

Abstract
Terminal amine isotopic labeling of substrates (TAILS), our recently introduced platform for quantitative N-terminome analysis, enables wide dynamic range identification of original mature protein N-termini and protease cleavage products. Modifying TAILS by use of isobaric tag for relative and absolute quantification (iTRAQ)-like labels for quantification together with a robust statistical classifier derived from experimental protease cleavage data, we report reliable and statistically valid identification of proteolytic events in complex biological systems in MS2 mode. The statistical classifier is supported by a novel parameter evaluating ion intensity-dependent quantification confidences of single peptide quantifications, the quantification confidence factor (QCF). Furthermore, the isoform assignment score (IAS) is introduced, a new scoring system for the evaluation of single peptide-to-protein assignments based on high confidence protein identifications in the same sample prior to negative selection enrichment of N-terminal peptides. By these approaches, we identified and validated, in addition to known substrates, low abundance novel bioactive MMP-2 targets including the plasminogen receptor S100A10 (p11) and the proinflammatory cytokine proEMAP/p43 that were previously undescribed.
Proteolytic processing is a posttranslational modification that affects every protein at least once in its life cycle. Rather than protein degradation, specific limited processing controls fundamental cellular processes such as apoptosis (1), angiogenesis (2), and indispensable physiological responses like blood coagulation (3). Thereby, the initial cleavage of key mediators by limited proteolysis triggers a cascade of proteolytic events leading to final execution of the overall process. These events have to be tightly controlled to maintain homeostasis of the proteolytic potential of the system (4–6), and its perturbation contributes either to the development or progression of diseases such as Alzheimer disease (7) and cancer (8, 9).
To better understand limited proteolysis and its disturbance in disease, it is crucial to reliably monitor proteolytic processing of proteins in complex proteomes. This can be achieved by specifically analyzing protein N-termini that together form the N-terminome of the sample (10). Specific proteolytic modifications of the proteome, for example by incubation with a test protease, will generate new N-termini (neo-N-termini) and thus reveal the substrate repertoire, also known as the substrate degradome (11), of the protease under study. In recent years, several techniques for proteomics analysis of the N-terminome have been described (for a review, see Ref. 12). Currently the most expansive approach is combined fractional diagonal chromatography (COFRADIC) that relies on HPLC separation of chemically acetylated N-terminal peptides (13). More recently, combined fractional diagonal chromatography was extended by isotopic labeling and was successfully used in various studies to identify proteolytic cleavage events (14–17). However, a major drawback of this technology is sequential HPLC runs and thus a high number of fractions, up to 150, to be analyzed by mass spectrometry (18). Other approaches use chemical or enzymatic modification of the protein N-terminus with biotinylated affinity probes to positively select for N-terminal peptides (19, 20) or acetylation for negative selection upon removal of non-N-termini by affinity capture (21, 22). As an alternative, one-dimensional gel-based approaches (23) have recently advanced with visually informative bioinformatics analyses (protein topography and migration analysis platform (PROTOMAP)) (24) and rely on shifts in migration of proteolytically processed proteins in SDS-PAGE but do not reveal the exact protease cleavage site. Subtraction of control or background proteolysis products can also be achieved in two-dimensional gel-based techniques using DIGE (25), but like one-dimensional analyses, they suffer from limited resolution of cleavage fragments differing by one or a few amino acids such as can be generated by amino and carboxyl peptidases (26), dipeptidyl peptidases (27), and many matrix metalloproteinase (MMP)1 cleavage sites (28).
Recently, our laboratory introduced a novel technique we call terminal amine isotopic labeling of substrates (TAILS) (29) that combines a negative selection procedure with differential isotopic labeling. TAILS uses reductive dimethylation with heavy and light formaldehyde for quantification of precursor ion doublets in survey scans at the MS1 level. Although very effective in substrate discovery, in fact 33 new substrates and 148 other cleavage sites in previously known but not characterized substrates were discovered for MMP-2 (also known as gelatinase A), any MS1 quantification such as isotopic acetylation, dimethylation, and stable isotope labeling with amino acids in cell culture (SILAC) leads to higher sample complexity because of the additional heavy and light labeled peptide pairs and thus lower proteome coverage and fewer identifications of low abundance proteins by mass spectrometry-based proteomics (30). An alternative is the use of isobaric mass tags that produce isobaric labeled peptides and only upon fragmentation differential reporter ions in tandem mass spectra for quantification (31). These labels are commercially available as iTRAQTM and have been used to discover >30 substrates of proteases in the cellular context in typical iTRAQ labeling shotgun proteomics experiments (32) and in only one other quantitative substrate analysis (33). However, in neither was there physical enrichment of protein N-termini, leading to low numbers (∼20) of neo-N-termini identified in the latter study. In addition to reducing sample complexity, iTRAQ reagents are available in four and recently eight different isotopic variants, allowing the comparison of up to eight different conditions in a single experiment (34). However, the reagents are expensive unlike dimethylation, which can be performed for ∼$1 per reaction.
Although TAILS identified ∼50% of the substrates by two or more peptides (29) corresponding to the original mature N-terminus and/or one or more cleavage site-generated neo-N-terminal peptides, these cannot be averaged for quantification as each peptide represents independent biological events. Hence, accurate quantification is a challenge in single peptide-based N-terminome analyses. This is particularly difficult for low intensity peptide ion identifications, and thus reliable measures are needed to evaluate quantification confidences within and between experiments. Furthermore, in all quantitative degradomics studies described to date, the critical peptide abundance ratio cutoff for determining protease-generated peptides (32, 35, 36) or a neo-N-terminus was either based on empirical assumptions (33) or calculated from the ratios of peptides from known substrates (32, 35), or from the general experimental variation (15, 37) but not derived from actual cleavage events. As a consequence, those approaches are mostly considered as “screens” needing secondary validation of the results. However, quantitative N-terminome analyses of complex biological systems generate vast amounts of data and require reliable probability scores for their interpretation.
Here, we used an inexpensive in-house synthesized isobaric iTRAQ-like reagent, CLIP-TRAQ, for quantification of enriched protein N-termini. For quality control, we established an ion-intensity based quantification confidence model for objective evaluation of quantification confidences by calculating a quantification confidence factor (QCF) for each peptide. Moreover, we introduced a statistical classifier derived from experimental data of protease cleavage events that allows for reliable differentiation between unprocessed N-termini and those generated by proteolytic cleavage. Finally, a new scoring system for the assignment confidence of single peptides to similar proteins and isoforms based on an isoform assignment score (IAS) was established, an issue that has been mainly ignored before. We validated this approach first by using Glu-C, a protease with canonical cleavage site specificity that readily enables manual inspection of the data to determine the accuracy of the model, and second by analyzing the well characterized but broad specificity MMP-2 substrate degradome. This widely encompassing statistically based bioinformatics platform readily enables processing of large amounts of N-terminome data from complex data sets. Thereby, it identifies with high confidence proteins from their N-terminal peptides and the sequence and modifications of the protein N-termini. From this information, hypotheses can be more accurately generated regarding their biological roles in vivo and for their proteolytically processed forms.
EXPERIMENTAL PROCEDURES
Cell Culture and Secretome Preparation
Mmp2−/− mouse embryonic fibroblasts used in this study were derived and cultured as described previously (32). To collect secreted proteins, ∼70% confluent cells were first washed four times with 20 ml of PBS each and then placed in serum-free Dulbecco's modified essential medium lacking phenol red. Following a 48-h incubation at 37 °C in a 5% CO2 atmosphere, the medium was collected by decanting, and the cells were discarded. The medium was first clarified by centrifugation (5 min at 500 × g) to remove any potentially carried over cells. After addition of protease inhibitors (0.5 mm PMSF and 1 mm EDTA), the supernatant was further centrifuged at 4 °C for 30 min at 8,000 × g to remove any smaller debris. The resulting supernatants were sterile filtered (0.22 μm) and concentrated at 4 °C by ultrafiltration with 5-kDa-cutoff membranes (Amicon), and the medium was exchanged to 50 mm HEPES buffer (pH 8). Protein concentration was determined by Bradford assay (Bio-Rad) and then adjusted with 1 m HEPES (pH 8) to 2 mg/ml protein and 250 mm HEPES. Resulting secretome preparations were aliquoted in 0.5-mg amounts and stored at −80 °C until further use.
In Vitro Protease Digestion
Glu-C was purchased from New England Biolabs, and MMP-2 was expressed and purified in its latent form as described previously (38). Glu-C was added to the secretome (1:100, w/w) and incubated for 16 h at 37 °C. Pro-MMP-2 was activated with 1 mm p-aminophenylmercuric acetate for 30 min and incubated with fibroblast secretome proteins at a 1:100 enzyme/substrate (w/w) ratio for 16 h at 37 °C in the presence of 10 mm CaCl2 and 100 mm NaCl. As a positive control, CCL7 was added (1 μg/100 μg of secretome) to all MMP-2 digests. Controls (without protease treatment) received an equivalent volume of buffer only. For each individual experiment, identical secretome aliquots from the same batch were used for both conditions. Repeat experiments performed with secretomes from the same batch were defined as technical replicates. Experiments with secretomes collected on different dates from different passage cells were considered biological replicates.
CLIP-TRAQ-TAILS
Typical CLIP-TRAQ-TAILS analysis utilized 0.5 mg of secretome/condition (channel). Prior to labeling, the protease-treated secretomes were first denatured, and cysteine residues were reduced and alkylated. Briefly, 8 m guanidinium chloride and 1 m HEPES (pH 8) were used to achieve a final concentration of 2.5 m and 250 mm of guanidinium chloride and HEPES, respectively. After 15-min incubation at 65 °C, the samples were reduced by tris(2-carboxyethyl)phosphine (1 mm final) for 45 min at 65 °C. Next, alkylation was performed using 5 mm iodoacetamide for 30 min at 65 °C in the dark followed by brief cooling to room temperature.
