

Abstract
We demonstrate a new approach for internal mass calibration on an electron transfer dissociation-enabled linear ion trap-orbitrap hybrid mass spectrometer. Fluoranthene cations, a byproduct of the reaction used for generation of electron transfer dissociation reagent anions, are co-injected with the analyte cations in all orbitrap mass analysis events. The fluoranthene cations serve as a robust internal calibrant with minimal impact on scan time (<20 ms) or spectral quality. Following external mass calibration, 60 replicate LC-MS/MS runs of a complex peptide mixture were collected over the course of ∼136 h (almost 6 days). Using only standard external mass calibration, the mass accuracy for a typical analysis was −3.31 ± 0.93 ppm (σ) for precursors and −2.32 ± 0.89 ppm for products. After application of internal recalibration, mass accuracy improved to +0.77 ± 0.71 ppm for precursors and +0.17 ± 0.67 ppm for products. When all 60 replicate runs were analyzed together without internal mass recalibration, the mass accuracy was −1.23 ± 1.54 ppm for precursors and −0.18 ± 1.42 ppm for products, nearly a 2-fold drop in precision relative to an individual run. After internal mass recalibration, this improved to +0.80 ± 0.70 ppm for precursors and +0.16 ± 0.67 ppm for products, roughly equivalent to that obtained in a single run, demonstrating a near complete elimination of mass calibration drift.
Mass accuracy is one of the most fundamental parameters of mass spectrometer performance (1). The debut of hybrid linear ion trap-Fourier transform mass spectrometers (2, 3) in 2004 was a major advancement in this field as peptide precursor masses could routinely be measured with low part-per-million accuracy. This introduction was non-coincidentally paired with automatic gain control, the ability to roughly control the number of ionic charges in a mass analyzer, which minimizes space-charge effects (4–7) and allows Fourier transform mass spectrometers to perform at their full potential (2, 8, 9). Such measurement capability can greatly improve the specificity of identification without significantly diminishing throughput. Another such turning point for mass accuracy in proteomics is likely approaching as the increasing speed, sensitivity, and efficiency of newer FTMS instruments (10, 11) make the acquisition of high-resolution tandem mass spectra more practical (12–20).
Achievement of higher mass measurement accuracy (21, 22) is one of the most straightforward ways to add specificity to database searching (1, 23, 24), allows more efficient de novo sequencing (25–28), and could perhaps facilitate alternative data acquisition and analysis strategies such as those that do not require collection of tandem mass spectra (29–33). Additionally, mass calibration drift, due to variations in the environment and electronics of the instrument, is always a concern that must be accounted for, particularly in large scale experiments.
Most of the approaches presented thus far for linear ion trap-Fourier transform hybrid mass spectrometers have focused on the use of internal calibrants, which are co-detected with analytes, as opposed to external calibration in which separate spectra are acquired that contain the calibrants. Internal mass calibration yields better accuracy (34, 35) but tends to be more experimentally demanding. A compromise between the two is external mass calibration paired with a single internal calibrant, or “lock mass,” to provide minor adjustments.
Several reports describing internal calibration under chromatographic conditions utilized a dual ESI source to simultaneously introduce calibrants and analytes to the mass spectrometer (36–40), although at least one report utilized a flow injection method where calibrants are combined with the chromatographic effluent prior to ionization (41). These approaches produce a reliable flux of calibrant ions but require a modified ion source or chromatographic setup. Depending upon the arrangement, competition for ionization between analytes and calibrants can be cause for concern. Other internal calibration techniques have been described that make use of information about analytes available under certain conditions, rather than calibrants, such as the same species present at different charge states (42, 43).
One of the most extensive studies of mass accuracy in shotgun proteomics performed to date was that of Haas et al. (44), who utilized a linear ion trap-Fourier transform ICR mass spectrometer in which internal mass calibration was performed using five common polydimethylcyclosiloxane ions present in ambient laboratory air that are often observed in LC-MS data. Additionally, the standard ICR calibration equation was modified to include an extra term accounting for the total ion current of the spectrum to adjust for space-charge effects. The accuracy observed for precursor masses was low part per million (−0.25 ± 1.46 ppm), only a few times worse than obtained by selected ion monitoring (SIM)1 scans (−0.41 ± 0.44 ppm).
A similar study was performed by Olsen et al. (45) on a linear ion trap-Fourier transform orbitrap mass spectrometer. In this work, the c-trap, a radio frequency storage and injection quadrupole, was used to store calibrant species after isolation in the linear ion trap prior to the normal analyte scan sequence. The six-residue polydimethylcyclosiloxane ion was used as the internal calibrant for MS1 spectra, whereas its methane neutral loss ion was used for MS2. To further improve mass accuracy, MS1 spectra were averaged over the precursor chromatographic elution profile. Low to sub-part-per-million mass accuracy was obtained in this study as well for precursors as well as products.
