

Abstract
JC virus (JCV) is the aetiological agent of progressive multifocal leukoencephalopathy (PML), a fatal, demyelinating disease of the brain affecting people with AIDS. Although immunosuppression is involved in infection of the brain by JCV, a direct influence of human immunodeficiency virus type 1 (HIV-1) has also been established. The Tat protein of HIV-1 has been implicated in activation of the cytokine transforming growth factor (TGF)-β in HIV-1-infected cells and in stimulating JCV gene transcription and DNA replication in oligodendroglia, the primary central nervous system cell type infected by JCV in PML. This study demonstrated that Tat can cooperate with SMAD proteins, the intracellular effectors of TGF-β, at the JCV DNA control region (CR) to stimulate JCV gene transcription. Tat stimulated JCV early gene transcription in KG-1 oligodendroglial cells when expressed via transfection or added exogenously. Using chromatin immunoprecipitation, it was shown that exogenous Tat enhanced binding of SMAD2, -3 and -4 and their binding partner Fast1 to the JCV CR in living cells. When SMAD2, -3 and -4 were expressed together, Tat, expressed from plasmid pTat, stimulated transcription from both early and late gene promoters, with the early promoter exhibiting stimulation of >100-fold. Tat, SMAD4 and JCV large T-antigen were all visualized in oligodendroglial cells at the border of an active PML lesion in the cerebral frontal lobe. These results revealed a positive reinforcement system in which the SMAD mediators of the TGF-β system act cooperatively with Tat to stimulate JCV gene transcription.
INTRODUCTION
JC virus (JCV) is the aetiological agent of progressive multifocal leukoencephalopathy (PML), a brain infection afflicting approximately 4 % of people with AIDS in the USA. Although highly active antiretroviral therapy has reduced the overall mortality due to opportunistic infections in AIDS, the pathology of PML has altered, and the disease remains debilitating and frequently fatal (Cinque et al., 2003). Immunosuppression is a factor in the development of PML, but the relatively high frequency of PML in AIDS, compared with other states of immunosuppression, has led to studies indicating a direct influence of HIV on activation of JCV in the brain (Chowdhury et al., 1990, 1992; Daniel et al., 2001; Gallia et al., 2000; Johnson, 2003; Krachmarov et al., 1996). Both HIV-1 and JCV infect the brain in AIDS patients with PML, although each virus primarily infects different cell types. Several studies have focused on the HIV-1 protein Tat, as Tat can be transported from one cell type to another (Conant et al., 1996; Daniel et al., 2001; Ensoli et al., 1990; Ezhevsky et al., 1997; Frankel & Pabo, 1988). Oligodendroglia, the central nervous system cell type lytically infected by JCV, are particularly adept at nuclear accumulation of exogenously introduced Tat (Daniel et al., 2004; Enam et al., 2004).
Two major pathways have been identified through which Tat can influence activation of JCV in the brain. First, Tat interacts with two well-characterized protein complexes: Cyclin T1/Cdk9 (Fujinaga et al., 1999; Garber et al., 1998b; Gold et al., 1998; Romano et al., 1999; Wei et al., 1998) and Purα (Chepenik et al., 1998; Daniel et al., 2001; Gallia et al., 1999; Krachmarov et al., 1996; Wortman et al., 2000), together with their respective partners. Purα is a sequence-specific DNA- and RNA-binding protein with affinity for, and strand displacement activity of, a purine-rich element (Bergemann et al., 1992; Darbinian et al., 2001a; Wortman et al., 2005). Purα functionally associates with multiple Cyclin/Cdk complexes (Barr & Johnson, 2001; Itoh et al., 1998) and is capable of recruiting such complexes to specific nucleic acid sites (Liu et al., 2005). One such site is a loop in the transactivation response element (TAR) RNA of the HIV-1 transcript (Chepenik et al., 1998; Johnson et al., 2006). Purα and Tat, together with JCV T-antigen (Gallia et al., 1998), bind to PUR elements in the JCV regulatory region to stimulate both JCV late gene transcription (Krachmarov et al., 1996) and DNA replication (Daniel et al., 2001).
A second pathway by which Tat influences JCV infection of the brain is via alteration of production and activities of immunomodulatory cytokines, particularly transforming growth factor (TGF)-β (Rasty et al., 1996; Reinhold et al., 1999; Thatikunta et al., 1997). The expression of TGF-β is upregulated in HIV-1-infected cells, which can include monocytic cells, microglial cells and astrocytes in the brain (Sawaya et al., 1998). In addition, Tat secreted by cells in the blood can cross the blood–brain barrier (Banks et al., 2005). During early development, TGF-β affects differentiation and apoptosis of oligodendroglia (Bottner et al., 2000; McKinnon et al., 1993; Schmierer & Hill, 2007; Schuster et al., 2002; Silvestri et al., 2008). The HIV-1 Tat protein has been implicated in stimulating transcription of the TGFB gene (Rasty et al., 1996; Sawaya et al., 1998).
TGF-β binding to transmembrane receptors triggers downstream phosphorylation and activation of SMAD proteins (Derynck et al., 1998; Feng & Derynck, 2005; Shi & Massague, 2003). SMAD proteins mediate the effects of the respective TGF-β family member on cellular gene expression. TGF-β1, for example, binds to receptors that catalyse Ser/Thr phosphorylation of SMAD2 and -3 (Shi & Massague, 2003). These proteins, thus activated, associate with SMAD4, which enters the nucleus and, together with cellular partner proteins, including Fast1 (FoxH1), stimulate expression of multiple genes (Feng & Derynck, 2005; Schmierer & Hill, 2007; Shi & Massague, 2003). Recent reports indicate that SMAD proteins stimulate transcription by recruiting histone acetyl transferases, thus achieving effects through chromatin remodelling (Ross et al., 2006). It has been demonstrated that SMAD proteins stimulate transcription of both early (E) and late (L) JCV genes (Enam et al., 2004), and it has been shown that Tat, together with its DNA-binding partner Purα, stimulates JCV gene expression multi-fold (Krachmarov et al., 1996). We therefore wondered whether Tat could cooperate with the SMAD proteins at the JCV regulatory region. Here, we provide evidence that Tat enhances binding of SMAD2, -3 and -4 and Fast1 to the JCV control region (CR) and stimulates the JCV transcriptional response to SMAD proteins in oligodendroglial cells. These findings, together with the observation that Tat stimulates TGF-β production, have revealed a novel signal reinforcement system of potential importance in the development of PML.
METHODS
Cell lines and culture.
KG-1 oligodendroglioma cells are a glial cell line positive for S-100 protein and negative for glial fibrillary acidic protein (Tanaka et al., 1983). U-87MG cells are human glioblastoma cells. 293-T cells are human embryonic kidney cells containing a partial adenovirus 5 genome and also expressing simian virus 40 (SV-40) large T-antigen. All cells were grown in Dulbecco's minimal essential medium with 10 % fetal bovine serum, 1 % penicillin/streptomycin and 1 % l-glutamine. Cells were replated at 70–85 % confluence. After replating, cells were allowed to adhere for 24 h before transfection.