Whole protein CLIP-TRAQ labeling was performed using a 1:5 protein/CLIP-TRAQ weight ratio and 50% DMSO (final) as a solvent as described in the accompanying paper (39). Thus, for 0.5-mg secretome reactions, 2.5 mg of an individual CLIP-TRAQ isotopic variant was first resuspended in 100% DMSO using an amount equal to the volume of each reaction after alkylation. After mixing and 30-min incubation at room temperature, any excess CLIP-TRAQ reagent was quenched with 100 mm ammonium bicarbonate for an additional 15 min. After labeling, individual reactions were combined at a 1:1 ratio, thoroughly mixed, and cleaned by acetone/methanol precipitation. Briefly, ice-cold acetone/methanol (8:1 volume ratio) was used to precipitate 1 volume of sample at −80 °C for 2–3 h. The pellet was sedimented by centrifugation at 4 °C for 30 min at maximum rpm. After discarding the supernatant, the sample was resuspended in ice-cold methanol to remove any precipitated guanidinium chloride and centrifuged again (two washes in total). The final pellet was briefly air-dried, first resuspended in 50 μl of 100 mm NaOH, and then adjusted to 1 mg/ml protein, 100 mm HEPES (pH 8).
A tryptic digest of the samples was performed using sequencing grade TrypsinGold (Promega) at a 1:100–1:200 ratio overnight at 37 °C. Digestion efficiency was assessed by SDS-PAGE and silver staining. Routinely, a aliquot (∼100 μg) of the sample would be removed and stored at −80 °C until further analysis if needed.
Following the tryptic digest, the samples were enriched for their N-termini using a highly soluble dendritic hyperbranched polyglycerol (HPG) aldehyde polymer (synthesis and characterization are described in detail elsewhere (29)). The pH of the reactions was first adjusted with concentrated HCl to 6–7 followed by addition of 30 mm (final) sodium cyanoborohydride and the polymer. To ensure complete capture of all internal peptides, HPG polymer was used in a 3–5-fold excess (w/w). The reaction was allowed to proceed overnight at 37 °C. After coupling, the polymer-bound peptides were removed by filtration using centrifugation tubes with 3-kDa-cutoff membranes (Amicon). When concentrated to ∼0.1–0.2 ml, the polymer was washed with 0.2 ml of 100 mm ammonium bicarbonate. Polymer-bound internal peptides on the filter were discarded, and the flow-through containing N-terminal peptides was frozen at −80 °C until further analysis.
Off-line High Performance Liquid Chromatography
Prior to LC-MS/MS analysis, the N-terminal peptides were separated on an Agilent Technologies 1200 series HPLC system (Agilent Technologies) and using a PolySULFOETHYL A 100 × 4.6-mm, 5-μm, 30-Å column (PolyLC Inc.). Solvent A consisted of 10 mm potassium phosphate and 25% acetonitrile (pH 2.7). Solvent B included 10 mm potassium phosphate, 25% acetonitrile, and 1 m potassium chloride (pH 2.7). Separation was monitored by absorbance at 214 and 280 nm. Peptides were separated and eluted at 1 ml/min using the following 65-min gradient. Peptides were bound to the column and washed first for 15 min with 100% solvent A. To elute peptides, solvent B was gradually increased to 30% from 15 to 37 min followed by an increase to 40% by 43 min. Solvent B increased to 100% at 45 min and was kept at 100% for 8 min more. Then, the mobile phase was switched back to solvent A from 53 to 55 min, and the column was equilibrated with 100% solvent A for an additional 10 min. Peptide-containing fractions were collected every 1.5 min, concentrated to 0.1 ml under vacuum, and desalted using C18 OMIX tips.
In-line Liquid Chromatography and Mass Spectrometry Analysis
Peptide separations by nano-LC (C18 150-mm × 100 μm-column at a flow rate of 100–200 nl/min) were performed in line with tandem MS/MS analysis. MS data were collected with a QStar XL Hybrid ESI (Applied Biosystems, MDS Sciex, Concord, Canada) mass spectrometer. Sample volumes of 2–10 μl were analyzed by LC-MS/MS. After loading the samples onto a trapping column, the column was washed with 5% acetonitrile containing 0.1% formic acid (v/v). Elution and separation of peptides were achieved with a 40–100-min linear 5–40% acetonitrile gradient (containing 0.1% formic acid) at a flow rate of 150–200 nl/min. MS data were acquired automatically with the software Analyst QS version 1.1 (Applied Biosystems, MDS Sciex). An information-dependent acquisition method consisted of a 1-s TOF MS survey scan of mass range 400–1500 amu and three 3-s product ion scans of mass range 75–1500 amu. The three most intense peaks over 20 counts with a charge state of 2+ to 4+ were selected for fragmentation.
Synthesis of Isobaric Tag Labeling Reagents
Isobaric reagents CLIP-TRAQ-113 and CLIP-TRAQ-114 were chemically synthesized as described elsewhere.2 Briefly, identical synthetic procedures were used for both CLIP-TRAQ-113 and CLIP-TRAQ-114 with the exception of the ethyl bromoacetate precursor used, which varied in the site of an incorporated 13C atom. To 1 ml of ice-cold diethyl ether, 100 μl of 1-methylpiperazine was added. To this solution, 60 μl of either ethyl bromo[1-13C]acetate (for 113 synthesis) or ethyl bromo[2-13C]acetate (for 114 synthesis) was slowly added. The reaction was incubated overnight at 4 °C. The reaction mixture was then centrifuged, and the supernatant was collected and dried under vacuum to obtain ethyl 2-(4-methylpiperazin-1-yl)acetate. The sample was then suspended in 1 ml of 10% HCl and heated to >85 °C overnight. The reaction mixture was dried under vacuum, and the chloride salt of the product was crystallized from hot 95% methanol. To generate the final products (CLIP-TRAQ-113 and CLIP-TRAQ-114), the N-hydroxysuccinimide esters of the respective 2-(4-methylpiperazin-1-yl)acetic acid precursors were synthesized. 70 mg of the precursor was dissolved in 3 ml of dry dimethylformamide to which were added 1.2 eq of dicyclcarbodiimide and 1.1 eq of N-hydroxysuccinimide. After 4 h, the mixture was filtered, and the filtrate was dried under vacuum. The dried reaction mixture was suspended in a minimal volume of dichloromethane/methanol/hexane (5:1:1) and purified by silica gel chromatography (using the identical solvent mixture). Purified products were collected and dried under vacuum, and 0.5–1 mg aliquots were stored at −80 °C until further use in protein labeling reactions.
Substrate Cleavage Assays
MMP-2 was activated with p-aminophenylmercuric acetate (1 mm; 45 min) and incubated with the candidate substrates in 50 mm Tris-HCl, 200 mm NaCl, 5 mm CaCl2, and 0.025% NaN3 for 16 h at 37 °C. Reaction products were analyzed by 15% Tris-Tricine SDS-PAGE and silver-stained.
MS2 Peptide Assignments and CLIP-TRAQ Quantification
Acquired MS2 scans were searched against a mouse International Protein Index protein database (v.3.24) supplemented with the sequences for human CCL7 and MMP-2 (52,415 protein entries total) by Mascot version 2.2 (Matrix Science) and the X! Tandem (2007.07.01 release). Searches were performed with the following parameters: semi-Arg-C cleavage specificity with up to two missed cleavages; cysteine carbamidomethyl and peptide lysine CLIP-TRAQ set as fixed modifications; N-terminal CLIP-TRAQ, N-terminal acetylation, and methionine oxidation set as variable modifications; peptide tolerance and MS/MS tolerance both set at 0.4 Da; and the scoring scheme set as ESI-QUAD-TOF. Search results were further evaluated on the Trans-Proteomic Pipeline (v.4.2, rev 0, Build 200811181145) (40, 41) using PeptideProphet (42) without using the number of tryptic termini model for peptide identification and Libra for quantification of CLIP-TRAQ-113 and -114 reporter ions. As the cutoff for accepting individual MS/MS spectra, a PeptideProphet minimum probability threshold was used that corresponds to an error rate for incorrect peptide assignments of <5%.
For the compilation of protein lists that were used to calculate IAS scans from individual MMP-2 experiments, were searched with Mascot using the same parameters as above. Searches were evaluated by PeptideProphet (using the number of tryptic termini model for prepullout but not for pullout analyses), and subsequently, data from both experiments were combined in a single peptide list using the iProphet algorithm (43). Finally, this list was processed by ProteinProphet (44) without assembling protein groups and filtered for proteins with ProteinProphet probability >0.9.
Statistical Data Analysis, Peptide Annotation, and Generation of Heat Maps and Sequence Logos
For generation of MA plots, histograms, probability densities, distribution, and non-linear curve fitting, the R statistical environment was used (version 2.8.0). Receiver operating characteristic (ROC) curve analysis was performed using the ROCR package (45). To calculate standard deviations by a sliding window approach, for spectrum merging and weighted averaging, and for peptide isoform assignments and positional annotation, we used in-house Perl scripts using appropriate BioPerl packages. For identifier mapping, the Protein Information and Property Explorer system (46) was used. For active site mapping, amino acid occurrences were calculated as described previously (47), and heat maps generated using TM4:MeV. Protein sequence logos were generated using the iceLogo software package (48) with random sampling of the reference database.