Both of these methods suffer from the fact that the signal of background ions fluctuates widely over the course of an analysis and from laboratory to laboratory. Additionally, in the method of Olsen et al. (45), isolation and transfer of the background ions adds significantly to the scan duration. Here we describe a simple, fast, and robust alternative for achieving sub-part-per-million mass accuracy on an ETD-enabled linear ion trap-orbitrap mass spectrometer. The method utilizes a single internal calibrant that is generated with high flux and steady signal over days and weeks in the ETD chemical ionization source. Using simple post-acquisition mass recalibration, we achieved a precursor mass accuracy comparable to the best reported for shotgun proteomics experiments with substantially less data manipulation and impact on scan time. Product mass accuracy was also sub-part per million.
EXPERIMENTAL PROCEDURES
Instrumentation
All data were collected using an ETD-enabled LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Fluoranthene cations are generated as a byproduct of the chemical ionization (CI) process that is used to produce radical fluoranthene anions, which are used in the ETD reaction. To enable the co-injection of fluoranthene calibrant cations with the analyte cation population, the instrument firmware was modified.
Whether the analysis took the form of an MS1 or MS2 scan, following injection of the analyte ions into the c-trap from the linear ion trap, all the ion optics between the CI source and c-trap were set to transmit positive ions. Then, using the second lens of the CI source as a gate, ions were transmitted from the CI source into the c-trap. Both the analyte ions and internal calibrant ions were stored simultaneously in the c-trap. Following injection of the fluoranthene cations, all the ion optics were restored to their normal settings, and the scan continued as normal with injection of the ion population from the c-trap into the orbitrap for m/z analysis.
Various experiments were performed to optimize the fluoranthene cation injection process. Typically, these involved ramping a particular voltage (e.g. the c-trap lens direct current offsets, the reagent transfer multipole radio frequency amplitude, etc.) while tracking the intensity of the fluoranthene cation. Additionally, other parameters such as ion flight time were measured. Only the absolute minimum necessary time was allotted to these processes. As a result, injection of the fluoranthene cations never took longer than 20 ms (the actual time varied slightly depending upon source performance) and was often just a few milliseconds.
Sample Preparation
Approximately 2 × 107 human embryonic stem cells (H9 cell line) were harvested and washed with PBS buffer. The cells were resuspended with 500 μl of lysis buffer (50 mm Tris, pH 7.8, 100 mm NaCl, and 8 m urea) and lysed by three steps of sonication (Misonix XL-2000, Farmingdale, NY) with a 90-s break on ice after each step: level 5 for 30 s, level 7 for 30 s, and level 10 for 30 s. The cell debris were removed by centrifugation at 14,000 × g for 3 min. The cell lysate was then incubated with 10 mm DTT at room temperature for 30 min followed by 20 mm iodoacetamide alkylation in the dark at room temperature for 30 min. Endoproteinase Lys-C (Wako, Osaka, Japan) was added at an enzyme:substrate ratio of 1:150. The digestion proceeded for 8 h in the dark at 30 °C. The sample was desalted by SepPak (Waters, Milford, MA) before LC-MS/MS analysis.
Liquid Chromatography-Tandem Mass Spectrometry
A Waters nanoAcquity UPLC system (Framingham, MA) was connected to the ETD-enabled LTQ Orbitrap XL. About 1.5 μg of the H9 total cell lysate peptide mixture was loaded onto a precolumn (5-μm C18, 5 cm, 75-μm inner diameter) and separated on an analytical column (5-μm C18, 12 cm, 50-μm inner diameter) using a 90-min gradient from 5 to 40% acetonitrile in 0.2% formic acid (46).
Before the start of data collection, the LTQ Orbitrap XL was calibrated externally twice using the standard calibration mixture (caffeine, peptide MRFA, and Ultramark 1621). The mass spectrometer was operated in data-dependent double-play mode in which the six most intense ions from the survey MS1 scan (m/z 150–1500) were chosen for ETD fragmentation at an activation time of 70 ms. Singly and doubly charged precursors were excluded as well as precursors with unassigned charge states. Dynamic exclusion was activated after one fragmentation event for 40 s with a maximum peak list of 500. The automatic gain control target values for the orbitrap were set to be 1 × 106 for FT MS1 scans and 5 × 105 for FT MSn scans. The resolving power of the orbitrap was set at 60,000 for all scans. The fluoranthene cation internal calibrant peak at m/z 202.07770 was introduced throughout data collection.
Custom Software
Custom software was developed in C# with Microsoft Visual Studio 2005 and/or 2008 using the Microsoft .NET Framework 2.0 and/or 3.5. The XRawfile COM library (XRawfile2.dll) provided with Xcalibur was used to access the proprietary Thermo Scientific .raw data file format.
Data Reduction
Input files for database searching were generated by the DTA Generator software described previously (47), modified to allow internal mass recalibration. ETD spectral pre-processing was performed on all MS/MS spectra to remove interfering peaks from unreacted precursor, charge-reduced precursors (ETnoD), and neutral losses from ETnoD. Calibrant peaks were also removed. A signal-to-noise ratio (S/N) threshold of 1.5 was applied to all MS/MS peaks. S/N was calculated from the FT scan label data as follows.