Proteins, antibodies and immunoblotting.
Purified HIV-1 Tat protein (clade B, 86 aa) was obtained from ProSpec. Antibodies recognizing SMAD2/3 (rabbit polyclonal), SMAD4 (mouse monoclonal) and Fast-1 (rabbit polyclonal) were from Santa Cruz Biotechnology. The anti-Tat antibody was a rabbit polyclonal from the NIH AIDS Research and Reference Reagent Program, NIAID, USA. The anti-polyomavirus large T-antigen antibody was a mouse monoclonal from Calbiochem. Secondary antibodies for gel visualization were from Li-Cor, labelled with IRDye 680 or IRDye 800CW near-infrared fluorophores. Imaging of immunoblots used a Li-Cor Odyssey infrared detection system.
Immunohistochemistry.
Brain samples from patients with PML, HIV-1 encephalitis (HIVE), PML and HIVE, and HIV with no CNS disease were obtained from the Manhattan HIV Brain Bank. Brain samples from HIV-negative individuals without neurological disease served as negative controls. Samples for this study were all formalin-fixed, paraffin-embedded tissues from frontoparietal lobes (cortex and underlying white matter) and were sectioned onto slides at 5 μm thickness. Samples were obtained under institutional review board-approved protocols. Deparaffinization, rehydration and H2O2 treatment of slides were as described previously (Johnson et al., 2006). All sections were given a 5 min block with normal horse serum, incubated for 60 min with primary antibody and 30 min with secondary antibody, and visualized by using a Vecta ABC Elite kit (Vector Laboratories) followed by diaminobenzidine. For dual labelling of large T-antigen and Tat, T-antigen staining (diaminobenzidine) was followed by Tat staining (Permanent Red; Dako). Digital micrographs were taken with a Nikon Coolscope microscope.
Chromatin immunoprecipitation (ChIP).
KG-1 cells were transiently transfected with pBLCAT3-Mad1 plasmid, containing a 388 bp segment from the JCV Mad-1 strain comprising the origin of replication, for 72 h (Daniel et al., 2001). The JCV sequence used was obtained from the ATCC and is identified as gbJ02226.1. ChIP was performed as described previously (Kinoshita & Johnson, 2004). Briefly, formaldehyde treatment of live cells was used to cross-link proteins to DNA, followed by sonication to fragment the chromatin and immunoprecipitation of specific proteins to obtain DNA segments. After reversal of the cross-linking, the DNA was analysed by quantitative real-time PCR using a Bio-Rad iCycler. Amplification of the JCV CR was performed in triplicate using CR-specific primers and quantified using a standard curve of JCV Mad-1 DNA. The primers employed were 5′-CTTCTGAGTAAGCTTGGAGGC-3′ and 5′-GTTCCCTTGGCTGCTTTCCAC-3′, representing nt 5103–5123 and 212–232 of the JCV Mad-1, which amplified a sequence of 260 bp from the JCV vector described above.
SMAD and Tat mammalian expression vectors.
SMAD2, -3 and -4 expression regions were in the pRK-5 vector (Genentech) and were FLAG-tagged and under the control of the human cytomegalovirus (CMV) immediate-early promoter (Zhang et al., 1996). JCV E and L promoters were on the pGL3 luciferase expression vector backbone (Promega). Plasmid pTAT expressed Tat (86 aa) from the HIV-1 long terminal repeat (LTR) promoter.
Transfection.
Cells were transfected using Roche FuGene-6 according to the manufacturer's instructions at a 3 : 2 FuGene to plasmid (total) ratio. KG-1 cells were co-transfected with JCV E or L promoter pGL3 luciferase constructs plus the pRL Renilla expression vector (Promega) for normalization. These were co-transfected with or without SMAD2, -3 and/or -4 mammalian expression vectors. Empty vectors were transfected to standardize the total amount of transfected DNA.
Luciferase reporter system.
Cells (2.5×104 per well), transfected as described above, were cultured in 12-well plates. At 48 and 72 h post-transfection, the cells were washed twice with PBS and lysed, and luciferase and Renilla activities were detected by using the Promega Dual-Luciferase Reporter Assay system.
RESULTS
Transfection of pTat stimulates both E and L JCV gene transcription in KG-1 oligodendroglial cells
Expression of the HIV-1 protein Tat stimulates not only HIV-1 gene transcription (Dingwall et al., 1990; Karn, 1991; Rice & Mathews, 1988; Sadaie et al., 1988), but also transcription controlled by the E and L promoters of JCV (Gallia et al., 1998, 2000; Krachmarov et al., 1996). For HIV-1 transcription, Tat acts primarily at the TAR RNA element together with the cellular proteins Purα (Chepenik et al., 1998), cyclin T1 and Cdk9 (Fujinaga et al., 1999; Garber et al., 1998a; Romano et al., 1999; Wei et al., 1998). Tat is not itself a DNA-binding protein. Therefore, at the JCV CR, Tat requires at least one cellular DNA-binding partner. We have shown that Tat cooperates with Purα, which has multiple DNA-binding sites in the CR, including one in a TAR-like element of the late-gene promoter (Haas et al., 1995; Krachmarov et al., 1996). In the brain, Tat is expressed primarily in HIV-infected monocytes, macrophages, microglial cells and astrocytes, whereas JCV primarily infects oligodendroglial cells. We have found that exogenous Tat can be incorporated effectively by oligodendroglia (Daniel et al., 2004). Tat is highly transportable (Daniel et al., 2001, 2004; Ensoli et al., 1993; Ezhevsky et al., 1997; Frankel & Pabo, 1988), and it is likely that transmigrating Tat can be passed among brain cells that are in close proximity (Ma & Nath, 1997). In HIV-infected brain, Tat is highly concentrated in certain oligodendroglial cells, as determined by immunohistochemistry (Daniel et al., 2004; Enam et al., 2004). We sought to compare the effects of expressed Tat on JCV E and L promoters in oligodendroglia and to determine the extent to which these effects are emulated by exogenous, e.g. potentially transmigrating, Tat. Fig. 1 shows the effects of Tat, expressed in KG-1 oligodendroglioma cells from plasmid pTat, expressed under the direction of the HIV-1 LTR promoter, on JCV E and L promoters. In this experiment, all values were relative to the same aliquot of Renilla-transfected KG-1 cells, so that the activities of the JCV E and L promoters could be compared directly. The E and L promoters both employ exactly the same segment of the JCV CR in reversed orientations. It was found that, in this system, the basal activity of the E promoter was approximately 1.7 relative light units (RLU) and that of the L promoter was approximately 35 RLU. Both were stimulated by expression of pTat. E gene expression was stimulated almost 2-fold by maximal amounts of pTat. L gene expression was only moderately stimulated by the same amount of pTat. It should be noted that KG-1 cells express significant amounts of endogenous Purα, which functions together with Tat in these cells (Daniel et al., 2001, 2004).
Fig. 1.