RESULTS
CLIP-TRAQ-TAILS
The overall workflow for terminal amine isotopic labeling of substrates using iTRAQ-like reagents is outlined in Fig. 1A. Incubation of a complex proteome with a test protease generates neo-N-termini that are absent from the control sample. To identify these, all protein N-termini as well as amine-reactive lysine side chains are differentially isotopically labeled with iTRAQ-like reagents synthesized inexpensively in house (CLIP-TRAQ-113 and -114: Unimod (49) accession number 525).2 These reagents follow the same principles as their commercially available counterparts (31) but present reporter ions of 113 and 114 Da, respectively. Subsequently, samples are pooled and trypsin-digested. Next, the mixture is depleted of internal tryptic peptides in a negative selection step using an amine-reactive HPG aldehyde (ALD) polymer that reacts with the newly generated and hence unblocked N-terminal amines of the trypsin-generated peptides (29). Consequently, the resulting sample is enriched for neo-N-termini present only in the protease-treated sample, original mature protein N-termini including naturally acetylated and cyclized peptides, and N-terminal peptides derived from proteases other than the test protease present in both samples (basal proteolysis) (29). Upon two-dimensional LC-MS/MS analysis, original mature protein N-termini and N-termini derived from basal proteolysis present reporter ion intensities in a 1:1 ratio, whereas neo-N-termini only generate a single reporter ion (singletons). The latter are neo-N-termini of substrates cleaved specifically by the test protease. In some cases, a low ratio singleton indicates proteolytic loss of a protein's N-terminal peptide spanning the cleavage site that is only present in the control sample but absent in the protease-treated sample. Negative selection preserves identification of naturally blocked (e.g. acetylated or cyclized N-terminal residues) N-terminal peptides and also their quantification provided they contain a lysine residue. Furthermore, this procedure allows the concomitant analysis of prepullout samples for higher confidence in protein identification. Because of fast and efficient protein labeling with iTRAQ-like reagents, easy cleanup, and polymer pullout as well as benefits in terms of minimal sample loss because of the absence of unspecific binding to the polymer, the protocol is performed in 2 days. If desired, off-line strong cation exchange chromatography peptide fractionation can be performed prior to LC-MS/MS analysis although this was not needed before (29) (Fig. 1B).
Fig. 1.

CLIP-TRAQ-TAILS. A, schematic representation of the CLIP-TRAQ-TAILS workflow. Proteins from protease-treated and control proteomes are labeled on N-terminal and lysine side chain amines with isotopically distinct CLIP-TRAQ reagents. Proteins are then tryptically digested, and internal tryptic peptides are removed via their free N-terminal amino group by the amine-reactive HPG-ALD polymer. Upon polymer removal, enriched protein N-termini are in the flow-through fraction and are subjected to two-dimensional LC-MS/MS analysis. In tandem mass spectra, the N-termini of unprocessed proteins present CLIP-TRAQ reporter ions in a 1:1 intensity ratio, whereas protease-derived neo-N-termini (red) are identified by a single reporter ion. Naturally blocked N-termini (indicated by gray diamonds) are also susceptible to quantification provided they harbor a lysine within their sequence. B, rapid 2-day experimental workflow. Denaturation, reductive alkylation, and isotopic labeling are all performed in the same tube prior to mixing and sample cleanup, minimizing chances of differential sample losses. Aliquots before (containing N-terminal, internal, and C-terminal tryptic peptides) and after polymer pullout (N-terminal peptides) can be analyzed from the same sample for higher confidence in protein identification (see text for details). GdnHCl, guanidinium hydrochloride; TCEP, tris(2-carboxyethyl)phosphine; SCX, strong cation exchange.
Spectrum Merging and Ion Intensity-dependent QCF
Around 30–50% of N-terminal peptides in CLIP-TRAQ-TAILS experiments are identified by more than one MS2 spectrum either through multiple CIDs of the same precursor or importantly by identification of the same peptide in multiple oxidation and/or charge states. This enhances reliability in both peptide identification and quantification (29, 50). However, simple averaging of spectra for reporter ion ratio determination at the peptide level does not normalize for differences in intensity-dependent variability. In fact, we considered that spectra with high reporter ion intensities should be weighted to contribute more to spectrum-averaged peptide quantifications than low intensity spectra, whereby the intensity variation correlation follows a non-linear function (51).
To address this, we prepared a sample from cell culture supernatants of Mmp2−/− mouse embryonic fibroblasts (32), labeling half with CLIP-TRAQ-113 and the other half with CLIP-TRAQ-114 without prior protease treatment and then performing TAILS. For peptide identification stringency, we used two database search engines (Mascot and X! Tandem) and the PeptideProphet algorithm (42) for secondary statistical validation. For quantification of CLIP-TRAQ reporter ion intensities, Libra, as part of the Trans-Proteomic Pipeline (52), was used. Thereby, 2,488 spectra of 1,202 peptides identified by both search engines and validated with an error <5% by PeptideProphet were further analyzed (supplemental Table 1).
First, we removed spectra with reporter ion intensity values of less than 30 in both channels (see below) and spectra assigned to unlabeled peptides or those having a non-tryptic C-terminus. Next, we calculated M values (log2(113/114)) of reporter ion intensities for the 2,204 spectra fulfilling these criteria. After further removal of obvious outliers more than 3-fold above or below the expected ratio of 1:1 (∼1%) and median centering to account for experimental handling errors, we plotted median-centered M values against corresponding A values (0.5 × log2(113 × 114)) in an MA plot known from intensity-dependent ratio analysis in microarray experiments (Fig. 2A). As expected, the deviation from the expected ratio of 1:1 (M = 0) was highest for low intensity reporter ion quantifications and lowest for high A values. To reward spectra with highest confidence based on intensity, we derived an intensity-dependent variability function. First we calculated A value-dependent standard deviations by random sampling and averaging in a sliding window approach with a window size of 1.5 and incremental steps of 0.1. By non-linear curve fitting, the following exponential decay function was derived,
where x = A value, a = 3.315, b = 0.4578, and c = 0.1428. This equation was used to determine intensity-dependent errors (v) and quantification confidences (1/v) for individual spectra. Quantification ratios of ≥7.3 or ≤1/7.3 (see below) were considered as high and low singletons, respectively, and the A value was calculated using the high intensity channel for both ion intensity values. Quantification confidences also served as weights for calculating weighted means of reporter ion intensity ratios for peptides identified by multiple MS2 spectra. In weighted means, individual errors were propagated by Gaussian error propagation to calculate a combined quantification confidence. For both intra- and interexperimental comparisons, we mean-centered log2-transformed confidences and adjusted the highest value to 10 according to the following formula.
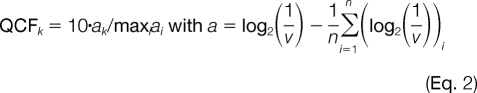 |
The resulting value we term the QCF of the peptide k. QCFs >0 represent peptide quantifications above the experimental mean confidence, and factors <0 represent peptide quantifications below the mean. It is important to note that even QCF values of 0 are considered good. As expected, the QCF grew exponentially with the number of MS2 spectra per peptide averaged for quantification as demonstrated for both the Glu-C and MMP-2 data sets described below (Fig. 2B and supplemental Fig. 3).
Fig. 2.

QCF and substrate ratio cutoff models. A, MA plot for reporter ion intensity ratios from a CLIP-TRAQ-TAILS experiment of secretomes from Mmp2−/− fibroblasts mixed at a 1:1 ratio without protease incubation. Black dots represent A-value dependent standard deviations calculated by averaging of 100 random sample means (n = 50) and a sliding window with size 1.5 and increment 0.1. Error bars are S.D. of the 100 random sample means. The dashed line represents a curve fitted to those values by non-linear curve fitting. This curve was used to calculate confidence factors and weights for weighted averaging of ratios for peptides identified by multiple spectra. B, increase of quantification confidence with the number of spectra used for peptide quantification. The averaged quantification confidence factor is plotted against the number of spectra used for peptide quantification. Error bars represent S.D. Data were derived from the validation experiment using Glu-C as test protease. C, distribution of abundance ratios (Glu-C/control) of spectra assigned to Glu-C-generated neo-N-termini (n = 155). The solid line represents the probability density, and the dashed line represents a fitted normal distribution with mean = 3.79 and S.D. = 0.95. The mean is an estimate for the dynamic range of CLIP-TRAQ quantification. D, ROC curve analysis of substrate classifier performance. A peptide abundance ratio (Glu-C/control) of 7.3 for cleavage events provides maximum sensitivity (86%) at a minimum false discovery rate (15%). The actual false positive and true positive rates for a ratio of 14 are ∼5 and ∼53%, respectively. The dashed line represents the same analysis for a classifier calculated from weighted averaged ratios if peptides were identified by multiple spectra. ctrl, control.
Derivation of Ratio-dependent Classifier and Cutoff for Protease Substrates
To determine a statistical reporter ion intensity cutoff ratio for protease-generated neo-N-terminal peptides, we used the endoproteinase Glu-C (Staphylococcus aureus protease V8) to partially digest Mmp2−/− embryonic fibroblast-conditioned medium supernatants under conditions to maintain the native state of the proteome components. The Glu-C-incubated and control samples were labeled with CLIP-TRAQ-113 and CLIP-TRAQ-114, respectively, and subjected to TAILS. Glu-C has a strict specificity for glutamate and to a lesser extent aspartate residues in the P1 position under the pH and buffer conditions used (47, 53). Again, only spectra of peptides identified by both search engines and with a PeptideProphet error of <5% were included in the analysis (1,712 spectra; supplemental Table 2). We also removed spectra corresponding to non-quantifiable peptides and peptides to which the preceding residue could not be unambiguously assigned following analysis of positions in all known isoforms (246 of 1,712 spectra). As expected for Glu-C activity, 47% (687 of 1,466) of all spectra were assigned to peptides with Glu or Asp as preceding amino acids (P1 position) after TAILS negative selection. The remaining spectra were assigned to peptides corresponding to original mature protein N-termini and internal N-termini resulting from basal proteolysis in the sample.