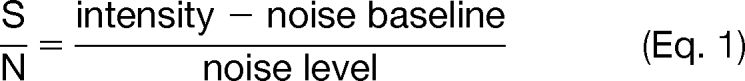 |
Internal mass recalibration was performed post-acquisition in DTA Generator for both MS1 and MS2 spectra based on the fluoranthene radical cation internal calibrant at a theoretical m/z of 202.07770 using either a linear or proportional m/z correction. The linear mass recalibration equation for one internal calibrant used was as follows.
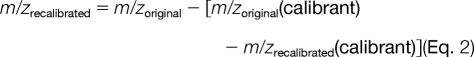 |
The proportional mass recalibration equation for one internal calibrant used was as follows.
Database Searching
Peptides were identified by searching with the Open Mass Spectrometry Search Algorithm (48) (OMSSA, version 2.1.4) with average precursor mass tolerance of ±5.0 Da, monoisotopic product mass tolerance of ±0.01 Da, carbamidomethylation of cysteine (+57.02146 Da) as a fixed modification, and oxidization of methionine (+15.99491 Da) as a variable modification. Spectra were searched against the human International Protein Index (49) FASTA database (version 3.57) concatenated with the reversed version of all protein sequences for target-decoy searching (50).
False Discovery Rate Optimization
The number of unique peptides identified at approximately a 1% false discovery rate (FDR) was optimized for each LC-MS/MS replicate run with custom software. Expectation value (e-value) score threshold and maximum precursor mass error were iteratively tested to determine the maximum number of target hits satisfying the allowed FDR. After the optimal parameters were found, only the spectrum with the best e-value score per unique peptide sequence was used for the next stage of analysis to prevent multiple spectra identifying the same peptide sequence from biasing the results.
Mass Accuracy Analysis
Spectra were analyzed with a custom application to evaluate mass accuracy. The software evaluated each unique peptide sequence identified with OMSSA at 1% FDR for each replicate LC-MS/MS run. First, the program retrieved data from the corresponding MS1 and MS2 spectra. The peak selected for isolation was located in the MS1 spectrum and converted to neutral mass using the known precursor charge state. This mass was then corrected for isotopic shift by subtracting an integer multiple of 1.003355 Da (13C − 12C) to yield the mass closest to the theoretical neutral monoisotopic mass. The difference between this a posteriori experimental monoisotopic mass and theoretical monoisotopic mass was taken to be the precursor mass error.
For MS2 spectra, each centroid peak was considered at the charge state recorded in the FT scan label data. Peaks with a charge state of zero, where the charge state could not be confidently assigned, were skipped. A S/N of 1.5 or higher is required for each peak (as was applied prior to database searching). Calibrant and isotopic peaks were not considered as well as peaks removed by ETD spectral pre-processing. Any peak within the very wide tolerance of ±100 ppm of a theoretical fragment peak was counted as a match.
Mass errors were histogrammed with a bin width of 0.25 ppm. A Gaussian function was then fit to the histogram using the Levenberg-Marquardt algorithm for nonlinear least-squares curve fitting, and the mean and S.D. of the normal distribution is reported as the mass accuracy. These parameters for the center and width of the distribution can be thought of as the accuracy and precision, respectively, of the mass measurement.
RESULTS
Internal Calibrant Source
The internal calibrant scan sequence is depicted in Fig. 1. The CI reaction that produces radical fluoranthene anions for ETD simultaneously produces a robust flux of fluoranthene cations. These cations can be rapidly transferred to the c-trap for storage, and the analyte scan sequence proceeds normally. Transfer and detection in the orbitrap also ensue as usual.
Fig. 1.

Schematic depiction of fluoranthene cation internal calibrant scan event. First, analyte cations (blue) are injected through the source region into the linear ion trap, isolated and fragmented (in the case of MS/MS), and then transferred to the c-trap. Next, calibrant fluoranthene cations (red) from the CI source are transferred through a multipole and the collision cell into the c-trap. Finally, all the ions are transferred together to the orbitrap for high-resolution and high-mass accuracy analysis.
Internal Calibrant Robustness
To study the effects of internal mass recalibration over a long period of time, 60 replicate LC-MS/MS runs of an unfractionated complex mixture of peptides, generated with Lys-C, were acquired. Roughly 2000 unique peptides were identified following ETD with FT MS1 and MS2 detection in each of the 60 analyses. To evaluate the robustness of the fluoranthene cation signal, mass chromatograms were extracted for both analytes and calibrant. Fig. 2a shows a typical MS1 base peak chromatogram. Note that although the analyte signal fluctuates as peptides elute throughout the LC gradient the calibrant signal remains essentially constant. Fig. 2b displays an example MS1 spectrum with the internal calibrant peak highlighted. At ∼202 Th, the peak is at a sufficiently low mass-to-charge ratio to avoid interfering with most analytes of interest.
Fig. 2.