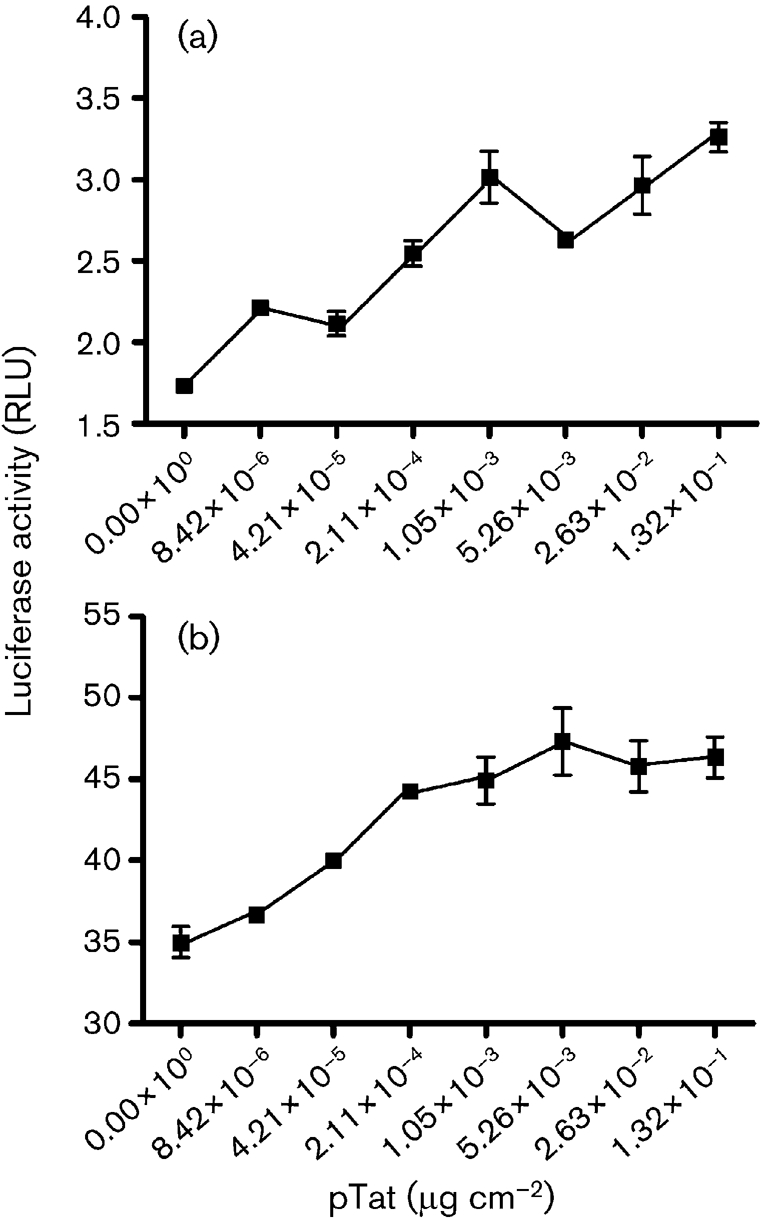
Effects of Tat, expressed from transfected plasmid pTat, on JCV E (a) and JCV L (b) promoters in KG-1 oligodendroglial cells. KG-1 cells were plated in 12-well plates and transfected with pTat expressed as μg (cm of well base area)−2. The JCV E and L plasmids contained the JCV CR in opposite orientations. Luciferase assays were carried out as described in Methods, and results are expressed as relative light units (RLU) for luciferase relative to Renilla. Results are means±sem of triplicate results for a representative experiment.
Exogenous Tat at low concentrations stimulates JCV E gene transcription in KG-1 cells
We compared the effects of expressed, transfected pTat with those of purified Tat added exogenously to the culture medium. Importation of such Tat could mimic the transmigration of Tat in the brain. It was thus important to determine the concentrations of exogenous Tat, if any, that were effective at the JCV E and L promoters. In Fig. 2, as in Fig. 1, the basal level of transcription of the E promoter was <2 RLU, whilst that of the E promoter was approximately 10 RLU. Differences in the exact basal RLU numbers between Figs 1 and 2 were probably due to minor differences in the numbers of cells cultured. In this case, stimulation of the E promoter was more than 7-fold at the optimum concentration of Tat, whilst stimulation of the L promoter was about 1.5-fold. Stimulation of the L promoter might have been higher if the basal level was not already so high. However, we felt it was important to compare these two promoters at exactly the same level of transfected plasmids. The important message from this experiment was that the optimum effects of exogenous Tat occurred at extremely low concentrations. The optimum effect of Tat on the E promoter was at 10−13 M. These results can be compared with the effects of exogenous Tat on JCV DNA replication, which were optimum at concentrations of less than 10−13 M (Daniel et al., 2004). Given that Tat in cells of the brain can be visualized by immunohistochemistry, these concentrations are entirely consistent with the potential for transmigration of Tat among cells of the brain followed by its activation of the JCV promoters. Another important aspect of this experiment is that the results generally emulated those using transfected pTat. Stimulation of the E promoter was significantly higher than that of the L promoter under the conditions employed here. It is interesting that stimulation of the E promoter by the added, purified Tat protein was higher than that with the transfected pTat plasmid. It is conceivable that exogenously added Tat enters certain relevant cell compartments more readily than internally synthesized protein.
Fig. 2.

Effects of exogenous Tat on JCV E (a) and L (b) gene promoters in KG-1 cells. The experiment was carried out as described in the legend to Fig. 1, but instead of transfection, purified Tat (Tat 86, clade B) was added exogenously to the medium at the concentrations indicated.
SMAD effectors of TGF-β bind to the JCV E and L gene transcriptional CR in KG-1 cells, as determined by ChIP
Because Tat stimulates production of TGF-β in HIV-infected cells, we hypothesized that the SMAD effectors of this cytokine might well act on the JCV CR. We have reported previously that Tat stimulates both E and L gene transcription in JCV (Gallia et al., 2000; Krachmarov et al., 1996), but no mechanism had been detailed. Here, we sought to demonstrate that SMAD proteins bind directly to the viral CR. We employed a ChIP assay, which captures the status of the protein–DNA interaction in living KG-1 cells. The amounts of JCV CR we obtained by ChIP from approximately 2×107 cells were in an easily measurable range (Fig. 3). The Tat/Purα complex binds to the CR, and the SMAD proteins, with their partner Fast1, bind to the CR. Does Tat influence the binding of the SMAD proteins? We started to address this by examining the binding of the SMAD proteins to the CR in the presence or absence of exogenous Tat. Fig. 3 is a non-quantitative visualization of the ChIP results by gel electrophoresis of the 260 bp CR segment. All three SMAD proteins and Fast1 were detected on the JCV CR by ChIP in the absence of Tat. SMAD4 was barely detectable, as shown, but was seen more easily following longer exposures. The gel clearly shows that binding of all three proteins to the CR was stimulated in the presence of 10−12 M Tat. Fig. 3 also shows that the specificity of ChIP followed by PCR is suitable for analysis by real-time PCR. All molar values obtained were between those of the negative IgG control (10−13 M) and the positive lysate control (5×10−8 M). Thus, all three endogenous cellular proteins were bound to the JCV CR.