Ratios of 113 to 114 reporter ion intensities of spectra assigned to peptides with Glu or Asp as preceding amino acids should be very high because corresponding peptides are expected to be only present in the Glu-C-treated (CLIP-TRAQ-113 labeled) sample. For this quantitative analysis only, we excluded spectra from peptides with Glu and/or Asp residues within their sequence (520 of 687 spectra) because they might sometimes also be internally cleaved by Glu-C and thus display an aberrant 113/114 ratio that is independent of the initial generation of the original Glu-C neo-N-terminus. Because acetylation of protease-generated neo-N-termini after cleavage is a possible but uncommon event, spectra assigned to peptides with acetylated N-termini were also omitted for the analysis (12 of 167 spectra). Expected high 113/114 reporter ion intensity ratios for 155 spectra from peptides with Glu or Asp as the preceding amino acid and no Glu and/or Asp as internal residues were confirmed by histogram analysis of log2(113/114) ratios (Fig. 2C). The majority of these ratios for the Glu-C-cleaved peptides appeared to be normally distributed with a mean of log2(113/114) = 3.79 for the maximum of the probability density (Fig. 2C, solid line) following a normal distribution with a standard deviation of 0.95 (Fig. 2C, dashed line). The value for the mean corresponds very well to previous reports on the dynamic range of iTRAQ quantification in complex mixtures (54) and verifies that spectra with 113/114 ratios of ≥23.79 (≈14) indeed represent high ratio singleton peptides only found in the Glu-C-treated sample. Therefore, we used ratios of 14 and 1/14 = 0.071 as upper and lower quantifiable limit thresholds to define the high and low ratios that mark a peptide as being a true singleton peptide. That is, these would unequivocally correspond to protease-generated neo-N-terminal peptides and peptides lost upon proteolysis, respectively.
Nonetheless, the stringent ratio cutoff of 14 for Glu-C-derived singletons would ignore true positives present in the left tail of the normal distribution. Therefore, we estimated the optimal 113/114 ratio cutoff for protease cleavage events by establishing a scoring classifier based on Glu or Asp residues in the P1 position as class labels and reporter intensity ion ratios as predictors on all spectra assigned to peptides without Glu and/or Asp as internal residues (288 spectra). This classifier could reliably separate cleavage event spectra assigned to peptides with Glu or Asp in the P1 position from non-substrate spectra based on 113/114 reporter ion intensities. It showed excellent performance in ROC curve analysis with an area under the ROC curve of 0.916 (Fig. 2D). From this curve, we determined a 113(Glu-C)/114(control) reporter ion intensity ratio of 7.3 as the optimal ratio cutoff for cleavage event spectra with a true positive rate of 86% and a false positive rate of 15%.The actual false positive rate for a ratio of 14 is ∼5%, but using this ratio alone would reduce the true positive rate for the entire experiment to 53%. It has to be remembered that these discovery rates are referring only to the probability of identifying bona fide substrates from peptides that have already been identified with high confidence by both two search engines and PeptideProphet.
Because variability of quantification is much higher in spectra with low reporter ion intensities, we wondered whether false positives within spectra above a reporter ion ratio cutoff of 7.3 are correlated to low absolute intensities of the 113 reporter ion. However, ROC analysis with 113 reporter ion intensities as predictors and amino acids Glu or Asp in the P1 position as class labels did not verify such a correlation (supplemental Fig. 1). Because the lowest absolute 113 reporter ion intensity value for a true positive spectrum was 28, we set the minimum intensity threshold for further analyses to be 30 in at least one channel. Thereby, this enables background noise and isotope contamination to be negated.
This analysis determined a statistical reporter ion ratio cutoff for cleavage events on the basis of single spectrum peptide identifications. However, around 50% of peptides in this experiment were identified by multiple spectra, which can be exploited for enhancing identification confidence (29) and quantification accuracy. To test the validity of our cutoff model for peptides identified by a single or by multiple spectra, we calculated intensity-weighted means of reporter ion ratios as described above and performed the same ROC analysis on the peptide level. ROC curve and optimal cutoff were almost identical to the spectrum-based analysis (Fig. 2D, gray dashed line), verifying our model for accurate quantification of single and multiple spectrum peptides as obtained by typical TAILS experiments. Hence, although the cutoff and false positive rate for peptides identified multiple times is the same as for peptides identified once, the quantification confidence is inherently much higher.
Validation of Statistical Models Using Test Protease with Canonical Cleavage Specificity
Finally, we tested our intensity-dependent quantification confidence factor and reporter ion ratio singleton cutoff models in an independent experiment. Again, Glu-C was used as a test protease because of its known cleavage specificity but on a biologically different Mmp2−/− cell secretome. As before, Glu-C-treated and control sample were labeled with CLIP-TRAQ-113 and CLIP-TRAQ-114, respectively. Thereby, we identified 961 peptides by 1,581 spectra matching our peptide identification and reporter ion intensity threshold criteria (supplemental Tables 3 and 4). 54% (514 of 950) of all quantifiable peptides were original mature protein or basal proteolysis neo-N-terminal peptides in the N-terminome, whereas 46% (436 of 950) had unambiguously assigned Glu or Asp in the P1 position, and 70% (306 of 436) of these had a ratio above the substrate cutoff of 7.3. This number increased to 78% (72 of 92) when excluding peptides with internal Glu and/or Asp residues that are affected by internal Glu-C cleavage. When only including peptides with quantification confidence factors above the experimental mean quantification confidence (see above), 81% (42 of 52) of peptides had the expected 113(Glu-C)/114(control) reporter ion intensity ratio, indicating a substrate cleavage event. Validating our intensity-dependent quality control model, the QCF grew exponentially with the number of MS2 spectra per peptide averaged for quantification (Fig. 2B).
Taken together, these results demonstrate that our combination of (i) statistical substrate ratio cutoff, (ii) intensity-weighted averaging for quantification of multiple spectrum peptide identifications, and (iii) intensity-dependent quantification confidence lead to reliable determination of protease cleavage events in complex proteomes. Being confident in this is very important for analyzing proteases with unknown or broad specificity where manual parsing of the data cannot be done because true positives in such non-canonical protease experiments are unknown.
Identification of MMP-2 Substrates
Unlike proteases such as Glu-C, caspases, and granzymes that have canonical specificity, most proteases have unknown or broad cleavage site preferences and so present serious challenges to N-terminome platforms that lack quantification capabilities. We tested our criteria and models on a well characterized protease with broad specificity, MMP-2, as an important cancer protease (55) that has been extensively studied in our laboratory (2, 32, 37). As such, we have a large in-house database of substrates and cleavage sites with which to compare the data derived from our new statistical models. Taken together, MMP-2 is an ideal representative test case of a “difficult” protease with loose cleavage site preference (47) to validate our statistical and bioinformatics approach.
Conditioned medium supernatants from Mmp2−/− murine fibroblasts as naïve proteomes never exposed to MMP-2 were incubated with active recombinant human MMP-2, labeled with CLIP-TRAQ-113, and compared with an undigested control of the same secretome labeled with CLIP-TRAQ-114. As a control substrate to monitor MMP-2 activity, we spiked 5 μg of human CCL7 (MCP-3), a well characterized MMP-2 chemokine substrate (56), into both samples. Applying our rigorous identification criteria and a minimum reporter ion intensity cutoff of 30 in at least one channel, we identified by two search engines 1,219 peptides by 1,708 spectra in a first experiment and 1,416 peptides by 1,994 spectra in a second independent experiment. Thereby, 833 peptides were identified in both experiments, resulting in 1,802 different peptides in total. By histogram and probability density analysis of log2(113(MMP-2)/114(control)) reporter ion ratios of spectra assigned to quantifiable peptides, we verified the high quantification reproducibility of both experiments (Fig. 3B). Based on these results, we combined the data sets but only included peptides identified at least either in both biological replicates or by two search engines (Fig. 3A) to increase the confidence in peptide identification (29). This enhanced the number of peptides to 2,101 identified by 4,774 spectra (supplemental Table 5).
Fig. 3.

CLIP-TRAQ-TAILS analysis of the MMP-2 substrate degradome. A, four-way Venn diagram of peptide identifications by two search engines (Mascot and X! Tandem) in experiments 1 and 2. Double identified peptides (2,101) are those identified at least either in both biological replicates or by two search engines. B, distribution of abundance ratios (MMP-2/control) of N-terminal peptides (4,774 spectra) in MMP-2-treated and control samples. The solid line represents the probability density of combined data from two highly reproducible experiments. The right peak resembles the substrate peak from the Glu-C experiment in Fig. 2C. The left dashed vertical line indicates the substrate ratio cutoff of 7.3. C, frequency distribution of natural N-termini. D, distribution of abundance ratios (MMP-2/control) of 179 quantifiable natural N-termini (553 spectra). The dashed line indicates a fitted normal distribution (norm. distr.) with mean = −0.05 and S.D. = 0.63.
The distribution of log2(113/114) reporter ion ratios for 4,725 spectra of 2,079 quantifiable peptides revealed two separate peaks (Fig. 3B) with the right peak presenting a maximum at log2(113/114) = 4.27 that closely follows the substrate event ratio peak observed in the Glu-C experiment described above. This suggests that this peak comprises a distribution of ratios of spectra assigned to peptides derived from substrate cleavages. The left peak has a maximum of close to log2(113/114) = 0 and should comprise ratios of spectra assigned to either original mature protein or neo-N-termini derived from basal proteolysis in proteins present in both the MMP-2-treated and control sample.