Mass chromatograms and single scan spectra from replicate LC-MS/MS run 30. Although the analyte signal varies greatly over the chromatographic gradient for both MS1 (a) and MS2 (c) scan events, the internal calibrant signal remains consistent and robust in both MS1 (b) and MS2 (d) spectra.
A typical MS2 total ion current chromatogram is presented in Fig. 2c. Both analyte and calibrant chromatograms show more variation than their MS1 counterparts; but again, the calibrant signal remains quite strong and fairly independent of the analyte signal. The primary reason for the added fluctuation in the calibrant signal was the varying m/z scan range, which is dependent upon precursor charge state. Source performance and the stability of the electronics played a minor role. Fig. 2d presents a typical MS2 spectrum. This spectrum was confidently matched (e-value score of 6.2 × 10−15) to the peptide with sequence AQLRELNITAAK. Again, the calibrant signal at 202 Th minimally interferes with product ion peaks as it is a lower m/z value than most fragments containing two amino acid residues, i.e. c2 and z2· fragments. This particular example also shows a benefit of high resolution MS2 analysis as two pairs of fragment ions in this spectrum, c4/z5· and c7/z8·, have isotopic peaks separated by only 0.018 Th that could only be differentiated at ≥27,000 and ≥47,000 resolving power (assuming equal peak intensities), respectively, substantially clarifying the peptide identification.
Mass Accuracy Analyses
Mass error distributions for a single run with external mass calibration and internal mass recalibration using either a linear or proportional correction are shown for precursors in Fig. 3a and for products in Fig. 3b. This example comes from the 60th and final replicate LC-MS/MS analysis, which started ∼134 h (∼5.5 days) following external mass calibration.
Fig. 3.

Mass error distributions for 1934 precursors (a) and ∼24,000 products (b) detected in orbitrap during replicate LC-MS/MS run 60. The data were analyzed with standard external mass calibration (dark gray), linear internal mass recalibration (light gray), and proportional internal mass recalibration (medium gray). This run started about 134 h (∼5.5 days) after external mass calibration. Both internal mass recalibration strategies show improvement in the center and width of the distribution, although proportional internal mass recalibration is superior.
Relying solely on external mass calibration, 1934 precursors had a mean mass accuracy of −3.31 ppm and S.D. of 0.93 ppm. Linear mass recalibration with the ETD internal calibrant yielded a distribution centered closer to zero with a mean of −1.68 ppm; however, the S.D. increased slightly to 0.98 ppm. Proportional mass recalibration yielded the best results with a mean of +0.77 ppm and S.D. of 0.71 ppm, both an improvement over external mass calibration and linear internal mass recalibration.
In comparing the mass accuracy obtained here with previous work, it is instructive to determine a mass tolerance that will include at least three standard deviations of mass error. Assuming a normal distribution, this criterion will encompass ∼99.7% of true positive precursor and fragment masses. This is likely close to the optimal mass tolerance used in peptide identification algorithms that ultimately determines the utility of higher mass accuracy.
Previous work by Haas et al. (44) demonstrated that a precursor mass accuracy of −0.41 ± 0.44 ppm could be obtained with SIM precursor scans detected in the ICR cell of a hybrid linear ion trap-ICR MS (LTQ FT) using external calibration. A precursor tolerance of ±2.0 ppm would be sufficient to capture ±3σ of mass deviations, whereas ±3.0 ppm would be necessary with the approach presented here.
A more direct comparison, however, is with precursors detected by non-SIM scans in the ICR cell presented in the same study. This experiment has a higher throughput so as not to negatively affect the number of peptide identifications in contrast to SIM precursor scans. The mass accuracy realized with this approach, which used internal mass calibration with space-charge correction, was considerably worse at −0.25 ± 1.46 ppm. A mass tolerance of ±5.0 ppm would be required to capture ±3σ of mass deviations.
Note that the data manipulation required for the approach presented here is relatively straightforward in contrast to the work of Haas et al. (44) because of the relative simplicity of correction for a single internal calibrant. Mass calibration with multiple ions requires repeating the step of nonlinear curve fitting of the calibration equation, which is computationally more intensive than a simple adjustment. Another significant consideration is that this step requires frequency data instead of m/z values, which are not typically stored in the raw data and therefore must be back-calculated from the calibration coefficients.
The approach presented here does add an additional 4–20 ms to each scan for internal calibrant injection that is not incurred by the strategy of Haas et al. (44) because background ions already present are used. However, in exchange for the minor penalty in throughput, the CI source provides a much more reliable supply of internal calibrant ions not subject to ambient laboratory conditions.