Fig. 3.

Effects of HIV-1 Tat on binding of SMAD proteins to the JCV CR as determined by ChIP. KG-1 cells were transfected to express SMAD2 and -3, SMAD4 or Fast1 in the presence or absence of added exogenous Tat at 10−12 M. ChIP was performed using antibodies against SMAD2/3, SMAD4 or Fast1 as described in Methods. The results of ChIP are presented as acrylamide gel bands of a PCR segment amplified from the immunoprecipitate using primers from the JCV CR. T, Transfected cells; Unt, untransfected cells; T+Tat, transfection in the presence of exogenous Tat. L, B and G1 are negative controls representing no immunoprecipitation, precipitation with protein G agarose beads alone, and beads with an irrelevant IgG1, respectively.
HIV-1 Tat stimulates binding of SMAD2, -3 and -4 and Fast1 to the JCV CR
Analyses of PCR gel bands, as in Fig. 3, are not quantitative. Analyses employing real-time PCR are presented in Fig. 4. Fig. 4(a) demonstrates that we were able, using very practical numbers of cycles, to amplify concentrations of the JCV CR as low as 10−16 M. These results were obtained with the same primers used to generate the single band in Fig. 3 and thus demonstrated our capacity to assess quantitatively the results of ChIP of the JCV CR. Such quantitative results are presented in Fig. 4(b), in which ChIP was performed using antibodies against SMAD2/3, SMAD4 or Fast1 in the presence or absence of purified 10−12 M Tat. Points shown are the means of triplicate results ±sem, which was uniformly very small. The results of Fig. 4(b) clearly demonstrated the degree to which Tat stimulates binding of SMAD2/3, SMAD4 and Fast1 to the JCV CR. In the case of SMAD2/3 and -4, this stimulation was 100- to 1000-fold.
Fig. 4.

Detection of endogenous SMAD proteins bound to the JCV CR by ChIP and real-time PCR. Real-time PCR was performed using primers to amplify the JCV CR as shown in Fig. 3. Melting curves showed a single PCR DNA peak. (a) Real-time PCR for detection of small amounts of CR DNA, as shown in a plasmid pJCV-Mad1 DNA titration. Note that Ct values for amounts as low as 10−16 mol were obtained with fewer than 30 cycles. RFU, Relative fluorescent units. (b) ChIP was performed as described in Methods and in the legend to Fig. 3. ▵, No Tat; ○, +Tat.
SMAD3 and -4 are transiently expressed at high levels after transfection, and Tat has little effect on endogenous or transfected SMAD cellular protein levels
It was necessary to determine whether the effects of Tat on SMAD binding to the JCV CR could be due to the effects of Tat on SMAD intracellular protein levels. We performed immunoblotting on equal aliquots of KG-1 cell lysates to compare endogenous SMAD2/3 with transfected SMAD3, and endogenous SMAD4 with transfected SMAD4. Lysates were equalized according to cell number. Endogenous SMAD2, -3 and -4 were all detected in the lysates (Fig. 5). The antibody used for SMAD3 detected SMAD2/3. Transiently expressed SMAD3 and -4 were detected at very high levels relative to their endogenous counterparts. There was little, if any, change in the levels of SMAD3 and -4, either endogenous or transfected, due to the presence of Tat. We have frequently observed that only very low levels of endogenous SMAD4 are detected by immunoblotting. This could be due to SMAD4 breakdown. As can be seen in the transfected SMAD4 lanes, there were numerous bands detected at lower molecular masses than that of full-length SMAD4. These are likely to be breakdown products for two reasons. First, no bands were detected that were larger than full-length SMAD4. Secondly, SMAD4 was expressed from a cDNA, and thus there would be no smaller bands due to alternative RNA splice products. Interestingly, Tat caused a modest decrease in the number and intensity of these breakdown products. At higher exposures, this Tat effect could also be seen for endogenous SMAD4 and for transfected SMAD3. As Tat had little effect on full-length SMAD protein levels, however, it is unlikely that the effects of Tat on the JCV CR can be ascribed solely to its effects on SMAD protein levels.
Fig. 5.

Immunoblot showing endogenous (End.) and overexpressed (transfected, Tr.) SMAD3 and -4 in KG-1 cells in the presence or absence of exogenously added Tat. Tat was added to a final concentration of 10−12 M. Protein detection was carried out using a Li-Cor Odyssey infrared system as described in Methods. The antibody against SMAD3 also detected SMAD2. SMAD3 expressed from the transfection vector migrated slower than the endogenous protein due to the presence of the FLAG tag.
Tat stimulates both JCV E and L gene transcription in the presence of SMAD2, -3 and -4
We wanted to compare the consequences of the interaction between Tat and SMAD proteins at the JCV CR in another line of glial cells and in a cell line expressing large T-antigen. U-87MG cells are an astroglioma cell line, and 293T cells are human embryonic kidney cells that express the SV-40 large T-antigen. Fig. 6 presents the results of an experiment in which U-87MG and 293T cells were transfected or not with pTat, and luciferase activity was measured as controlled by either the JCV E or L promoter. Of the two lines, 293T cells had the highest transcriptional activity, although the basal activity of the E promoter was not as high as expected, given that these cells reportedly express T-antigen. The levels of T-antigen present under the conditions of our experiments were not determined. The L promoter was highly significantly stimulated by Tat in each cell line. The JCV L promoter was stimulated more than 4-fold by Tat in U-87MG cells. The E promoter was also significantly stimulated by Tat in U-87MG and 293T cells. In contrast to the oligodendroglial KG-1 cells of Fig. 1, promoter stimulation in the astroglial U-87MG cells by Tat was several-fold higher for the L promoter than for the E promoter (Fig. 6).
Fig. 6.
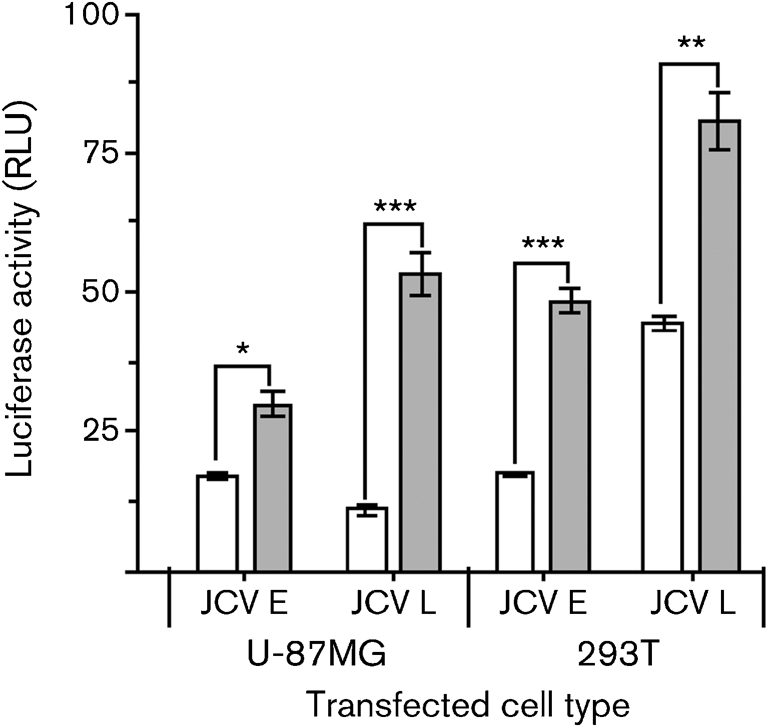
Effects of Tat on JCV E and L gene promoters in U-87MG astrocytes and 293T embryonic kidney cells. Transfections were carried out as described in the legends to Figs 1 and 2. Empty bars, no Tat; shaded bars, +Tat (exogenous Tat was added at a concentration of 10−12 M). Results are means±sem of quadruplicates. *, P<0.05; **, P<0.005; ***, P<0.0005.