To test this hypothesis, we annotated all peptides with their position in the corresponding original mature protein and extracted 184 natural N-termini (identified by 567 spectra) that are either acetylated or unblocked and are assigned to proteins with or without the initiator methionine, signal peptide, or propeptide (Fig. 3C). Of these, 179 peptides (553 spectra) were quantifiable, and for these, we plotted a histogram of log2(113/114) ratios (Fig. 3D). Indeed, the probability density revealed one major peak that could be approximated by a normal distribution with mean = −0.05 and S.D. = 0.63 (Fig. 3D, dashed line). The left skewness of the overall density estimate is due to an overlaying distribution of original mature protein N-termini that are internally cleaved by MMP-2 and so have a log2(113/114) ratio <0. Hence, analysis of such lost peptides can indirectly reveal cleaved substrates, complimenting the direct identification of cleaved substrates as described by high confidence, high ratio peptides (29).
These results were further validated by a label swap experiment when we labeled the MMP-2-treated sample with CLIP-TRAQ-114 and the control sample with CLIP-TRAQ-113. Histogram analysis of log2(113/114) ratios of 1,346 quantifiable peptides identified by 2,034 spectra (supplemental Table 6) revealed the same bimodal distribution, but the substrate event ratio peak mirrored to the left with a maximum at log2(113/114) = −4.7 (supplemental Fig. 2A), whereas ratios for 94 quantifiable original mature protein N-termini (215 spectra) were normally distributed with a mean of log2(113/114) = 0.09 (supplemental Fig. 2B).
To demonstrate the effectiveness of our negative enrichment strategy for N-terminal peptides, we performed the same analysis on identical samples before depletion of internal tryptic peptides by coupling to the HPG-ALD polymer. Thereby, we identified only 47 original mature N-termini by 113 spectra compared with 184 by 567 spectra after the pullout.
As indicated in Fig. 3B (dashed vertical line), our substrate ratio cutoff of 7.3 derived from the Glu-C experiment is a statistically determined value to reliably separate substrate from non-substrate peptides with high confidence and low false discovery. Next, we calculated 113/114 reporter ion ratios for peptides using our intensity-dependent weighted averaging and cleavage event ratio cutoff models. Finally, we calculated a QCF for each peptide. As a further validation for our quantification confidence model, QCF values again grew exponentially with the number of independent spectra assigned to quantified peptides (supplemental Fig. 3).
Protein Identification and IAS
Although ∼50% of proteins can be identified in a typical TAILS experiment by two or more different and unique peptides (29), N-terminome analysis must also rely on protein identification by only one peptide. However, it is not always possible to unambiguously assign this peptide to one protein and particularly to a specific isoform. The TAILS negative selection procedure has the advantage of also easily providing high confidence multiple peptide protein identifications by analysis of the prepullout sample in the same workflow (Fig. 1B, dotted arrow line). In addition, the isotopic labeling of primary amines results in trypsin cleavage that skips the blocked lysines, generating longer peptides with higher protein assignment confidences in the samples. Exploiting these unique advantages of TAILS, we compiled lists of 1,037 protein assignments from prepullout (ppo) (supplemental Table 7) and 744 from pullout (po) (supplemental Table 8) analyses combined from both samples and with high confidence protein identifications as indicated by ProteinProphet (44) probabilities of >0.9 and error rates of 0.9 and 1.3%, respectively. These lists were used to calculate assignment scores for all isoforms assigned to all 2,101 identified peptides after negative enrichment of N-terminal peptides. First, we checked whether an assigned isoform was found in either one or both of our compiled high confidence protein lists. Thereby, we defined factors Fpo and Fppo and set a value of 1 if the isoform in question was present and 0 if it was not present in the corresponding protein list. Next, we divided these factors by the number of proteins indistinguishable from this isoform (npo and nppo) by ProteinProphet analysis to calculate an isoform score (Siso) according to Equation 3.
 |
To calculate the final IAS for an isoform assigned to an identified peptide, we added Siso for every additional isoform also assigned to the same peptide to the divisor, resulting in Equation 4,
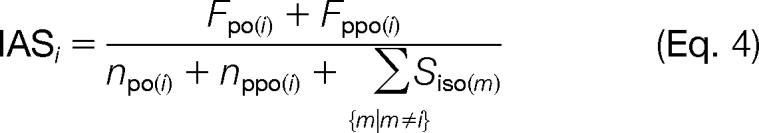 |
where isoform m is another isoform assigned to the same peptide and Siso(m) is the isoform score of this isoform. According to this formula, the highest IAS of 1.0 will be assigned to isoforms that have been identified as a unique high confidence protein by ProteinProphet analysis in either the prepullout (Fppo = 1, nppo = 1), the pullout (Fpo = 1, npo = 1), or both samples (Fppo = 1, nppo = 1, Fpo = 1, npo = 1) and with either no other isoform assigned to the same peptide or no isoforms present in either high confidence protein list (ΣSiso(m) = 0). The IAS decreases with the number of ambiguously identified isoforms and their confidence in identification. Applying this concept to the analysis of our 2,101 high confidence protein N-termini yielded 1,057 (50%) that had an IAS of 1.0 and hence represent peptides unambiguously assigned to unique protein isoforms with high probability protein assignments (supplemental Table 9). Notably, the known MMP-2-generated neo-N-terminus of the spiked-in control substrate human CCL7 was identified with the highest QCF of all substrates and an IAS of 1.0 and thus further validates both our cleavage event ratio cutoff, quantification confidence, and isoform assignment models.
Analysis of MMP-2 Substrates
To validate N-terminomics platforms, comparisons need to be made with existing data. From the 2,101 peptides, we identified 1,183 separate MMP-2 cleavage events in 608 proteins (supplemental Table 10) with 600 cleavage events in 272 proteins having an IAS of 1.0. Hence, using the IAS, ∼50% of all substrates could be unambiguously assigned to a specific protein isoform. In addition, there was an ∼70% overlap of substrate proteins identified previously using dimethylation TAILS (29). Moreover, >70% of these had exactly the same cleavage site. In addition, 33% of these substrates have also been described in previous ICAT- and iTRAQ-based MMP-2 substrate screens that did not enrich for the cleaved neo-N-terminus, and so the exact cleavage site was not determined (2, 32).
Lastly, Table I shows known MMP-2 substrates identified by CLIP-TRAQ labeling in TAILS, further validating our statistical models and bioinformatics procedure. Specific cleavage sites such as the GPXG↓L motif in collagen α1 have been demonstrated previously (57), and cleavage of cystatin C after position 9 in the mature protein matches with Edman sequencing for both the mouse and the human homologs (29, 32).
Table I. Known MMP-2 substrates identified by CLIP-TRAQ-TAILS.
Numbers indicate cleavage site positions in the unprocessed protein precursor. Note that the spiked-in known MMP-2 substrate CCL7 has the highest QCF. IGFBP, insulin-like growth factor-binding protein; PCPE, procollagen C-proteinase enhancer; SPARC, secreted protein acidic & rich in cystein.

* Spiked-in control.
Biochemical Validation of S100A10 and proEMAP/p43 as Novel MMP-2 Substrates
Particularly high confidence protein identification is assigned to protein substrates that have been identified by their natural N-terminus in both MMP-2 and control samples and in addition by a neo-N-terminus in the MMP-2 treated sample. This was the case for two important extracellular mediators, the key plasminogen receptor S100A10 (p11) and the inflammatory cytokine proEMAP/p43 that have not been previously identified as MMP-2 substrates. The sequence of the natural N-terminal peptide (Fig. 4A) of murine S100A10 (Swiss-Prot accession number P08207) confirms removal of the initiator methionine that has been predicted by similarity as indicated in the Swiss-Prot annotation. As expected, the ratio of 113/114 CLIP-TRAQ reporter ion peaks for this peptide was close to 1.0 (Fig. 4B), indicating the equal presence of the S100A10 original mature N-terminus in the MMP-2-treated and the control samples. A second peptide identified for the same protein presented a CLIP-TRAQ reporter ion only in the 113 (MMP-2) channel (Fig. 4B), identifying it as a neo-N-terminus derived from MMP-2 cleavage at position 40–41. Notably, both peptides could be unambiguously assigned to this protein as indicated by an IAS of 1.0. In addition, the cleavage site is valid for both the human and the mouse proteins as shown by identical sequences 8 residues on either side of the scissile bond (supplemental Fig. 4A). To validate proteolytic processing, we incubated recombinant human full-length S100A10 with MMP-2 and confirmed cleavage by SDS-PAGE analysis (Fig. 4C). Furthermore, Edman sequencing confirmed the MMP-2-dependent generation of a C-terminally truncated fragment of S100A10 that increased in stain intensity on SDS-PAGE gels after incubation with MMP-2.
Fig. 4.

Processing of human S100A10 (p11) by MMP-2. A, amino acid sequence of S100A10 showing the natural N-terminus of the mature protein (green) and an MMP-2-generated neo-N-terminus (blue) identified by CLIP-TRAQ-TAILS. B, mass spectra assigned to the natural N-terminus and neo-N-terminus of S100A10, respectively. Insets show a close-up of the CLIP-TRAQ reporter ion region indicating the presence of the original mature N-terminal peptide in both samples and the neo-N-terminus only in the MMP-2-treated (CLIP-TRAQ-113) sample. C, MMP-2 cleavage of S100A10 analyzed by 15% Tris-Tricine SDS-PAGE. Arrows show full-length and cleaved S100A10. Both bands were sequenced by Edman degradation and shown to be the N-terminal part of either unprocessed or C-terminally truncated protein as indicated by the N-terminal sequence (asterisks). Notably, the lower band was consistently increased in amount after incubation with MMP-2.