Another similar approach for optimizing mass accuracy was shown by Olsen et al. (45) that, like this study, utilized the c-trap of a hybrid linear ion trap-orbitrap MS (LTQ Orbitrap) for ion storage. Analysis of the supplementary data (45) provided with the same methods described here (mass measurement errors were provided for slightly over 200 unique peptides) yielded a precursor mass accuracy of −0.26 ± 1.06 ppm, which improved to −0.24 ± 0.57 ppm after averaging over the elution of the peptide. Although this mass accuracy is slightly superior to what is presented here (requiring a precursor tolerance of ±2.0 ppm to capture ±3σ of mass deviations), throughput is an important consideration. For fluoranthene cation internal recalibration, the time penalty is minimal because a sufficient population of calibrant ions can be transferred to the c-trap very rapidly. With the on-the-fly lock mass recalibration approach, which is implemented as an option in the LTQ Orbitrap firmware, the calibrant ions are trapped and isolated in the LTQ prior to injection into the c-trap. These isolation and transfer steps result in an additional ∼50 ms added to each scan relative to the fluoranthene internal calibrant.
The mass error distribution of products is shown in Fig. 3b. Without internal mass recalibration, ∼24,000 products had a mean error of −2.32 ppm and S.D. of 0.89 ppm. With linear internal mass recalibration, both improved slightly to a mean of −1.62 ppm and S.D. of 0.81 ppm. Once again, proportional internal mass recalibration gave the best performance with a mean of +0.17 ppm and S.D. of 0.67 ppm.
The same analysis was performed on all 60 replicate LC-MS/MS runs. The mean with error bars of ±1 S.D. was plotted as a function of time after external mass calibration for precursors in Fig. 4a and for products in Fig. 4b. Without correction, drift leads to mass accuracy distributions that are centered several parts per million away from the ideal of zero and fluctuate by more than a part per million per day. Similar plots have been shown by Olsen et al. (45) and Young et al. (40). Linear internal mass recalibration helps to moderate these fluctuations but does not eliminate them. Proportional internal mass recalibration, on the other hand, shows a virtual complete elimination of mass calibration drift.
Fig. 4.

Mass calibration drift for precursors (a) and products (b) over 60 replicate LC-MS/MS runs performed after external mass calibration. A Gaussian fit was applied to the mass error distribution for each run before (red) and after linear (green) and proportional (blue) internal mass recalibration; error bars represent ±1 S.D. For both MS1 and MS2 scans, proportional internal mass recalibration virtually eliminated drift and reduced the width of the distributions.
These analyses demonstrate that a proportional correction is preferable to a linear correction for internal mass recalibration because the former eliminates drift, whereas the latter only reduces it. Furthermore, the proportional adjustment can significantly reduce the width of the mass error distributions (typically about 25%), whereas the linear adjustment usually does not and sometimes actually worsens it. This is not surprising considering that only a single internal calibrant is used, which can primarily correct the effects of systematic but not random error. The main reason for seeing this modest improvement is likely the reduction of mass calibration drift within a single run.
An interesting phenomenon displayed in Fig. 4 is that even for LC-MS/MS analyses performed directly after two external mass calibrations there is significant systematic error. For precursors, this initial mass error is about −2.6 ppm compared with −1.3 ppm for products. This demonstrates another important reason for the use of internal calibration, not only the removal of drift but also to correct for errors in the external calibration. Internal mass calibration appears to be impervious to these initial inaccuracies.
One concern that arises from Fig. 4 is a significant, consistent systematic error evident even after proportional internal mass recalibration. For precursors, shown in Fig. 4a, the shift is about +0.8 ppm, whereas for products, shown in Fig. 4b, it is about +0.2 ppm. Significant effort was made to determine the source of these systematic errors without success. We note, however, that because the deviation is quite reproducible, it could easily be corrected for by simply subtracting the constant from any observed mass errors to yield a distribution centered on zero.
The cumulative effects of mass accuracy drift were then examined by constructing mass error distributions for the combined analysis of precursors and products from all 60 replicate runs simultaneously. The distribution for ∼120,000 precursor masses is shown in Fig. 5a and for ∼1.6 million product masses is shown in Fig. 5b.
Fig. 5.

Mass error distributions for ∼120,000 precursors (a) and 1.6 million products (b) measured over 60 replicate LC-MS/MS runs. The data were treated with external mass calibration (dark gray), linear internal mass recalibration (light gray), and proportional internal mass recalibration (medium gray). Although the center of the mass error distributions did not improve much after mass recalibration, drift correction led to substantially narrower peaks.
Although the mass error distributions for individual analyses have roughly the same widths, drift leads to dispersion of these distributions over time. These distributions sum together to yield a substantially widened overall distribution. The net result is an approximate doubling of the width of the mass error distribution without recalibration as compared with proportional internal mass recalibration where drift is minimized. By contrast, after proportional internal mass recalibration, the cumulative mass accuracy distributions do not widen relative to a single analysis. For precursor mass errors, a single replicate showed a width of 0.71 versus 0.70 ppm for the cumulative analysis; for product mass errors, both had a width of 0.67 ppm. The results of all mass accuracy analyses are summarized in Table I.