To test the effects of SMAD in the KG-1 cells, we decided to use SMAD expression vectors to be certain that all three SMAD proteins were present at functional levels. Fig. 7 shows the effects of SMAD2, -3 and -4, with or without Tat, on the JCV E and L promoters. For this experiment, KG-1 cells were transfected simultaneously with E and L promoter luciferase constructs together with expression vectors for SMAD2, -3 and -4 under the control of the CMV promoter. Vectors for SMAD2 and -3 were transfected together. All of these plasmids were transfected either without pTat or simultaneously with pTat. Empty vectors were used to standardize the total amounts of each plasmid DNA transfected. At 48 and 72 h, Tat either had little effect on or was slightly inhibitory to the effects of SMAD2/3 and -4 on the JCV E promoter. When SMAD2, -3 and -4 were expressed together, however, Tat was highly stimulatory to the E promoter at both 48 and 72 h (Fig. 7; empty bars). At 72 h, the stimulation of JCV early transcription by Tat and SMAD2/3/4 was more than 100-fold. The L promoter was also stimulated by Tat in the presence of SMAD2/3 and SMAD2/3/4, but not to the extent of the E promoter. It was notable that SMAD2/3 had a slight or inhibitory effect on the E promoter in the presence of Tat, whereas they had a significant stimulatory effect on the L promoter. It should also be noted that the basal levels of transcription in the experiment in Fig. 7, i.e. without SMAD proteins, of approximately 2.5 RLU (data not shown) were considerably lower than in Fig. 6 (approximately 25 RLU). We attribute this to the fact that there were multiple vectors transfected in Fig. 7, using three different promoters (JCV, HIV-1 LTR and CMV). This could reduce the effective concentrations of certain basal transcription factors. Regardless of this, the results of Fig. 7 clearly demonstrated that SMAD2, -3 and -4 together greatly magnified the stimulatory effects of Tat on the JCV E promoter, but much less so on the JCV L promoter.
Fig. 7.
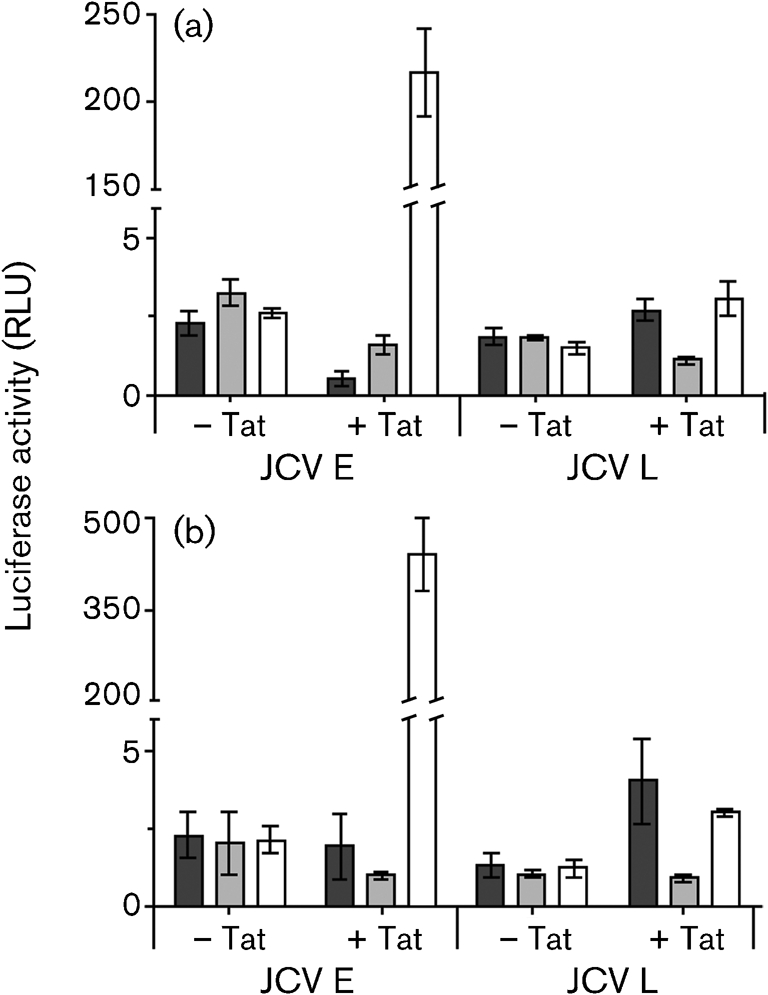
Effects of SMAD proteins (SMAD2/3, dark-shaded bars; SMAD4, light-shaded bars; SMAD2/3/4, empty bars) on JCV E and L promoters in KG-1 cells in the presence or absence of added Tat after (a) 48 h and (b) 72 h. Transfections and luciferase assays were carried out as described in the legends to Figs 1 and 2. Purified Tat was added at a concentration of 10−12 M.