As a second example, proEMAP/p43, a member of the small inducible cytokine family, was identified by an acetylated N-terminus after removal of the initiator methionine (Fig. 5, A and B). Although the corresponding Swiss-Prot entry (P31230) lacks this information, it was also predicted by the TermiNator algorithm (58) with 83% confidence. Importantly, the TAILS negative selection strategy allows the identification of acetylated N-termini that are proposed for up to 70% of all proteins (59) that positive selection strategies cannot detect. In addition, isotopic labeling of lysine side chains preserves quantification data provided the naturally blocked N-terminus harbors a lysine in its sequence as demonstrated for this example. Because CLIP-TRAQ labeling is at the protein level with trypsinization performed afterward, cleavage only occurs at arginine residues, preserving the label and increasing the size of the peptide and thus the probability for high confidence peptide identification. Again, the original mature N-terminal peptide presented CLIP-TRAQ reporter ions with equal intensities in the 113 (MMP-2) and the 114 (control) channels (Fig. 5B), whereas a second peptide assigned to the proEMAP/p43 sequence gave rise only to a reporter ion in the 113 channel (Fig. 5B). Therefore, the latter is a neo-N-terminus revealing MMP-2 cleavage of proEMAP/p43 at position 170 in the mature protein. Here too, high overall amino acid sequence homology and identity of the cleavage site sequence for the mouse and the human proEMAP/p43 proteins validated processing by MMP-2 for both species (supplemental Fig. 4B). Cleavage of proEMAP/p43 by MMP-2 was confirmed by SDS-PAGE analysis of recombinant murine proEMAP/p43 incubated with the protease (Fig. 5C) that could be inhibited by the broad spectrum MMP inhibitor Marimastat (60).
Fig. 5.

Processing of murine proEMAP/p43 by MMP-2. A, amino acid sequence of proEMAP/p43 showing the acetylated natural N-terminus of the mature protein (green) and an MMP-2-generated neo-N-terminus (blue) identified by CLIP-TRAQ-TAILS. B, mass spectra assigned to the natural N-terminus and neo-N-terminus of proEMAP/p43, respectively. Insets show a close-up of the CLIP-TRAQ reporter ion region indicating the presence of the natural N-terminal peptide in both samples and the neo-N-terminus only in the MMP-2-treated (CLIP-TRAQ-113) sample. C, MMP-2 cleavage of proEMAP/p43 analyzed by 15% Tris-Tricine SDS-PAGE. Arrows show full-length and cleavage products of proEMAP/p43.
Mapping MMP-2 Active Site Specificity
Because of the large number of cleavage events identified by CLIP-TRAQ-TAILS analysis, it can also be used to map protease active sites using native protein rather than peptide substrates (47, 61). We derived consensus sequences for 4 amino acids upstream of the identified cleavage site referred to as P4 to P1 (62) for all 1,183 cleavage events by mapping to all matching isoforms as described previously (47). Unambiguous prime side amino acids (P1′ to P4′) were derived from the actual neo-N-terminal peptide sequences. As indicated by the heat map (Fig. 6A) and protein sequence logo analysis (Fig. 6B) of the active site, our results are in very good agreement with a previous study using proteome-derived peptide libraries (47). Most prominent are the MMP characteristic preferences for proline in P3 (21%) and leucine in P1′ (40%) positions. In addition, we identified the same preference for alanine (19%), glycine (12%), and serine (14%) in P2; alanine (14%) and glycine (12%) in P1; and alanine (16%), glycine (14%), and serine (15%) in P3′ positions, adding further weight to their being bona fide cleavage sites.
Fig. 6.

MMP-2 active site mapping. A, heat map for the amino acid occurrences in P4–P4′ for all identified MMP-2-generated neo-N-termini (n = 1,183). CLIP-TRAQ-TAILS analysis confirms the predominant MMP-2 preferences for leucine in P1′ and proline in P3 position. B, protein sequence logo calculated from the same data set applying correction for natural amino acid abundance. The logo was generated using the iceLogo software package (48).
The high number of cleavage events now also allows statistical analysis of subsite cooperativity based on data derived from whole protein cleavages. When analyzing only cleavage sites with leucine in the P1′ position (464 events), the number of cleavage sites with proline in P3 dropped from 21 to 16%. Consistently, 30 instead of 41% of cleavage sites revealed leucine in the P1′ position in an analysis of cleavage sites with fixed proline in P3 (231 events). This negative frequency change indicates that leucine and proline in these positions are not cooperative, consistent with our own observations using the proteomic identification of protease cleavage site (PICS) approach (47). Rather, both Pro and Leu are each rather strong elements of specificity that either alone can drive substrate recognition with high probability.
DISCUSSION
With TAILS, we introduced a robust assay system for the system-wide identification of the N-terminome, mature protein N-termini and their modifications, and protease substrates and their cleavage sites in complex biological samples (29). Extending TAILS by the use of iTRAQ reagents instead of reductive dimethylation for quantification brings many advantages to the approach (39). To capitalize on the quantification advantages of isobaric tags, we describe a flow of novel statistical and bioinformatics procedures for protein assignments from single peptides and to discriminate mature original N-terminal peptides from cleaved substrate neo-N-terminal peptides with high fidelity.
A major limitation in proteome coverage and particularly in identification of low abundance proteins by mass spectrometry-based proteomics is sample complexity (30). This results in large quantities of precursor peptides at the MS1 level that are not accessible to MS2 fragmentation and subsequent identification because of technical limits of current mass spectrometers. A major disadvantage of MS1-based isotopic quantification, such as stable isotope labeling with amino acids in cell culture (16, 37), 18O/16O labeling (15), and reductive dimethylation (63), is that the number of precursor ions is doubled, heightening the undersampling problem. TAILS addresses the undersampling problem by massive reductions in sample complexity through selective removal of internal tryptic and C-terminal peptides (29). Nonetheless, MS2 quantification and further reduction of sample complexity by off-line strong cation exchange chromatography fractionation of the peptide sample should result in higher proteome coverage and more identifications of low abundance proteins. Indeed, we identified 3 times more cleavage events for MMP-2 by CLIP-TRAQ labeling and TAILS than by dimethylation and TAILS using the same cell secretomes and an Applied Biosystems QStar XL mass spectrometer that has a longer duty cycle than the Thermo LTQ-Orbitrap used previously for dimethylation TAILS (29). As evidence of this, CLIP-TRAQ-TAILS analysis determined the low abundance cytokine pro-EMAP/p43 as a novel MMP-2 substrate; it is a protein that has not even been detected in previous shotgun iTRAQ analyses of similar samples (2, 32). A further advantage of quantification with iTRAQ-like reagents is the availability of peptide identification and quantification information from the same tandem MS spectrum (31). This reduces false positive identifications of mature original N-termini or neo-N-termini selected by TAILS by wrong assignments of corresponding precursor doublets in MS1 spectra, particularly when not using high accuracy mass spectrometers. However, it should be noted that contaminating overlapping spectra might also contribute to false positive signals in MS2 reporter ion regions of tandem mass spectra (30). Although from our own experience, the analysis of MS2 quantification data requires less manual curation than MS1 quantification results where peaks more frequently overlap, an advantage for automated high throughput analyses.
In traditional quantitative proteomics, multiple peptides of the same protein are separately quantified but then averaged to determine the relative abundance of the protein. Thereby, outliers are discarded and removed from the analysis. Interestingly, those outliers are most likely the result of differential proteolytic processing that we already successfully exploited to narrow down the location of protease cleavage sites by a process termed “peptide mapping” (32). The mark of a successful N-terminal peptide selection strategy is the generation of single peptides per protein for analysis. However, quantitative N-terminome analysis relies not only on identification of proteins by only one peptide, an issue we recently addressed by stringent statistical criteria for high confidence peptide identification (29), but also on accurate quantification of single peptides. This is a particular problem in MS2-based quantification because data are only available for discrete CID events and not derived from integration of peptide elution peaks as in MS1 quantification. In many cases, these CIDs are not taken at the maximum of the precursor elution peak, resulting in lower intensity reporter ions and thus higher quantification variability. We addressed this issue by deriving experiment-based intensity-dependent variations (Fig. 2A) and calculating a corresponding quantification confidence for every spectrum. Combined confidences are then normalized to allow direct comparisons of peptides within the same and between different experiments by calculating QCFs for every peptide. This strategy is validated by the highest QCF that was assigned to the neo-N-terminal peptide of our spiked-in MMP-2 test substrate CCL7 (Table I).
The critical factor that defines a neo-N-terminus and thereby both a protease substrate and the proteolytic cleavage site is the relative abundance of the corresponding peptide in the protease-treated and the control samples. Thereby, the highest confidence is assigned to peptides that are only present in the protease-treated sample and therefore referred to as “singletons.” However, in previous studies, the actual measured ratio defining a singleton was mostly based on empirical estimation of the dynamic range of the quantification method used (32, 33). The few examples that use statistics to determine a critical cutoff ratio do this based on experimental quantification variation for expected non-events like natural N-termini rather than on actual cleavage events themselves (15). Thereby, they still consider the appearance of a neo-N-terminus as a binary event dependent on the presence of a specific proteolytic activity. Here, we statistically determine for the first time both the dynamic range of the quantification method and the cleavage event (singleton) ratio cutoff from experimental data using a test protease (Glu-C) with canonical specificity. This was enabled by a large enough sample space fulfilling our criteria due to the high number of high confidence peptide identifications. Thereby, we calculated a dynamic range of 14 for CLIP-TRAQ-based quantification of N-terminal peptides (Fig. 2C) that is in agreement with previous studies using iTRAQ for quantification of proteins in complex proteomes (54). It can be reasonably assumed that singleton peptides present an abundance ratio below this value mostly because of background underestimation in the low intensity channel. Often, this background is determined arbitrarily, and reporter ion intensities below a certain threshold are then treated as zero. However, this can easily result in a high number of false positive singleton assignments particularly for low intensity peptides. Again, we overcame this problem by experimentally generating proteolytically derived singleton peptides and defining a cutoff on the actual cleavage event without any arbitrary modification to the data. Thereby, the use of receiver operating characteristics curve analysis allowed the calculation of an optimal cutoff ratio (protease/control) of 7.3 for maximum sensitivity (86%) and minimal false discovery rate (15%) (Fig. 2D). Depending on the properties of individual proteases and the aims of a particular experiment, a lower false discovery rate can of course be selected but at the cost of lower sensitivity and hence coverage. Although this value might be generally valid for MS2 quantification of protease-generated neo-N-termini, it should be individually determined for the particular isotopic label and mass spectrometer used.