Table I. Summary of mass accuracy analyses of a typical single LC-MS/MS run (replicate 60) and combined analysis of all 60 replicate runs for precursors and products using no mass recalibration, linear mass recalibration, and proportional mass recalibration.
| Analysis | Precursors/products | Mass recalibration type | Count | Mean | S.D. |
|---|---|---|---|---|---|
| ppm | ppm | ||||
| Replicate 60 | Precursors | None | 1,934 | −3.31 | 0.93 |
| Replicate 60 | Precursors | Linear | 1,934 | −1.68 | 0.98 |
| Replicate 60 | Precursors | Proportional | 1,934 | +0.77 | 0.71 |
| Replicate 60 | Products | None | 23,988 | −2.32 | 0.89 |
| Replicate 60 | Products | Linear | 23,988 | −1.62 | 0.81 |
| Replicate 60 | Products | Proportional | 23,990 | +0.17 | 0.67 |
| Combined | Precursors | None | 119,184 | −1.23 | 1.54 |
| Combined | Precursors | Linear | 119,184 | −0.35 | 1.09 |
| Combined | Precursors | Proportional | 119,184 | +0.80 | 0.70 |
| Combined | Products | None | 1,622,070 | −0.18 | 1.42 |
| Combined | Products | Linear | 1,621,500 | −0.10 | 1.10 |
| Combined | Products | Proportional | 1,621,440 | +0.16 | 0.67 |
DISCUSSION
Mass accuracy was something of an afterthought throughout the early days of shotgun proteomics. Because of new hybrid instruments and more advanced data analysis software, this viewpoint has been steadily changing over the past few years to the point where accurate precursor mass is a critical component of contemporary shotgun proteomics experiments. Preliminary reports are beginning to demonstrate that accurate product masses are also extremely beneficial, and acquisition of high resolution tandem mass spectra will likely be the default strategy in the near future.
With this increasing importance of mass accuracy of MS1 and MS2 spectra in proteomics, it is critical to have a mass calibration strategy that is robust, easy to implement, minimally interferes with spectral quality and throughput, and consistently provides sub-part-per-million mass accuracy. Internal mass recalibration with fluoranthene cations, using a proportional adjustment, meets all these criteria. Fluoranthene cations are already produced by the CI source in very high numbers, so an adequate population can be rapidly accumulated for co-detection with analyte cations. Because the ions are produced by separate sources in different parts of the instrument, there is no competition for ionization. Post-acquisition recalibration can be accomplished with simple software that detects the internal calibrant peak in each spectrum and adjusts every other peak accordingly. For these reasons, we believe this approach is the ideal choice for obtaining optimal mass accuracy in shotgun proteomics on an ETD-enabled linear ion trap-orbitrap hybrid mass spectrometer.
Acknowledgments
We thank Ryan Lynch, Rachael Larson, and A. J. Bureta for figure illustrations as well as Jens Griep-Raming, Oliver Lange, Mike Senko, John Syka, Jae Schwarz, and George Stafford of Thermo Fisher Scientific for helpful discussions.
* This work was supported, in whole or in part, by National Institutes of Health Grant R01GM080148 (to J. J. C.) and a predoctoral fellowship (Biotechnology Training Program Grant 5T32GM08349).
1 The abbreviations used are:
- SIM
- selected ion monitoring
- ETD
- electron transfer dissociation
- CI
- chemical ionization
- ETnoD
- electron transfer no dissociation
- S/N
- signal-to-noise ratio
- FDR
- false discovery rate
- OMSSA
- Open Mass Spectrometry Search Algorithm
- Th
- thomson.
REFERENCES
- 1.Zubarev R., Mann M. (2007) On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 6, 377–381 [DOI] [PubMed] [Google Scholar]
- 2.Syka J. E., Marto J. A., Bai D. L., Horning S., Senko M. W., Schwartz J. C., Ueberheide B., Garcia B., Busby S., Muratore T., Shabanowitz J., Hunt D. F. (2004) Novel linear quadrupole ion trap/FT mass spectrometer: Performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J. Proteome Res 3, 621–626 [DOI] [PubMed] [Google Scholar]
- 3.Makarov A., Denisov E., Kholomeev A., Balschun W., Lange O., Strupat K., Horning S. (2006) Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem 78, 2113–2120 [DOI] [PubMed] [Google Scholar]
- 4.Jeffries J. B., Barlow S. E., Dunn G. H. (1983) Theory of space-charge shift of ion-cyclotron resonance frequencies. Int. J. Mass Spectrom 54, 169–187 [Google Scholar]
- 5.Francl T. J., Sherman M. G., Hunter R. L., Locke M. J., Bowers W. D., Mciver R. T. (1983) Experimental determination of the effects of space charge on ion cyclotron resonance frequencies. Int. J. Mass Spectrom 54, 189–199 [Google Scholar]
- 6.Ledford E. B., Jr., Rempel D. L., Gross M. L. (1984) Space-charge effects in Fourier transform mass spectrometry. Mass calibration. Anal. Chem 56, 2744–2748 [DOI] [PubMed] [Google Scholar]
- 7.Easterling M. L., Mize T. H., Amster I. J. (1999) Routine part-per-million mass accuracy for high-mass ions: space-charge effects in MALDI FT-ICR. Anal. Chem 71, 624–632 [DOI] [PubMed] [Google Scholar]