SMAD4, HIV-1 Tat and JCV T-antigen are present in JCV-infected cells at the border of an active PML lesion
We sought to determine whether the SMAD proteins and Tat and T-antigen were present in cells infected with JCV in PML. Fig. 8(a) shows the border of a PML lesion in the cerebral hemisphere of a patient with AIDS, stained with anti-SMAD4 antibody. The top left corner of the inset is located at the border of the lesion, which extends in Fig. 8(a) from top right to bottom left, with lesional, noticeably vacuolated tissue superior to this. SMAD4 staining is red–brown. The non-lesional (non-PML) tissue, at the bottom right of Fig. 8(a), stained more intensely blue with haematoxylin. The PML lesion [Fig. 8(a), upper left] was characterized by the presence of large phagocytic cells and a relative paucity of blue-staining myelin. On both sides of the apparent border, but particularly on the PML side, many cells stained positively for SMAD4. Further away from the PML lesion [Fig. 8(a), bottom right], fewer SMAD4-positive cells were seen. Throughout the non-PML regions of the sample, there were only scattered cells positive for SMAD4, whereas at the border of the lesion, most cells with the appearance of oligodendrocytes were positive for SMAD4 (arrows). These cells were characteristic of JCV-infected cells, as indicated by large, distended nuclei with intranuclear inclusions [Fig. 8(a), inset]. These cells showed intense nuclear staining for SMAD4, whereas nearby phagocytic or astrocytic cells showed weak cytoplasmic staining [Fig. 8(a), inset]. Confirmation that the enlarged oligodendrocytes were infected with JCV is presented in Fig. 8(b), which shows that they stained positively for T-antigen. Fig. 8(c, d) show that the same type of cells also stained positively for HIV-1 Tat. The arrows in Fig. 8(a–d) and the inset indicate cells characteristic of JCV-infected oligodendrocytes. There is no evidence that oligodendrocytes are co-infected with HIV-1 and JCV. Therefore, the Tat protein in these cells has most likely been transported from cells in close proximity that produce HIV-1 proteins. Cells consistent with astrocytes producing Tat are denoted in Fig. 8(c) by the letter A. The presence of Tat in enlarged oligodendrocytes in PML has been observed previously (Enam et al., 2004), and the ability of oligodendrocytes to avidly incorporate Tat has been documented (Daniel et al., 2004). Fig. 8(d) shows a white matter region from a patient with both PML and HIVE, stained with anti-Tat antibody. A variety of positively staining cells could be visualized, including oligodendrocytes (arrows). There was considerable apoptosis in this region, as indicated by multiple cells with fragmented nuclei. Fig. 8(e, f) represents two 5 μm serial sections of a cerebral region of a patient with PML. Fig. 8(e) was dually stained with antibodies against T-antigen and Tat, with corresponding secondary antibodies staining red-brown and red, respectively. Fig. 8(f) shows staining for Tat (red) only. Many of the same cells could be seen in each section, notably the large, distended JCV-infected oligodendrocyte at the centre of each panel. Fig. 8(e, f) clearly demonstrates the oligodendroglial co-localization of Tat and T-antigen, and hence of Tat and JCV, in PML. The results showed that Tat is present at high levels in JCV-bearing cells expressing T-antigen. The results of Fig. 8, taken together, demonstrated that Tat, T-antigen and SMAD4 are all localized in JCV-infected oligodendroglial cells at the borders of PML lesions.
Fig. 8.

Immunohistochemical localization of SMAD4, JCV large T-antigen and HIV-1 Tat at the borders of PML lesions. Immunohistochemistry was performed as described in Methods. Multiple cerebral lesions were examined from four PML patients. These were compared with similar brain regions from three HIV patients without PML, four HIVE patients and three HIV-negative control patients. All samples were from autopsies. Micrographs from representative PML lesions are shown. (a) Staining for SMAD4. The border of a PML lesion is shown, which is visible as a change in intensity of haematoxylin staining and vacuolization running from upper right to lower left. The PML lesion is at the left, superior to this border. The upper left corner of the inset is placed at the border. Inset: magnification of a region near that in the main panel. (b) Staining of the same region as in (a) for JCV large T-antigen. (c) Staining of the same region as in (a) for HIV-1 Tat. (d) Staining for Tat at the border of a PML lesion from a patient with both PML and HIVE. (e) Co-localization of HIV-1 Tat and JCV T-antigen. Secondary antibodies are labelled red (Tat) and red-brown (T-antigen). (f) A serial section adjacent to that in (e). The same distended oligodendrocyte is seen near the centre of (e) and (f); (f) is stained for Tat only. Magnification: (a) ×100; (b–f) ×400. (a–d) are counterstained with haematoxylin, whilst (e) and (f) have no counterstain. Arrows point to cells characteristic of JCV-infected oligodendrocytes.
DISCUSSION
SMAD proteins are produced endogenously in oligodendroglial cells (Fig. 5), and their activity in the nucleus is activated by TGF-β. TGF-β1 is produced at high levels in the brain in PML and can be localized to JCV-infected oligodendrocytes (Enam et al., 2004). In microglial cells, the gene encoding TGF-β1 is induced by the HIV-1 protein Tat (Cupp et al., 1993). We have therefore revealed a powerful synergistic mechanism for the potential activation of JCV by HIV-1 in the brain. HIV-1 encodes Tat, which induces the production of TGF-β in infected brain cells. TGF-β and Tat then both transmigrate to oligodendroglial cells, where they act together to stimulate both E and L gene transcription. TGF-β activates the SMAD proteins, and Tat acts with these proteins to enhance their binding to the JCV CR. E gene transcription is synergistically stimulated by Tat and SMAD proteins more than 100-fold (Fig. 7). Tat does not bind directly to JCV DNA, but complexes with the cellular protein Purα to bind to the CR (Krachmarov et al., 1996). Further work will be necessary to determine whether this complex plays a role in recruiting SMAD proteins to the CR. There are no canonical SMAD-binding elements containing the sequence 5′-AGAC-3′ in the JCV CR. In addition, Tat alone does not bind in this DNA region. It is known that both SMAD proteins and Tat require cellular co-factors to bind nucleic acids. It is conceivable that Purα could fulfil this role as an adaptor, as there are numerous PUR elements in the CR. The CR does contain many sites that are similar to SMAD-binding elements, and SMAD proteins may bind to these.
The TGF-β system is widely believed to mediate immunosuppression. HIV-1 has most likely evolved to benefit from that. In turn, immunosuppression plays a role in the activation of JCV in PML. The present results, however, delineate a role for the TGF-β system that is not clearly related to immunosuppression. In stimulating JCV E and L gene transcription, the SMAD mediators of TGF-β facilitate infection and death of brain cells, oligodendroglia, which are not known to be involved in any aspect of immunity. It should be noted, however, that rearranged JCV sequences in the bone marrow of a patient with rheumatoid arthritis and PML have recently been reported (Marzocchetti et al., 2008). Thus, cells of the immune system may not only be infected with JCV, but may also be a conduit for trafficking JCV to the brain.
Our immunohistochemical studies revealed that SMAD4, Tat and T-antigen are all present in oligodendroglial cells at the borders of PML lesions. Fig. 8 presents a co-localization of Tat and T-antigen in an oligodendrocyte. T-antigen in the brain is specifically located in JCV-infected oligodendroglial cells. There is no reported co-infection of these cells with HIV-1 and JCV. Therefore, the Tat protein present in these cells almost certainly comes from an external source. Further work must be carried out to ascertain the cells of origin of the Tat protein co-localizing with T-antigen in oligodendrocytes.
The results presented describe a mechanism by which HIV-1 can help activate JCV in the brain by its effects on a cytokine, TGF-β. The results raise the question: how did the interplay between these two viruses originate? There is little evolutionary advantage to HIV-1 from such a relationship because JCV in the brain ultimately kills the host harbouring HIV-1. Conversely, JCV would not have evolved to benefit from HIV-1, as HIV-1 has infected humans relatively recently. Thus, the relationship is likely to be incidental. It probably involves benefits to both viruses by the interaction of their distinct viral proteins with the same set of host-cell proteins. For example, the HIV-1 Tat protein interacts with cellular proteins to stimulate HIV-1 transcription and replication as well as JCV transcription and replication. In doing so, Tat relies on some of the same cellular proteins, including Purα, that interact with JCV T-antigen. Tat activation of the TGF-β system culminates in activation of the SMAD proteins described here, which promote HIV-1 immunosuppression and incidentally stimulate JCV gene transcription.