Because of limited sequence coverage, a common problem in mass spectrometry-based proteomics is the unambiguous discrimination between very similar proteins or isoforms of the same protein. This is even more complicated for N-terminome analyses when proteins are usually identified by only one peptide. Previous studies either ignored this problem by reporting only one and mostly the best annotated isoform (20) or listing all matching isoforms without any confidence ranking (16). Thereby, preference was given only to isoforms identified by more than one peptide as a result of multiple cleavages (16). Here, we make use of high confidence multiple peptide protein identifications that are statistically validated by the ProteinProphet algorithm (44) from analysis of the same sample before and after N-termini enrichment, a particular advantage of the TAILS negative selection workflow, to establish a scoring factor, the IAS, for protein/isoform assignment confidences. Applying this scoring system, we could unambiguously assign unique isoforms to around 50% of all 2,101 identified N-terminal peptides.
To finally validate our novel statistical models for the identification of protease-generated neo-N-termini in complex proteomes, we used MMP-2, a protease with non-canonical cleavage specificity that had been extensively studied in our laboratory, thereby providing us a large in-house database against which to validate (2, 32). In fact, we were able to identify many known MMP-2 substrates and their cleavage sites with high reliability (Table I). In addition and demonstrating the high sensitivity of iTRAQ-TAILS analysis, we determined and biochemically verified S100A10 and proEMAP/p43 as novel bioactive MMP-2 substrates (Figs. 4 and 5). Both could be identified after TAILS with high confidence by both the natural N-terminus and an MMP-2-generated neo-N-terminal peptide. Importantly, the negative selection strategy of TAILS preserves quantifiability of the naturally acetylated proEMAP/p43 N-terminal peptide via its internal CLIP-TRAQ-labeled lysine side chain. Although species mismatch in cleavage specificity might occur between human and murine MMP-2, these enzymes show 97% identity with only eight substitutions occurring in the catalytic domain. Hence, it is unlikely that human and murine MMP-2 show cleavage specificity differences. Nonetheless, although some differences are to be expected in cleavage sites found in murine substrate proteins identified by TAILS and the human proteins used in biochemical validation (Table I), this was not the case for S100A10 and proEMAP/p43.
S100A10, also known as p11, is found on the cell surface of many cancer cells as a heterotetrameric complex with annexin A2 (64–66). There it functions as plasminogen receptor and regulates, via its C-terminal lysines, the stimulation of tissue plasminogen activator-dependent plasminogen activation (67), an important step in activation of the serine protease plasmin to facilitate tumor cell invasion and metastasis (68). Therefore, its cleavage by MMP-2 might have an impact on the cross-talk between both proteolytic systems. ProEMAP/p43, also known as small inducible cytokine subfamily E member 1, is a proinflammatory cytokine that has been associated with antitumorigenic and antiangiogenic activities (69). Although its proform already exhibits cytokine function, the mature cytokine EMAP-II (from position 145 to 312) is generated upon secretion by proteolytic cleavage. Currently, several proteases mediating this processing are under debate including cathepsin L and MMP-9 (70). However, MMP-9 cleaves at position 108, whereas cathepsin L seems to be the major factor in generating the mature protein by cleavage after aspartate 144. Interestingly, our analysis shows MMP-2 cleavage after serine 171 and so might inactivate EMAP-II and thereby contribute to proangiogenic activities of MMP-2. However, the consequences of MMP-2 processing of S100A10 and proEMAP/p43 have still to be determined in appropriate biological assay systems.
In conclusion, CLIP-TRAQ-TAILS is a robust platform for the system-wide quantitative analysis of N-terminomes. The use of in-house synthesized iTRAQ-like reagents brings the advantages of isobaric tags at greatly reduced cost comparable with isotopic labeling by dimethylation. However, the use of commercial iTRAQ reagents allows multiplex system-wide quantitative comparisons of N-terminomes that are reported in the accompanying paper by Prudova et al. (39). We also report here for the first time the use of statistical models for the derivation of normalized QCFs and critical peptide abundance ratio cutoffs for the reliable detection of protease cleavage events. Furthermore, our novel analysis pipeline provides an IAS for confidence in protein assignment, a particular problem of N-terminome single peptide approaches. These are major steps toward a probability-based decision system, evolving TAILS from a validation-dependent screen to a system-level N-terminome analysis platform.
Supplementary Material
Acknowledgments
We thank Dr. Wei Chen from the University of British Columbia Centre for Blood Research Mass Spectrometry Suite for excellent mass spectrometry analyses and Prof. Jayachandran N. Kizhakkedathu (University of British Columbia) for kindly providing the HPG-ALD polymer.
Footnotes
* This work was supported in part by a grant from the Canadian Institutes of Health Research, a program project grant in Breast Cancer Metastases from the Canadian Breast Cancer Research Alliance with funds from the Canadian Breast Cancer Foundation and the Cancer Research Society, and an infrastructure grant from the Michael Smith Foundation for Health Research.
 This article contains Figs. 1–4 and Tables 1–10.
This article contains Figs. 1–4 and Tables 1–10.
2 Fahlman R., Chen W., Overall C. M., submitted manuscript.
1 The abbreviations used are:
- MMP
- matrix metalloproteinase
- CCL7
- chemokine (CC motif) ligand 7
- HPG
- hyperbranched polyglycerol
- ALD
- aldehyde
- IAS
- isoform assignment score
- iTRAQ
- isobaric tag for relative and absolute quantification
- QCF
- quantification confidence factor
- ROC
- receiver operating characteristic
- TAILS
- terminal amine isotopic labeling of substrates
- EMAP
- endothelial monocyte-activating polypeptide
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1.Salvesen G. S. (2002) Caspases and apoptosis. Essays Biochem 38, 9–19 [DOI] [PubMed] [Google Scholar]
- 2.Dean R. A., Butler G. S., Hamma-Kourbali Y., Delbé J., Brigstock D. R., Courty J., Overall C. M. (2007) Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/Connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell. Biol 27, 8454–8465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riddel J. P., Jr., Aouizerat B. E., Miaskowski C., Lillicrap D. P. (2007) Theories of blood coagulation. J. Pediatr. Oncol. Nurs 24, 123–131 [DOI] [PubMed] [Google Scholar]
- 4.auf dem Keller U., Doucet A., Overall C. M. (2007) Protease research in the era of systems biology. Biol. Chem 388, 1159–1162 [DOI] [PubMed] [Google Scholar]
- 5.Overall C. M., Dean R. A. (2006) Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev 25, 69–75 [DOI] [PubMed] [Google Scholar]
- 6.Overall C. M., Kleifeld O. (2006) Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 7.Cole S. L., Vassar R. (2008) The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem 283, 29621–29625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 9.Folgueras A. R., Pendás A. M., Sánchez L. M., López-Otín C. (2004) Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int. J. Dev. Biol 48, 411–424 [DOI] [PubMed] [Google Scholar]
- 10.Doucet A., Butler G. S., Rodríguez D., Prudova A., Overall C. M. (2008) Metadegradomics: toward in vivo quantitative degradomics of proteolytic post-translational modifications of the cancer proteome. Mol. Cell. Proteomics 7, 1925–1951 [DOI] [PubMed] [Google Scholar]
- 11.López-Otín C., Overall C. M. (2002) Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol 3, 509–519 [DOI] [PubMed] [Google Scholar]
- 12.Prudova A., Auf dem Keller U., Overall C. M. (2008) Identification of protease dubstrates by mass spectrometry approaches—2, in The Cancer Degradome: Proteases and Cancer Biology (Edwards D., Hoyer-Hansen G., Blasi F., Sloane B. F. eds) pp. 83–100, Springer, New York [Google Scholar]
- 13.Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G. R., Vandekerckhove J. (2003) Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol 21, 566–569 [DOI] [PubMed] [Google Scholar]
- 14.Aivaliotis M., Gevaert K., Falb M., Tebbe A., Konstantinidis K., Bisle B., Klein C., Martens L., Staes A., Timmerman E., Van Damme J., Siedler F., Pfeiffer F., Vandekerckhove J., Oesterhelt D. (2007) Large-scale identification of N-terminal peptides in the halophilic archaea Halobacterium salinarum and Natronomonas pharaonis. J. Proteome Res 6, 2195–2204 [DOI] [PubMed] [Google Scholar]
- 15.Van Damme P., Martens L., Van Damme J., Hugelier K., Staes A., Vandekerckhove J., Gevaert K. (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2, 771–777 [DOI] [PubMed] [Google Scholar]
- 16.Van Damme P., Maurer-Stroh S., Plasman K., Van Durme J., Colaert N., Timmerman E., De Bock P. J., Goethals M., Rousseau F., Schymkowitz J., Vandekerckhove J., Gevaert K. (2009) Analysis of protein processing by N-terminal proteomics reveals novel species-specific substrate determinants of granzyme B orthologs. Mol. Cell. Proteomics 8, 258–272 [DOI] [PubMed] [Google Scholar]