- 8.Belov M. E., Rakov V. S., Nikolaev E. N., Goshe M. B., Anderson G. A., Smith R. D. (2003) Initial implementation, of external accumulation liquid chromatography/electrospray ionization Fourier transform ion cyclotron resonance with automated gain control. Rapid Commun. Mass Spectrom 17, 627–636 [DOI] [PubMed] [Google Scholar]
- 9.Belov M. E., Zhang R., Strittmatter E. F., Prior D. C., Tang K., Smith R. D. (2003) Automated gain control and internal calibration with external ion accumulation capillary liquid chromatography-electrospray ionization-Fourier transform ion cyclotron resonance. Anal. Chem 75, 4195–4205 [DOI] [PubMed] [Google Scholar]
- 10.Second T. P., Blethrow J. D., Schwartz J. C., Merrihew G. E., MacCoss M. J., Swaney D. L., Russell J. D., Coon J. J., Zabrouskov V. (2009) Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Anal. Chem 81, 7757–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damoc E., Denisov E., Blethrow J., Second T. P., Zabrouskov V., Griep-Raming J., Makarov A., Moehring T. (2009) Overcoming undersampling in proteomic experiments with the help of a novel hybrid linear trap-orbitrap mass spectrometer, in Proceedings of the 57th Conference on Mass Spectrometry and Allied Topics, Philadelphia, May 31–June 4, 2009, American Society for Mass Spectrometry, Santa Fe, NM [Google Scholar]
- 12.Mann M., Kelleher N. L. (2008) Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U.S.A 105, 18132–18138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parks B. A., Jiang L., Thomas P. M., Wenger C. D., Roth M. J., Boyne M. T., 2nd, Burke P. V., Kwast K. E., Kelleher N. L. (2007) Top-down proteomics on a chromatographic time scale using linear ion trap Fourier transform hybrid mass spectrometers. Anal. Chem 79, 7984–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth M. J., Parks B. A., Ferguson J. T., Boyne M. T., 2nd, Kelleher N. L. (2008) “Proteotyping”: population proteomics of human leukocytes using top down mass spectrometry. Anal. Chem 80, 2857–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scherl A., Shaffer S. A., Taylor G. K., Hernandez P., Appel R. D., Binz P. A., Goodlett D. R. (2008) On the benefits of acquiring peptide fragment ions at high measured mass accuracy. J. Am. Soc. Mass Spectrom 19, 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenger C. D., Boyne M. T., 2nd, Ferguson J. T., Robinson D. E., Kelleher N. L. (2008) Versatile online-offline engine for automated acquisition of high-resolution tandem mass spectra. Anal. Chem 80, 8055–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyne M. T., Garcia B. A., Li M., Zamdborg L., Wenger C. D., Babai S., Kelleher N. L. (2009) Tandem mass spectrometry with ultrahigh mass accuracy clarifies peptide identification by database retrieval. J. Proteome Res 8, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson J. T., Wenger C. D., Metcalf W. W., Kelleher N. L. (2009) Top-down proteomics reveals novel protein forms expressed in Methanosarcina acetivorans. J. Am. Soc. Mass Spectrom 20, 1743–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., Horning S. (2009) A dual pressure linear ion trap orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAlister G. C., Phanstiel D., Wenger C. D., Lee M. V., Coon J. J. (2010) Analysis of tandem mass spectra by FTMS for improved large-scale proteomics with superior protein quantification. Anal. Chem 82, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clauser K. R., Baker P., Burlingame A. L. (1999) Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS MS and database searching. Anal. Chem 71, 2871–2882 [DOI] [PubMed] [Google Scholar]
- 22.Liu T., Belov M. E., Jaitly N., Qian W. J., Smith R. D. (2007) Accurate mass measurements in proteomics. Chem. Rev 107, 3621–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng F., Cargile B. J., Miller L. M., Forbes A. J., Johnson J. R., Kelleher N. L. (2001) Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol 19, 952–957 [DOI] [PubMed] [Google Scholar]
- 24.Bakalarski C. E., Haas W., Dephoure N. E., Gygi S. P. (2007) The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal. Bioanal. Chem 389, 1409–1419 [DOI] [PubMed] [Google Scholar]
- 25.Horn D. M., Zubarev R. A., McLafferty F. W. (2000) Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 97, 10313–10317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spengler B. (2004) De novo sequencing, peptide composition analysis, and composition-based sequencing: a new strategy employing accurate mass determination by Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom 15, 703–714 [DOI] [PubMed] [Google Scholar]
