] [Google Scholar]
- Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med. 2008;14:731–740. doi: 10.2119/2008-00050.Beauchamp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Bozym RA, Thompson RB, Stoddard AK, Fierke CA. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol. 2006;1:103–111. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- Cima RR, Dubach JM, Wieland AM, Walsh BM, Soybel DI. Intracellular Ca(2+) and Zn(2+) signals during monochloramine-induced oxidative stress in isolated rat colon crypts. Am J Physiol Gastrointest Liver Physiol. 2006;290:G250–G261. doi: 10.1152/ajpgi.00501.2004. [DOI] [PubMed] [Google Scholar]
- Cortese MM, Suschek CV, Wetzel W, Kroncke KD, Kolb-Bachofen V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic Biol Med. 2008;44:2002–2012. doi: 10.1016/j.freeradbiomed.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Faget KY. Zinc takes the center stage: its paradoxical role in Alzheimer’s disease. Brain Res Brain Res Rev. 2003;41:44–56. doi: 10.1016/s0165-0173(02)00219-9. [DOI] [PubMed] [Google Scholar]
- DeWitt DS, Prough DS. Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. J Neurotrauma. 2003;20:795–825. doi: 10.1089/089771503322385755. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Allen JD. Functional dissociation of the prefrontal cortex and the hippocampus in timing behavior. Behav Neurosci. 1998;112:1043–1047. doi: 10.1037//0735-7044.112.5.1043. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Busto R, Ginsberg MD. Widespread metabolic depression and reduced somatosensory circuit activation following traumatic brain injury in rats. J Neurotrauma. 1994;11:629–640. doi: 10.1089/neu.1994.11.629. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Devinney MJ, Zeak JA, Rintoul GL, Reynolds IJ. Glutamate mobilizes [Zn2+] through Ca2+-dependent reactive oxygen species accumulation. J Neurochem. 2008;106:2184–2193. doi: 10.1111/j.1471-4159.2008.05536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KE, Votyakova TV, Reynolds IJ. Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J Neurochem. 2003;85:563–570. doi: 10.1046/j.1471-4159.2003.01678.x. [DOI] [PubMed] [Google Scholar]
- Doering P, Danscher G, Larsen A, Bruhn M, Sondergaard C, Stoltenberg M. Changes in the vesicular zinc pattern following traumatic brain injury. Neuroscience. 2007;150:93–103. doi: 10.1016/j.neuroscience.2007.09.066. [DOI] [PubMed] [Google Scholar]
- Ellis E, McKinney J, Willoughby K, Liang S, Povlishock J. A new model for rapid stretch-induced injury of cells in culture: characterization of the model using astrocytes. J Neurotrauma. 1995;12:325–329. doi: 10.1089/neu.1995.12.325. [DOI] [PubMed] [Google Scholar]
- Enriquez P, Bullock R. Molecular and cellular mechanisms in the pathophysiology of severe head injury. Curr Pharm Des. 2004;10:2131–2143. doi: 10.2174/1381612043384060. [DOI] [PubMed] [Google Scholar]
- Fekete A, Vizi ES, Kovacs KJ, Lendvai B, Zelles T. Layer-specific differences in reactive oxygen species levels after oxygen-glucose deprivation in acute hippocampal slices. Free Radic Biol Med. 2008;44:1010–1022. doi: 10.1016/j.freeradbiomed.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Floyd CL, Golden KM, Black RT, Hamm RJ, Lyeth BG. Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J Neurotrauma. 2002;19:303–316. doi: 10.1089/089771502753594873. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Maret W, Cuajungco MP. Zinc and excitotoxic brain injury: a new model. Neuroscientist. 2004;10:18–25. doi: 10.1177/1073858403255840. [DOI] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, Satin LS. Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. J Neurotrauma. 2004;21:719–732. doi: 10.1089/0897715041269704. [DOI] [PubMed] [Google Scholar]
- Grady CL, McIntosh AR, Craik FI. Age-related differences in the functional connectivity of the hippocampus during memory encoding. Hippocampus. 2003;13:572–586. doi: 10.1002/hipo.10114. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase H, Maret W. A differential assay for the reduced and oxidized states of metallothionein and thionein. Anal Biochem. 2004;333:19–26. doi: 10.1016/j.ab.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Hall ED, Braughler JM, McCall JM. New pharmacological treatment of acute spinal cord trauma. J Neurotrauma. 1988;5:81–89. doi: 10.1089/neu.1988.5.81. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, O’Dell DM, Pike BR, Lyeth BG. Exposure to environmental complexity promotes recovery of cognitive function after traumatic brain injury. J Neurotrauma. 1996;13:41–47. doi: 10.1089/neu.1996.13.41. [DOI] [PubMed] [Google Scholar]
- Hao Q, Maret W. Imbalance between pro-oxidant and pro-antioxidant functions of zinc in disease. J Alzheimers Dis. 2005;8:161–170. doi: 10.3233/jad-2005-8209. [DOI] [PubMed] [Google Scholar]
- Hellmich HL, Eidson K, Cowart J, Crookshanks J, Boone DK, Shah S, Uchida T, DeWitt DS, Prough DS. Chelation of neurotoxic zinc levels does not improve neurobehavioral outcome after traumatic brain injury. Neurosci Lett. 2008;440:155–159. doi: 10.1016/j.neulet.2008.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich HL, Frederickson CJ, DeWitt DS, Saban R, Parsley MO, Stephenson R, Velasco M, Uchida T, Shimamura M, Prough DS. Protective effects of zinc chelation in traumatic brain injury correlate with upregulation of neuroprotective genes in rat brain. Neurosci Lett. 2004;355:221–225. doi: 10.1016/j.neulet.2003.10.074. [DOI] [PubMed] [Google Scholar]