- 17.Vande Walle L., Van Damme P., Lamkanfi M., Saelens X., Vandekerckhove J., Gevaert K., Vandenabeele P. (2007) Proteome-wide identification of HtrA2/Omi substrates. J. Proteome Res 6, 1006–1015 [DOI] [PubMed] [Google Scholar]
- 18.Staes A., Van Damme P., Helsens K., Demol H., Vandekerckhove J., Gevaert K. (2008) Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 19.Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmer J. C., Enoksson M., Wildfang E., Zhu W., Igarashi Y., Denault J. B., Ma Y., Dummitt B., Chang Y. H., Mast A. E., Eroshkin A., Smith J. W., Tao W. A., Salvesen G. S. (2007) Profiling constitutive proteolytic events in vivo. Biochem. J 407, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald L., Beynon R. J. (2006) Positional proteomics: preparation of amino-terminal peptides as a strategy for proteome simplification and characterization. Nat. Protoc 1, 1790–1798 [DOI] [PubMed] [Google Scholar]
- 22.McDonald L., Robertson D. H., Hurst J. L., Beynon R. J. (2005) Positional proteomics: selective recovery and analysis of N-terminal proteolytic peptides. Nat. Methods 2, 955–957 [DOI] [PubMed] [Google Scholar]
- 23.Guo L., Eisenman J. R., Mahimkar R. M., Peschon J. J., Paxton R. J., Black R. A., Johnson R. S. (2002) A proteomic approach for the identification of cell-surface proteins shed by metalloproteases. Mol. Cell. Proteomics 1, 30–36 [DOI] [PubMed] [Google Scholar]
- 24.Dix M. M., Simon G. M., Cravatt B. F. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bredemeyer A. J., Lewis R. M., Malone J. P., Davis A. E., Gross J., Townsend R. R., Ley T. J. (2004) A proteomic approach for the discovery of protease substrates. Proc. Natl. Acad. Sci. U.S.A 101, 11785–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomis-Rüth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 27.Thornberry N. A., Gallwitz B. (2009) Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best. Pract. Res. Clin. Endocrinol. Metab 23, 479–486 [DOI] [PubMed] [Google Scholar]
- 28.Cox J. H., Overall C. M. (2008) Cytokine substrates: MMP regulation of inflammatory mediator signalling, in The Cancer Degradome: Proteases and Cancer Biology (Edwards D., Hoyer-Hansen G., Blasi F., Sloane B. F. eds) pp. 519–538, Springer, New York [Google Scholar]
- 29.Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 30.Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 31.Zieske L. R. (2006) A perspective on the use of iTRAQTM reagent technology for protein complex and profiling studies. J. Exp. Bot 57, 1501–1508 [DOI] [PubMed] [Google Scholar]
- 32.Dean R. A., Overall C. M. (2007) Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611–623 [DOI] [PubMed] [Google Scholar]
- 33.Enoksson M., Li J., Ivancic M. M., Timmer J. C., Wildfang E., Eroshkin A., Salvesen G. S., Tao W. A. (2007) Identification of proteolytic cleavage sites by quantitative proteomics. J. Proteome Res 6, 2850–2858 [DOI] [PubMed] [Google Scholar]
- 34.Phanstiel D., Unwin R., McAlister G. C., Coon J. J. (2009) Peptide quantification using 8-plex isobaric tags and electron transfer dissociation tandem mass spectrometry. Anal. Chem 81, 1693–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler G. S., Dean R. A., Tam E. M., Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol 28, 4896–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tam E. M., Morrison C. J., Wu Y. I., Stack M. S., Overall C. M. (2004) Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. U.S.A 101, 6917–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gioia M., Foster L. J., Overall C. M. (2009) Cell-based identification of natural substrates and cleavage sites for extracellular proteases by SILAC proteomics. Methods Mol. Biol 539, 131–153 [DOI] [PubMed] [Google Scholar]
- 38.Butler G. S., Tam E. M., Overall C. M. (2004) The canonical methionine 392 of matrix metalloproteinase 2 (gelatinase A) is not required for catalytic efficiency or structural integrity: probing the role of the methionine-turn in the metzincin metalloprotease superfamily. J. Biol. Chem 279, 15615–15620 [DOI] [PubMed] [Google Scholar]
- 39.Prudova A., auf dem Keller U., Butler G. S., Overall C. M. (2010) Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics, 9, 894–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedrioli P. G. (2010) Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol. Biol 604, 213–238 [DOI] [PubMed] [Google Scholar]
- 42.Keller A., Kolker E., Aebersold R., Nesvizhskii A. I. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 43.Shteynberg D., Deutsch E. W., Lam H., Aebersold R., Nesvizhskii A. I. (2008) iProphet: improved validation of peptide identification in shotgun proteomics, in HUPO 7th Annual World Congress, Amsterdam, August 16–20, 2008, Abstr. P-TUE-181, Human Proteome Organisation, Montreal [Google Scholar]
- 44.Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 45.Sing T., Sander O., Beerenwinkel N., Lengauer T. (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 [DOI] [PubMed] [Google Scholar]
- 46.Ramos H., Shannon P., Aebersold R. (2008) The protein information and property explorer: an easy-to-use, rich-client web application for the management and functional analysis of proteomic data. Bioinformatics 24, 2110–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schilling O., Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 48.Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 49.Creasy D. M., Cottrell J. S. (2004) Unimod: Protein modifications for mass spectrometry. Proteomics 4, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 50.Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zougman A., Pilch B., Podtelejnikov A., Kiehntopf M., Schnabel C., Kumar C., Mann M. (2008) Integrated Analysis of the Cerebrospinal Fluid Peptidome and Proteome. J. Proteome Res 7, 386–399 [DOI] [PubMed] [Google Scholar]
- 52.Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol 1, 2005.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Houmard J., Drapeau G. R. (1972) Staphylococcal protease: a proteolytic enzyme specific for glutamoyl bonds. Proc. Natl. Acad. Sci. U.S.A 69, 3506–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bantscheff M., Boesche M., Eberhard D., Matthieson T., Sweetman G., Kuster B. (2008) Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol. Cell. Proteomics 7, 1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minn A. J., Gupta G. P., Siegel P. M., Bos P. D., Shu W., Giri D. D., Viale A., Olshen A. B., Gerald W. L., Massagué J. (2005) Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McQuibban G. A., Gong J. H., Tam E. M., McCulloch C. A., Clark-Lewis I., Overall C. M. (2000) Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 [DOI] [PubMed] [Google Scholar]
- 57.Aimes R. T., Quigley J. P. (1995) Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem 270, 5872–5876 [DOI] [PubMed] [Google Scholar]
- 58.Frottin F., Martinez A., Peynot P., Mitra S., Holz R. C., Giglione C., Meinnel T. (2006) The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteomics 5, 2336–2349 [DOI] [PubMed] [Google Scholar]
- 59.Meinnel T., Giglione C. (2008) Tools for analyzing and predicting N-terminal protein modifications. Proteomics 8, 626–649 [DOI] [PubMed] [Google Scholar]
- 60.Overall C. M., López-Otín C. (2002) Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer 2, 657–672 [DOI] [PubMed] [Google Scholar]
- 61.Turk B. E., Huang L. L., Piro E. T., Cantley L. C. (2001) Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol 19, 661–667 [DOI] [PubMed] [Google Scholar]
- 62.Schechter I. (2005) Mapping of the active site of proteases in the 1960s and rational design of inhibitors/drugs in the 1990s. Curr. Protein Pept. Sci 6, 501–512 [DOI] [PubMed] [Google Scholar]
- 63.Boersema P. J., Foong L. Y., Ding V. M., Lemeer S., van Breukelen B., Philp R., Boekhorst J., Snel B., den Hertog J., Choo A. B., Heck A. J. (2010) In-depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immunoaffinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 9, 84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siever D. A., Erickson H. P. (1997) Extracellular annexin II. Int. J. Biochem. Cell Biol 29, 1219–1223 [DOI] [PubMed] [Google Scholar]
- 65.Tressler R. J., Updyke T. V., Yeatman T., Nicolson G. L. (1993) Extracellular annexin II is associated with divalent cation-dependent tumor cell-endothelial cell adhesion of metastatic RAW117 large-cell lymphoma cells. J. Cell. Biochem 53, 265–276 [DOI] [PubMed] [Google Scholar]
- 66.Yeatman T. J., Updyke T. V., Kaetzel M. A., Dedman J. R., Nicolson G. L. (1993) Expression of annexins on the surfaces of non-metastatic and metastatic human and rodent tumor cells. Clin. Exp. Metastasis 11, 37–44 [DOI] [PubMed] [Google Scholar]
- 67.Kassam G., Le B. H., Choi K. S., Kang H. M., Fitzpatrick S. L., Louie P., Waisman D. M. (1998) The p11 subunit of the annexin II tetramer plays a key role in the stimulation of t-PA-dependent plasminogen activation. Biochemistry 37, 16958–16966 [DOI] [PubMed] [Google Scholar]
- 68.Ulisse S., Baldini E., Sorrenti S., D'Armiento M. (2009) The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr. Cancer Drug Targets 9, 32–71 [DOI] [PubMed] [Google Scholar]
- 69.van Horssen R., Eggermont A. M., ten Hagen T. L. (2006) Endothelial monocyte-activating polypeptide-II and its functions in (patho)physiological processes. Cytokine Growth Factor Rev 17, 339–348 [DOI] [PubMed] [Google Scholar]
- 70.Liu J., Schwarz M. A. (2006) Identification of protease-sensitive sites in human endothelial-monocyte activating polypeptide II protein. Exp. Cell Res 312, 2231–2237 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.