Acknowledgments
Work was supported by NIH grants to E. M. J., K. K., J. G., J. R. and S. M. All brain tissue samples were generously provided by the Manhattan HIV Brain Bank (U01MH083501/R24MH59724), directed by S. M. We thank the NIH AIDS Research and Reference Reagent Program, NIAID, USA, for anti-Tat antibodies.
References
- Banks, W. A., Robinson, S. M. & Nath, A. (2005). Permeability of the blood-brain barrier to HIV-1 Tat. Exp Neurol 193, 218–227. [DOI] [PubMed] [Google Scholar]
- Barr, S. M. & Johnson, E. M. (2001). Ras-induced colony formation and anchorage-independent growth inhibited by elevated expression of Purα in NIH3T3 cells. J Cell Biochem 81, 621–638. [DOI] [PubMed] [Google Scholar]
- Bergemann, A. D., Ma, Z.-W. & Johnson, E. M. (1992). Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol Cell Biol 12, 5673–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner, M., Krieglstein, K. & Unsicker, K. (2000). The transforming growth factor-βs: structure, signaling, and roles in nervous system development and functions. J Neurochem 75, 2227–2240. [DOI] [PubMed] [Google Scholar]
- Chepenik, L. G., Tretiakova, A. P., Krachmarov, C. P., Johnson, E. M. & Khalili, K. (1998). The single-stranded DNA binding protein, Pur-α, binds HIV-1 TAR RNA and activates HIV-1 transcription. Gene 210, 37–44. [DOI] [PubMed] [Google Scholar]
- Chowdhury, M., Taylor, J. P., Tada, H., Rappaport, J., Wong-Staal, F., Amini, S. & Khalili, K. (1990). Regulation of the human neurotropic virus promoter by JCV-T antigen and HIV-1 Tat protein. Oncogene 5, 1737–1742. [PubMed] [Google Scholar]
- Chowdhury, M., Taylor, J. P., Chang, C. F., Rappaport, J. & Khalili, K. (1992). Evidence that a sequence similar to TAR is important for induction of the JC virus late promoter by human immunodeficiency virus type 1 Tat. J Virol 66, 7355–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque, P., Koralnik, I. J. & Clifford, D. B. (2003). The evolving face of human immunodeficiency virus-related progressive multifocal leukoencephalopathy: defining a consensus terminology. J Neurovirol 9 (Suppl. 1), 88–92. [DOI] [PubMed] [Google Scholar]
- Conant, K., Ma, M., Nath, A. & Major, E. O. (1996). Extracellular human immunodeficiency virus type 1 Tat protein is associated with an increase in both NF-κB binding and protein kinase C activity in primary human astrocytes. J Virol 70, 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp, C., Taylor, J. P., Khalili, K. & Amini, S. (1993). Evidence for stimulation of the transforming growth factor β1 promoter by HIV-1 Tat in cells derived from CNS. Oncogene 8, 2231–2236. [PubMed] [Google Scholar]
- Daniel, D. C., Wortman, M. J., Schiller, R. J., Liu, H., Gan, L., Mellen, J. S., Chang, C. F., Gallia, G. L., Rappaport, J. & other authors (2001). Coordinate effects of human immunodeficiency virus type 1 protein Tat and cellular protein Purα on DNA replication initiated at the JC virus origin. J Gen Virol 82, 1543–1553. [DOI] [PubMed] [Google Scholar]
- Daniel, D. C., Kinoshita, Y., Khan, M. A., Del Valle, L., Khalili, K., Rappaport, J. & Johnson, E. M. (2004). Internalization of exogenous human immunodeficiency virus-1 protein, Tat, by KG-1 oligodendroglioma cells followed by stimulation of DNA replication initiated at the JC virus origin. DNA Cell Biol 23, 858–867. [DOI] [PubMed] [Google Scholar]
- Darbinian, N., Gallia, G. L. & Khalili, K. (2001a). Helix-destabilizing properties of the human single-stranded DNA- and RNA-binding protein Purα. J Cell Biochem 80, 589–595. [PubMed] [Google Scholar]
- Derynck, R., Zhang, Y. & Feng, X. H. (1998). Smads: transcriptional activators of TGF-β responses. Cell 95, 737–740. [DOI] [PubMed] [Google Scholar]
- Dingwall, C., Ernberg, I., Gait, M. J., Green, S. M., Heaphy, S., Karn, J., Lowe, A. D., Singh, M. & Skinner, M. A. (1990). HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J 9, 4145–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enam, S., Sweet, T. M., Amini, S., Khalili, K. & Del Valle, L. (2004). Evidence for involvement of transforming growth factor β1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch Pathol Lab Med 128, 282–291. [DOI] [PubMed] [Google Scholar]
- Ensoli, B., Barillari, G., Salahuddin, S. Z., Gallo, R. C. & Wong-Staal, F. (1990). Tat protein of HIV-1 stimulates growth of cells derived from Kaposi's sarcoma lesions of AIDS patients. Nature 345, 84–86. [DOI] [PubMed] [Google Scholar]
- Ensoli, B., Buonaguro, L., Barillari, G., Fiorelli, V., Gendelman, R., Morgan, R. A., Wingfield, P. & Gallo, R. C. (1993). Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein in cell growth and viral transactivation. J Virol 67, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhevsky, S. A., Nagahara, H., Vocero-Akbani, A. M., Gius, D. R., Wei, M. C. & Dowdy, S. F. (1997). Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci U S A 94, 10699–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, X. H. & Derynck, R. (2005). Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 21, 659–693. [DOI] [PubMed] [Google Scholar]
- Frankel, A. D. & Pabo, C. O. (1988). Cellular uptake of the Tat protein from human immunodeficiency virus. Cell 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- Fujinaga, K., Taube, R., Wimmer, J., Cujec, T. P. & Peterlin, B. M. (1999). Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc Natl Acad Sci U S A 96, 1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallia, G. L., Safak, M. & Khalili, K. (1998). Interaction of the single-stranded DNA-binding protein Purα with the human polyomavirus JC virus early protein T-antigen. J Biol Chem 273, 32662–32669. [DOI] [PubMed] [Google Scholar]
- Gallia, G. L., Darbinian, N., Tretiakova, A., Ansari, S. A., Rappaport, J., Brady, J., Wortman, M. J., Johnson, E. M. & Khalili, K. (1999). Association of HIV-1 Tat with the cellular protein, Purα, is mediated by RNA. Proc Natl Acad Sci U S A 96, 11572–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallia, G. L., Johnson, E. M. & Khalili, K. (2000). Purα: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res 28, 3197–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber, M. E., Wei, P. & Jones, K. A. (1998a). HIV-1 Tat interacts with cyclin T1 to direct the P-TEFb CTD kinase complex to TAR RNA. Cold Spring Harb Symp Quant Biol 63, 371–380. [DOI] [PubMed] [Google Scholar]