- Katayama T, Honda Y, Yamasaki H, Kitamura S, Okano Y. Serum zinc concentration in acute myocardial infarction. Angiology. 1990;41:479–485. doi: 10.1177/000331979004100610. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Hopkins RO. Mnemonic functions of the hippocampus: a comparison between animals and humans. Biol Psychol. 2006;73:3–18. doi: 10.1016/j.biopsycho.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Berger RP, Bayir H, Wagner AK, Jenkins LW, Clark RS. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr Opin Crit Care. 2008;14:135–141. doi: 10.1097/MCC.0b013e3282f57564. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Krezel A, Maret W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem. 2006;11:1049–1062. doi: 10.1007/s00775-006-0150-5. [DOI] [PubMed] [Google Scholar]
- Krezel A, Maret W. Different redox states of metallothionein/thionein in biological tissue. Biochem J. 2007;402:551–558. doi: 10.1042/BJ20061044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim YJ, Kim TY, Koh JY, Kim YH. Essential role for zinc-triggered p75NTR activation in preconditioning neuroprotection. J Neurosci. 2008;28:10919–10927. doi: 10.1523/JNEUROSCI.3421-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maret W. Human metallothionein metallomics. J Anal At Spectrom. 2008;23:1055–1062. [Google Scholar]
- Li Y, Maret W. Transient fluctuations of intracellular zinc ions in cell proliferation. Exp Cell Res. 2009;315:2463–2470. doi: 10.1016/j.yexcr.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Rosselet A, Schaller MD, Markert M, Perret C, Feihl F. Nonselective versus selective inhibition of inducible nitric oxide synthase in experimental endotoxic shock. J Infect Dis. 1998;177:127–132. doi: 10.1086/513813. [DOI] [PubMed] [Google Scholar]
- Maret W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc Natl Acad Sci USA. 1994;91:237–241. doi: 10.1073/pnas.91.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W. Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc. Neurochem Int. 1995;27:111–117. doi: 10.1016/0197-0186(94)00173-r. [DOI] [PubMed] [Google Scholar]
- Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal. 2006;8:1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- Maret W. Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc. Exp Gerontol. 2008;43:363–369. doi: 10.1016/j.exger.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Maret W, Jacob C, Vallee BL, Fischer EH. Inhibitory sites in enzymes: zinc removal and reactivation by thionein. Proc Natl Acad Sci USA. 1999;96:1936–1940. doi: 10.1073/pnas.96.5.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W, Li Y. Coordination Dynamics of Zinc in Proteins. Chem Rev. 2009;109:4882–4707. doi: 10.1021/cr800556u. [DOI] [PubMed] [Google Scholar]
- Maret W, Vallee BL. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc Natl Acad Sci USA. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew BP, Dewitt DS, Bryan RMJ, Bukoski RD, Prough DS. Traumatic brain injury reduces myogenic responses in pressurized rodent middle cerebral arteries. J Neurotrauma. 1999;16:1177–1186. doi: 10.1089/neu.1999.16.1177. [DOI] [PubMed] [Google Scholar]
- McAllister TW. Neuropsychiatric sequelae of head injuries. Psychiatr Clin North Am. 1992;15:395–413. [PubMed] [Google Scholar]
- McClain CJ, Twyman DL, Ott LG, Rapp RP, Tibbs PA, Norton JA, Kasarskis EJ, Dempsey RJ, Young B. Serum and urine zinc response in head-injured patients. J Neurosurg. 1986;64:224–230. doi: 10.3171/jns.1986.64.2.0224. [DOI] [PubMed] [Google Scholar]
- Rzigalinski BA, Weber JT, Willoughby KA, Ellis EF. Intracellular free calcium dynamics in stretch-injured astrocytes. J Neurochem. 1998;70:2377–2385. doi: 10.1046/j.1471-4159.1998.70062377.x. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci. 2009;10:780–791. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- Spahl DU, Berendji-Grun D, Suschek CV, Kolb-Bachofen V, Kroncke KD. Regulation of zinc homeostasis by inducible NO synthase-derived NO: nuclear metallothionein translocation and intranuclear Zn2+ release. Proc Natl Acad Sci USA. 2003;100:13952–13957. doi: 10.1073/pnas.2335190100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix CM, Wasserloos KJ, Dineley KE, Reynolds IJ, Levitan ES, Pitt BR. Nitric oxide-induced changes in intracellular zinc homeostasis are mediated by metallothionein/thionein. Am J Physiol Lung Cell Mol Physiol. 2002;282:L185–L192. doi: 10.1152/ajplung.00267.2001. [DOI] [PubMed] [Google Scholar]
- Suh SW, Chen JW, Motamedi M, Bell B, Listiak K, Pons NF, Danscher G, Frederickson CJ. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000;852:268–273. doi: 10.1016/s0006-8993(99)02095-8. [DOI] [PubMed] [Google Scholar]
- Turan B, Fliss H, Desilets M. Oxidants increase intracellular free Zn2+ concentration in rabbit ventricular myocytes. Am J Physiol. 1997;272:H2095–H2106. doi: 10.1152/ajpheart.1997.272.5.H2095. [DOI] [PubMed] [Google Scholar]
- Yang Y, Maret W, Vallee BL. Differential fluorescence labeling of cysteinyl clusters uncovers high tissue levels of thionein. Proc Natl Acad Sci USA. 2001;98:5556–5559. doi: 10.1073/pnas.101123298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B, Ott L, Kasarskis E, Rapp R, Moles K, Dempsey RJ, Tibbs PA, Kryscio R, McClain C. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13:25–34. doi: 10.1089/neu.1996.13.25. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS. Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science. 1996;274:1921–1923. doi: 10.1126/science.274.5294.1921. [DOI] [PubMed] [Google Scholar]