- 27.Olson M. T., Epstein J. A., Yergey A. L. (2006) De novo peptide sequencing using exhaustive enumeration of peptide composition. J. Am. Soc. Mass Spectrom 17, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 28.Hubler S. L., Jue A., Keith J., McAlister G. C., Craciun G., Coon J. J. (2008) Valence parity renders z(center dot)-type ions chemically distinct. J. Am. Chem. Soc 130, 6388–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitelegge J. P., Gundersen C. B., Faull K. F. (1998) Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci 7, 1423–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gómez S. M., Nishio J. N., Faull K. F., Whitelegge J. P. (2002) The chloroplast grana proteome defined by intact mass measurements from liquid chromatography mass spectrometry. Mol. Cell. Proteomics 1, 46–59 [DOI] [PubMed] [Google Scholar]
- 31.Conrads T. P., Anderson G. A., Veenstra T. D., Pasa-Toliæ L., Smith R. D. (2000) Utility of accurate mass tags for proteome-wide protein identification. Anal. Chem 72, 3349–3354 [DOI] [PubMed] [Google Scholar]
- 32.Smith R. D., Anderson G. A., Lipton M. S., Pasa-Tolic L., Shen Y., Conrads T. P., Veenstra T. D., Udseth H. R. (2002) An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2, 513–523 [DOI] [PubMed] [Google Scholar]
- 33.Cargile B. J., Stephenson J. L., Jr. (2004) An alternative to tandem mass spectrometry: isoelectric point and accurate mass for the identification of peptides. Anal. Chem 76, 267–275 [DOI] [PubMed] [Google Scholar]
- 34.Muddiman D. C., Oberg A. L. (2005) Statistical evaluation of internal and external mass calibration laws utilized in Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 77, 2406–2414 [DOI] [PubMed] [Google Scholar]
- 35.Zhang L. K., Rempel D., Pramanik B. N., Gross M. L. (2005) Accurate mass measurements by Fourier transform mass spectrometry. Mass Spectrom. Rev 24, 286–309 [Google Scholar]
- 36.Hannis J. C., Muddiman D. C. (2000) A dual electrospray ionization source combined with hexapole accumulation to achieve high mass accuracy of biopolymers in Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom 11, 876–883 [DOI] [PubMed] [Google Scholar]
- 37.Eckers C., Wolff J. C., Haskins N. J., Sage A. B., Giles K., Bateman R. (2000) Accurate mass liquid chromatography/mass spectrometry on orthogonal acceleration time-of-flight mass analyzers using switching between separate sample and reference sprays. 1. Proof of concept. Anal. Chem 72, 3683–3688 [DOI] [PubMed] [Google Scholar]
- 38.Nepomuceno A. I., Muddiman D. C., Bergen H. R., 3rd, Craighead J. R., Burke M. J., Caskey P. E., Allan J. A. (2003) Dual electrospray ionization source for confident generation of accurate mass tags using liquid chromatography Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 75, 3411–3418 [DOI] [PubMed] [Google Scholar]
- 39.Lee S. W., Berger S. J., Martinoviæ S., Pasa-Toliæ L., Anderson G. A., Shen Y., Zhao R., Smith R. D. (2002) Direct mass spectrometric analysis of intact proteins of the yeast large ribosomal subunit using capillary LC/FTICR. Proc. Natl. Acad. Sci. U.S.A 99, 5942–5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young N. L., Sisto M. C., Young M. N., Grant P. G., Killilea D. W., LaMotte L., Wu K. J., Lebrilla C. B. (2007) Steady-state asymmetric nanospray dual ion source for accurate mass determination within a chromatographic separation. Anal. Chem 79, 5711–5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charles L. (2003) Flow injection of the lock mass standard for accurate mass measurement in electrospray ionization time-of-flight mass spectrometry coupled with liquid chromatography. Rapid Commun. Mass Spectrom 17, 1383–1388 [DOI] [PubMed] [Google Scholar]
- 42.Bruce J. E., Anderson G. A., Brands M. D., Pasa-Tolic L., Smith R. D. (2000) Obtaining more accurate Fourier transform ion cyclotron resonance mass measurements without internal standards using multiply charged ions. J. Am. Soc. Mass Spectrom 11, 416–421 [DOI] [PubMed] [Google Scholar]
- 43.Cox J., Mann M. (2009) Computational principles of determining and improving mass precision and accuracy for proteome measurements in an orbitrap. J. Am. Soc. Mass Spectrom 20, 1477–1485 [DOI] [PubMed] [Google Scholar]
- 44.Haas W., Faherty B. K., Gerber S. A., Elias J. E., Beausoleil S. A., Bakalarski C. E., Li X., Villén J., Gygi S. P. (2006) Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell. Proteomics 5, 1326–1337 [DOI] [PubMed] [Google Scholar]
- 45.Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 46.Martin S. E., Shabanowitz J., Hunt D. F., Marto J. A. (2000) Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 72, 4266–4274 [DOI] [PubMed] [Google Scholar]
- 47.Good D. M., Wenger C. D., McAlister G. C., Bai D. L., Hunt D. F., Coon J. J. (2009) Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom 20, 1435–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 49.Kersey P. J., Duarte J., Williams A., Karavidopoulou Y., Birney E., Apweiler R. (2004) The International Protein Index: an integrated database for proteomics experiments. Proteomics 4, 1985–1988 [DOI] [PubMed] [Google Scholar]
- 50.Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]