- Garber, M. E., Wei, P., KewalRamani, V. N., Mayall, T. P., Herrmann, C. H., Rice, A. P., Littman, D. R. & Jones, K. A. (1998b). The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 12, 3512–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold, M. O., Yang, X., Herrmann, C. H. & Rice, A. P. (1998). PITALRE, the catalytic subunit of TAK, is required for human immunodeficiency virus Tat transactivation in vivo. J Virol 72, 4448–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, S., Thatikunta, P., Steplewski, A., Johnson, E. M., Khalili, K. & Amini, S. (1995). A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J Cell Biol 130, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, H., Wortman, M. J., Kanovsky, M., Uson, R. R., Gordon, R. E., Alfano, N. & Johnson, E. M. (1998). Alterations in Purα levels and intracellular localization in the CV-1 cell cycle. Cell Growth Differ 9, 651–665. [PubMed] [Google Scholar]
- Johnson, E. M. (2003). The Pur protein family: clues to function from recent studies on cancer and AIDS. Anticancer Res 23, 2093–2100. [PubMed] [Google Scholar]
- Johnson, E. M., Kinoshita, Y., Weinreb, D. B., Wortman, M. J., Simon, R., Khalili, K., Winckler, B. & Gordon, J. (2006). Role of Purα in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res 83, 929–943. [DOI] [PubMed] [Google Scholar]
- Karn, J. (1991). Control of human immunodeficiency virus replication by the tat, rev, nef and protease genes. Curr Opin Immunol 3, 526–536. [DOI] [PubMed] [Google Scholar]
- Kinoshita, Y. & Johnson, E. M. (2004). Site-specific loading of an MCM protein complex in a DNA replication initiation zone upstream of the c-MYC gene in the HeLa cell cycle. J Biol Chem 279, 35879–35889. [DOI] [PubMed] [Google Scholar]
- Krachmarov, C. P., Chepenik, L. G., Barr-Vagell, S., Khalili, K. & Johnson, E. M. (1996). Activation of the JC virus Tat-responsive transcriptional control element by association of the Tat protein of human immunodeficiency virus 1 with cellular protein Purα. Proc Natl Acad Sci U S A 93, 14112–14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H., Barr, S. M., Chu, C., Kohtz, D. S., Kinoshita, Y. & Johnson, E. M. (2005). Functional interaction of Purα with the Cdk2 moiety of cyclin A/Cdk2. Biochem Biophys Res Commun 328, 851–857. [DOI] [PubMed] [Google Scholar]
- Ma, M. & Nath, A. (1997). Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol 71, 2495–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzocchetti, A., Wuthrich, C., Tan, C. S., Tompkins, T., Bernal-Cano, F., Bhargava, P., Ropper, A. H. & Koralnik, I. J. (2008). Rearrangement of the JC virus regulatory region sequence in the bone marrow of a patient with rheumatoid arthritis and progressive multifocal leukoencephalopathy. J Neurovirol 14, 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon, R. D., Piras, G., Ida, J. A., Jr & Dubois-Dalcq, M. (1993). A role for TGF-β in oligodendrocyte differentiation. J Cell Biol 121, 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasty, S., Thatikunta, P., Gordon, J., Khalili, K., Amini, S. & Glorioso, J. C. (1996). Human immunodeficiency virus tat gene transfer to the murine central nervous system using a replication-defective herpes simplex virus vector stimulates transforming growth factor β1 gene expression. Proc Natl Acad Sci U S A 93, 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold, D., Wrenger, S., Kahne, T. & Ansorge, S. (1999). HIV-1 Tat: immunosuppression via TGF-β1 induction. Immunol Today 20, 384–385. [DOI] [PubMed] [Google Scholar]
- Rice, A. P. & Mathews, M. B. (1988). Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature 332, 551–553. [DOI] [PubMed] [Google Scholar]
- Romano, G., Kasten, M., De Falco, G., Micheli, P., Khalili, K. & Giordano, A. (1999). Regulatory functions of Cdk9 and of cyclin T1 in HIV Tat transactivation pathway gene expression. J Cell Biochem 75, 357–368. [PubMed] [Google Scholar]
- Ross, S., Cheung, E., Petrakis, T. G., Howell, M., Kraus, W. L. & Hill, C. S. (2006). Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J 25, 4490–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie, M. R., Benter, T. & Wong-Staal, F. (1988). Site-directed mutagenesis of two trans-regulatory genes (tat-III,trs) of HIV-1. Science 239, 910–913. [DOI] [PubMed] [Google Scholar]
- Sawaya, B. E., Thatikunta, P., Denisova, L., Brady, J., Khalili, K. & Amini, S. (1998). Regulation of TNFα and TGFβ-1 gene transcription by HIV-1 Tat in CNS cells. J Neuroimmunol 87, 33–42. [DOI] [PubMed] [Google Scholar]
- Schmierer, B. & Hill, C. S. (2007). TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8, 970–982. [DOI] [PubMed] [Google Scholar]
- Schuster, N., Bender, H., Philippi, A., Subramaniam, S., Strelau, J., Wang, Z. & Krieglstein, K. (2002). TGF-β induces cell death in the oligodendroglial cell line OLI-neu. Glia 40, 95–108. [DOI] [PubMed] [Google Scholar]
- Shi, Y. & Massague, J. (2003). Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700. [DOI] [PubMed] [Google Scholar]
- Silvestri, C., Narimatsu, M., von Both, I., Liu, Y., Tan, N. B., Izzi, L., McCaffery, P., Wrana, J. L. & Attisano, L. (2008). Genome-wide identification of Smad/Foxh1 targets reveals a role for Foxh1 in retinoic acid regulation and forebrain development. Dev Cell 14, 411–423. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Nagao, S., Tohgo, A., Sekiguchi, F., Kohno, M., Ogawa, H., Matsui, T. & Matsutani, M. (1983). Effects of human fibroblast interferon on human gliomas transplanted into nude mice. Gann 74, 308–316. [PubMed] [Google Scholar]
- Thatikunta, P., Sawaya, B. E., Denisova, L., Cole, C., Yusibova, G., Johnson, E. M., Khalili, K. & Amini, S. (1997). Identification of a cellular protein that binds to Tat-responsive element of TGF β-1 promoter in glial cells. J Cell Biochem 67, 466–477. [PubMed] [Google Scholar]
- Wei, P., Garber, M. E., Fang, S. M., Fischer, W. H. & Jones, K. A. (1998). A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462. [DOI] [PubMed] [Google Scholar]
- Wortman, M. J., Krachmarov, C. P., Kim, J. H., Gordon, R. G., Chepenik, L. G., Brady, N. N., Gallia, G. L., Khalili, K. & Johnson, E. M. (2000). Interaction of HIV-Tat with Pur-α in nuclei of human glial cells: characterization of RNA-mediated protein–protein binding. J Cell Biochem 77, 65–74. [DOI] [PubMed] [Google Scholar]
- Wortman, M. J., Johnson, E. M. & Bergemann, A. D. (2005). Mechanism of DNA binding and localized strand separation by Purα and comparison with Pur family member, Purβ. Biochim Biophys Acta 1743, 64–78. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Feng, X., We, R. & Derynck, R. (1996). Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 383, 168–172. [DOI] [PubMed] [Google Scholar]